
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glibenclamide–sulfonylurea receptor 1 antagonist alleviates LPS-induced BV2 cell activation through the p38/MAPK pathway†
Zhiming Xu† a,
Yingliang Liu†a,
Dianxu Yanga,
Fang Yuana,
Jun Dinga,
Liping Wangb,
Meijie Qub,
Guoyuan Yangbc and
Hengli Tian*a
a,
Yingliang Liu†a,
Dianxu Yanga,
Fang Yuana,
Jun Dinga,
Liping Wangb,
Meijie Qub,
Guoyuan Yangbc and
Hengli Tian*a
aDepartment of Neurosurgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China. E-mail: tianhlsh@126.com
bDepartment of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
cNeuroscience and Neuroengineering Research Center, Med-X Research, Shanghai Jiao Tong University, Shanghai, China
First published on 24th May 2017
Abstract
We investigated the anti-neuroinflammatory activity and mechanism of glibenclamide, sulfonylurea receptor 1 (Sur1) antagonist, against LPS-induced microglial activation in vitro. BV2 microglia cells were exposed to LPS (100 ng mL−1). iNOS and COX-2 levels, proinflammatory cytokine mRNA expression, and the p38/MAPK signaling pathway were analyzed by RT-PCR and Western blotting. Pretreatment with glibenclamide (2.5, 10, and 40 μM) inhibited the LPS-induced overexpression of iNOS and COX-2 in BV2 microglia cells. Blocking Sur1 reduced intracellular reactive oxygen species (ROS) levels. Glibenclamide dose-dependently (2.5, 10 μM) decreased LPS-induced over-expression of TNF-α, IL-6, and IL-1β, and alleviated the intracellular calcium accumulation in LPS-treated BV2 microglia cells. Moreover, glibenclamide diminished the LPS-induced phosphorylation of p38/MAPK, SB203580, a selective p38/MAPK inhibitor, significantly potentiated glibenclamide-caused inhibition of the expression of iNOS and COX-2 in LPS-exposed BV2 cells. Glibenclamide–Sur1 antagonist exerts anti-inflammatory activity in murine microglia in vitro by inhibiting the p38/MAPK signaling pathways and proinflammatory responses. Glibenclamide may be developed as a novel agent for suppressing inflammatory responses in the central nervous system.
Introduction
Sulfonylurea receptor 1 (Sur1), a member of the ATP-binding cassette (ABC) superfamily, is best known for its role in KATP channels.1,2 Two nucleotide-binding folds contained in Sur1 provide its high affinity for therapeutic sulfonylurea drugs. More recently, Sur1 has also been found to have a role in the formation of Sur1-regulated NCCa-ATP channels.3 Sur1-regulated channel activity can be strongly inhibited by drugs, such as glibenclamide (glyburide) and repaglinide. Previous studies have shown that this receptor is involved in cerebral ischemia,3 subarachnoid hemorrhage (SAH),4 spinal cord injury,5 traumatic brain injury (TBI),6 multiple sclerosis (MS),7 intestinal ischemia and reperfusion injury,8 and many pathophysiological processes. Previous studies have suggested that Sur1 was associated with cerebral edema, including central nervous system (CNS) impairment induced cerebral edema and cerebral edema caused by hepatic encephalopathy9 and cerebral edema caused by hepatic encephalopathy.10 It has also been shown that Sur1 played a role in blood–brain tumor barrier permeability, as permeability of the barrier decreased significantly after blocking the receptor.11Increasing numbers of studies have found that the CNS inflammation has a key role in ischemic brain injury, TBI, Alzheimer's disease, and Parkinson's disease (PD).12 Microglial activation is an important part of inflammation, which can impair neuronal survival and blood–brain barrier (BBB) integrity by generating oxidative stress and cytokines.13 Thus, the activation of microglia has important theoretical and therapeutic significance. To date, although studies have suggested that the Sur1 is related to the inflammatory response after stroke and that the receptor blocker glibenclamide can alleviate inflammation in brain tissue after ischemia, there is no reported study regarding the role of Sur1, if any, in the activation of microglia.14 Thus, there is an important need to clarify the role of Sur1 in microglial activation and to examine the biological effects of Sur1 inhibitors on microglia.
Glibenclamide, known as glyburide in the US, is a widely prescribed drug that has been used for the treatment of diabetes mellitus type 2 (DM II) since the 1960s.15 Glibenclamide binds to sulfonylurea receptors and stimulates the closure of adenosine triphosphate (ATP)-sensitive potassium channels, which, in turn, encourages the secretion of insulin from pancreatic β-cells. As a member of the sulfonylurea class of drugs, glibenclamide inhibits sulfonylurea receptor 1 (Sur1) and KATP (Sur1-Kir6.2) channels on β cells in the pancreatic islet. This leads to enhanced insulin release, which is beneficial for patients with DM II.16–19 Laboratory investigations have also shown that subjects with various CNS pathologies, including focal cerebral ischemia,3 neonatal encephalopathy due to prematurity,20 and metastatic brain tumors,21 benefit from the administration of glibenclamide.
In this study, we used microglial BV2 cells as a cellular model to determine the effects of glibenclamide on microglial activation. Our study suggests that glibenclamide can profoundly affect the activation levels of the cells.
Experimental section
Reagents
Glibenclamide (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in water containing 25% Cremophor EL (Sigma-Aldrich) and 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich) diluted in culture medium before use (the concentration of DMSO was < 0.001% after dilution). LPS was purchased from Sigma-Aldrich. SB203580 (a p38/MAPK pathway-specific inhibitor) was purchased from Cell Signaling Technology (Danvers, MA, USA).Lactate dehydrogenase (LDH) assay
BV2 cells were lysed for 20 min in lysing buffer (0.04% Triton-X, 2 mM HEPES, 0.2 mM dithiothreitol, 0.01% bovine serum albumin, and 0.1% phenol red, pH 7.5). Cell lysates (50 μL) were mixed with 150 μL 500 mM potassium phosphate buffer (pH 7.5) containing 1.5 mM NADH and 7.5 mM sodium pyruvate, and the A340-nm change was monitored over 90 s with a spectrophotometer (BioTek, Winooski, VT, USA).Cell culture
The BV2 microglia cell line was obtained from the Institute of Neurology, Ruijin Hospital (Shanghai, China) and maintained in Dulbecco's Modified Eagle's Medium (DMEM; Gibco Laboratories; Grand Island, NY, USA), supplemented with 5% fetal bovine serum (FBS), 100 μg mL−1 streptomycin, and 100 μg mL−1 penicillin. Cultures were then transferred to an anaerobic chamber and infused with a gas mixture containing 5% CO2 and 95% N2.Pharmacological treatment
At 24 h after seeding, the cells were exposed to LPS (100 ng mL−1). The Sur1 antagonist glibenclamide (2.5, 10, 40 μM) and SB203580 (10 μM) were added to the medium 1 h before exposure to LPS until the end of the experiments.Measurement of reactive oxygen species (ROS) levels
Intracellular reactive oxygen species (ROS) levels were monitored using a fluorescent probe with dihydroethidium (DHE; Beyotime, China). Ethidium can be combined with RNA or DNA to produce a red fluorescence. Under the condition of high level of intracellular superoxide anion, ethidium would exhibit strong red fluorescence. BV2 cells treated in 96-well plates were incubated with fresh working solution containing 5 mM DHE in PBS for 30 min in an anaerobic chamber. Then, the cells were washed twice with ice-cold PBS, and subsequently visualized using a Leica (Germany) inverted fluorescence microscope. The red fluorescence intensities were quantitated by ELISA plate reader (BioTek, Winooski, VT, USA).Real-time PCR
Total RNA samples from BV2 cells were extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed to cDNA using a PrimeScript RT reagent kit (TaKaRa). The cDNA was amplified by PCR using the specific primers in Table 1. Quantitative real-time PCR was performed with an ABI 7900HT by using SYBR Premix Ex Taq (TaKaRa). Data were analyzed by using the comparative threshold cycle (Ct) method, results are expressed as fold differences, normalized to GAPDH.| Gene target | Primer sequence |
|---|---|
| iNOS | F: CCTTGGTGAAGGGACTGAGC |
| R: CAACGTTCTCCGTTCTCTTGC | |
| IL-1β | F: GCAACTGTTCCTGAACTCAACT |
| R: ATCTTTTGGGGCGTCAACT | |
| IL-6 | F: TCTATACCACTTCACAAGTCGGA |
| R: GAATTGCCATTGCACAACTCTTT | |
| TNF-α | F: GGAACACGTCGTGGGATAATG |
| R: GGCAGACTTTGGATGCTTCTT | |
| GAPDH | F: AGGTCGGTGTGAACGGATTTG |
| R: TGTAGACCATGTAGTTGAGGTCA |
Determination of intracellular free calcium ion concentrations
To determine the intracellular free Ca2+ concentration levels in BV2 cells, Fluo-3 AM (1–5 mmol L−1) was used. BV2 cells were pre-incubated with glibenclamide (2.5, 10, and 40 μM) for 1 h, then stimulated with 100 ng mL−1 LPS for a 24 h incubation period. Cells were washed with HBSS three times and then loaded with Fluo-3 AM for 10–60 min. Fluo-3 AM immunofluorescent intensity was quantified by ELISA plate reader (BioTek, Winooski, VT, USA).Western blot analysis
For Western blot analyses, samples were lysed in radioimmunoprecipitation assay (RIPA; Millipore, Bedford, MA, USA) supplemented with 1 mmol L−1 PMSF (Thermo; Waltham, MA, USA), a protease inhibitor cocktail (Thermo), and a phosphatase inhibitor (Thermo). After denaturing, the same amount of proteins were loaded onto the resolving gel (Promoton; Shanghai, China) for electrophoresis and the proteins were transferred onto a nitrocellulose membrane (Whatman; Piscataway, NJ, USA). After blocking with 5% non-fat milk, the membranes were incubated with primary antibodies overnight at 4 °C at the following dilutions: p-ERK1/2, ERK1/2, p-JNK, JNK, p-p38, p38, and COX-2 (1:1000; Cell Signaling Technology), iNOS (1:10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; Proteintech Group, Rosemont, IL, USA); Sur1 (1:1000; Abcam, Cambridge, MA, USA); and β-tubulin, and β-actin (1:1000, Santa Cruz Technology; Santa Cruz, CA, USA). After washing, the membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature and then reacted with an enhanced chemiluminescence substrate (Pierce; Rockford, IL, USA). The results were recorded using the Quantity One imaging software (Bio-Rad; Hercules, CA, USA), and relative intensities were calculated with the Gel-Pro Analyzer software (Media Cybernetics; Bethesda, MD, USA).
000; Proteintech Group, Rosemont, IL, USA); Sur1 (1:1000; Abcam, Cambridge, MA, USA); and β-tubulin, and β-actin (1:1000, Santa Cruz Technology; Santa Cruz, CA, USA). After washing, the membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature and then reacted with an enhanced chemiluminescence substrate (Pierce; Rockford, IL, USA). The results were recorded using the Quantity One imaging software (Bio-Rad; Hercules, CA, USA), and relative intensities were calculated with the Gel-Pro Analyzer software (Media Cybernetics; Bethesda, MD, USA).
Statistical analysis
All results were presented as mean ± SD. Student's t test was used for comparison between the two groups. P < 0.05 was considered statistically significant. All data analyses were performed with SPSS 20.0 (SPSS Inc., Chicago, IL), GraphPad Prism 6 software (GraphPad Software; San Diego, CA) was used to create graphic representations of the data.Results and discussion
To study the role of Sur1 in the development of cellular activation, we analyzed the expression of Sur1 protein in LPS-induced BV2 microglial cells. Protein samples were collected at 24 h after after LPS treatment, expression of Sur1 significantly increased compared with control group (Fig. 1B and C). In order to confirm whether glibenclamide could acted as a neuroinflammatory modulator, we used the BV2 microglial cell line as a model in vitro. First, we assessed its cytotoxicity to BV2 cells after a 24 h incubation with three different concentrations of glibenclamide. As shown in Fig. 1, the cell viability following treatment with 2.5, 10, and 40 μM of glibenclamide did not significantly differ from that of the control. Proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, play central roles in microglia-mediated inflammation. Thus, the effects of glibenclamide on these proinflammatory cytokines were analyzed. BV2 cells were incubated with glibenclamide (2.5, 10, and 40 μM) from 1 h before exposure to LPS (100 ng mL−1) until the end of the experiment. RT-PCR results demonstrated that the mRNA levels of these cytokines were elevated 6 h after LPS treatment. Pre-treatment with glibenclamide significantly inhibited LPS-induced iNOS, TNF-α, IL-1β, and IL-6 production versus the LPS-treated cells (Fig. 2). Statistical analysis showed that LPS-stimulated proinflammatory cytokine mRNA levels (iNOS, TNF-α, IL-1β, and IL-6) decreased significantly following glibenclamide treatment (Fig. 2A–D; P < 0.05 and P < 0.01, vs. the LPS group). Thus, these results indicated that glibenclamide inhibited the expression of cytokines involved in the inflammatory process. At the concentrations indicated (2.5, 10, and 40 μM), glibenclamide significantly attenuated LPS-induced proinflammatory cytokines (TNF-α, IL-1β, and IL-6) levels compared with LPS-treated cells (Fig. 2). Furthermore, we detected protein levels of iNOS and COX-2 by Western blotting. iNOS and COX-2 protein levels increased significantly in LPS-exposed BV2 cells compared with the controls (Fig. 3A and B, P < 0.01, vs. the LPS group). Pre-treatment with glibenclamide at various concentrations (2.5, 10, and 40 μM) reduced the LPS-stimulated increase in iNOS and COX-2 levels significantly (Fig. 3A and B; P < 0.01, vs. the LPS group). Western blot analysis showed that glibenclamide at 2.5 and 10 μM inhibited iNOS and COX-2 levels in a dose-dependent manner, whereas 40 μM glibenclamide did not show a stronger inhibitory effect (P < 0.01, vs. the control group).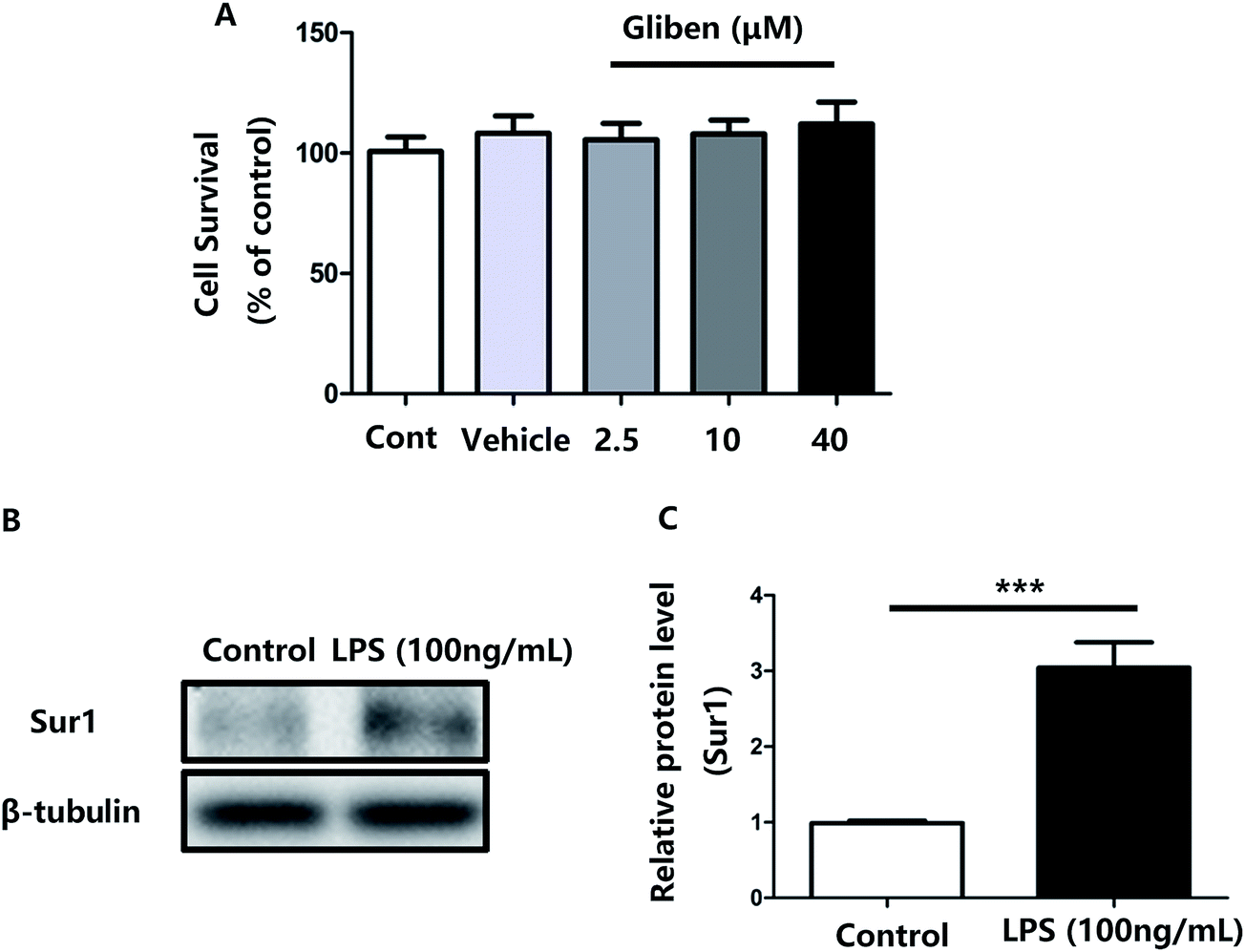 | ||
| Fig. 1 Effect of glibenclamide on the cell viability of BV2 microglia, and Sur1 protein expression in LPS-exposed BV2 cells. Cells were treated with indicated concentrations of glibenclamide for a 24 h incubation period. Cell viability was assessed by LDH assay (A). BV2 cells were exposed to LPS (100 ng mL−1), and Sur1 protein expression was measured 24 h later (B). A significant increase in Sur1 protein expression was observed following treatment with LPS (100 ng mL−1) versus the control condition (C). Data were subjected to two-sample Student's t-test. ***P < 0.001, vs. control (Cont). Error bars, mean ± SE. | ||
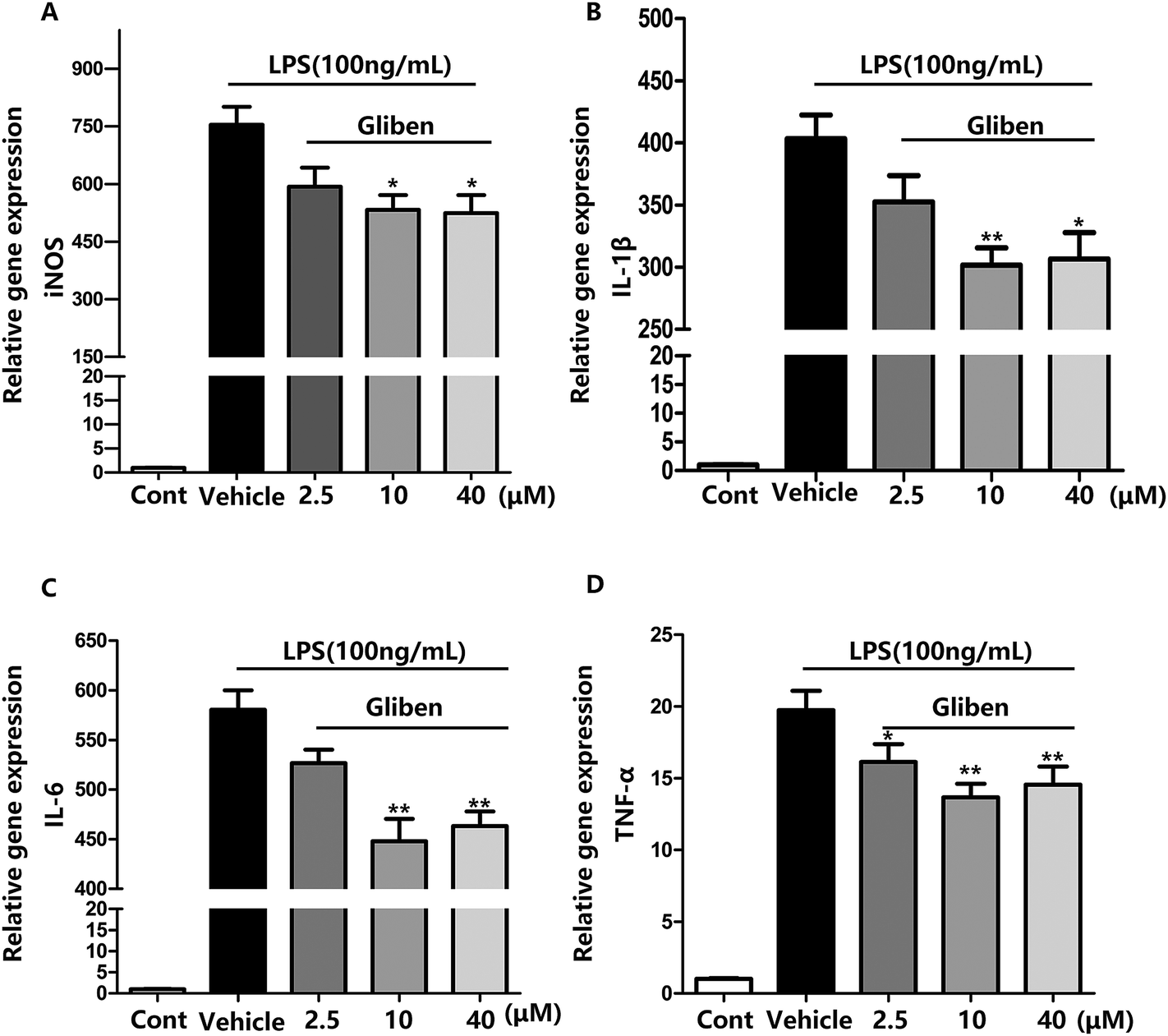 | ||
| Fig. 2 Blocking Sur1 suppressed expression of iNOS, TNF-α, IL-1β, and IL-6 in BV2 cells. BV2 microglial cells were incubated with glibenclamide (Gliben, 2.5, 10, 40 μM) or vehicle for 1 h and were then stimulated with LPS (100 ng mL−1). After 6 h, mRNA levels of iNOS (A), IL-1β (B), IL-6 (C), and TNF-α (D) were assessed by real-time PCR. Values represent the mean ± SEM from at least three independent experiments. Control was abbreviated as Cont. Cont *P < 0.05, vs. LPS alone, **P < 0.01, vs. LPS alone. | ||
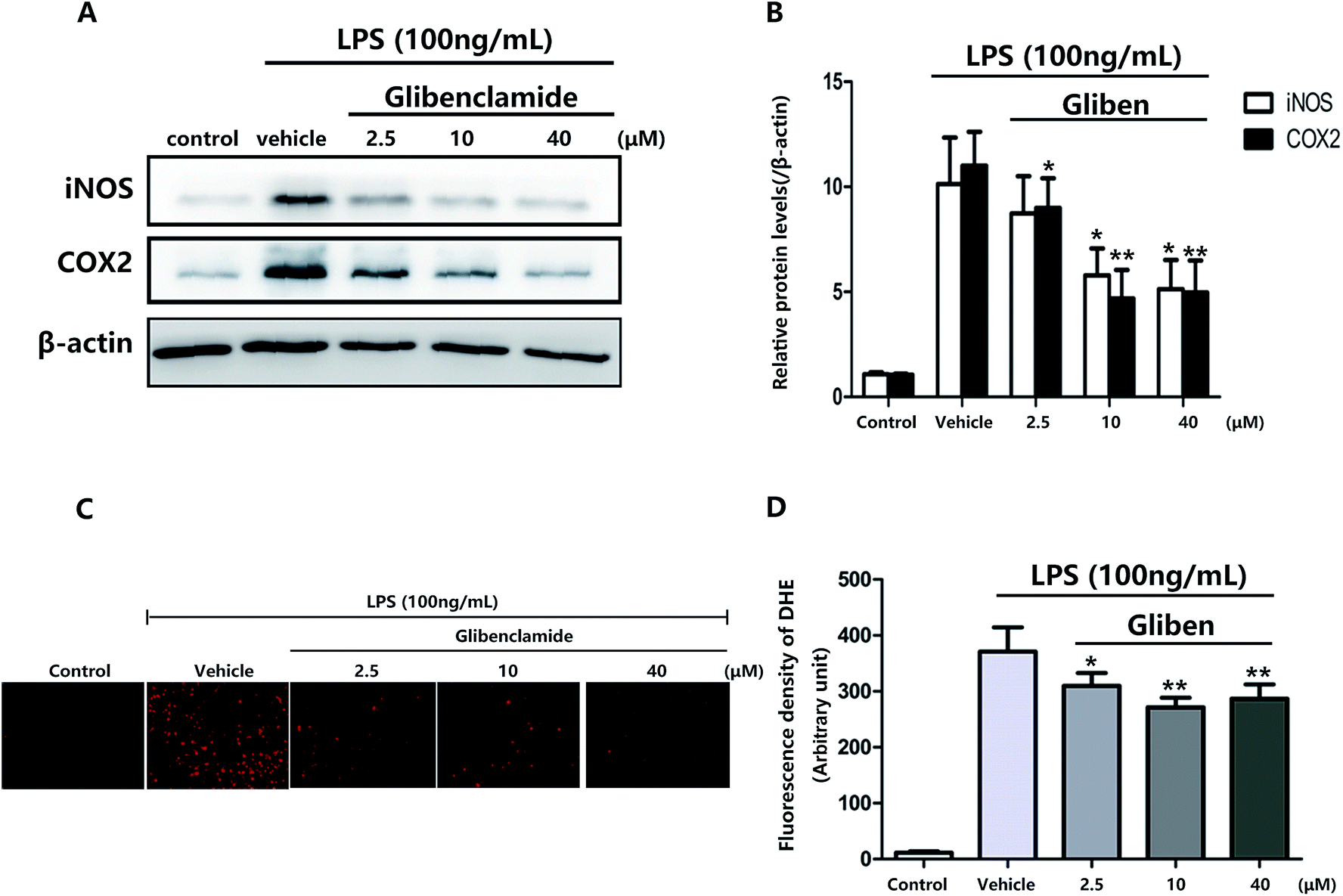 | ||
| Fig. 3 The oxidative stress-induced inflammation in LPS-exposed BV2 cells were reduced by blocking Sur1. Levels of the inflammatory proteins iNOS and COX-2 (A) in LPS-exposed BV2 cells, pretreated with glibenclamide (2.5, 10, and 40 μM) and vehicle, were determined by Western blot analysis (B). β-Actin was used as a control. BV2 cells were pretreated with glibenclamide (Gliben, 2.5, 10, and 40 μM) or vehicle and exposed to 100 mg mL−1 LPS for 24 h. Intracellular ROS production was (C) visualized with the fluorescent dye, DHE was detected by fluorescence microscopy, and fluorescence intensity was quantified with a spectrophotometer (D). *P < 0.05, vs. cells treated with only LPS alone. | ||
Increased oxidative stress induces excessive ROS that can directly inflict DNA damage and activate cellular signaling pathways.22 Pre-treatment with glibenclamide (2.5, 10, and 40 μM) restrained ROS levels induced by LPS in BV2 cells. Dihydroethidium (DHE) was used to detect intracellular superoxide anion levels. Glibenglamide at 2.5 and 10 μM of glibenclamide decreased ROS levels in a dose-dependent manner (Fig. 3C and D); 10 and 40 μM glibenclamide had significantly different effects on levels of ROS.
Pretreatment with 100 ng mL−1 LPS elevated Ca2+ levels in BV cells .All three concentrations of glibenclamide reduced intracellular Ca2+ concentrations, although 40 μM glibenclamide was not more effective than 10 μM (Fig. 4C).
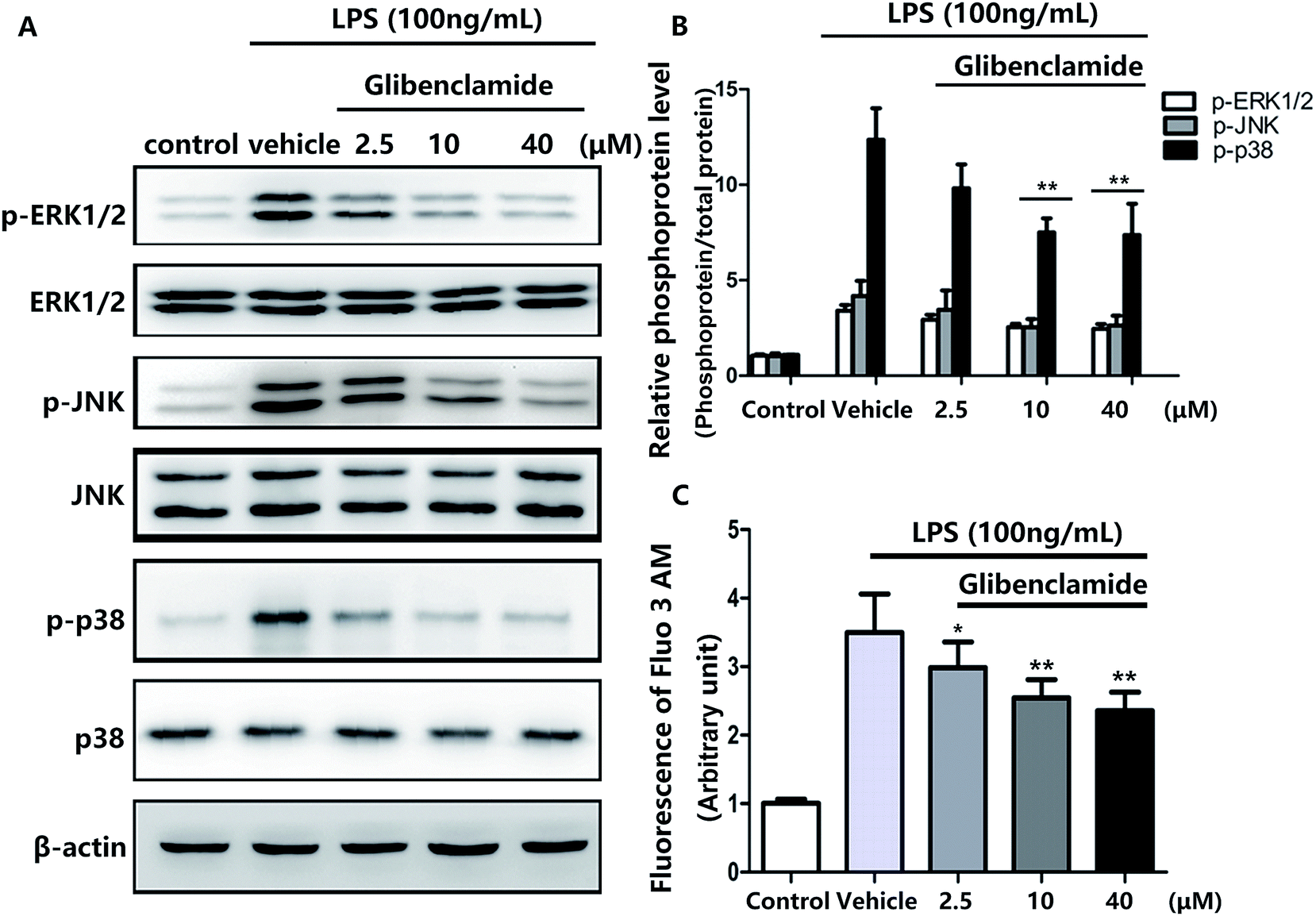 | ||
| Fig. 4 Glibenclamide attenuated LPS-induced increased concentrations of Ca2+ and phosphorylation of the p38/MAPK pathway in BV2 cells. Phosphorylation of ERK, JNK, and p38 was analyzed (A) and quantified by Western blot analysis (B). Fluo-3 AM levels were quantified with an ELISA plate reader (C). **P < 0.01, vs. with LPS alone. | ||
p38/MAPK pathway components, including ERK1/2, JNK, and p38, are phosphorylated in various stress responses.23 The phosphorylation of p38/MAPK induces the expression of proinflammatory molecules and is one of the most important signaling pathways in the inflammation response.24 Consequently, we used Western blotting to assess the effects of glibenclamide on stress-mediated cellular p38/MAPK signaling pathways after incubating BV2 cells with LPS for 24 h. Glibenclamide at different concentrations (2.5, 10, and 40 μM) suppressed LPS-induced phosphorylation of ERK1/2, p38, and JNK (Fig. 4). This finding suggested that glibenclamide was capable of modulating the p38/MAPK pathway which was activated by LPS treatment and decreasing the production of proinflammatory mediators.
We showed that LPS exposure increased p38/MAPK phosphorylation, and this induction was decreased after glibenclamide treatment (Fig. 3C and D). To further explore the mechanism(s) of glibenclamide in neuroinflammation, we analyzed iNOS and COX-2 levels. To determine whether glibenclamide-induced downregulation of iNOS and COX-2 was p38/MAPK-dependent, we analyzed the effect of glibenclamide at the cellular level, using LPS-induced BV2 cell activation to mimic in vivo neuroinflammation. In previous tests, we showed that different concentrations of glibenclamide (2.5, 10, and 40 μM) could reduce LPS-induced iNOS and COX-2 overexpression in BV2 cells; 10 μM glibenclamide showed the maximum inhibitory effect (40 μM did not show a greater effect).
MAPKs, including p38, JNK, and ERK1/2, which could be activated by LPS, are an important kinase family involved in various cellular processes, such as apoptosis, differentiation, stress responses and immune defense.25 This kinase family modulates cytokine production and expression of pro-inflammatory enzymes, such as NF-κB, iNOS, COX-2 and IL-1β.26,27 Therefore, MAPKs are crucial elements in the inflammatory process and essential targets for anti-inflammatory molecules. We found that pretreatment with glibenclamide reduced upregulation of phospho-p38, phospho-ERK1/2 and phospho-JNK in activated BV2 cells. Thus, MAPKs may involve in the protective effects of glibenclamide in activated BV2 cells. Previous studies reported that attenuating phosphorylation of p38 MAPK relieved the level of microglia activation,28 phospho-ERK and phospho-JNK is predominantly localized to astrocytes or neurons, and minimally localized to microglia cells in the spinal cord injury, however.29,30
To assess whether the p38/MAPK signaling pathway was involved, a selective p38/MAPK inhibitor, SB203580, was used. The effects of glibenclamide on the inhibition of iNOS and COX-2 expression were consistent with the effect of a specific p38/MAPK inhibitor SB203580 when examined simultaneously in LPS-stimulated BV2 cells (Fig. 5, P < 0.01, vs. the LPS group). Furthermore, the effect of glibenclamide on the inhibition of iNOS and COX-2 was consistent with the effect SB203580 when examined simultaneously in LPS-stimulated BV2 cells. Experiments were also carried out and indicated that the SB203580 alone decreased LPS-induced iNOS and COX-2 overepression on BV2 cells (Fig. 5, P < 0.01, P < 0.01, vs. the LPS group). Thus, we concluded that glibenclamide diminished neuroinflammation via the p38/MAPK signaling pathway.
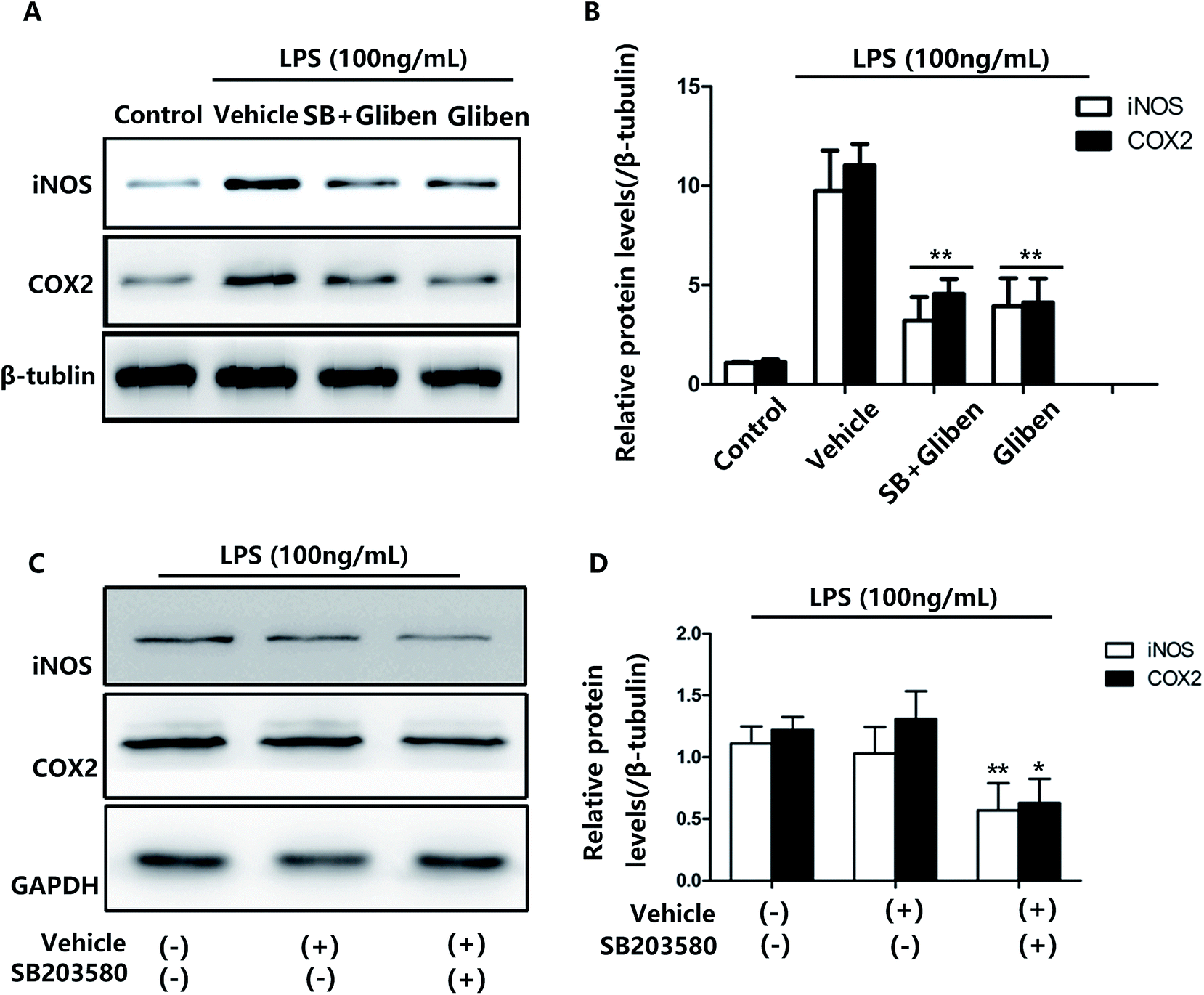 | ||
| Fig. 5 Comparison of the effects of glibenclamide and the p38/MAPK signal pathway inhibitor (SB203580) on LPS-induced inflammatory proteins in BV2 cells. Cells were pre-treated with SB203580 (SB, 10 μM) and glibenclamide (Gliben, 10 μM) for 1 h before LPS (100 ng mL−1) treatment. Level of iNOS and COX-2 proteins were determined by Western blotting (A). Quantification data are shown in (B). Cells were pre-treated with SB203580 (10 μM) alone for 1 h before LPS (100 ng mL−1) exposure. Level of iNOS and COX-2 proteins were determined by Western blotting (C). Quantification data are shown in (D). Data represent the mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01, vs. the LPS-alone group. | ||
In the present study, we demonstrated that glibenclamide, ATP-sensitive potassium channel (KATP) blocker, and a first-line sulfonylurea pharmacotherapy option for the treatment of DM II,31 has a significant inhibitory effect against inflammation, blocking the synthesis of a proinflammatory intermediary in LPS-stimulated BV2 microglia cells. A growing body of evidence suggests that neuroinflammation plays a pivotal role in numerous processes, such as TBI, ischemia, MS, SAH.32 Microglial activation is a key process in the propagation of inflammation to neighboring tissue, which may lead to neuronal impairment in the surrounding area as well as BBB damage as a result of the generation of cytokines and ROS.13,33,34 Activated BV2 microglia cells, induced by lipopolysaccharide (LPS) in an in vitro environment, provide a good model to study neuroinflammation.35 Neurotoxic molecules, such as iNOS, and COX-2, and cytokines, such as TNF-α, IL-1β, and IL-6 are synthesized and liberated during microglial activation.36,37 Because of the protective role of glibenclamide in CNS pathophysiological, glibenclamide was investigated further for its effects on iNOS and COX-2 expression and its effect on the release of proinflammatory cytokines. We observed that glibenclamide reduced the mRNA levels of TNF-α, IL-1β, and IL-6, Three concentrations of glibenclamide were tested, higher concentration (40 μM) of glibenclamide did not function better. Glibenclamide was also observed to dose-dependently reduce mRNA levels of proinflammatory cytokines and protein levels of iNOS and COX-2 at concentrations of 2.5 and 10 μM. Moreover, the three concentrations (2.5, 10, and 40 μM) of glibenclamide tested reduced LPS (100 ng mL−1)-stimulated microglial cell phosphorylation in the p38/MAPK pathway and the intracellular free calcium ion concentration. Our results are consistent with a report on LPS-induced morphological changes in microglial cells.38
It has been reported that blocking Sur1 by glibenclamide not only treats DM II but also reduces cerebral edema caused by hepatic encephalopathy and alleviates intestinal ischemia-reperfusion injury.8,18 Recently, studies have indicated that Sur1 mediates cell death in a variety of animal models of diseases, including TBI, SAH, and MS.7,39,40 Studies has also indicate that Sur1 plays a pivotal role in neuroinflammation induced by CNS impairment and that glibenclamide can attenuate injury due to an inflammatory reaction.4 Sur1 inhibition has also been shown to decrease secondary brain injury in animal models of TBI.41 The intracellular calcium concentration influences multiple cellular functions, including enzyme activities; microglia respond to CNS damage by upregulating functions that involve Ca2+ signaling.42 Consistent with previous studies,43 the present study demonstrated that LPS exposure significantly induced dissociative calcium ions in microglial cells, whereas glibenclamide could abolish LPS-induced increases in free calcium and mRNA levels of proinflammatory cytokines, indicating that glibenclamide's effects on free calcium ions resulting from microglia activation may contribute to its anti-inflammatory actions. It has also been noted that de novo expression of Sur1-regulated NCCa-ATP channels in endothelial cells is important in the temporal and spatial evolution of progressive hemorrhagic necrosis after spinal cord injury.5 However, no direct correlation between changes in intracellular Ca2+ concentrations and the anti-inflammatory actions of glibenclamide have yet been documented.
The p38/MAPK signaling pathway has been implicated in the development of many neurodegenerative diseases, such as AD, PD and amyotrophic lateral sclerosis (ALS).44,45 It has been shown that the production of inflammatory molecules depends partly on the hyperphosphorylation of p38/MAPK pathway components.46 Moreover, several studies have shown that the p38/MAPK pathway is an important target for therapeutic drugs intended to control inflammation in the CNS.47,48 Recent evidence also suggests that the p38/MAPK pathway regulates microglial responsiveness in TBI.49
We found expression of Sur1 along with the BV2 cell activation; glibenclamide significantly attenuated the LPS-induced intracellular Ca2+ concentration in a dose-dependent manner (2.5, 10 μM). Previous research showed Ca2+-dependent activation of p38/MAPK pathway in several cell types,50,51 including immune cells.52–54 The recently characterized Sur1-mediated Ca2+ aggregation in LPS-induced BV2 cells can be blocked by glibenclamide. Thus, calcium overloading leading to p38/MAPK signaling pathway phosphorylation could likely be reduced.
Conclusions
In conclusion, the results of this study suggest that pre-treatment with glibenclamide alleviated the effects of LPS-induced iNOS, and COX-2 protein and cytokine mRNA levels in the progression of microglia BV2 cell activation. Glibenclamide may exhibit anti-inflammatory effects by suppressing LPS-induced production of pro-inflammatory mediators through inhibition of p38 MAPK pathway in BV2 microglial cells.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81471245, 81671207, 81501048) and the Shanghai Jiaotong University Medicine-engineering Research Fund (YG2016QN20).References
- S. Seino, Annu. Rev. Physiol., 1999, 61, 337–362 CrossRef CAS PubMed.
- J. Bryan, A. Munoz, X. Zhang, M. Dufer, G. Drews, P. Krippeit-Drews and L. Aguilar-Bryan, Eur. J. Appl. Physiol., 2007, 453, 703–718 CrossRef CAS PubMed.
- J. M. Simard, M. Chen, K. V. Tarasov, S. Bhatta, S. Ivanova, L. Melnitchenko, N. Tsymbalyuk, G. A. West and V. Gerzanich, Nat. Med., 2006, 12, 433–440 CrossRef CAS PubMed.
- J. M. Simard, Z. Geng, S. K. Woo, S. Ivanova, C. Tosun, L. Melnichenko and V. Gerzanich, J. Cereb. Blood Flow Metab., 2009, 29, 317–330 CrossRef CAS PubMed.
- J. M. Simard, O. Tsymbalyuk, A. Ivanov, S. Ivanova, S. Bhatta, Z. Geng, S. K. Woo and V. Gerzanich, J. Clin. Invest., 2007, 117, 2105–2113 CrossRef CAS PubMed.
- A. D. Patel, V. Gerzanich, Z. Geng and J. M. Simard, J. Neuropathol. Exp. Neurol., 2010, 69, 1177–1190 CrossRef CAS PubMed.
- B. Schattling, K. Steinbach, E. Thies, M. Kruse, A. Menigoz, F. Ufer, V. Flockerzi, W. Bruck, O. Pongs, R. Vennekens, M. Kneussel, M. Freichel, D. Merkler and M. A. Friese, Nat. Med., 2012, 18, 1805–1811 CrossRef CAS PubMed.
- K. Pompermayer, F. A. Amaral, C. T. Fagundes, A. T. Vieira, F. Q. Cunha, M. M. Teixeira and D. G. Souza, Eur. J. Pharmacol., 2007, 556, 215–222 CrossRef CAS PubMed.
- K. Zweckberger, C. Eros, R. Zimmermann, S. W. Kim, D. Engel and N. Plesnila, J. Neurotrauma, 2006, 23, 1083–1093 CrossRef PubMed.
- A. R. Jayakumar, V. Valdes, X. Y. Tong, N. Shamaladevi, W. Gonzalez and M. D. Norenberg, Transl. Stroke Res., 2014, 5, 28–37 CrossRef CAS PubMed.
- N. S. Ningaraj, M. K. Rao and K. L. Black, Cancer Res., 2003, 63, 8899–8911 CAS.
- W. Xia, J. Han, G. Huang and W. Ying, Clin. Exp. Pharmacol. Physiol., 2010, 37, 253–258 CrossRef CAS PubMed.
- Y. Liu, G. Tang, Y. Li, Y. Wang, X. Chen, X. Gu, Z. Zhang, Y. Wang and G. Y. Yang, J. Neuroinflammation, 2014, 11, 177 CrossRef PubMed.
- F. J. Ortega, J. Gimeno-Bayon, J. F. Espinosa-Parrilla, J. L. Carrasco, M. Batlle, M. Pugliese, N. Mahy and M. J. Rodriguez, Exp. Neurol., 2012, 235, 282–296 CrossRef CAS PubMed.
- A. Marble, Drugs, 1971, 1, 109–115 CrossRef CAS PubMed.
- F. M. Ashcroft, Horm. Metab. Res., 1996, 28, 456–463 CrossRef CAS PubMed.
- J. M. Feldman, Am. J. Med., 1985, 79, 102–108 CrossRef CAS PubMed.
- W. Kramer, G. Muller, F. Girbig, U. Gutjahr, S. Kowalewski, D. Hartz and H. D. Summ, Diabetes Res. Clin. Pract., 1995,(28 Suppl), S67–S80 CrossRef CAS.
- U. Panten, M. Schwanstecher and C. Schwanstecher, Exp. Clin. Endocrinol. Diabetes, 1996, 104, 1–9 CrossRef CAS PubMed.
- C. Tosun, M. T. Koltz, D. B. Kurland, H. Ijaz, M. Gurakar, G. Schwartzbauer, T. Coksaygan, S. Ivanova, V. Gerzanich and J. M. Simard, Brain Sci., 2013, 3, 215–238 CrossRef CAS PubMed.
- E. M. Thompson, G. L. Pishko, L. L. Muldoon and E. A. Neuwelt, Neoplasia, 2013, 15, 535–543 CrossRef CAS PubMed.
- H. Jaeschke, J. Gastroenterol. Hepatol., 2011, 1(26 Suppl), 173–179 CrossRef PubMed.
- T. Uto, N. Suangkaew, O. Morinaga, H. Kariyazono, S. Oiso and Y. Shoyama, Am. J. Chin. Med., 2010, 38, 985–994 CrossRef PubMed.
- J. Da Silva, B. Pierrat, J. L. Mary and W. Lesslauer, J. Biol. Chem., 1997, 272, 28373–28380 CrossRef CAS PubMed.
- G. Pearson, F. Robinson, T. Beers Gibson, B. E. Xu, M. Karandikar, K. Berman and M. H. Cobb, Endocr. Rev., 2001, 22, 153–183 CAS.
- K. M. Rao, J. Leukocyte Biol., 2001, 69, 3–10 CAS.
- N. Rajapakse, M. M. Kim, E. Mendis and S. K. Kim, Immunology, 2008, 123, 348–357 CrossRef CAS PubMed.
- Y. W. Gu, D. S. Su, J. Tian and X. R. Wang, Neurosci. Lett., 2008, 431, 129–134 CrossRef CAS PubMed.
- Z. Y. Zhuang, P. Gerner, C. J. Woolf and R. R. Ji, Pain, 2005, 114, 149–159 CrossRef PubMed.
- Z. Y. Zhuang, Y. R. Wen, D. R. Zhang, T. Borsello, C. Bonny, G. R. Strichartz, I. Decosterd and R. R. Ji, J. Neurosci., 2006, 26, 3551–3560 CrossRef CAS PubMed.
- M. C. Riddle, J. Clin. Endocrinol. Metab., 2003, 88, 528–530 CrossRef PubMed.
- A. Y. Lai and K. G. Todd, Can. J. Physiol. Pharmacol., 2006, 84, 49–59 CrossRef CAS PubMed.
- B. Liao, W. Zhao, D. R. Beers, J. S. Henkel and S. H. Appel, Exp. Neurol., 2012, 237, 147–152 CrossRef CAS PubMed.
- F. Yuan, Z. M. Xu, L. Y. Lu, H. Nie, J. Ding, W. H. Ying and H. L. Tian, J. Neurochem., 2016, 136, 581–593 CrossRef CAS PubMed.
- Y. Li, H. Nie, D. Wu, J. Zhang, X. Wei and W. Ying, Neurosci. Lett., 2013, 544, 36–40 CrossRef CAS PubMed.
- S. Lund, K. V. Christensen, M. Hedtjarn, A. L. Mortensen, H. Hagberg, J. Falsig, H. Hasseldam, A. Schrattenholz, P. Porzgen and M. Leist, J. Neuroimmunol., 2006, 180, 71–87 CrossRef CAS PubMed.
- V. H. Perry, J. A. Nicoll and C. Holmes, Nat. Rev. Neurol., 2010, 6, 193–201 CrossRef PubMed.
- A. Sakai, K. Takasu, M. Sawada and H. Suzuki, PLoS One, 2012, 7, e32268 CAS.
- J. M. Simard, M. Kilbourne, O. Tsymbalyuk, C. Tosun, J. Caridi, S. Ivanova, K. Keledjian, G. Bochicchio and V. Gerzanich, J. Neurotrauma, 2009, 26, 2257–2267 CrossRef PubMed.
- C. Tosun, D. B. Kurland, R. Mehta, R. J. Castellani, J. L. deJong, M. S. Kwon, S. K. Woo, V. Gerzanich and J. M. Simard, Stroke, 2013, 44, 3522–3528 CrossRef CAS PubMed.
- K. Zweckberger, K. Hackenberg, C. S. Jung, D. N. Hertle, K. L. Kiening, A. W. Unterberg and O. W. Sakowitz, Neuroscience, 2014, 272, 199–206 CrossRef CAS PubMed.
- K. Farber and H. Kettenmann, Glia, 2006, 54, 656–665 CrossRef PubMed.
- L. Yuan, S. Liu, X. Bai, Y. Gao, G. Liu, X. Wang, D. Liu, T. Li, A. Hao and Z. Wang, J. Neuroinflammation, 2016, 13, 77 CrossRef PubMed.
- M. Lyman, D. G. Lloyd, X. Ji, M. P. Vizcaychipi and D. Ma, Neurosci. Res., 2014, 79, 1–12 CrossRef CAS PubMed.
- E. K. Kim and E. J. Choi, Biochim. Biophys. Acta, 2010, 1802, 396–405 CrossRef CAS PubMed.
- M. R. Guimaraes, F. R. Leite, L. C. Spolidorio, K. L. Kirkwood and C. Rossa Jr, Arch. Oral Biol., 2013, 58, 1309–1317 CrossRef CAS PubMed.
- D. Gomez-Nicola, B. Valle-Argos and M. Nieto-Sampedro, Glia, 2010, 58, 264–276 CrossRef PubMed.
- S. Saccani, S. Pantano and G. Natoli, Nat. Immunol., 2002, 3, 69–75 CrossRef CAS PubMed.
- A. D. Bachstetter, R. K. Rowe, M. Kaneko, D. Goulding, J. Lifshitz and L. J. Van Eldik, J. Neurosci., 2013, 33, 6143–6153 CrossRef CAS PubMed.
- P. R. Blanquet, Neuroscience, 2000, 95, 705–719 CrossRef CAS PubMed.
- S. Dehez, L. Daulhac, A. Kowalski-Chauvel, D. Fourmy, L. Pradayrol and C. Seva, FEBS Lett., 2001, 496, 25–30 CrossRef CAS PubMed.
- S. Kreideweiss, C. Ahlers, A. Nordheim and A. Ruhlmann, Eur. J. Biochem., 1999, 265, 1075–1084 CrossRef CAS PubMed.
- D. J. Elzi, A. J. Bjornsen, T. MacKenzie, T. H. Wyman and C. C. Silliman, Am. J. Physiol.: Cell Physiol., 2001, 281, C350–C360 CAS.
- Y. Shigemoto-Mogami, S. Koizumi, M. Tsuda, K. Ohsawa, S. Kohsaka and K. Inoue, J. Neurochem., 2001, 78, 1339–1349 CrossRef CAS PubMed.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |