
A NASBA on microgel-tethered molecular-beacon microarray for real-time microbial molecular diagnostics
Y.
Ma
a,
X.
Dai
ab,
T.
Hong
c,
G. B.
Munk
c and
M.
Libera
 *a
*a
aDept of Chemical Engr & Materials Science, Stevens Institute of Technology, Hoboken, NJ 07030, USA. E-mail: mlibera@stevens.edu
bCurrently with Oxford Nanopore, New York, NY 10013, USA
cHackensack University Medical Center, Hackensack, NJ 07601, USA
First published on 24th November 2016
Abstract
Despite their many advantages and successes, molecular beacon (MB) hybridization probes have not been extensively used in microarray formats because of the complicating probe–substrate interactions that increase the background intensity. We have previously shown that tethering to surface-patterned microgels is an effective means for localizing MB probes to specific surface locations in a microarray format while simultaneously maintaining them in as water-like an environment as possible and minimizing probe–surface interactions. Here we extend this approach to include both real-time detection together with integrated NASBA amplification. We fabricate small (∼250 μm × 250 μm) simplex, duplex, and five-plex assays with microarray spots of controllable size (∼20 μm diameter), position, and shape to detect bacteria and fungi in a bloodstream-infection model. The targets, primers, and microgel-tethered probes can be combined in a single isothermal reaction chamber with no post-amplification labelling. We extract total RNA from clinical blood samples and differentiate between Gram-positive and Gram-negative bloodstream infection in a duplex assay to detect RNA- amplicons. The sensitivity based on our current protocols in a simplex assay to detect specific ribosomal RNA sequences within total RNA extracted from S. aureus and E. coli cultures corresponds to tens of bacteria per ml. We furthermore show that the platform can detect RNA- amplicons from synthetic target DNA with 1 fM sensitivity in sample volumes that contain about 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 DNA molecules. These experiments demonstrate an alternative approach that can enable rapid and real-time microarray-based molecular diagnostics.
000 DNA molecules. These experiments demonstrate an alternative approach that can enable rapid and real-time microarray-based molecular diagnostics.
1. Introduction
The effective treatment of an infection requires microbial identification in order to prescribe an appropriate antimicrobial therapy. Conventional diagnostic approaches culture body fluids or tissue samples to determine if microbes are present. Phenotypes, such as morphology or antimicrobial susceptibility, are then examined to identify the specific pathogen. While effective, this approach is slow and typically requires 3–5 days to fully complete. During that time a patient-specific antimicrobial therapy is insufficiently informed, which both threatens patient wellbeing and further burdens the healthcare system. There is consequently substantial interest in developing molecular-diagnostic (MDx) techniques for microbial identification.2–5 MDx approaches are both specific and fast. They are able to identify microbial species and strains based on biospecific markers and, in specific cases, can do so in times as short as one hour.Despite their attractiveness, the throughput associated with many of the current MDx approaches is relatively low. Throughput is constrained by complexity associated with the target amplification process, the nature and extent of multiplexing, and the detection mechanism, all of which are closely coupled to each other as well as to the instrumentation needed to coordinate, control, and monitor amplification and detection. Improvements in throughput have been achieved by microarraying and coupling these microarrays with PCR amplification of target nucleotides.
A further simplification can be made by eliminating the thermal cycling associated with PCR by using an isothermal amplification method such as Nucleic Acid Sequence Based Amplification (NASBA).6,7 NASBA is driven by the concurrent activity of avian myleoblastosis virus (AMV) reverse transcriptase, RNase H, and T7 RNA polymerase to amplify RNA targets at 41 °C (Fig. 1). The NASBA amplicons are single-stranded RNAs (ssRNAs) that can be detected by hybridization probes. Notably, Kurg et al.8,9 and Ruhe et al.10 have both developed integrated NASBA on microarray platforms, where RNA targets are amplified in solution immediately above the microarray spots. Kurg et al., for example, incorporated amino-allyl UTP (aaUTP) into the NASBA amplification mixture so that RNA amplicons hybridized to microarrayed linear probes could be fluorescently labelled for an end-point detection process. They used this approach in a simplex assay to detect S. pneumoniae. They subsequently designed highly specific hybridization probes for S. pneumoniae tmRNA and report a sensitivity equivalent to 0.1 CFU.9 Ruhe et al. expanded this approach to a five-plex NASBA-on-microarray assay in a breast-cancer model.10 A mixture of gel precursors and DNA probes was spotted on PMMA slides and polymerized to generate individual ∼200 μm diameter spots with the probes covalently grafted to the gel network. Their NASBA process produced RNA amplicons containing biotinylated UTP, which enabled post-hybridization labelling by fluorescently tagged streptavidin for end-point detection. They estimated the sensitivity as sufficient to possibly detect an individual HeLa cell, and subsequent work expanded the multiplexing to 14.11
