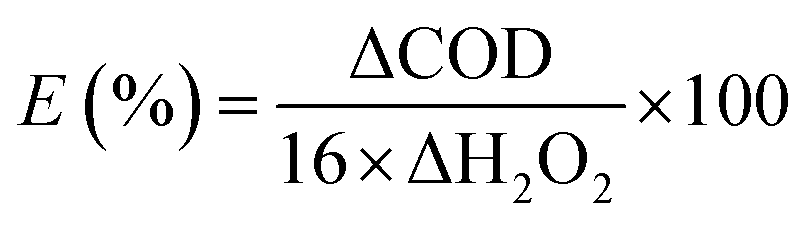DOI:
10.1039/C7RA04312K
(Paper)
RSC Adv., 2017,
7, 26983-26991
Unveiling the mechanism of electron transfer facilitated regeneration of active Fe2+ by nano-dispersed iron/graphene catalyst for phenol removal†
Received
17th April 2017
, Accepted 7th May 2017
First published on 22nd May 2017
Abstract
Nano-dispersed Fe0 and Fe3O4 on reduced graphene oxide (Fe0/Fe3O4-RGO) was prepared and characterized. The prepared Fe0/Fe3O4-RGO was used as a magnetically separable Fenton-like catalyst and showed superior catalytic activity compared to Fe3O4-RGO and Fe3O4 as well as other Fenton-like catalysts for the removal of phenol. The Fe0/Fe3O4-RGO achieved 100% removal efficiency for phenol within 30 min. Free radical inhibition experiments and Electron Paramagnetic Resonance (EPR) showed that the main reactive species was ˙OH rather than FeIV. High resolution TEM results revealed that nanoscale Fe0 and Fe3O4 were uniformly dispersed and distributed on RGO without agglomeration, furnishing more active sites. The catalyst featured a unique mechanism of electron transfer-facilitated regeneration of active Fe2+ by nano-dispersed iron/graphene. RGO served as an effective mediator to facilitate the electron transfer from Fe0 to ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ for the regeneration of Fe2+, which was critical in the catalytic process. This electron transfer-facilitated regeneration of active Fe2+ resulted in a reusable catalyst with high catalytic activity for the removal of phenol. The nano-dispersed Fe0/Fe3O4-RGO could be easily separated and recovered by magnetic field. The Fe0/Fe3O4-RGO catalyst was reusable and the removal efficiency of phenol after 5 catalytic cycles was as high as 93%. The Fe0/Fe3O4-RGO could be an effective Fenton-like catalyst for the treatment of waste water containing refractory phenol and phenol type pollutants.
Fe3+ for the regeneration of Fe2+, which was critical in the catalytic process. This electron transfer-facilitated regeneration of active Fe2+ resulted in a reusable catalyst with high catalytic activity for the removal of phenol. The nano-dispersed Fe0/Fe3O4-RGO could be easily separated and recovered by magnetic field. The Fe0/Fe3O4-RGO catalyst was reusable and the removal efficiency of phenol after 5 catalytic cycles was as high as 93%. The Fe0/Fe3O4-RGO could be an effective Fenton-like catalyst for the treatment of waste water containing refractory phenol and phenol type pollutants.
1. Introduction
The Fenton reaction (Fe2+/H2O2) is one of most commonly used advanced oxidation processes, which can generate a highly reactive hydroxyl radical (˙OH). ˙OH is the second strongest oxidizing agent next to fluorine, which can oxidize organic pollutants into H2O and CO2 or low molecular weight organic compounds.1,2 However, its application for degradation of refractory organic pollutants is limited by difficult separation and low recovery of the catalyst, and further treatments of the dissolved iron and sludge.3 To overcome these limitations, various solid catalysts have been developed, such as supported noble metal nanoparticles (NPs),4–6 iron-based clays, silica and zeolites,7–10 and iron-based magnetic NPs,11–15 as heterogeneous Fenton-like catalysts. Among those, iron-based catalysts have received increasing attention due to their unique advantages including (1) they are inexpensive and relatively non-toxic; (2) the magnetic properties of Fe-based catalysts have the advantage of easy separation by external magnetic field; and (3) desirable catalytic activity in comparison to other catalysts.
Fe3O4 was shown to be an efficient catalyst for the heterogeneous Fenton-like reaction due to the presence of FeII species in the magnetite structure initiating the Fenton reaction.11,13,14 The octahedral structure of Fe3O4 can accommodate both Fe2+ and Fe3+, allowing the iron species to be reversibly oxidized and reduced through electron transfer between Fe2+ and Fe3+. In addition, Fe3O4 can be easily separated from the reaction system by an external magnetic field. The mechanism of the heterogeneous Fenton-like reaction was established in the literature.11,13,16–18 The process involves the redox recycling of Fe2+/Fe3+ on the surface of catalysts, analogous to the Haber–Weiss mechanism.2 Fe2+ can react with H2O2 to generate ˙OH, as shown in reaction (1) (Fe2+ stands for Fe2+ sites on the catalyst surface). Fe2+ can be regenerated through the reaction between Fe3+ and H2O2/HO2˙ as depicted in reaction (2) and (3). However, in most cases, the concentration of HO2˙ is too low, so the conversion of Fe3+ to Fe2+ is a rate-determining step and is quite limited. Therefore, the slow reduction of Fe3+ to Fe2+ is critical for the Fenton-like reaction.
| | |
Fe2+ + H2O2 → Fe3+ + HO− + ˙OH k1 = 63 M s−1
| (1) |
| | |
Fe3+ + H2O2 → Fe2+ + H+ + HO2˙ k2 = 2 × 10−3 M s−1
| (2) |
| | |
Fe3+ + HO2˙ → Fe2+ + H2O2 k4 = 1.3 × 106 M s−1
| (3) |
Previous reports have shown that Fe0 as an electrons donor could reduce Fe3+ into Fe2+ on the surface of Fe3O4 through electron transfer.19–21 However, Fe0 and Fe3O4 NPs are prone to aggregate and form large particles due to strong anisotropic dipolar interactions, specifically in the aqueous phase.15,22 This dramatically reduced specific surface area and exposed active site, which eventually limited the catalytic activity. Therefore, it is essential to anchor and immobilize Fe0 and Fe3O4 NPs onto supports to prevent aggregation. Furthermore, it would be highly desirable if a support could facilitate rapid interfacial electron transfer between Fe0 and Fe3O4 NPs.
Graphene, a single layer of two-dimensional versatile carbon material with a hexagonal packed lattice, has exhibited promising applications as a 2-D catalyst support22–25 due to its high surface area,26 superior mechanical properties,27 and excellent mobility of charge carriers.28 Graphene oxide (GO) consists of abundant oxygenated functional groups, such as hydroxyl and epoxides on the basal plane with carbonyl and carboxyl groups at the edges.29 These oxygenated functional groups can anchor and immobilize metal and metal oxides on its surface and can effectively prevent agglomeration.24,30 The combination of graphene-based material and inorganic NPs can also prevent the aggregation of graphene sheets. Furthermore, theoretically, graphene can serve as an effective mediator to facilitate the electron transfer from Fe0 to Fe3+ for the regeneration of Fe2+. In our previous study,31 graphene-based material showed excellent absorption ability for bisphenol A owing to the strong π–π interaction between graphene and the aromatic ring of bisphenol A. This significantly promoted the accessibility of active sites, leading to improved mass transfer and catalytic efficiency.
With diminishing fossil fuels such as oil and gas, the coal-based chemical industry is rejuvenating around the world. As a result, increasing amounts of industrial waste water containing refractory phenol and phenol type pollutants has become an intractable environmental concern. Along this line, uniformly dispersed nanoscale Fe0 NPs and Fe3O4 NPs on reduced graphene oxide (Fe0/Fe3O4-RGO) was designed, prepared and used as a heterogeneous Fenton-like catalyst for enhanced removal of phenol. The catalyst achieved 100% removal efficiency for phenol within 30 min. Nano-dispersed Fe0/Fe3O4 on RGO showed superior catalytic activity compared to other Fenton-like catalysts for phenol removal ever reported in the literature. The catalyst featured a unique mechanism of electron transfer-facilitated regeneration of active Fe2+ by nano-dispersed iron/grapheme. Moreover, the effects of various influential parameters, such as the concentration of H2O2 and the pH value, on the degradation efficiency of phenol were systematically investigated. The reusability of the catalyst, COD removal, iron leaching and H2O2 decomposition are also discussed in detail.
2. Materials and methods
2.1 Materials
Natural graphite powder (200 mesh, 99.9% purity), phenol (99% purity) was purchased from Alfa Aesar Chemical Co., Ltd. All other chemicals were analytical grade and were purchased from Tianjin Zhiyuan Chemical Co., Ltd. All of the chemicals were used without further purification unless notified. Deionized water was used throughout the experiments.
2.2 Preparation of the catalysts
GO was synthesized from graphite powder according to a reported procedure.29,31 (For detailed synthesis procedure, see ESI†). The Fe0/Fe3O4-RGO composites were prepared in two steps. Firstly, the nano-dispersed Fe3O4 on RGO (Fe3O4-RGO) was prepared by solvothermal method32 as follows: 1.0 g of graphite oxide flakes was exfoliated and dispersed in 200 mL of ethylene glycol (EG)/diethylene glycol (DEG) (EG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEG = 1:19 by volume) through ultrasonic treatment for 1 h. 15 g of sodium acetate, 15 g of sodium acrylate, and 5.4 g of FeCl3·6H2O, were then added to the suspension of GO under magnetic stirring at 50 °C for 30 min. The resulting homogeneous solution was transferred into a 100 mL Teflon-lined stainless-steel autoclave and sealed for solvothermal reaction at 200 °C for 10 h. The product (Fe3O4-RGO) was washed with deionized water and ethanol three times. The obtained Fe3O4-RGO was dispersed in 600 mL of oxygen-free deionized water for further use. Secondly, the Fe0/Fe3O4-RGO was synthesized by in situ reduction method. 1.5 g of FeSO4·7H2O was dissolved in 50 mL of deionized water. The solution was slowly added to the dispersion of Fe3O4-RGO with stirring for 1 h. In the next step, a stoichiometric amount (1.0 g) of NaBH4 was added dropwise to 50 mL of the solution prepared in the previous step and stirred for 1 h. The ferrous iron was reduced to iron zero according to reaction (4). The product Fe0/Fe3O4-RGO was obtained as black particles and was washed with deionized water and ethanol five times, and was then dried in a vacuum oven at 40 °C for 24 h.
:DEG = 1:19 by volume) through ultrasonic treatment for 1 h. 15 g of sodium acetate, 15 g of sodium acrylate, and 5.4 g of FeCl3·6H2O, were then added to the suspension of GO under magnetic stirring at 50 °C for 30 min. The resulting homogeneous solution was transferred into a 100 mL Teflon-lined stainless-steel autoclave and sealed for solvothermal reaction at 200 °C for 10 h. The product (Fe3O4-RGO) was washed with deionized water and ethanol three times. The obtained Fe3O4-RGO was dispersed in 600 mL of oxygen-free deionized water for further use. Secondly, the Fe0/Fe3O4-RGO was synthesized by in situ reduction method. 1.5 g of FeSO4·7H2O was dissolved in 50 mL of deionized water. The solution was slowly added to the dispersion of Fe3O4-RGO with stirring for 1 h. In the next step, a stoichiometric amount (1.0 g) of NaBH4 was added dropwise to 50 mL of the solution prepared in the previous step and stirred for 1 h. The ferrous iron was reduced to iron zero according to reaction (4). The product Fe0/Fe3O4-RGO was obtained as black particles and was washed with deionized water and ethanol five times, and was then dried in a vacuum oven at 40 °C for 24 h.| | |
Fe3O4-RGO–Fe2+ + 2BH4− + 6H2O → Fe0/Fe3O4-RGO + 2B(OH)3 + 7H2
| (4) |
The Fe0-RGO was prepared by the following procedure: 0.20 g of graphite oxide flakes was exfoliated and dispersed in 120 mL of deionized water through ultrasonic treatment for 1 h. Then 0.30 g of FeSO4·7H2O was dissolved in 10 mL of deionized water. The solution was slowly added to the dispersion of GO with stirring for 1 h. Then, a stoichiometric amount (0.20 g) of NaBH4 was added dropwise to 10 mL of the solution prepared in previous step and stirred for 1 h. The obtained product was washed with deionized water and ethanol five times, and was then dried in a vacuum oven at 40 °C for 24 h.
2.3 Characterization of the catalyst
X-ray diffraction (XRD) spectra were recorded on a Bruker D8 Advance X-ray diffractometer equipped with a diffracted-beam monochromator set for Cu Kα radiation (λ = 1.5418 Å). Fourier transform infrared (FT-IR) spectra were collected with a Bruker VERTEX-70 spectrometer in the range of 4000–500 cm−1. The morphology and particle size of the prepared catalysts were analyzed using transmission electron microscopy (TEM) (Tecnai G2 F20, FEI, USA). X-ray energy dispersive spectroscopy (EDS) was used to analyze the element composition and distribution. The zeta-potential of the Fe3O4-RGO was measured using a Malvern Zetasizer Ultra ZS90. The Brunauer–Emmett–Teller surface area (SBET) of the catalysts was obtained by N2 adsorption–desorption using an Automated Gas Sorption Analyzer (Quadrasorb IQ, Quantachrome Instrument Corp). The Raman spectra were recorded from 40 to 4000 cm−1 on a micro laser Raman spectrometer (Horiba Scientific, France). The surface element compositions were measured using X-ray photoelectron spectroscopy (XPS) with a Thermo ESCALAB 250XI spectrometer using monochromatic 150 W Al Kα radiations. The magnetic properties of the catalysts at room temperature were measured using a vibrating-sample magnetometer (MPMS XL-7, Quantum Design, USA).
2.4 Degradation experiments
The phenol degradation experiments were carried out in a 50 mL conical flask placed in a rotary shaker in the dark. The rotate speed was set at 150 rpm. Typically, 25 mg of catalyst was added to 25 mL of 50 mg L−1 phenol solution whose pH value was adjusted by addition of H2SO4 or NaOH. H2O2 was added to the solution to initiate the reaction. Then, 1.0 mL of the suspension was taken at given time intervals using a syringe, separated and quenched with an excessive amount of methanol. To test the stability of catalyst, the catalyst was collected, washed, dried under vacuum, and reused in a fresh solution of phenol and H2O2 for multiple catalytic cycles.
2.5 Sample analysis
The concentrations of phenol were analyzed via high-performance liquid chromatography (Ultimate 3000, Dionex) equipped with UV absorbance detector and C18 column (4.6 mm × 250 mm). The H2O2 concentration was analyzed by iodometric method.33 Chemical Oxygen Demand (COD) was determined by a known procedure.33 The concentration of total dissolved iron was measured with 1,10-phenanthroline after adding hydroxylamine hydrochloride at 510 nm on a UV/Vis spectrophotometer.34,35 (For detailed procedure, see ESI†).
2.6 Electron paramagnetic resonance (EPR) studies
The EPR spectra were obtained on a Bruker E500 spectrometer with a microwave bridge at room temperature. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was used as spin-trapping agent (for detailed procedure, see ESI†).
3. Results and discussion
3.1 Synthesis methods and characterization
The typical synthesis of Fe0/Fe3O4-RGO is depicted in Fig. 1. Firstly, the pre-synthesized graphite oxide was transformed to exfoliated graphene oxide sheets via ultrasonic dispersion. Then, the nano-dispersed Fe3O4 on RGO composite was prepared by solvothermal method followed by reduction to RGO. Zeta potential analysis (Fig. S1†) illustrated that the surface of Fe3O4-RGO was electronegative. The Fe3O4-RGO captured Fe2+ ions in ferrous sulfate solution. Finally the Fe2+ ions captured on the Fe3O4-RGO surface were reduced to Fe0 NPs by NaBH4 and were deposited on the Fe3O4-RGO surface.
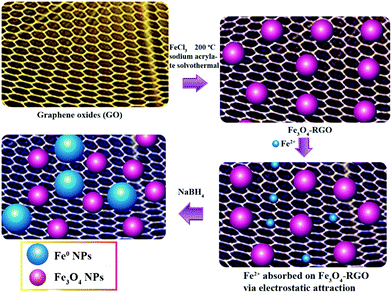 |
| | Fig. 1 Schematic illustration of the synthesis of nano-dispersed Fe0/Fe3O4-RGO. | |
The transmission electron microscope (TEM) images of the Fe3O4-RGO composites revealed that the nanoscale Fe3O4 NPs with a size of 8–15 nm were uniformly anchored on the surface of the graphene sheets (Fig. 2a and b). As shown in Fig. 2c, the high-resolution TEM (HRTEM) image of Fe3O4-RGO showed the presence of crystal lattices of Fe3O4. The lattice fringes of Fe3O4-RGO were observed clearly with interlayer spacing of 0.25 nm, which matched well with the (311) lattice planes of Fe3O4. The TEM images of the Fe0/Fe3O4-RGO displayed distinctive differences compared to that of Fe3O4-RGO. As shown in Fig. 2d and e, more iron NPs were evenly deposited on the graphene sheet, which was ascribed to the formation of Fe0 NPs on Fe3O4-RGO. HRTEM revealed that one could even distinguish the two different iron phases (Fe0 and Fe3O4). The HRTEM image of Fe0/Fe3O4-RGO (Fig. 2f) explicitly showed lattice fringes with interlayer spacing of 0.25 nm and 0.21 nm, which were ascribed to the (311) lattice plane of Fe3O4 and the (110) lattice plane of Fe0, respectively.
 |
| | Fig. 2 TEM images of Fe3O4-RGO (a and b) and Fe0/Fe3O4-RGO (d and e). HRTEM images of Fe3O4-RGO (c) and (f). | |
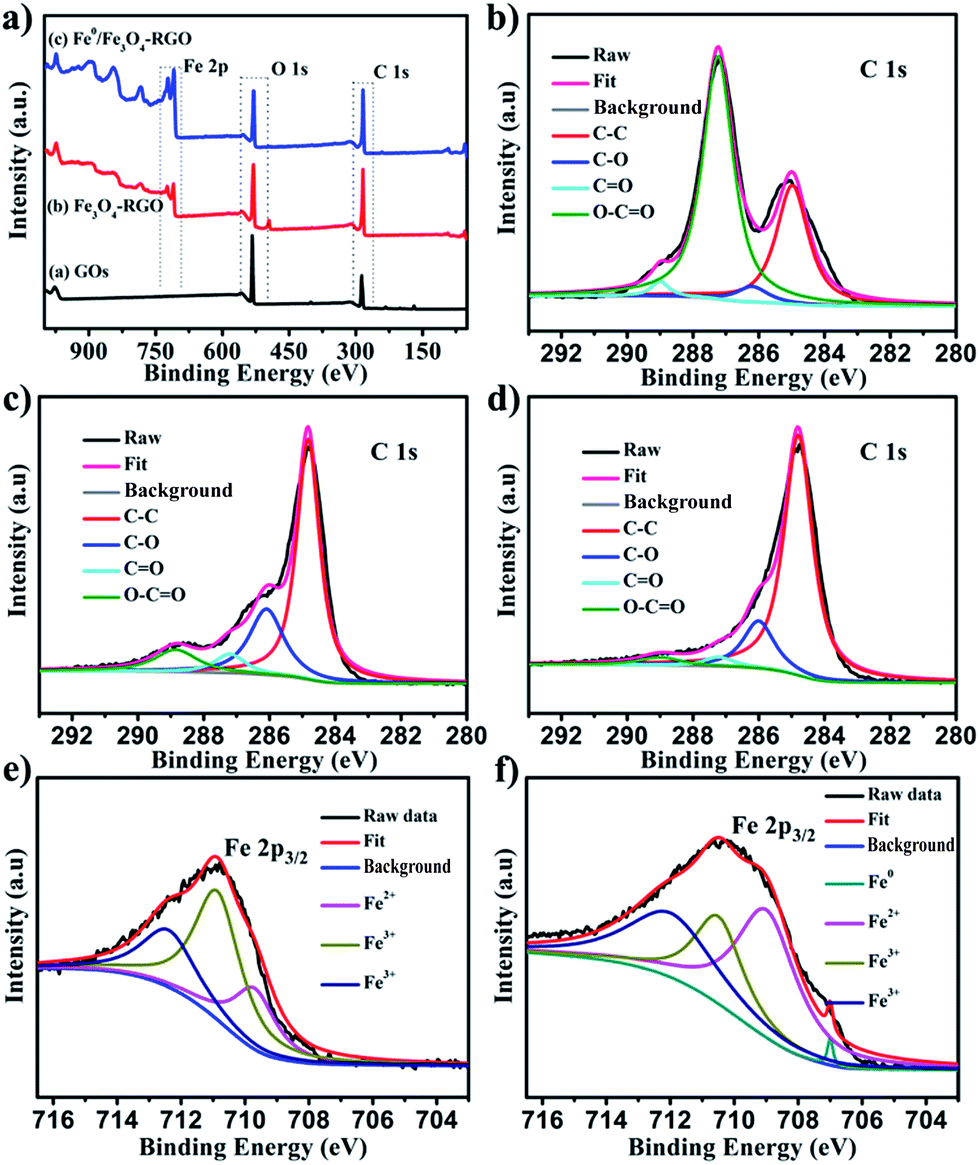
XPS measurements were conducted to analyze the surface element composition and chemical states of Fe3O4-RGO and Fe0/Fe3O4-RGO. The XPS survey spectra of Fe3O4-RGO and Fe0/Fe3O4-RGO are shown in Fig. 3a. The peaks at 285 eV, 531 eV, 711 eV and 725 eV were assigned to C 1s, O 1s, Fe 2p3/2 and Fe 2p1/2, respectively. The survey spectra revealed the presence of C, O, and Fe elements on the surface of Fe3O4-RGO and Fe0/Fe3O4-RGO. Moreover, as shown in Fig. 3b, four different peaks at 284.5, 286.2, 287.2 and 289 eV were observed in high-resolution scans in the C 1s region. Those peaks could be distinguished by deconvolution and were assigned to the non-oxygenated ring C, epoxy/hydroxyls (C–O), the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and the carboxylate carbon (O–CO).29,36 For Fe3O4-RGO and Fe0/Fe3O4-RGO, the intensity of the C 1s peaks, especially the peak assigned to oxidized carbon, decreased dramatically, implying that the GO was reduced to RGO (Fig. 3c and d). In addition, three different peaks at 709.5, 710.7 and 712.2 eV could be distinguished by deconvolution using data from the high-resolution scans in the Fe 2p3/2 region of Fe3O4-RGO (Fig. 3e). Those peaks were assigned to the binding energies for Fe2+–O, and Fe3+–O.37 Similarly, those characteristic peaks were observed in Fe 2p3/2 spectra of Fe0/Fe3O4-RGO (Fig. 3f). In addition, the presence of a Fe0 peak with weak intensity at 707 eV was further evidence for the presence of Fe0 in nano-dispersed Fe0/Fe3O4-RGO (Fig. 3f).
O), and the carboxylate carbon (O–CO).29,36 For Fe3O4-RGO and Fe0/Fe3O4-RGO, the intensity of the C 1s peaks, especially the peak assigned to oxidized carbon, decreased dramatically, implying that the GO was reduced to RGO (Fig. 3c and d). In addition, three different peaks at 709.5, 710.7 and 712.2 eV could be distinguished by deconvolution using data from the high-resolution scans in the Fe 2p3/2 region of Fe3O4-RGO (Fig. 3e). Those peaks were assigned to the binding energies for Fe2+–O, and Fe3+–O.37 Similarly, those characteristic peaks were observed in Fe 2p3/2 spectra of Fe0/Fe3O4-RGO (Fig. 3f). In addition, the presence of a Fe0 peak with weak intensity at 707 eV was further evidence for the presence of Fe0 in nano-dispersed Fe0/Fe3O4-RGO (Fig. 3f).
 |
| | Fig. 3 (a) XPS survey spectra of GO, Fe3O4-RGO and Fe0/Fe3O4-RGO. (b–d) C 1s peaks of GO, Fe3O4-RGO and Fe0/Fe3O4-RGO. (e and f) Fe 2p3/2 peaks of Fe3O4-RGO and Fe0/Fe3O4-RGO. | |
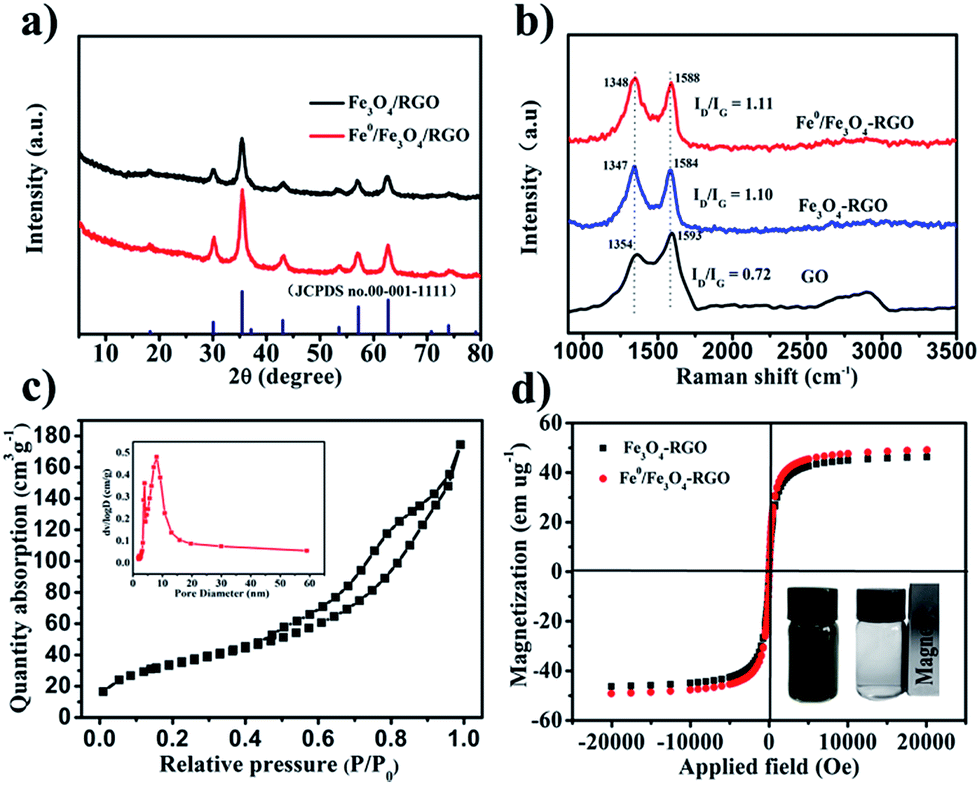
XRD measurements were carried out to investigate the phase structure of the nano-dispersed Fe0/Fe3O4-RGO. Fig. 4a shows the XRD patterns of Fe3O4-RGO and Fe0/Fe3O4-RGO.
 |
| | Fig. 4 (a) XRD patterns of Fe3O4-RGO and Fe0/Fe3O4-RGO. (b) Raman spectra of GO, Fe3O4-RGO and Fe0/Fe3O4-RGO. (c) Nitrogen adsorption–desorption isotherms and pore size distribution curve (insert pattern) of Fe0/Fe3O4-RGO composite. (d) Magnetization curves of Fe3O4-RGO and Fe0/Fe3O4-RGO composites. | |
The diffraction patterns for Fe3O4-RGO showed seven broad peaks at 18.2°, 30.4°, 35.6°, 43.3°, 53.2°, 56.9° and 62.7°, corresponding to (111), (220), (311), (400), (422), (511) and (440) of Fe3O4 (JCPDS no. 001-1111), respectively. The typical peak of Fe3O4 was also observed in Fe0/Fe3O4-RGO. However, the peak of Fe0 at 44.5° was not found in the XRD pattern of Fe0/Fe3O4-RGO. Similar phenomenon have been observed and reported in the literature.9,38,39
Raman spectra of the GO, Fe3O4-RGO and Fe0/Fe3O4-RGO are shown in Fig. 4b. The peaks at ∼1350 and ∼1590 cm−1 are the characteristic peaks of the D and G bands from graphene. Peak shift was observed for both D and G bands, indicating a charge transfer between the graphene sheet and the Fe0 and Fe3O4 NPs.40,41 The charge transfer between graphene and Fe3O4 could be beneficial for conversion of Fe3+ to Fe2+. In addition, the spectra of Fe3O4-RGO and Fe0/Fe3O4-RGO were compared to that of GO. There was an obvious increase of the intensity ratio (ID/IG), implying the reduction of GO to RGO,36,42,43 which was consistent with the results of XPS.
The nitrogen adsorption–desorption curve was obtained to evaluate the specific surface area and pore size distribution. The N2 adsorption–desorption isotherm of Fe0/Fe3O4-RGO is shown in Fig. 4c. The SBET of the Fe0/Fe3O4-RGO was obtained as 124 m2 g−1. The curve belongs to the Type IV isotherm classified by IUPAC with distinct hysteresis loops close to H3 type, suggesting that the Fe0/Fe3O4-RGO composite has a characteristic lamellar stacking. Fig. 4c (inset) shows the pore size distribution of Fe0/Fe3O4-RGO. The total pore volume and average pore diameter of Fe0/Fe3O4-RGO were 0.22 cm3 g−1 and 7.26 nm, respectively.
The magnetic properties of the Fe3O4-RGO and Fe0/Fe3O4-RGO are shown in Fig. 4d. The magnetic hysteresis loops were S-like curves. The saturation magnetization of Fe3O4-RGO (46.3 emu g−1) was smaller than that of Fe0/Fe3O4-RGO (49.1 emu g−1), which could be attributed to the formation of Fe0. The magnetic remanence of Fe3O4-RGO and Fe0/Fe3O4-RGO was nearly zero, suggesting that Fe3O4-RGO and Fe0/Fe3O4-RGO exhibit superparamagnetic behavior. Moreover, the magnetic response of the Fe0/Fe3O4-RGO was also examined by an external magnet (Fig. 4d inset). The results showed that Fe0/Fe3O4-RGO could be easily separated and recovered. This could be an advantage for the recycling and reuse of the catalyst in real applications.
3.2 Catalytic activity of the Fe0/Fe3O4-RGO composite
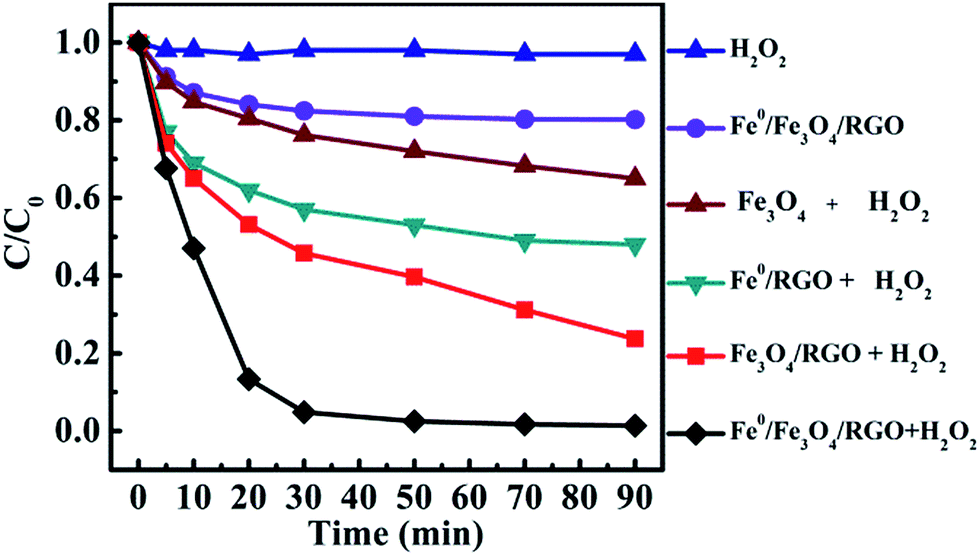
Batch experiments were conducted to compare the removal efficiencies of phenol by various processes. As shown in Fig. 5, H2O2 without catalyst only led to a limited removal efficiency of 2.8% within 90 min, implying that the oxidation ability of H2O2 for phenol was poor. Interestingly, in the absence of H2O2, Fe0/Fe3O4-RGO achieved removal efficiency 20% for phenol in 30 min. This is owing to the surface adsorption of phenol on the Fe0/Fe3O4-RGO via π–π interaction.31,44 Noticeably, the removal efficiency of phenol using the Fe3O4-RGO composite as the heterogeneous Fenton-like catalyst in the presence of H2O2 was significantly higher (76%) than that of a commercial Fe3O4 catalyst (35%), which could be explained by various aspects. Firstly, Fe3O4 NPs were highly dispersed on the substrate, furnishing a high density of active sites. Secondly, the strong π–π interaction between RGO and the aromatic group of phenol promoted the absorption of phenol and thus increased the effective concentration near the surfaces of the Fe3O4-RGO composite,45 resulting in the improved catalytic activity of Fe3O4-RGO.
 |
| | Fig. 5 Comparison of the removal efficiency of phenol with different catalytic systems. Reaction conditions: [phenol]0 = 50 mg L−1, [Fe3O4] = 1 g L−1, [Fe3O4-RGO] = 1 g L−1, [Fe0/Fe3O4-RGO] = 1 g L−1, [H2O2]0 = 5 mM, pH 3.0 and T = 25 °C; C0 and C are the initial phenol concentration and its concentration at any time during the reaction, respectively. | |
Interestingly, the Fe0-RGO with H2O2 led to 65% removal efficiency for phenol within 90 min. This was probably due to the reason that Fe0 was oxidized producing Fe2+ via two electron transfer mechanisms from the particle surface to H2O2 (reaction (5)).46 The oxidants responsible for the removal of phenol were generated by Fenton reaction (reactions (6) and (7)).
| | |
Fe0 + H2O2 + 2H+ → Fe2+ + 2H2O
| (5) |
| | |
Fe2+ + H2O2 → Fe3+ + ˙OH + OH−
| (6) |
| | |
Fe2+ + H2O2 → Fe(IV) (e.g., FeO2+) + H2O
| (7) |
Noticeably, with the Fe0/Fe3O4-RGO composite as the heterogeneous Fenton-like catalyst, the phenol was completely removed within 30 min. The catalytic activity of Fe0/Fe3O4-RGO was the highest compared to all other Fenton-like catalysts for phenol removal ever reported in the literature. These results are summarized in Table 1 (for detailed information, see ESI†). By comparing the catalytic performance with a series of control systems, it was concluded that the catalytic activity of the Fe3O4-RGO was significantly enhanced by the introduction of Fe0. It was proposed that there was a synergistic effect between Fe0, Fe3O4 and RGO in phenol removal using Fe0/Fe3O4-RGO as a heterogeneous Fenton-like catalyst. A possible mechanism of enhanced removal of phenol using Fe0/Fe3O4-RGO as a heterogeneous Fenton-like catalyst is discussed in detail in Section 3.6.
Table 1 Comparison of phenol removal using different Fenton-like catalysts
| Catalyst |
Catalyst dose (g L−1) |
[Phenol]0 mM |
[H2O2]0 mM |
Degradation (%) |
Reaction time (h) |
Reference |
| Fe3O4 |
5 |
1.00 |
1200 |
95 |
6 |
14 |
| Au/HO-npD |
N/A |
1.06 |
5.88 |
93 |
24 |
6 |
| Fe/AC |
0.5 |
1.06 |
15 |
100 |
4 |
47 |
| Fe-ZSM-5 |
1.5 |
0.691 |
90 |
81 |
3 |
48 |
| FeAlSi-ox |
3.0 |
0.500 |
50 |
32 |
8 |
34 |
| FeSi-ox |
3.0 |
0.500 |
50 |
44 |
8 |
34 |
| Magnetite (Fe3O4) |
3.0 |
0.266 |
150 |
42 |
24 |
49 |
| Fe–Al-pillared clay |
0.6 |
0.213 |
4.00 |
100 |
2.5 |
50 |
| Fe0/Fe3O4-RGO |
1.0 |
0.531 |
5.00 |
100 |
0.5 |
This work |
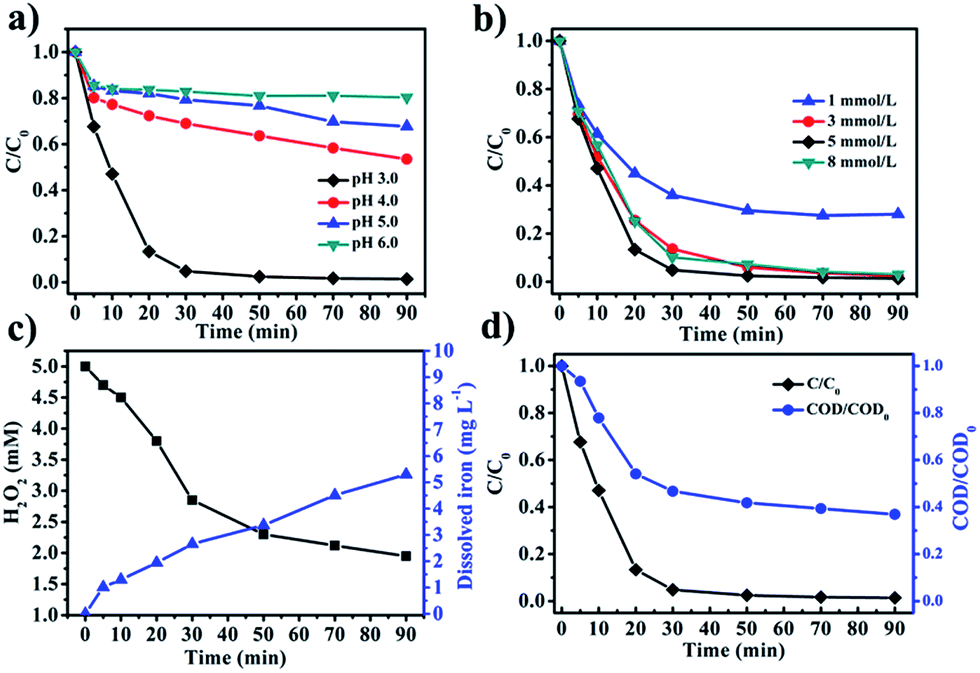
3.3 Effects of pH and H2O2 concentration on phenol removal
The effect of pH on the removal of phenol using Fe0/Fe3O4-RGO is shown in Fig. 6a. The results showed that the catalytic activity of Fe0/Fe3O4-RGO for phenol removal was highly pH-dependent. The removal efficiency of phenol was only 20% after 90 min at pH 6.0, while the removal efficiency of phenol dramatically increased with decreasing pH. The phenol was almost totally removed after 30 min at pH 3.0. The higher removal efficiency at lower pH was ascribed to the higher oxidation potential of ˙OH under acidic conditions.51,52 It was also proposed that acidic conditions were favourable for the stability of H2O2 and were beneficial for the generation of ˙OH and the formation of metal oxide-pollutant inner-sphere complexes.14 The effect of the H2O2 concentration on removal of phenol using Fe0/Fe3O4-RGO was also investigated (Fig. 6b). It was observed clearly that the removal efficiency of phenol increased with increasing H2O2 concentration from 1 mM to 5 mM. Hydrogen peroxide is the precursor in the reaction with Fe2+ generating ˙OH as described in reaction (1). With insufficient H2O2 concentration (below 5 mM), the amount of ˙OH generated will be limited, leading to a low removal efficiency of phenol. Interestingly and noticeably, the removal efficiency of phenol decreased with increasing H2O2 concentration from 5 mM to 8 mM. This was probably because the excess H2O2 could be a scavenger of ˙OH, as described in reaction (8).42,53 Although other oxidative species, such as HOO˙ and O2˙−, are generated, as described in reactions (8) and (9), ˙OOH and O2˙− have much lower oxidation potentials than ˙OH.11,54| | |
Fe2+ + H2O2 → Fe3+ + HO− + ˙OH
| (1) |
| | |
˙OH + H2O2 → H2O + HOO˙
| (8) |
| | |
HOO˙ ↔ O2˙− + H+ (pKa 4.8)
| (9) |
 |
| | Fig. 6 (a) The effect of initial pH on phenol removal. (b) The effect of H2O2 concentration on phenol removal. (c) The concentration of H2O2 and dissolved iron during phenol removal over time. (d) The phenol removal and COD removal over time [phenol]0 = 50 mg L−1, [Fe0/Fe3O4-RGO] = 1 g L−1, [H2O2]0 = 5 mM, pH 3.0 and T = 25 °C. | |
3.4 Iron leaching and H2O2 decomposition
The concentrations of H2O2 and total dissolved iron during phenol removal using the Fe0/Fe3O4-RGO composite were investigated as well (Fig. 6c). It was observed that the concentration of total dissolved iron increased with the reaction time. The total dissolved iron could be the result of ferrous and ferric ions leaching from the Fe0/Fe3O4-RGO composite. The concentration of total dissolved iron after 90 min was 5.2 mg L−1, which was only 0.8% of the total iron of the 1.0 g L−1 catalyst used. It was observed that the H2O2 concentration decreased rapidly within the first 30 min then slowed down gradually from 30 min to 90 min during the removal of phenol. This was consistent with the removal efficiency of phenol over time.
The COD of the phenol solution during the reaction was monitored in order to further investigate the utilization efficiency of H2O2 (Fig. 6d). The maximum COD removal (63%) was achieved after 90 min, indicating that a residual amount of organic compounds remained in solution, presumably generated by catalytic oxidation reaction. The stoichiometry utilization efficiency of H2O2 was defined as the ratio of the amount of H2O2 used for the degradation of phenol against the total amount of H2O2 consumed in the reaction.55,56 Based on reactions (11)–(13), the utilization efficiency of H2O2 can be calculated as 86% through eqn (10). The utilization efficiency of H2O2 was notably higher than previously reported results.13,53
| |
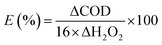 | (10) |
where ΔCOD is the change of COD value (mg L
−1), ΔH
2O
2 is the amount of H
2O
2 (mol L
−1) consumed and 16 is the conversion factor.
| | |
C6H5OH + 14H2O2 → 6CO2 + 17H2O
| (11) |
| | |
C6H5OH + 7O2 → 6CO2 + 3H2O
| (12) |
3.5 The reusability of Fe0/Fe3O4-RGO
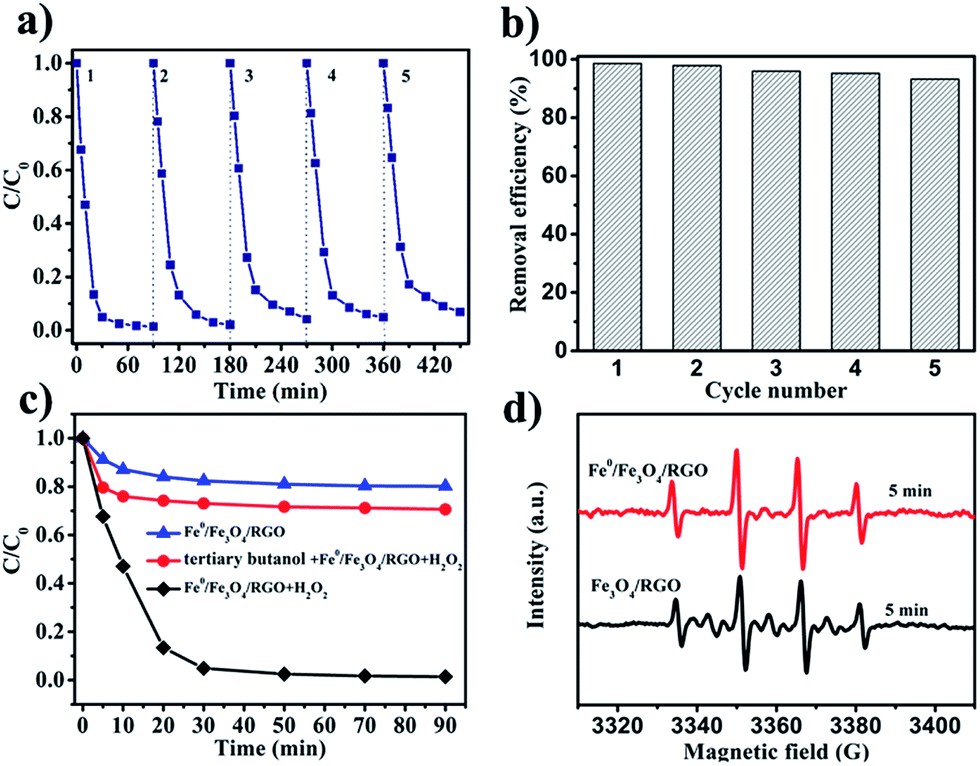
Reusability is one of the most important factors in evaluating a catalyst's performance in practical applications.57 Five successive catalytic cycles were carried out under the same conditions to test the reusability of the Fe0/Fe3O4-RGO. The used catalyst was collected and washed with water before next use. As shown in Fig. 7a, the kinetic rate of phenol removal only decreased slightly after five successive batch experiments. This was attributed to iron leaching from the catalyst surface into solution, as discussed in the previous section. Another possible reason could be the poisoning of the active catalytic sites by adsorbed organic species. Impressively, the removal efficiency of phenol remained as high as 93% after 90 min reaction and reuse in five catalytic cycles (Fig. 7b). The removal efficiency of phenol after each cycle was 99%, 98%, 96%, 95% and 93%, sequentially, demonstrating the excellent catalytic activity and decent reusability of Fe0/Fe3O4-RGO.
 |
| | Fig. 7 Reusability of Fe0/Fe3O4-RGO catalyst for phenol removal: (a) the kinetic rate of removal for phenol; (b) the removal efficiencies of phenol after 90 min reaction; (c) effect of radical scavengers on phenol removal; (d) DMPO spin-trapping EPR spectra of Fe0/Fe3O4-RGO/H2O2 system and Fe3O4-RGO/H2O2 system [catalyst] = 1.0 g L−1, [DMPO] = 0.2 M, [tertiary butanol] = 300 mM, pH 3 and T = 25 °C. | |
3.6 Catalytic mechanism of Fe0/Fe3O4-RGO for phenol removal
Free radical inhibition experiments are known to be effective to identify reactive species in Fenton or Fenton-like systems.11,58 Therefore, tertiary butanol was selected as a scavenger of ˙OH. As shown in Fig. 7c, the removal efficiency of phenol was 30% after 90 min in the presence of 300 mM tertiary butanol. For Fe0/Fe3O4-RGO/H2O2 without adding tertiary butanol, the removal efficiency of phenol was as high as 100%. These results imply that the ˙OH is the main reactive species responsible for phenol removal rather than high oxidative state iron species (FeIV).34 To further identify free radical species generated in the catalytic system, EPR technique with spin-trapping agent DMPO was used. As shown in Fig. 7d, four characteristic peaks of the DMPO–˙OH adduct with an intensity ratio of 1:2:2:1 were observed in the EPR spectra of both Fe3O4-RGO/H2O2 and Fe0/Fe3O4-RGO/H2O2 in the presence of 0.1 M DMPO. This result was further evidence for the existence of ˙OH. Furthermore, the peak intensity of DMPO–OH in the Fe0/Fe3O4-RGO/H2O2 system was higher than that of Fe3O4-RGO/H2O2, indicating that the concentration of ˙OH generated in the Fe0/Fe3O4-RGO/H2O2 system was higher. The result was consistent with the enhanced removal efficiency of phenol using Fe0/Fe3O4-RGO.
It was reported that the heterogeneous Fenton-like reaction and ˙OH production mostly occur on the surface of the solid catalyst.22,42,54 In addition, ˙OH was found to have a very short half-life (<1 μs).59 According to this, the tight adsorption of phenol on the catalyst surface would increase the probability of the reaction between phenol and hydroxyl radicals formed at the surface of the catalyst.60 Therefore, the effective absorption of phenol on the catalyst surface could actually facilitate the catalytic process of phenol removal.11
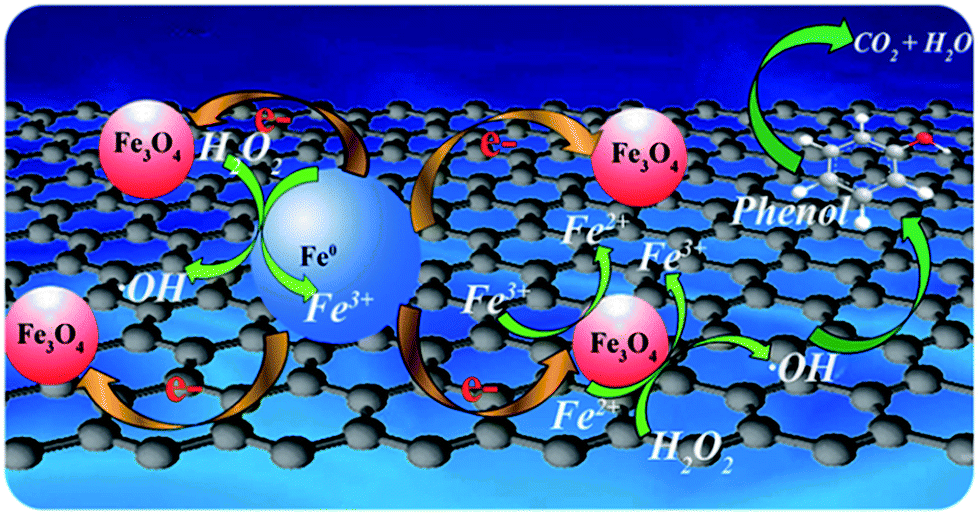
Based on previous literature and our experimental results, a possible mechanism of enhanced removal of phenol using Fe0/Fe3O4-RGO as a heterogeneous Fenton-like catalyst was proposed (Fig. 8). With our approach, the Fe0 NPs and Fe3O4 NPs were uniformly dispersed and distributed on RGO, furnishing more active sites. Fe2+ from the Fe3O4 and Fe0 of Fe0/Fe3O4-RGO reacted with H2O2 to generate ˙OH (reaction (1)). Not only was Fe2+ regenerated from Fe3+ by reacting with H2O2 (reaction (2) and (3)), but also Fe3+ could react with Fe0 via graphene-facilitated electron transfer to regenerate Fe2+. It has been reported that this reaction is thermodynamically favourable (reaction (14)).20 Interestingly, the Raman results for Fe3O4-RGO and Fe0/Fe3O4-RGO showed an electron transfer between the Fe0/Fe3O4 and RGO. This implied that RGO actually served as an effective mediator to promote electron transfer from Fe0 to Fe3+ of Fe3O4 for the generation of Fe2+.61 Thus, more ˙OH was generated on the surface of Fe0/Fe3O4-RGO to oxidize the phenol absorbed on Fe0/Fe3O4-RGO facilitated by π–π interaction between RGO and phenol. Overall, nano-dispersed Fe0/Fe3O4 on RGO as a Fenton-like catalyst for enhanced removal of phenol was successful.
| | |
Fe2+ + H2O2 → Fe3+ + HO− + ˙OH
| (1) |
| | |
Fe3+ + H2O2 → Fe2+ + H+ + HO2˙
| (2) |
| | |
Fe3+ + HO2˙ → Fe2+ + H2O2
| (3) |
| | |
Fe0 + Fe3+ → Fe2+ ΔE0 = 1.21 V
| (14) |
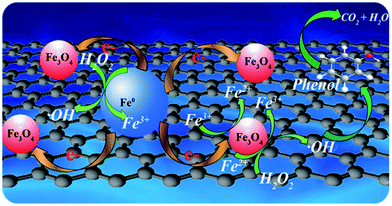 |
| | Fig. 8 The nano-dispersed Fe0/Fe3O4-RGO catalyst featured a unique mechanism of electron transfer-facilitated regeneration of active Fe2+ for catalytic phenol removal. | |
4. Conclusions
Nano-dispersed Fe0 and Fe3O4 on reduced graphene oxide (Fe0/Fe3O4-RGO) was prepared and characterized. The Fe0/Fe3O4-RGO composite as a heterogeneous Fenton-like catalyst achieved 100% removal efficiency for phenol within 30 min. The Fe0/Fe3O4-RGO catalyst was reusable and the removal efficiency of phenol after five catalytic cycles was as high as 93%. The Fe0/Fe3O4-RGO was also magnetically separable. The Fe0/Fe3O4-RGO could be an effective Fenton-like catalyst for the treatment of waste water containing refractory phenol and phenol-type pollutants. The nano-dispersed Fe0/Fe3O4-RGO catalyst featured a unique mechanism of electron transfer-facilitated regeneration of active Fe2+ for catalytic phenol removal. Nano-dispersed Fe0 and Fe3O4 NPs on RGO could furnish more active sites exposed on the catalyst surface. RGO served as an effective mediator to facilitate the electron transfer from Fe0 to Fe3+ for the regeneration of Fe2+, which resulted more ˙OH. The strong π–π interaction between RGO and the aromatic ring of phenol promoted the absorption of phenol and thus increased the probability of the reaction with proximate hydroxyl radicals formed on the surface of the catalyst. All these helped achieve the high catalytic activity.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21472235), Xinjiang Distinguished Youth Scholar Program (qn2015jq012), “One Thousand Talents” Program (Y32H291501) of China and the STS program of Chinese Academy of Sciences (2017).
Notes and references
- E. Brillas, I. Sires and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 CrossRef CAS.
- X. J. Yang, P. F. Tian, C. X. Zhang, Y. Q. Deng, J. Xu, J. L. Gong and Y. F. Han, Appl. Catal., B, 2013, 134, 145–152 CrossRef.
- X. Li, X. Liu, L. Xu, Y. Wen, J. Ma and Z. Wu, Appl. Catal., B, 2015, 165, 79–86 CrossRef CAS.
- S. Navalon, R. Martin, M. Alvaro and H. Garcia, Angew. Chem., Int. Ed., 2010, 49, 8403–8407 CrossRef CAS PubMed.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- M. Danish, X. Gu, S. Lu and M. Naqvi, Environ. Sci. Pollut. Res. Int., 2016, 23, 13298–13307 CrossRef CAS PubMed.
- L. Ma, H. He, R. Zhu, J. Zhu, I. D. R. Mackinnon and Y. Xi, Catal. Sci. Technol., 2016, 6, 6066–6075 CAS.
- A. Cihanoğlu, G. Gündüz and M. Dükkancı, Appl. Catal., B, 2015, 165, 687–699 CrossRef.
- M. Wang, G. Fang, P. Liu, D. Zhou, C. Ma, D. Zhang and J. Zhan, Appl. Catal., B, 2016, 188, 113–122 CrossRef CAS.
- F. Chen, S. Xie, X. Huang and X. Qiu, J. Hazard. Mater., 2017, 322, 152–162 CrossRef CAS PubMed.
- L. Xu and J. Wang, Appl. Catal., B, 2012, 123–124, 117–126 CrossRef CAS.
- S. Zhang, X. Zhao, H. Niu, Y. Shi, Y. Cai and G. Jiang, J. Hazard. Mater., 2009, 167, 560–566 CrossRef CAS PubMed.
- H. Ren, Y. Su, X. Han and R. Zhou, J. Chem. Technol. Biotechnol., 2017, 92, 1421–1427 CrossRef CAS.
- Z. W. Wan and J. L. Wang, Environ. Sci. Pollut. Res., 2017, 24, 568–577 CrossRef CAS PubMed.
- Z. Ma, L. Ren, S. Xing, Y. Wu and Y. Gao, J. Phys. Chem. C, 2015, 119, 23068–23074 CAS.
- X. Huang, X. Hou, J. Zhao and L. Zhang, Appl. Catal., B, 2016, 181, 127–137 CrossRef CAS.
- F. C. C. Moura, M. H. Araujo, R. C. C. Costa, J. D. Fabris, J. D. Ardisson, W. A. A. Macedo and R. M. Lago, Chemosphere, 2005, 60, 1118–1123 CrossRef CAS PubMed.
- R. C. C. Costa, F. C. C. Moura, J. D. Ardisson, J. D. Fabris and R. M. Lago, Appl. Catal., B, 2008, 83, 131–139 CrossRef CAS.
- H. Li, J. Wan, Y. Ma and Y. Wang, Chem. Eng. J., 2016, 301, 315–324 CrossRef CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz da Costa, Sci. Rep., 2014, 4, 4594 CrossRef PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- X. Yang, W. Chen, J. Huang, Y. Zhou, Y. Zhu and C. Li, Sci. Rep., 2015, 5, 10632 CrossRef PubMed.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- S. Latil and L. Henrard, Phys. Rev. Lett., 2006, 97, 036803 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- H. L. Wang, J. T. Robinson, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS PubMed.
- P. Wang, X. Zhou, Y. Zhang, L. Wang, K. Zhi and Y. Jiang, RSC Adv., 2016, 6, 102348–102358 RSC.
- H. Sun, L. Cao and L. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS.
- T. Wu and J. D. Englehardt, Environ. Sci. Technol., 2012, 46, 2291–2298 CrossRef CAS PubMed.
- A. L. T. Pham, C. Lee, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2009, 43, 8930–8935 CrossRef CAS PubMed.
- H. Tamura, K. Goto, T. Yotsuyanagi and M. Nagayama, Talanta, 1974, 21, 314–318 CrossRef CAS PubMed.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- S. Bae, S. Gim, H. Kim and K. Hanna, Appl. Catal., B, 2016, 182, 541–549 CrossRef CAS.
- X. Lv, J. Xu, G. Jiang, J. Tang and X. Xu, J. Colloid Interface Sci., 2012, 369, 460–469 CrossRef CAS PubMed.
- L. Tan, S. Lu, Z. Fang, W. Cheng and E. P. Tsang, Appl. Catal., B, 2017, 200, 200–210 CrossRef CAS.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- Z. W. Wan and J. L. Wang, J. Hazard. Mater., 2017, 324, 653–664 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed.
- Y. Li, J. Qu, F. Gao, S. Lv, L. Shi, C. He and J. Sun, Appl. Catal., B, 2015, 162, 268–274 CrossRef CAS.
- C. Lee, C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 4921–4926 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, A. F. Mohedano and J. J. Rodríguez, Appl. Catal., B, 2006, 65, 261–268 CrossRef CAS.
- K. Fajerwerg and H. Debellefontaine, Appl. Catal., B, 1996, 10, L229–L235 CrossRef CAS.
- K. Rusevova, F.-D. Kopinke and A. Georgi, J. Hazard. Mater., 2012, 241–242, 433–440 CrossRef CAS PubMed.
- M. Luo, D. Bowden and P. Brimblecombe, Appl. Catal., B, 2009, 85, 201–206 CrossRef CAS.
- N. Masomboon, C. Ratanatamskul and M.-C. Lu, Environ. Sci. Technol., 2009, 43, 8629–8634 CrossRef CAS PubMed.
- M. Panizza and G. Cerisola, Water Res., 2009, 43, 339–344 CrossRef CAS PubMed.
- L. Xu and J. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CrossRef CAS PubMed.
- X. Hu, B. Liu, Y. Deng, H. Chen, S. Luo, C. Sun, P. Yang and S. Yang, Appl. Catal., B, 2011, 107, 274–283 CrossRef CAS.
- W. Luo, L. Zhu, N. Wang, H. Tang, M. Cao and Y. She, Environ. Sci. Technol., 2010, 44, 1786–1791 CrossRef CAS PubMed.
- Y. W. Kang and K.-Y. Hwang, Water Res., 2000, 34, 2786–2790 CrossRef CAS.
- J. Ma, Q. Yang, Y. Wen and W. Liu, Appl. Catal., B, 2017, 201, 232–240 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ChemSusChem, 2011, 4, 1712–1730 CrossRef CAS PubMed.
- M. E. Snook and G. A. Hamilton, J. Am. Chem. Soc., 1974, 96, 860–869 CrossRef CAS.
- S. Karthikeyan, M. P. Pachamuthu, M. A. Isaacs, S. Kumar, A. F. Lee and G. Sekaran, Appl. Catal., B, 2016, 199, 323–330 CrossRef CAS.
- X. Li, L. Ai and J. Jiang, Chem. Eng. J., 2016, 288, 789–797 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04312k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc,
Liping Yangab,
Keke Zhiab,
Lulu Wangab,
Letao Zhanga and
Xinfeng Guoa
*abc,
Liping Yangab,
Keke Zhiab,
Lulu Wangab,
Letao Zhanga and
Xinfeng Guoa