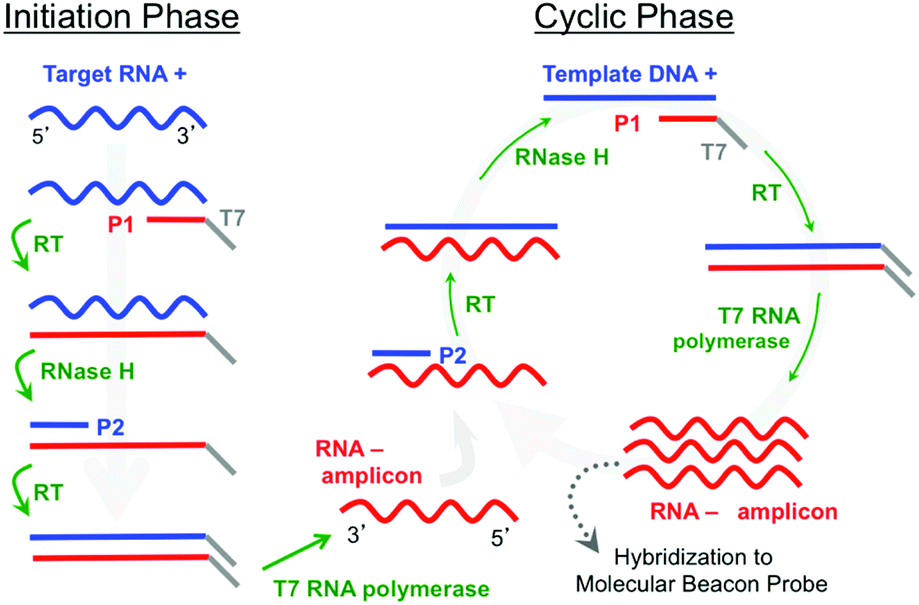 | ||
| Fig. 1 Schematic representation of NASBA including molecular beacon detection. The left downward arrow represents the initiation phase and the right circle represents the cyclic phase. The activities of reverse transcriptase, RNase-H and T7 polymerase as well as the primer-binding activities are indicated. The product, amplified in the cyclic phase, is complementary to that of the target nucleic acid. | ||
In contrast to the use of linear hybridization probes, which require some form of target labelling or probe–target hybrid labelling, structured probes such as molecular beacons are self-reporting and fluoresce upon hybridization.12,13 Hence, post-hybridization labelling is not required, and real-time detection is possible. NASBA has previously been combined with molecular-beacon probes in a traditional solution-based approach to detect viral RNA in a simplex assay14 and more recently in duplex and three-plex assays within a bloodstream-infection model.1
Despite their many successes in solution-based assays, molecular beacon probes have not been extensively used in microarray applications. Non-specific interactions with a hard substrate can lead to a relatively high background intensity. However, microgel tethering has provided a new means to effectively immobilize molecular beacon probes to a solid surface in a microarray format.15 Microgels based on, for example, poly(ethylene glycol) [PEG] can be patterned on solid surfaces using focused electron beams.16,17 They resist non-specific biomolecule interactions,18,19 and tethering sites can be introduced by using PEG with functional endgroups such as biotin (PEG-B).20–22 The concentration of tethering sites can be controlled by blending hydroxyl-terminated PEG (PEG-OH) with functional PEG-B to optimize the spatial distribution of molecular beacon probes on the microgel surface.23 In contrast to other work where biomolecular probes are physically trapped within the hydrogel interior,24,25 including that of Ruhe et al. involving the NASBA-on-microarray format,10 the unique structure of an e-beam-patterned microgel forces oligonucleotide probes to bind to the outermost portions of the microgel where they find themselves both physically separated from the hard substrate and tethered in the most water-like environment possible with minimal conformational constraints.
Here we present a platform in which MB probes are immobilized on a microgel substrate in a low-density two-dimensional array in direct contact with a NASBA amplification mixture. Amplification and detection occur simultaneously in a closed chamber fabricated on a glass microscope slide. We use the solution-based NASBA assay developed and validated by Zhao et al.1 but move the detection to surface-immobilized spots. In contrast to other microarray-based assays, the self-reporting character of the molecular beacon hybridization probes enables the array response to be monitored in real time. This advance brings significant advantages to the quantification and removal of the background signal as well as to the characterization of the noise associated with detection.
2. Experimental methods
E-beam-patterned microgel arrays
We created a variety of simplex and low-density multiplex microarrays consisting of thin-film pads of microgel-tethered molecular beacon probes patterned on glass substrates. We used previously established methods of electron-beam lithography to fabricate biotinylated poly(ethylene glycol) (PEG-B) microgel arrays on a glass microscope slide using a mixture of 60 wt% PEG-B (Mw = 5 kDa) and 40 wt% hydroxyl-terminated PEG (Mw = 6 kDa).15,26 The patterning was done using 2 keV electrons with a point dose of 10 fC and an interpixel spacing of 200 nm. The resulting array consisted of circular pads of microgel thin film (20 μm diameter, ∼60 nm dry thickness) with each pad separated from its nearest neighbour by ∼250 μm (Fig. 2). Between adjacent pads was the exposed surface of the glass substrate. Note that the flexibility of e-beam patterning enables the microgel pad size, shape, and inter-pad spacing to be easily controlled.15,16 Since a typical microarray in these experiments occupied an area of less than 500 μm × 500 μm, as many as 12 different microarrays were typically patterned on a single microscope slide. A 12-well polydimethylsiloxane (PDMS) gasket was subsequently attached to the dry slide using uncured PDMS (5:1 base/curing agent) as a glue followed by a brief curing treatment (4 hours in oven at 40 °C) so that each microarray was confined within its own reaction well (Fig. 2). Prior to MB functionalization, the biotinylated microgel pads were activated by immersion in a streptavidin (SA) solution (200 μg mL−1) for two hours at room temperature followed by rinsing in deionized (DI) water.
 | ||
| Fig. 2 The experimental layout. A PDMS gasket with multiple reaction wells is attached to a glass slide. A five-spot low-density microarray is patterned on the glass surface within each reaction chamber. Each spot within each microarray consists of a thin-film microgel pad to which molecular beacon probes are tethered via a streptavidin–biotin linkage. The system is maintained at 41 °C, and the fluorescence response of the microgel array is monitored in real time by an inverted microscope. | ||
Our system was designed to detect up to five different ssRNA-amplicons. The five sets of MB probes and primers (Table 1) were all previously validated and used in duplex and triplex solution-based assays to detect pan Gram positive/negative bacteria and pan fungi, Aspergillus, and Candida in blood stream infections.1 As many as nine different primers were combined in a NASBA amplification mixture. The NASBA reagents were purchased from Advanced Life Sciences Technologies (St Petersburg, FL) and used as specified by the manufacturer.
| a The italic portions in the P1 primers are the T7 polymerase promoter sequences. The bold portions in the molecular beacon (MB) probes indicate the MB-target binding regions. | |
|---|---|
| Pan Gram positive bacteria | |
| GP P2 | 5′TACGGGAGGCAGCAGT3′ |
| GP MB | 5′Alexa594-CGAGCTAGCAACGCCGCGTGAGTGAAGCTCG-BHQ2-Biotin3′ |
| CP DNA template | 5′TACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGGCGAAAGCCTGACGGAGCAACGCCGCGTGAGTGATGAAGGTCTTCGGATCGTAAAACTCTGTTATTAGGGAAGAACATATGTGTAAGTAACTGTGCACATCTTGACGGTACCTAATCAGAAAGCCACGGCTAACTACGTGCCAGCAGCCGCGGTAATAC3′ |
| Pan Gram negative bacteria | |
| GN P2 | 5′CCTGATGCAGCCATGCCGCGTG-3′ |
| GN MB | 5′ Alexa594-CGAGCTTGAAGAAGGCCTTCGGGTTGTAAAGAGCTCG-BHQ2-Biotin3′ |
| GN DNA template | 5′CCTGATGCAGCCATGCCGCGTGTATGAAGAAGGCCTTCGGGTTGTAAAGTACTTTCAGCGGGGAGGAAGGGAGTAAAGTTAATACCTTTGCTCATTGACGTTACCCGCAGAAGAAGCACCGGCTAACTCCGTGCCAGCAGCCGCGGTAATAC3′ |
| Common P1 GN/GP | 5′AATTCTAATACGACTCACTATAGGGGTATTACCGCGGCTGCTGGCAC3′ |
| Candida | |
| CAN P2 | 5′GGAATCCGCTAAGGAGTGTG3′ |
| CAN P11 | 5′AATTCTAATACGACTCACTATAGGGCCATCCATTTTCAGGGCTAGT3′ |
| CAN MB | 5′Alexa 594-CGCGATTAACAACTCACCGGCCGAATATCGCG-BHQ2-Biotin3′ |
| CAN DNA template | 5′GGAATCCGCTAAGGAGTGTGTAACAACTCACCGGCCGAATGAACTAGCCCTGAAAATGGATGG3′ |
| Aspergillus | |
| ASP P2 | 5′CAGCAGTTGGACATGGGTTA3′ |
| ASP P1 | 5′AATTCTAATACGACTCACTATAGGGGAGAATCCACATCCAGGTGC3′ |
| ASP MB | 5′Alexa 594-CGACCGGCATAGGGAAGTTCCGTTTGGTCG-BHQ2-Biotin3′ |
| ASP DNA template | 5′CAGCAGTTGGACATGGGTTAGTCGATCCTAAGGCATAGGGAAGTTCCGTTTGAAAGGCGCCCTCGTGCGCCGTGTGCCGAAAGGGAAGCCGGTTAACATTCCGGCACCTGGATGTGGATTCTC3′ |
| Fungi | |
| FUN P2 | 5′-CGGCTCTTCCTATCATACCG3′ |
| FUN P1 | 5′AATTCTAATACGACTCACTATAGGGCTAAACCCAGCTCACGTTCC3′ |
| FUN MB | 5′ Alexa 594-CGCGATATTCGGTAAGCGTTGGATTGATCGCG-BHQ2-Biotin-3′ |
| FUN DNA template | 5′CGGCTCTTCCTATCATACCGAAGCAGAATTCGGTAAGCGTTGGATTGTTCACCCACTAATAGGGAACGTGAGCTGGGTTTAG3′ |
We studied simplex (1 MB probe), duplex (2 MB probes), and five-plex (5 MB probes) arrays. The simplex arrays involved microgel pads all functionalized with the same MB. In this case, SA-activated microgel pads were functionalized simply by depositing 30 μl of 1 μM MB solution in 0.1× PBS (10 mM sodium phosphate, 15 mM sodium chloride, pH 7.2) for 1 hour at room temperature followed by washing in DI water. Duplex and five-plex microarrays required spatially resolved microgel-pad functionalization. A homemade microspotter coupled to an inverted optical microscope was thus used to differently functionalize different microgel pads within an array. The microspotter used an SMP3 (Arrayit) spotting pin to deposit ∼1 nL of 1 μM MB solution in spotting buffer (0.1× PBS with 20% glycerol v/v) onto site-specific locations on the microarray surface. This relatively simple system was able to differently functionalize microgel pads approximately 250 μm apart. After one-hour incubation at room temperature, the microarray was rapidly flushed with 5 mL of 0.1× PBS and then further washed by twice soaking in 0.1× PBS with 60 rpm gentle shaking for 5 minutes. After a water rinse to remove salt, the microarray slide was then blown dry with gently flowing nitrogen gas. A 12-well polydimethylsiloxane (PDMS) gasket was attached to the dry slide by PDMS glue so that each well confined a reaction volume over a single microgel array (Fig. 2). Prior to adding an amplification mixture consisting of either extracted RNA+ or synthetic DNA+ in buffer containing NASBA enzymes and amplification primers, each of the 12-well chambers was exposed to 1 μg μL−1 bovine serum albumin (BSA) and 1 unit per μL RNase inhibitor in 80 mM Tris-HCl, 10 mM MgCl2, 70 mM KCl at pH 8.0 to block nonspecific adsorption to the exposed glass and PDMS chamber. Note that we have previously shown that e-beam-patterned PEG microgel pads very effectively resist non-specific biomolecule adsorption.17,22,26
RNA/DNA target preparation
Our experiments involving bacterial culture used Gram-positive (Staphylococcus aureus, ATCC 12600) and Gram-negative (Escherichia coli, NEB #C2987). Bacteria were cultured in Tryptic Soy Broth (TSB) for 10 hours at 37 °C and then vortexed to break bacterial clusters. The concentration of bacteria was determined using a counting chamber (Hausser Scientific, PA). An aliquot containing 1 × 105 bacteria was centrifuged (1500 rpm) for 15 min at room temperature to separate a bacterial pellet. We then used an RNeasy mini kit (Qiagen) to isolate total bacterial RNA following the manufacturer's protocol. This process produced 100 μL of eluted total RNA solution in RNase-free water.We obtained positive blood culture samples from Hackensack University Medical Center (Hackensack, NJ). The use of human blood was performed in compliance with the institutional guidelines of both Hackensack University Medical Center and the Stevens Institute of Technology with committee approval from each institution. The blood cultures were performed using a BD BACTEC FX following standard procedures. 1 mL of a blinded sample was first lysed with 1 mL human cell lysis buffer (pH 11.7, 0.6% Brij-97, 0.4 M CAPS) for 2 minutes in a 15 mL centrifuge tube. Then the solution was centrifuged (1500 rpm for 15 minutes) to isolate bacterial cells in a pellet. The supernatant was removed, replaced with 2 mL of washing buffer (20 mM NaPO3, 0.05% Brij-97 and 0.45% NaCl) followed by vortexing. Bacteria were again isolated by centrifugation (1600 rpm for 6 minutes). The bacteria were subsequently lysed using 4 mg mL−1 lysozyme in Tris-EDTA buffer at pH 8.0. Total RNA was isolated using the procedure described above to produce 100 μL of eluted total RNA solution in RNase-free water.
Synthetic DNA+ targets used in this work were obtained from Integrated DNA Technologies (Coralville, Iowa). Table 1 describes the sequences used.
Total RNA/DNA target amplification and identification
Total RNA, isolated either from in vitro bacterial cultures or from culture-positive human blood, and synthetic DNA+ were used for integrated NASBA amplification and molecular-beacon detection. 1 μL of total RNA or DNA+ solution was added to 19 μl of reaction buffer containing primers and enzymes for NASBA amplification to create a reaction mixture. For a real-time experiment, this reaction mixture was injected into one of 12 independent PDMS wells (Fig. 2) surrounding an individual microarray patterned on glass. Prior to introduction of the reaction mixture, the slide with the PDMS wells was preheated in an oven for 5 min at 41 °C and then placed on a heating stage at 41 °C on a Nikon TiE inverted fluorescence microscope. Imaging was carried out using a CFI S Fluor ELWD 20× objective lens (NA = 0.45, WD = 8.2–6.9 mm) objective lens. Images were recorded at periodic intervals using a SensiCam high-sensitivity CCD Camera (Cooke). Digital image data were analyzed with ImageJ27 and Fiji.28 For an endpoint experiment, the amplification mixture was incubated at 41 °C for 1 hour in an Eppendorf tube. 20 μL of the incubated solution was deposited on an array and allowed to hybridize for 60–90 minutes in saturated-humidity air at room temperature. A cover slip was then placed over the microarray, and fluorescence images were collected using the same imaging protocol as the real-time experiments.3. Results and discussion
To establish the robustness of the NASBA on microgel-tethered microarray format we performed a duplex assay to differentiate between Gram-positive (GP) and Gram-negative (GN) infection of human blood. Ribosomal RNA+ within total RNA extracted from blood samples independently verified to be culture positive was amplified and then analyzed using a duplex assay involving microarray spots functionalized with molecular beacons to probe for pan-GP or pan-GN bacteria. Fig. 3A schematically describes the array format. The top row presents microgel pads functionalized with pan-GP MBs while the lower row presents pads functionalized with pan-GN MBs. In each experiment 1 μL of the 100 μL total RNA solution extracted from a clinical blood sample infected either by GP (Fig. 3C) or GN (Fig. 3D) bacteria was added to 19 μL of NASBA reaction mixture containing GP primer 2 (P2), GN P2, and the P1 common to both (Table 1). The mixture was then allowed to react for 60 min at 41 °C over the array, washed, and imaged. The mixed case (Fig. 3E) used 1 μL of total RNA solution from a mixed blood sample. The blood samples were independently determined to be culture positive with E. coli (GN) and MRSA (GP). We obtained very similar experimental results using clinical samples of human blood independently determined to be culture positive with Proteus or MSSA. | ||
| Fig. 3 Duplex assay of positive human blood culture. (A) Schematic array format; (B) no-target (negative) control; (C) Gram-positive blood; (D) Gram-negative blood; and (E) mixed GN + GP blood; (F) average and range (error bars) of fluorescence intensities for the 2 circular spots in images B–E. | ||
The fluorescence intensity of each circular microarray spot was quantified by measuring the total number of counts within an area included within the microgel pad (dashed circle 1, Fig. 3A) and subtracting from that the total number of counts from an identical area away from a microgel pad (dashed circle 2, Fig. 3A). We report the average intensity per pixel within the image area analyzed. For the image-collection and image-analysis conditions used in the present experiments, a typical spot area (dashed circle) contained about 4420 pixels. The average and range for the two circular spots in the experiments described by Fig. 3C–E are plotted in Fig. 3F, which shows that this assay is able to differentiate GP from GN culture-positive blood in a self-reporting fashion with an endpoint signal intensity exceeding the control intensity by as much as a factor of seven times.
Fig. 3 illustrates another unique aspect of the microgel-tethering approach to oligonucleotide probe immobilization. Spotting methods exclusively create array spots that are circular or, when defective, with some uncontrolled deviation from a circle. Because of the flexibility associated with electron-beam surface patterning,18,29 microgel pads can be made in user-defined sizes and shapes. In the case of Fig. 3, the plus and minus signs on the left side of the upper and lower rows are functionalized with GP and GN MB probes, respectively, and respond to the hybridization conditions in the same manner as the adjacent circular microgel pads. One can thus not only conceive of creating array spots that are controllably small, e.g. on the order of microns in size, but a microarray can be constructed that uses spot shape in addition to, or in place of, the traditional method of position to identify a spot with a particular probe functionalization.
Having established that the low-density array can function in an end-point assay using clinically relevant culture-positive blood specimens, we then used a simplex assay involving total RNA extracted from cultured S. aureus to explore target amplification and detection in real time thus exploiting the self-reporting properties of the MB probes. Fig. 4 shows a typical sequence of images from a pan-GP simplex array with five-fold redundancy, i.e. all five spots in the array were functionalized with pan GP MBs. In this experiment, total RNA was extracted from 105S. aureus cells and concentrated into 100 μL. 1 μL of the concentrate was added to 19 μL of NASBA reaction mixture to create a total RNA concentration corresponding to 50000 CFU ml−1. This sequence of images clearly shows that the fluorescence intensity of each spot increases with reaction time as RNA- amplicons are produced by the NASBA reaction and subsequently hybridize to molecular beacon probes tethered to the microgels comprising each spot.
 | ||
| Fig. 4 Time-resolved images of a simplex NASBA on a pan-Gram positive microarray with five-fold spot redundancy after exposure to S. aureus total RNA. | ||
The fluorescence intensity of each of the five spots can be quantified using digital image analysis, and Fig. 5A shows the real-time intensity profiles for each of the five microgel spots. The t = 75 min image in Fig. 4 illustrates the position in the array associated with each spot number. Prior to adding total RNA, images were collected every minute for the first nine minutes. Fig. 5B shows the fluorescence intensity for each of the five spots during this initial period. At t = 10 minutes, 1 μL of total RNA solution was added to the NASBA solution above the microarray, and images were subsequently collected every three minutes. Beyond this time point, the fluorescence intensity of each spot clearly increases for all five spots.
 | ||
| Fig. 5 (A) Time-resolved intensity profiles of a simplex NASBA on pan-GP microarray with five-fold spot redundancy after exposure to 50000 CFU ml−1S. aureus total RNA at t = 10 min. (B) Detail of the first 9 minutes illustrating the spot-specific background and noise. | ||
Intensity data from the first nine minutes of this experiment enable the spot-specific background signal to be characterized. The average of this pre-target signal corresponds to the background intensity. The background is due at least in part to the several percent quenching inefficiency associated with the fluorophore–quencher pair.23,30 However, the fact that the intensity differences between the five spots is greater at the end of the experiment than it is at the beginning suggests that there may also be slight variations in the number of molecular beacon probes being presented by each of the five microgel spots. While there is little variation in the majority of individual microgels,23 a spot that covers an area of 20 μm diameter and consists of ∼104 microgels is very likely to have been patterned from a portion of PEG precursor thin film that contains one or more spherulite boundaries or other film defects. At these defects the film thickness may deviate from the average and the e-beam patterning properties may be different, both of which could affect the number of binding sites to which molecular beacon probes can be tethered. We furthermore observe that certain spots, e.g. 2, 4, and 5, typically have a slightly higher background intensity than spots 1 and 3 suggesting that there may be slight variations to the electron dose each spot receives during the lithography process which, in turn, would slightly affect the microgel structure and properties.17
Intensity data from the first nine minutes of this experiment also enable the spot-specific characterization of the noise associated with the assay. This noise is very low. Fig. 5B shows solid lines of zero slope corresponding to the average of ten spot-specific intensity measurements as well as error bars that correspond to the standard deviation associated with each average. These error bars are barely visible in Fig. 5B, and the embedded table lists the standard deviations of the signal from each spot. These values are all less than one intensity count per pixel. Thus, while there is some variation in the background associated with different spots that have been redundantly functionalized, there is substantially less noise associated with intensity measurements made over time from one particular spot. Consequently, when following the signal from an individual spot, relatively low levels of hybridization are required to differentiate a real signal from the noise.
We used serial dilutions of total RNA extracted from cultured bacteria to assess the sensitivity of this assay. The results are show in Fig. 6A for E. coli and in Fig. 6B for S. aureus. Both sets of experiments used total RNA concentrations corresponding to 50000 CFU ml−1 down to 5 CFU ml−1. In each case we plot only the intensity of spot 5 in a five-fold redundant simplex assay. Except for variations in the background, the intensity profiles from each of the other four spots are similar. We subtracted from each dataset the appropriate spot-specific background intensity so the baseline corresponds to zero counts per pixel. The intensity profile associated with the 5 CFU ml−1 concentration decreases with time in both the GN and GP experiments. We attribute the decrease to incremental photobleaching associated with each image acquisition. This concentration sets a lower bound of sensitivity for this particular assay. There is substantial signal from all of the higher total RNA concentrations studied. The inset to each figure details the early stages of the intensity profiles associated with the 50 CFU ml−1 experiments. In these insets the horizontal dashed line corresponds to a threshold defined as five standard deviations of noise above the background. The insets in both the GN and GP experiments show that the signal intensity exceeds that threshold within minutes after the total RNA is added at a concentration corresponding to 50 CFU ml−1. Hence, the sensitivity of the assay, as configured in these experiments, is between 5 and 50 CFU ml−1. This value corresponds well to that determined by Zhao et al. in their solution based assay.1 They found a sensitivity of 1.3 CFU in a reaction volume of 20 μl, which corresponds to a sensitivity of about 65 CFU ml−1.
 | ||
| Fig. 6 Time-resolved intensity profiles of a simplex assay assessing the sensitivity of: (A) pan-GN spots exposed to E. coli total RNA; and (B) pan-GP microgel spots exposed to S. aureus total RNA. The insets show the early assay stages of the 50 CFU ml−1 dilution. The lines are smoothed spline-fits to the raw data. | ||
To further assess sensitivity from an alternate perspective, we performed a simplex assay to probe for Aspergillus using serial dilutions of DNA+ as the starting target. DNA+ can be used in the NASBA process without the initiation phase (Fig. 1) while still producing RNA- amplicons. The results of one such experiment are presented in Fig. 7. This figure shows background-subtracted time-resolved intensity profiles for target concentrations of 1 fM, 10 fM, and 100 fM together with a no-target control. We again attribute the slight decrease in intensity associated with the control specimen to photobleaching during data acquisition. The intensity profile from a sample with 0.1 fM DNA+ target, not shown here, overlaps with the no-target control. There is significant signal in the 1 fM sample, however, and there is again very little noise in this intensity profile. The standard deviation associated with the 10 pre-target intensity measurements is 0.4 counts, and the profile exceeds five times this value by the 15th minute, 5 minutes after DNA+ target introduction. Note that the total number of DNA+ target molecules in a 20 μl sample of 1 fM solution is about 12000.
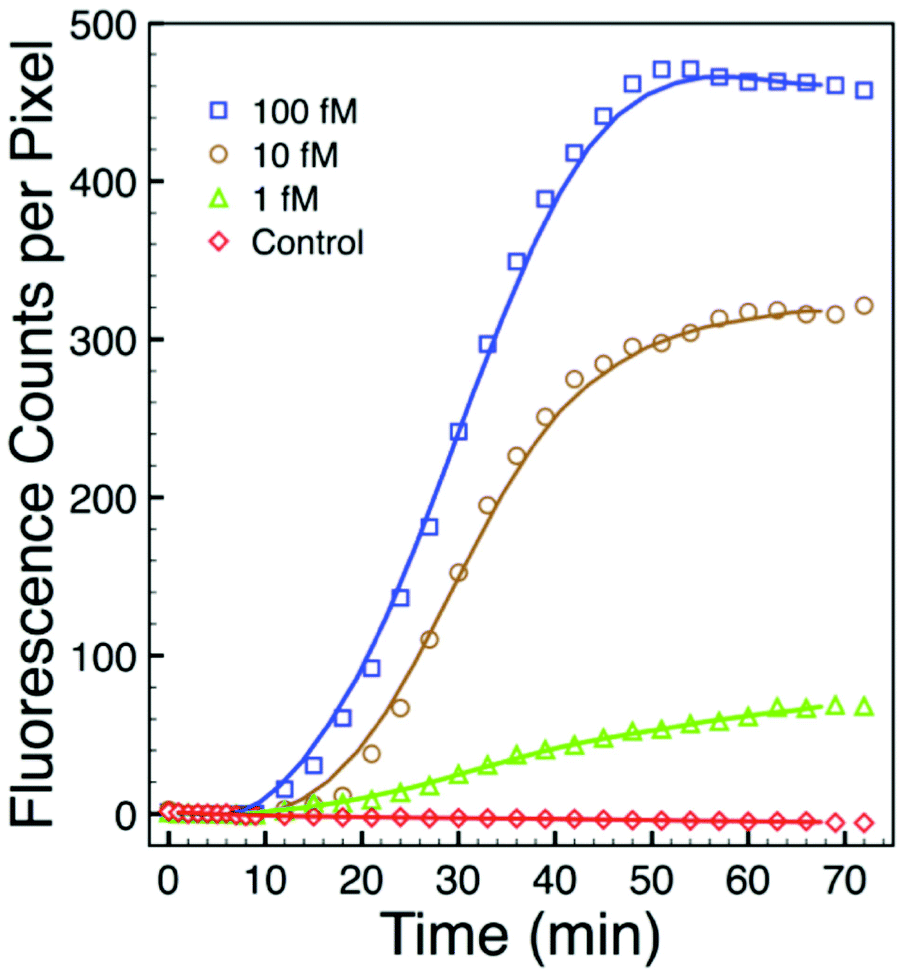 | ||
| Fig. 7 Background-subtracted time-resolved intensity profiles of a simplex NASBA on an Aspergillus microarray with five-fold spot redundancy after exposure to various concentrations of synthetic DNA target indicates a sensitivity exceeding 1 femtomolar. | ||
To explore the detection of different targets under controlled conditions, we performed an assay designed to detect RNA- amplicons produced from DNA+ templates using a universal NASBA amplification mixture. To fabricate this array, five different MB probes (Table 1) were spotted separately on five microgel pads patterned on glass. Importantly, since in the microarray format the MB probes are immobilized on a surface that is in contact with the bulk amplification mixture, the opportunity for non-specific interactions between the MB probes and the NASBA primers is reduced. In our experiment, nine different primers were combined in a NASBA amplification mixture: 0.25 μM concentrations of the three fungal primer pairs; 0.5 μM concentrations of the Gram-positive bacterial primer P2 and of the Gram-negative bacterial primer P2; and 1 μM concentration of the common bacterial primer P1. To test the ability of this mixture to selectively amplify templates of each of the five pathogens, we prepared six samples of the NASBA amplification mixture. To five of these we added one of the five DNA+ templates (Table 1) at a concentration of 1 pM and left the sixth with no template as a negative control. Each sample amplification mixture was then added to an individual reaction chamber presenting the five-plex microarray. As shown in Fig. 8A–E, the five end point (90 min) fluorescence images clearly indicate the successful amplification and detection of each of the five templates with high specificity relative to the negative control group (Fig. 8F). The RNA- amplicons generated by one template only light up the complementary MB probes in the appropriate microarray spot. Non-specific interactions between non-complementary MBs and the RNA- amplicons, which could otherwise cause false positive results, appear minimal by visual inspection. Note that the synthetic pan-fungi DNA+ template samples a short DNA sequence separate from that of either the Candida or Aspergillus templates. Consequently, the pan-fungi spot does not become positive simultaneously with the Candida (Fig. 8C) or Aspergillus (Fig. 8D) assays.
 | ||
| Fig. 8 Endpoint (90 min) fluorescence images from five-spot arrays probing for (A) Gram positive bacteria, (B) Gram negative bacteria, (C) Candida, (D) Aspergillus, and (E) Fungi. Each experiment involved 1 pM of a DNA+ target (1 pM) in a NASBA reaction mixture containing nine different primers. The negative control (F) shows no signal. | ||
4. Conclusions
The ability of microarray technology to ask many questions simultaneously about a biological system has had a significant impact on a number of basic scientific disciplines and clinically important applications. The current work has addressed issues associated with real-time microarray analysis by combining an isothermal oligonucleotide amplification method – NASBA – with self-reporting molecular beacon probes that fluoresce upon hybridization. Key to the success of this platform is the fact that the probes are tethered to their substrate – a glass microscope slide – by microgel pads patterned on the glass surface by electron-beam lithography. The unique structure of these microgels not only localizes the probes at particular surface positions, in user-specified sizes, patterns, and shapes, but maintains them in as water-like an environment as possible. This present set of experiments has demonstrated that the microgel-tethering platform can provide real-time diagnostic microarray capabilities with low noise and high sensitivity in a manner that mimics in many respects the now-common approach of solution-based assays that combine molecular beacon probes with an amplification method such as NASBA or, more commonly, PCR.The elements of the detection system are simple. A single-channel, wide-field, fluorescence microscope equipped with a constant-temperature slide-heating holder is able to automatically collect time-resolved fluorescence images at relatively low magnification (200×). Prior to introduction of the target a set of such images can be used to calibrate both the background signal and the measurement noise. Using nominally identical spots in a five-fold redundant simplex array, we have shown that the magnitude of the background varies from spot to spot, an effect we speculate is a consequence of the e-beam patterning process, but that for a given spot the background can be well characterized and subtracted to give a zero baseline. The measurement noise, characterized as the standard deviation of the spot intensity during the pre-target calibration period, is small and can be used to set a threshold intensity that defines whether a target is present or not. We used a spot intensity five standard deviations above the background to define such a threshold intensity.
By tethering molecular beacon probes specific to sequences within ribosomal RNA characteristic of Gram-positive bacteria and Gram-negative bacteria the assay was able to distinguish between human-blood samples independently determined to be culture positive with S. aureus and E coli, respectively. Using serial dilutions of total RNA extracted from cultured ATCC bacterial strains, we showed that this particular assay has a sensitivity on the order of tens of bacteria per mL. We furthermore showed that the platform can also effectively detect DNA. Synthetic DNA targets having a sequence characteristic of Aspergillus were detected in a simplex assay at femtomolar concentrations of the DNA target. Using a five-plex assay with a universal NASBA reaction mixture containing five primer pairs, we also demonstrated an endpoint assay that can differentiate between DNA characteristic of pan-Gram-positive bacteria, pan-Gram-negative bacteria, pan-fungi, Candida, and Aspergillus.
Acknowledgements
This work has been supported by the United States National Science Foundation (CBET-1402706) and by the Stevens Institute of Technology Innovation and Entrepreneurship Fellowship program.Notes and references
- Y. A. Zhao, S. Park, B. N. Kreiswirth, C. C. Ginocchio, R. Veyret, A. Laayoun, A. Troesch and D. S. Perlin, J. Clin. Microbiol., 2009, 47, 2067–2078 CrossRef CAS PubMed.
- A. Deshpande and P. S. White, Expert Rev. Mol. Diagn., 2012, 12, 645–659 CrossRef CAS PubMed.
- B. C. Millar, J. Xu and J. E. Moore, Curr. Issues Mol. Biol., 2007, 9, 21–40 CAS.
- K. L. Muldrew, Curr. Opin. Pediatr., 2009, 21, 102–111 CrossRef PubMed.
- O. Scheler, B. Glynn and A. Kurg, Expert Rev. Mol. Diagn., 2014, 14, 489–500 CrossRef CAS PubMed.
- J. Compton, Nature, 1991, 350, 91–92 CrossRef CAS PubMed.
- B. Deiman, P. van Aarle and P. Sillekens, Mol. Biotechnol., 2002, 20, 163–179 CrossRef CAS PubMed.
- O. Scheler, B. Glynn, S. Parkel, P. Palta, K. Toome, L. Kaplinski, M. Remm, M. Maher and A. Kurg, BMC Biotechnol., 2009, 9, 45 CrossRef PubMed.
- O. Scheler, L. Kaplinski, B. Glynn, P. Palta, S. Parkel, K. Toome, M. Maher, T. Barry, M. Remm and A. Kurg, BMC Biotechnol., 2011, 11 DOI:10.1186/1472-6750-11-17.
- A. Mader, U. Riehle, T. Brandstetter, E. Stickeler, A. zur Hausen and J. Ruhe, Anal. Bioanal. Chem., 2010, 397, 3533–3541 CrossRef CAS PubMed.
- A. Mader, U. Riehle, T. Brandstetter, E. Stickeler and J. Ruehe, Anal. Chim. Acta, 2012, 754, 1–7 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- W. Tan, K. Wang and T. J. Drake, Curr. Opin. Chem. Biol., 2004, 8, 547–553 CrossRef CAS PubMed.
- G. Leone, H. van Schijndel, B. van Gemen, F. R. Kramer and C. D. Schoen, Nucleic Acids Res., 1998, 26, 2150–2155 CrossRef CAS PubMed.
- X. Dai, W. Yang, E. Firlar, S. A. E. Marras and M. Libera, Soft Matter, 2012, 8, 3067–3076 RSC.
- Y. Wang, E. Firlar, X. Dai and M. Libera, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1543–1554 CAS.
- P. Krsko, M. Mansfield, S. Sukhishvili, R. Clancy and M. Libera, Langmuir, 2003, 19, 5618–5625 CrossRef CAS.
- C. M. Kolodziej and H. D. Maynard, Chem. Mater., 2012, 24, 774–780 CrossRef CAS.
- P. Krsko and M. Libera, Mater. Today, 2005, 8, 36–44 CrossRef CAS.
- K. L. Christman, E. Schopf, R. M. Broyer, R. C. Li, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2009, 131, 521–527 CrossRef CAS PubMed.
- I. Saaem, V. Papasotiropoulos, T. Wang, P. Soteropoulos and M. Libera, J. Nanosci. Nanotechnol., 2007, 7, 2623–2632 CrossRef CAS PubMed.
- Y. Hong, P. Krsko and M. Libera, Langmuir, 2004, 20, 11123–11126 CrossRef CAS PubMed.
- Y. Ma and M. Libera, Langmuir, 2016, 32, 6551–6558 CrossRef CAS PubMed.
- A. Y. Rubina, S. V. Pan'kov, E. I. Dementieva, D. N. Pen'kov, A. V. Butygin, V. A. Vasiliskov, A. V. Chudinov, A. L. Mikheikin, V. M. Mikhailovich and A. D. Mirzabekov, Anal. Biochem., 2004, 325, 92–106 CrossRef CAS PubMed.
- J. Zlatanova and A. Mirzabekov, Methods Mol. Biol., 2001, 170, 17–38 CAS.
- Y. Wang, E. Firlar, X. Dai and M. Libera, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1543–1554 CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- P. Krsko, I. Saaem, R. Clancy, H. Geller, P. Soteropoulos and M. Libera, Proceedings of SPIE – The International Society for Optical Engineering, Boston, MA, 2005.
- S. A. Marras, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30(21), e122 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |