
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A peptide-decorated and curcumin-loaded mesoporous silica nanomedicine for effectively overcoming multidrug resistance in cancer cells†
Xian Sun‡
a,
Yingping Luo‡a,
Liwei Huanga,
Bo-Yang Yu *a and
Jiangwei Tian*ab
*a and
Jiangwei Tian*ab
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, Nanjing 211198, P. R. China. E-mail: boyangyu59@163.com; jwtian@cpu.edu.cn; Fax: +86 25 86185158; Tel: +86 25 86185158
bCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals, Shandong Normal University, 250014, P. R. China
First published on 14th March 2017
Abstract
Multidrug resistance (MDR) is a major obstacle to the success of cancer chemotherapy, which is closely associated with the overexpression of P-glycoprotein (P-gp) and the endolysosomal capture of drug. Therefore, to explore a strategy that could down-regulate P-gp expression and promote drugs escaping from the endolysosome is highly desired for overcoming MDR in chemotherapeutic treatment. This work reports a peptide-functionalized mesoporous silica nanomedicine (MSN) for encapsulation of curcumin (CUR) and doxorubicin (DOX) to significantly reverse the MDR in consequence of rapid endolysosomal escape and the inhibition of P-gp function. The nanomedicine is modified with the peptide of GFLGHHHRRGDS to encapsulate CUR and DOX with high loading efficiency and content. It can be selectively uptaken by the DOX resistant MCF-7/ADR cells via the specific recognition between RGDS and αvβ3 integrin to enter into lysosomes. Afterwards, the GFLG is cleaved by the lysosomal cathepsin B to release CUR and DOX from MSN. Furthermore, the imidazole-rich HHHR peptide can induce effective lysosomal membrane permeability which is beneficial for drug escape from lysosome, CUR-mediated inhibition of P-gp, and nuclear import of DOX in MCF-7/ADR cells. Confocal fluorescence imaging, flow cytometry and MTT assays demonstrate that the combination of CUR and the peptide-functionalized MSN markedly enhances the intracellular DOX concentration and achieves a successful chemotherapeutic treatment to MCF-7 or MCF-7/ADR cells, thus this work offers an effective strategy to overcome MDR.
Introduction
Chemotherapy is an indispensable method against cancer in the clinic,1,2 but a major impediment to successful treatment is the emergence of multidrug resistance (MDR), which is a state of resilience against structurally and mechanistically unrelated drugs.3,4 The most established mechanism of resistance is related to the overexpression of P-glycoprotein (P-gp), an energy-dependent efflux pump belongs to the adenosine triphosphate (ATP)-binding cassette superfamily of proteins and is often associated with resistance to a broad spectrum of anticancer drugs.5 Other factors have also been suggested to contribute to MDR and many of them are connected to the endolysosomal compartment.6–8 MDR cells have increased membrane area of endolysosomal system and thereby they comprise a great capacity for sequestering large amounts of chemotherapeutic drugs, hindering their ability to exert a cytotoxic effect on the target sites.9,10 Due to the extreme complexity of the biological processes involved in MDR of cancer cells, recent progresses in overcoming MDR were only partially effective. Therefore, to explore a novel strategy which could down-regulate P-gp expression and function and promote the drugs escaping from endolysosome is an important objective in the chemotherapeutic treatment against MDR.Curcumin (CUR), a phenolic compound purified from the rhizome of Curcuma longa, exhibits a wide range of therapeutic activities including anti-oxidant,11 antitoxic,12 anti-inflammatory13,14 and antitumor15,16 effects and has low side effects to human organs.17,18 In addition, CUR is an effective chemosensitizer to downregulate P-gp expression and function.19–23 Researches show that combination of chemotherapy drug and CUR promotes synergism against cancer cells and suppresses drug resistance.24–26 However, a simple blend of chemotherapy drug and CUR is an inadvisable formulation for their combined use because CUR has low aqueous solubility and poor stability. Moreover, it's difficult to deliver two different pharmacologically active agents to tumors simultaneously after systemic administration. Consequently, their combinatorial and synergistic effects for conquering MDR of cancer cells remains to be improved.
Owing to the high loading capacity and easy surface modification, mesoporous silica nanoparticle (MSN) constitutes a multifunctional platform for drug delivery.27–29 By decorating with tumor-targeting moieties such as aptamers,30 vitamins,31 proteins32 and peptides,33 MSN can achieve site-specific delivery. Meanwhile, in an attempt to increase the retention of drugs in tumors, much research has been focused on stimuli-responsive drug delivery systems for therapy of tumor.34–37 Moreover, cumulative evidence indicated that nanocarriers incorporating histidine peptides possess a particular function of facilitating the endolysosomal escape, which was attributed to the proton buffering capacity of imidazole groups.38–40 The protonated imidazole groups could trigger an osmotic swelling and destabilization of endosome/lysosome compartments.41 These unique properties motivate us to design a multifunctional MSN release system for dual delivery of CUR and chemotherapeutic drug.
Herein, a peptide-decorated envelope-type MSN-based nanomedicine (DOX/CUR@MSN-Pep, Fig. 1) which co-encapsulated CUR and doxorubicin (DOX) has been fabricated. The peptide with the sequence of GFLGHHHRRGDS was selected as the capping agents and modified on the surface of MSN. After the targeting Arg-Gly-Asp-Ser (RGDS) motif was bonded to the αvβ3-positive tumor cells,42–44 the nanomedicine entered into the lysosome via receptor-mediated endocytosis, and then the cargo molecules were released from the MSN due to the cleavage of Gly-Phe-Leu-Gly (GFLG) by lysosomal cathepsin B.45,46 Remarkably, lysosomal membrane permeabilization occurred in response to protonation effect of His-His-His-Arg (HHHR) sequence which included imidazole side chains, leading to the drug escape from lysosome, CUR-mediated inhibition of P-gp, and nuclear import of DOX. The nanomedicine had greatly improved the intracellular DOX accumulation in resistant MCF-7/ADR cells compared with that of single DOX implementation. Therefore, this “magic bullet” approach provides a useful strategy for effectively overcoming MDR.
 | ||
| Fig. 1 Schematic illustration of the structure and function of DOX/CUR@MSN-Pep nanomedicine for effective overcoming MDR. | ||
Experimental
Materials and reagents
Copper(II) sulfate pentahydrate (CuSO4·5H2O), sodium ascorbate (NaAsc), propargyl bromide and γ-(2-aminoethyl)-aminopropyl trimethoxysilane (AAPTMS) were purchased from J&K Scientific Co. Ltd. (Beijing, China). CUR, chlorpromazine, colchicine, indometacin was purchased from Aladdin Industrial Corporation (Shanghai, China). Doxorubicin hydrochloride and cathepsin B active human recombinant proteins (active form, 25 units) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MSN was purchased from Nan-jing XF Nano Materials Tech Company Ltd. (Nanjing, China). MTT Cell Proliferation and Cytotoxicity Assay Kit was obtained from KeyGen Biotech. Co. Ltd. (Nanjing, China). LysoTracker Deep Red and Hoechst 33342 were obtained from Invitrogen (Carlsbad, CA, USA). Azido-GFLGHHHRRGDS were synthesized by ChinaPeptides Co., Ltd. (Shanghai, China). All the chemical reagents used in this experiment were analytical grade and used without further purification. Ultrapure water was prepared using a Millipore Simplicity System (Millipore, Bedford, USA).Apparatus
Absorption spectra were recorded on an UV-2550 UV-VIS spectrophotometer (Shimadzu Company, Japan). Fluorescence spectra were measured on an F-7000 spectrofluorometer (Hitachi, Japan). The morphology of the DOX/CUR@MSN-Pep nanomedicine was characterized at a JEOL JEM-200CX transmission electron micro-scope (TEM) operated at 200 kV. The size of DOX/CUR@MSN-Pep was obtained by dynamic light scattering (DLS) at 25 °C by means of a 90 Plus/BI-MAS equipment (Brookhaven, USA). Zeta potential measurement was performed at 25 °C on a Zetasizer (Nano-Z, Malvern, UK). Nitrogen adsorption and desorption analysis of MSN was conducted on a BET (Micromeritics TriStar 3000). MTT assay was performed using microplate reader (Biotek, USA). Confocal fluorescence imaging of cells was performed on a confocal laser scanning microscope (CLSM, LSM700, Zeiss, Germany). Flow cytometric assay was performed using MACSQuant Analyzer 10 (Miltenyi Biotec, Germany).Synthesis of DOX/CUR@MSN-Pep nanomedicine
The synthesis route of MSN-Pep was shown in Scheme S1.† Amine modified MSNs were synthesized using the soft template method as previously described in the literature.47 In a typical synthesis, MSNs (300.0 mg) were suspended in 20 mL of methanol. After sonication, 2 mL of AAPTMS was added and the reaction proceeded for 24 h at room temperature to obtain MSN-NH2 nanoparticles. The nanoparticles were then centrifuged (8500 rpm, 10 min), washed three times with methanol and dried under vacuum.Alkyne modification of the as synthesized MSN-NH2 was carried out to enable further surface functionalization. Briefly, MSN-NH2 (150 mg) was dispersed in methanol (30 mL) and reaction with propargyl bromide (1.5 mL) at room temperature for 24 h. The MSN-alkyne nanoparticles were obtained by centrifugation (8500 rpm, 10 min), washed with methanol three times, and finally dried under high vacuum.
For preparation of DOX/CUR@MSN-Pep, MSN-alkyne nanoparticles (4.0 mg) and CUR (0.6 mg) were dispersed in 5 mL of methanol. After 4 h of stirring at room temperature, DOX (0.4 mg) was added to the mixture. The resulting mixture was then allowed to stir in dark at room temperature overnight. Upon completion, azido-GFLGHHHRRGDS (4.0 mg), CuSO4·5H2O (5 mg), and NaAsc (7.9 mg) were added. The mixture was kept stirring under nitrogen atmosphere for 3 d. The azido-GFLGHHHRRGDS was conjugated to the surface of DOX/CUR@MSN-alkyne via the click reaction between azido and alkyne functional groups. The nanoparticles were then centrifuged (8500 rpm, 10 min), washed thoroughly with methanol, and dried under vacuum. Single drug-loaded formulation and MSN-Pep (without drugs) were prepared by using a similar procedure. For comparison, DOX/CUR@MSN-Pep nanoparticles without RGDS or HHH sequences were prepared under the same conditions. The content of DOX or CUR in the materials was determined by measuring the absorbance at 488 nm or 425 nm respectively, using UV-VIS-NIR spectrophotometer. The drug loading content (LC) and loading efficiency (LE) were expressed according to the following formulas:


In vitro cathepsin B-induced drug release
3 mg of DOX/CUR@MSN-Pep nanomedicine was dispersed in 3 mL of citrate buffer (pH 7.4, and pH 5.0) with or without 20 U mL−1 cathepsin B and then moved into a dialysis bag (Mn = 30 kDa, Millipore) against 50 mL of different buffer solution containing 0.1% Tween 80. Subsequently, the dialysis bag incubated at 37 °C in a shaking platform at 100 rpm. At predetermined time intervals, 500 μL aliquot of each release medium was taken out and replaced with equivoluminal fresh medium. The withdrawn samples were assayed for DOX or CUR content by measuring absorbance at 488 nm and 425 nm, respectively. Three specimens were measured for each sample.Cell culture
The human breast adenocarcinoma cell line MCF-7 and the adriamycin resistant breast cell line MCF-7/ADR were obtained from KeyGEN Biotech Co. Ltd. (Nanjing, China). All cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 100 μg mL−1 streptomycin and 100 U mL−1 penicillin at 37 °C in a humidified incubator containing 5% CO2 and 95% air. The medium was replenished every other day and the cells were subcultured after reaching confluence.Cellular uptake
The cellular uptake of DOX was investigated by flow cytometric assay.48 MCF-7/ADR cells were seeded in six-well plates at a density of 1 × 105 per well and incubated in complete medium for 24 h at 37 °C. Then, MCF-7/ADR cells were randomly divided into four groups for the following treatments: group 1, incubation with free DOX; group 2, incubation with DOX + CUR; group 3, incubation with DOX@MSN-Pep; group 4, incubation with DOX/CUR@MSN-Pep. The concentration of different nanoparticles was at an equivalent DOX dosage of 10 μM. After 4 h incubation, the different treatment cells were trypsinized, harvested, rinsed with PBS and resuspended, and subjected to flow cytometric assay using MACSQuant Analyzer 10. All experiments detected at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells and the data were analyzed with FlowJo V10.
000 cells and the data were analyzed with FlowJo V10.
For microscopic observation, MCF-7 and MCF-7/ADR cells were seeded into 35 mm confocal dishes (Glass Bottom Dish) at a density of 1 × 104 per dish and incubated with free DOX, DOX + CUR, DOX@MSN-Pep or DOX/CUR@MSN-Pep (10.0 μM DOX equiv.). After 4 h incubation, the cells were stained with 1.0 μM Hoechst 33342 for 15 min, rinsed three times with PBS (pH 7.4) to perform fluorescence imaging with a CLSM.
For specificity assays, MCF-7/ADR cells were randomly divided into three groups for the following treatments: group 1, incubation with DOX/CUR@MSN-Pep (without RGDS); group 2, incubation with DOX/CUR@MSN-Pep; group 3, adding 2.5 mg mL−1 free RGD for 30 min and then incubation with DOX/CUR@MSN-Pep. The concentration of DOX/CUR@MSN-Pep was 10 μM DOX equiv. After 4 h incubation, the cells were washed. Confocal fluorescence imaging was performed to visualize the intracellular DOX fluorescence.
Endocytic pathways
MCF-7/ADR cells (1 × 105 cells per well) were seeded in six-well plates and incubated in complete medium for 24 h at 37 °C. Then, the MCF-7/ADR cells were incubated at either 4 or 37 °C. Prior to incubation with nanoparticles at 37 °C, the cells were pretreated with different inhibitors including chlorpromazine (clathrin-mediated uptake, 10.0 μg mL−1), colchicine (macropinocytosis, 8.0 μg mL−1), and indometacin (caveolae-mediated uptake, 6.0 μg mL−1) in serum-free DMEM for 1 h. Afterwards, DOX/CUR@MSN-Pep were further added to the medium for another 4 h incubation. The applied equivalent amount of DOX for all groups was 10 μM. Finally, the cells were washed 3 times with PBS, treated with trypsin, centrifuged, then dispersed in 0.5 mL of PBS to measure DOX by fluorescence spectrophotometer (the excitation wavelength set at 488 nm and the emission wavelength set at 575 nm).49,50 The fluorescence intensity of cells treated without any cellular uptake inhibitors served as the control. The cells treated with various inhibitors was compared with the control group to obtain the percentage of cellular uptake. All experiments were performed in triplicate.Endo-lysosomal escape activity
MCF-7/ADR cells were treated with free DOX, DOX/CUR@MSN-Pep (without HHH) and DOX/CUR@MSN-Pep at a DOX dose of 5 μM at 37 °C for 2, 6 and 12 h. The cells were further stained with 1.0 μM LysoTracker Deep Red and 1.0 μM Hoechst 33342 for 15 min. The intracellular distribution of fluorescence was visualized using CLSM.To further observe the fate of internalized nanoparticles, MCF-7/ADR cells were treated with DOX/CUR@MSN-Pep (5.0 μM DOX equiv.) for periods ranging from 1 h to 24 h and performed confocal fluorescence imaging to visualize intracellular behavior.
Evaluation of P-gp expression
MCF-7 and MCF-7/ADR cells were seeded into 6-well plates at a density of 1 × 105 cells per well with 2 mL culture medium and allowed to attach overnight. The medium was replaced with fresh growth medium containing CUR@MSN-Pep with the concentrations of 10 μM and cultured for 24 h at 37 °C for flow cytometry analysis.51 Then cells were trypsinized, collected and resuspended in PBS (pH 7.4). The cells treated with only culture medium were used as control. PE-conjugated mouse anti-human monoclonal antibody against P-gp was used to label cells according to the manufacturer's instruction, and the nonspecific labeling was corrected by its isotype control.In vitro cytotoxicity
The in vitro cytotoxicity of various formulations against MCF-7 and MCF-7/ADR cells was determined by MTT assay. Briefly, MCF-7 or MCF-7/ADR cells were seeded into 96-well culture plates at a density of 1 × 103 cells per well and incubated in the complete medium at 37 °C for 24 h. These wells were then divided into different groups and treated with DOX, DOX + CUR, DOX@MSN-Pep or DOX/CUR@MSN-Pep at various drug concentrations for 24 h, followed by adding 20 μL of 5 mg mL−1 MTT solution in pH 7.4 PBS. The similar experimental conditions were applied to CUR and blank nanocarriers. After 4 h incubation, the culture medium was removed and the cells were mixed with 150 μL DMSO. After shaking for 10 min, cell viability was determined by the optical density (OD) at a wavelength of 550 nm and the following formula:
Results and discussion
Preparation and characterization of DOX/CUR@MSN-Pep
In this work, original MSN was reacted with AAPTMS to obtain MSN-NH2, resulting in the surface charge changing from negative (−18.42 mV) to positive (+21.15 mV) (Table S1†). The charge reversion suggested that amine modification was successful. Then MSN-NH2 was further reacted with propargyl bromide to generate MSN-alkyne and the surface charge turned to be +2.12 mV, because most of the ionized amine groups had changed into un-ionized amide groups. Next, the DOX and CUR were loaded into the pores of MSN by shaking the mixture of drugs and MSN-alkyne in methyl alcohol. The peptide with the sequence of azido-GFLGHHHRRGDS as a stopper was connected to the end-point of tether via “click chemistry” to yield DOX/CUR@MSN-Pep nanomedicine. From the FT-IR spectra (Fig. S1†), a typical absorption peak of alkyne moiety at 2123 cm−1 was clearly seen. After subsequent reaction, the absorbance at ∼2123 cm−1 disappeared owing to click reaction, proving that the multifunctional peptides were immobilized onto the MSN. Meanwhile, consistent with the result observed in FT-IR analysis, the absorption spectrum of MSN-alkyne exhibited the characteristic absorbance spectrum of alkyne group (absorbance max: 300 nm) and subsequently disappeared after click reaction (Fig. S2†), also verifying the successful peptide functionalization. Besides, the mesostructure of the MSN was measured by nitrogen adsorption/desorption isotherm (Fig. S3†), with a BET surface area of 698.02 m2 g−1 and an average pore size of 2.6 nm. While, after grafted with peptides, MSN-Pep exhibited reduced BET surface area of 88.51 m2 g−1 due to the blocked pore channels. Furthermore, the thermal gravimetric analysis (TGA) was performed in Fig. 2. When the temperature increased to 800 °C, weight loss of 11.2% was observed for MSNs-NH2, which could be attributed to the amine and alkane group on the surface. As for MSN-alkyne, an additional weight loss of 6.7% suggested the alkyne chains grafted on MSNs. Moreover, the increased weight loss of MSN-Pep (48.6%) further made it clear that the peptide was successfully conjugated onto the MSN. | ||
| Fig. 2 TGA curves of the MSN, MSN-NH2, MSN-alkyne, and MSN-Pep. | ||
The morphology of the blank MSN was observed by transmission electron microscopy (TEM), revealing a uniform spherical structure with an average diameter of ∼70 nm (Fig. S4A†). The DLS result showed that the average hydrodynamic diameter was 94 nm (Fig. S4B†), which was larger than TEM analysis due to the presence of hydration layers. After encapsulation with drugs and conjugation with peptide on the surface, the DOX/CUR@MSN-Pep nanomedicine still maintained uniform spherical morphology, while the average diameter of nanomedicine increased to be ∼90 nm from TEM image (Fig. 3A) and hydrodynamic diameter of 113 nm by DLS (Fig. 3B). The thickness of the peptide shell was approximately 20 nm, which further confirmed the successful preparation of the DOX/CUR@MSN-Pep nanomedicine.
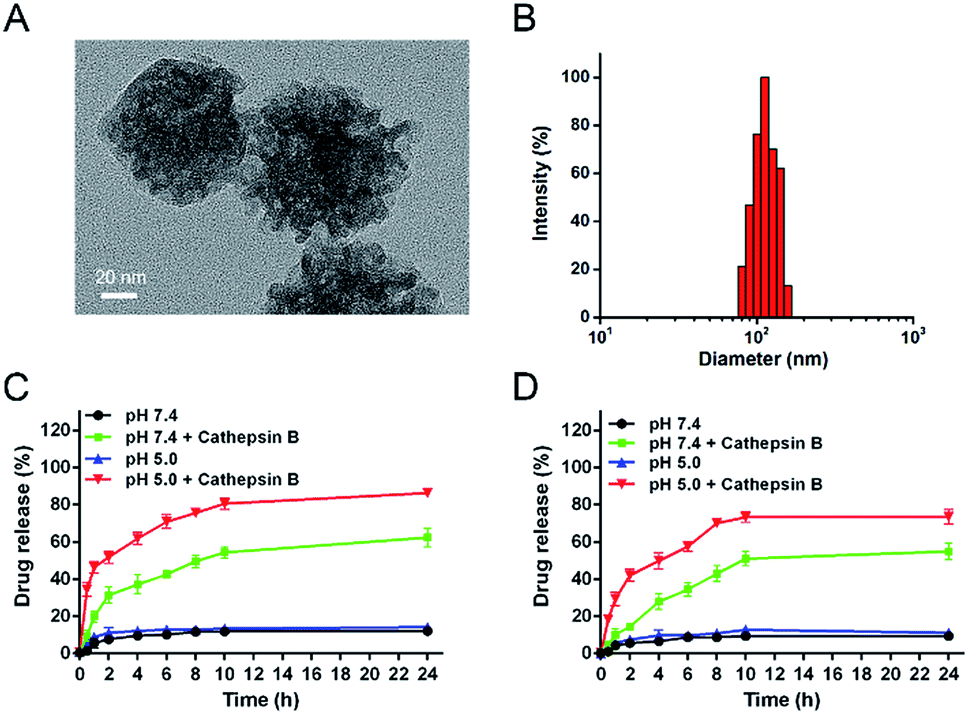 | ||
| Fig. 3 (A) TEM image of DOX/CUR@MSN-Pep. Scale bar: 20 nm. (B) Size distribution of DOX/CUR@MSN-Pep measured by DLS. In vitro release profiles of (C) DOX and (D) CUR from DOX/CUR@MSN-Pep in citrate buffer (pH 7.4 and 5.0) in the absence or presence of cathepsin B at 37 °C over time. The concentration of cathepsin B added in citrate was 20 U mL−1 (n = 3, mean ± SD). | ||
Drug loading and release
In order to investigate the cathepsin B-specific release behavior of our nanoparticle system, in vitro release behaviors were investigated in citrate buffer containing 10% FBS at a pH of 7.4 (physiological environment) and 5.0 (lysosomal environment). Herein, the LC of DOX and CUR in DOX/CUR@MSN-Pep nanoparticles was detected to be 3.78% and 4.46%, respectively, and the matched LE was determined to be 85% for DOX and 67% for CUR, which was calculated via absorbance standard curve method (Fig. S5†). Besides, the LC and LE values of DOX in DOX@MSN-Pep nanoparticles were 10.67% and 96%. Cumulative release profiles exhibit that the fast drug release in the presence of cathepsin B; approximately 86% of DOX (Fig. 3C) and 74% of CUR (Fig. 3D) were released from the DOX/CUR@MSN-Pep nanoparticles after incubation in pH 5.0 citrate buffer for 24 h. Importantly, almost no DOX and CUR was released in citrate buffer (pH = 7.4 or 5.0, without cathepsin B) within the same time scale, suggesting that DOX/CUR@MSN-Pep nanomedicine possessed high cathepsin B responsibility. Furthermore, 62% of DOX and 55% of CUR have been released from the DOX/CUR@MSN-Pep nanomedicine incubated in pH 7.4 for 24 h, which shown a slower DOX release rate than that at pH 5.0. This result was ascribed to the relatively high activity of cathepsin B under acidic environment.52 Therefore, we can speculate the DOX/CUR@MSN-Pep nanomedicine could rapidly release the CUR and DOX in lysosomal acidic environment which involved abundant cathepsin B, leading to high cytotoxicity to tumor cells.Cellular uptake efficiency of DOX
Using the intrinsic fluorescence of DOX, a comparative analysis of intracellular uptake efficiency of DOX were performed in the MCF-7/ADR cells treated with different groups including free DOX, DOX + CUR, DOX@MSN-Pep and DOX/CUR@MSN-Pep by flow cytometer. Free DOX presented the lowest cellular uptake efficiency of DOX (Fig. 4A), which confirmed that free DOX molecules could barely enter MCF-7/ADR cells. It was observed that DOX + CUR group showed higher cellular uptake efficiency than free DOX group. This could be explained that CUR can assist MCF-7/ADR cells uptaking DOX. Additionally, DOX@MSN-Pep treated group showed more cellular uptake than DOX group in MCF-7/ADR cells. More importantly, the result clearly exhibited that the DOX/CUR@MSN-Pep group had a much greater cellular uptake than either the DOX + CUR group or DOX@MSN-Pep group, suggesting that both CUR and the MSN-Pep nanocarrier played essential roles in cellular internalization. | ||
| Fig. 4 (A) Flow cytometric assays of cellular uptake efficiency of DOX in MCF-7/ADR cells after treated with different DOX formations. (B) Confocal fluorescence and bright field (BF) images of MCF-7/ADR cells incubated with free DOX, DOX + CUR, DOX@MSN-Pep and DOX/CUR@MSN-Pep for 4 h (10 μM DOX equiv.), and then stained with Hoechst 33342. Scale bar: 10 μm. | ||
Furthermore, cellular uptake of DOX were monitored by confocal fluorescence imaging. The red fluorescence with high brightness in Fig. S6† indicated that both the free DOX and the DOX/CUR@MSN-Pep were effective in treatment of MCF-7 cells. However, for the drug-resistant MCF-7/ADR cells, no obvious red DOX fluorescence was detected within the free DOX-treated cells. In comparison with DOX + CUR and DOX@MSN-Pep group, strong DOX fluorescence was appeared within the cells treated by DOX/CUR@MSN-Pep (Fig. 4B), which meant that the existence of CUR and MSN-Pep nanocarrier could reverse the MDR.
Previous research has shown that active targeting drug delivery promotes a high drug concentration in the targeted cells, achieving more effective tumor suppression.53 To study the effect of RGDS on the surface of DOX/CUR@MSN-Pep, the nanomedicine without RGDS motif was synthesized as a control. As shown in Fig. S7,† the MCF-7/ADR cells displayed strong fluorescence after a 4 h incubation with DOX/CUR@MSN-Pep. On the contrary, the cells incubated with DOX/CUR@MSN-Pep (without RGDS) exhibited weak fluorescence signal under the similar experimental condition. Besides, for the competition assay, RGD was utilized as the αvβ3 integrin-associated blocking reagent. Results showed that only a slight DOX fluorescence was observed within MCF-7/ADR cells after preincubation with RGD, verifying that the existence of RGD motif improved the enhancement of cell uptake. Therefore, it could be summarized that DOX/CUR@MSN-Pep effectively enhanced intracellular accumulation and retention of DOX in drug-resistant cancer cells.
Endocytic pathways
Endocytosis is one of the important entry means for the nanocarriers, which is connected to lysosomal drug sequestration and become a major barrier against the intracellular delivery in drug-resistant cells.54 To understand the mechanism of MCF-7/ADR cellular uptake of DOX/CUR@MSN-Pep, various endocytosis inhibitors, including indomethacin, chlorpromazine and colchicine were used to discriminate among distinct endocytosis pathway subcategories (Fig. 5). Compared with control group without inhibitors, the cellular uptake was significantly inhibited by chlorpromazine and indometacin. In addition, low temperature of 4 °C also induced a significant decrease of cellular uptake of DOX/CUR@MSN-Pep, reflecting that the cellular uptake was also energy-dependent. These results clearly indicated that clathrin and caveolin regulated the endocytosis of DOX/CUR@MSN-Pep.55 Therefore, the nanomedicine would be transported into endolysosomes inevitably after internalization. | ||
| Fig. 5 Endocytosis mechanism study of DOX/CUR@MSN-Pep in MCF7/ADR cells after incubation with different endocytosis inhibitors or at 4 °C. Data were normalized to the cells treated with only DOX/CUR@MSN-Pep at 37 °C. | ||
Intracellular endolysosomal escape
The intracellular endolysosomal escape behaviors of DOX/CUR@MSN-Pep were visualized in MCF-7/ADR cells by confocal fluorescence imaging. After incubation with the different preparations, the cells were stained with LysoTracker Deep Red to show the endolysosomes as green and Hoechst 33342 to show the nuclei as blue. As presented in Fig. 6, a large area of yellow fluorescence ascribing to the overlap of red DOX fluorescence and LysoTracker fluorescence was observed in DOX/CUR@MSN-Pep (without HHH) and DOX/CUR@MSN-Pep treated cells at 2 h, which indicated that the formulation was stranded in the endolysosomes. As prolonging incubation periods to 6 h, the DOX/CUR@MSN-Pep and DOX/CUR@MSN-Pep (without HHH) displayed greatly different intracellular distributions. For the DOX/CUR@MSN-Pep (without HHH), the visible superimposed signals remained bright yellow, suggesting that DOX is still trapped in endolysosomes. However, the DOX/CUR@MSN-Pep group demonstrated the DOX fluorescence signal was in the cytoplasm and nucleus which can be confirmed by the colocalization between DOX and the Hoechst 33342. Furthermore, according to the images of DOX/CUR@MSN-Pep at 12 h, DOX mostly entered the nuclei and the green fluorescence in the cells is practically extinguished because of the damage of endo-lysosomes. In addition, no obvious DOX fluorescence was detected within the MCF-7/ADR cells treated by the free DOX for 2 h. Even after 12 h incubation, the DOX signal of free DOX group still remained highly overlaying with the endolysosomal signal and could not be observed in the nuclei, indicating that it was very difficult for the free DOX to escape from the endolysosomes. The above results support the idea that the histidine-containing nanomedicine possessed the capability of endolysosomal escape due to the proton sponge effect of histidines. | ||
| Fig. 6 Colocalization of DOX/CUR@MSN-Pep or Free DOX in MCF-7/ADR cells with LysoTracker Deep Red (green) and Hoechst 33342 (blue). Cells were incubated with Free DOX, DOX/CUR@MSN-Pep (without HHH), or DOX/CUR@MSN-Pep (5.0 μM DOX equiv.) for 2 h, 6 h and 12 h, respectively. Scale bar: 10 μm. | ||
To further study the capability of intranuclear import for the endolysosomal escaped drugs, intracellular trafficking of DOX/CUR@MSN-Pep in MCF-7/ADR cells at different time was investigated. As shown in Fig. S8,† after addition of the DOX/CUR@MSN-Pep for 1 h and 2 h, the DOX signal remained highly overlaying with the endolysosomal signal and the yellow fluorescence dots were apparent in the merged images. It meant that the DOX had become highly concentrated within endolysosomes of the MCF-7/ADR cells. Subsequently, the DOX fluorescence from DOX/CUR@MSN-Pep became distinctly stronger for 4 h incubation owing to the enhanced uptaken of DOX. After further incubation, red fluorescent signal began to overlap with the blue fluorescent signal of nuclei at 6 h, illustrating that DOX entered the nuclei as the drug target site. The LysoTracker fluorescence signal gradually became weaker with extended incubation time, and the nuclear uptake of DOX was increased over 12 h. At 24 h, the DOX and Hoechst fluorescence is feeble due to the toxicity of DOX which led to apoptosis of the cells. These meant that DOX/CUR@MSN-Pep could significantly reverse the MDR in consequence of rapid endolysosomal escape and the enhanced nuclear drug accumulation.
Downregulation of P-gp expression
MDR generally involves the overexpression of P-gp as an efflux transporter.56 Thus, to evaluate the reversal effects, we detected the P-gp expression levels in MCF-7 and MCF-7/ADR cells after treated with CUR@MSN-Pep by flow cytometer using a fluorescence-labeled anti-MDR1 antibody. A similar fluorescence intensity compared with the isotype control group was found in drug sensitive MCF-7 cells (Fig. 7A), while drug resistant MCF-7/ADR cells exhibited a stronger fluorescence intensity to that labeled by the isotype control (Fig. 7B), which reflected that P-gp was over-expressed in MCF-7/ADR cells. Moreover, we found that the CUR@MSN-Pep treated group can significantly decrease the expression of P-gp compared to the CUR@MSN-Pep untreated group (Fig. 7B), indicating that CUR@MSN-Pep nanocarrier possessed a strong ability to downregulate the expression of P-gp in MCF-7/ADR cells. | ||
| Fig. 7 P-gp expression on MCF-7 (A) and MCF-7/ADR (B) cell membrane (black: control group; green: isotype control group; blue: P-gp expression on cells untreated with CUR@MSN-Pep; red: P-gp expression on cells after treated with CUR@MSN-Pep for 24 h). | ||
In vitro anticancer activity of DOX/CUR@MSN-Pep
To demonstrate the in vitro anticancer activity of DOX/CUR@MSN-Pep, we tested the half-maximal inhibitory concentration (IC50) value of DOX and its formulations against both MCF-7 and MCF-7/ADR cells. As presented in Fig. 8, the IC50 values of DOX for MCF-7 and MCF-7/ADR cells were measured to be 0.82 μM and 18.77 μM, respectively. Besides, the drug resistance index (DRI, the ratio of IC50 values between MCF-7/ADR and MCF-7 cells) of MCF-7/ADR cells was calculated to be 22.89, indicating that cultured MCF-7/ADR cells were highly resistant to DOX (Table 1). Intriguingly, the cytotoxicity of CUR towards MCF-7 and MCF-7/ADR cells was relatively low as the IC50 values was determined to be 22.93 μM and 41.68 μM, respectively (Fig. S9†). Nevertheless, DOX + CUR with a molar ratio of 1:2 had a much higher cytotoxicity against MCF-7/ADR cells (IC50 = 9.11 μM) than DOX alone, which proved that CUR had the capability to reverse MDR. Moreover, the IC50 of DOX@MSN-Pep and DOX/CUR@MSN-Pep was 7.64 μM and 2.19 μM, and the reversal index (RI, the DRI of DOX/the DRI of DOX's formulation) was calculated to be 2.46 and 8.57 (Table 1), respectively. These results certified that MSN-Pep nanocarrier could enhance the drug delivery into cells to improve the cytotoxicity of DOX and DOX + CUR towards MCF-7/ADR cells. Moreover, the blank MSN, surface modified MSN along with MSN-pep nanocarrier exhibited nontoxicity, showing good biocompatibility to both MCF-7 and MCF-7/ADR cells (Fig. S10†). Notably, the DOX/CUR@MSN-Pep nanomedicine remarkably enhanced cytotoxicity against MCF-7/ADR cells as their IC50 value was considerably lowered to 2.19 μM and their RI was as high as 8.57, which further confirmed the importance of CUR@MSN-Pep for conquering MDR.
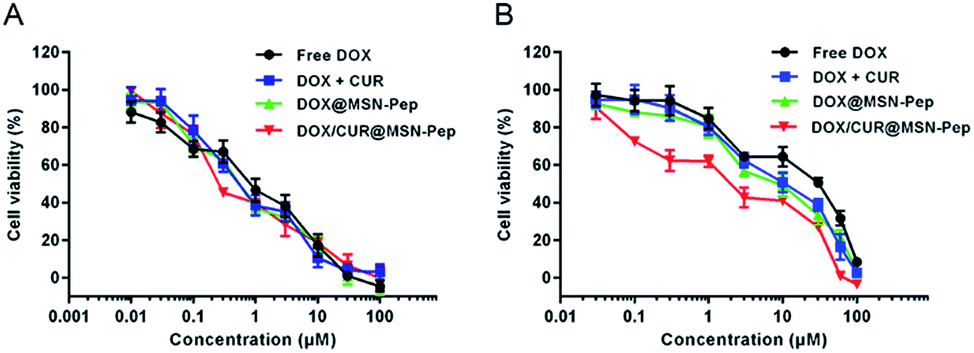 | ||
| Fig. 8 MTT assays of (A) MCF-7 cells and (B) MCF-7/ADR cells incubated with free DOX, DOX + CUR, DOX@MSN-Pep and DOX/CUR@MSN-Pep at different concentrations (DOX equiv.). Data are means ± SD (n = 5). | ||
| Treatmentc | IC50 valuea (μM) | DRId | RIe |
|---|---|---|---|
| a The IC50 value of DOX against MCF-7 cells was measured to be 0.82 μM.b Molar ratio of DOX:CUR was 1:2.c All formulas contain an equivalent amount of DOX.d The drug resistance index (DRI) of MCF-7/ADR cells was calculated as the IC50 for MCF-7/ADR cells/the IC50 for MCF-7 cells.e The reversal index (RI) was calculated as the DRI of DOX/the DRI of the formulation containing DOX. |
|||
| DOX | 18.77 | 22.89 | |
| DOX + CURb | 9.11 | 11.11 | 2.06 |
| DOX@MSN-Pep | 7.64 | 9.32 | 2.46 |
| DOX/CUR@MSN-Pep | 2.19 | 2.67 | 8.57 |
Conclusions
In summary, we have developed an effective DOX/CUR@MSN-Pep nanomedicine for overcoming MDR. After functionalized with the peptide of GFLGHHHRRGDS on the surface of MSN, the nanomedicine possesses a series of advantages such as targeting ability, controlled drug release and endolysosomal escape. It has been proved that the designed nanomedicine entered the cells via endocytosis pathway. Then CUR and DOX were simultaneously released correlated to specific cleavage at the GFLG linker by cathepsin B. More attractively, the nanomedicine could disrupt endolysosomes via protonation effect of HHH sequence, which greatly assists the DOX to avoid lysosomal sequestration and reach the site of action in MCF-7/ADR cells. On the other hand, combination of CUR and DOX, especially when administered in our histidine-containing nanocarrier, has a strong effect on down-regulation of P-gp and exhibits a highest therapeutic efficacy towards MCF-7/ADR cells. We believe that the DOX/CUR@MSN-Pep nanomedicine can provide a new guidance for the treatment of MDR cancer.Acknowledgements
This research was supported by National Natural Science Foundation of China (21505161), Natural Science Foundation of Jiangsu Province (BK20150701), Fundamental Research Funds for the Central Universities (2632017ZD10), Program for Changjiang Scholars and Innovative Research Team in University (IRT1193) and Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals (ZDSYS-KF201503).Notes and references
- H. Kitano, Nat. Rev. Cancer, 2004, 4, 227–235 CrossRef CAS PubMed.
- B. A. Chabner and T. G. Roberts Jr, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- A. Persidis, Nat. Biotechnol., 1999, 17, 94–95 CrossRef CAS PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- N. Altan, Y. Chen, M. Schindler and S. M. Simon, J. Exp. Med., 1998, 187, 1583–1598 CrossRef CAS PubMed.
- A. K. Larsen, A. E. Escargueil and A. Skladanowski, Pharmacol. Ther., 2000, 85, 217–229 CrossRef CAS PubMed.
- B. Zhitomirsky and Y. G. Assaraf, Drug Resist. Updates, 2016, 24, 23–33 CrossRef PubMed.
- M. Sehested, T. Skovsgaard, B. van Deurs and H. Winther-Nielsen, J. Natl. Cancer Inst., 1987, 78, 171–179 CrossRef CAS PubMed.
- M. Sehested, T. Skovsgaard, B. van Deurs and H. Winther-Nielsen, Br. J. Cancer, 1987, 56, 747–751 CrossRef CAS PubMed.
- A. Kuhad, S. Pilkhwal, S. Sharma, N. Tirkey and K. Chopra, J. Agric. Food Chem., 2007, 55, 10150–10155 CrossRef CAS PubMed.
- J. Piper, Int. J. Biochem. Cell Biol., 1998, 30, 445–456 CrossRef CAS PubMed.
- M. T. Huang, T. Lysz, T. Ferraro, T. F. Abidi, J. D. Laskin and A. H. Conney, Cancer Res., 1991, 51, 813–819 CAS.
- K. Sugimoto, H. Hanai, K. Tozawa, T. Aoshi, M. Uchijima, T. Nagata and Y. Koide, Gastroenterology, 2002, 123, 1912–1922 CrossRef CAS PubMed.
- R. A. Sharma, H. R. McLelland, K. A. Hill, C. R. Ireson, S. A. Euden, M. M. Manson, M. Pirmohamed, L. J. Marnett, A. J. Gescher and W. P. Steward, Clin. Cancer Res., 2001, 7, 1894–1900 CAS.
- S. Aggarwal, Y. Takada, S. Singh, J. N. Myers and B. B. Aggarwal, Int. J. Cancer, 2004, 111, 679–692 CrossRef CAS PubMed.
- R. A. Sharma, S. A. Euden, S. L. Platton, D. N. Cooke, A. Shafayat, H. R. Hewitt, T. H. Marczylo, B. Morgan, D. Hemingway, S. M. Plummer, M. Pirmohamed, A. J. Gescher and W. P. Steward, Clin. Cancer Res., 2004, 10, 6847–6854 CrossRef CAS PubMed.
- M. M. Yallapu, B. K. Gupta, M. Jaggi and S. C. Chauhan, J. Colloid Interface Sci., 2010, 351, 19–29 CrossRef CAS PubMed.
- A. Goel and B. B. Aggarwal, Nutr. Cancer, 2010, 62, 919–930 CrossRef CAS PubMed.
- X. L. Hou, K. Takahashi, K. Tanaka, K. Tougou, F. Qiu, K. Komatsu, K. Takahashi and J. Azuma, Int. J. Pharm., 2008, 358, 224–229 CrossRef CAS PubMed.
- N. Romiti, R. Tongiani, F. Cervelli and E. Chieli, Life Sci., 1998, 62, 2349–2358 CrossRef CAS PubMed.
- S. Anuchapreeda, P. Leechanachai, M. M. Smith, S. V. Ambudkar and P. N. Limtrakul, Biochem. Pharmacol., 2002, 64, 573–582 CrossRef CAS PubMed.
- P. Limtrakul, S. Anuchapreeda and D. Buddhasukh, BMC Cancer, 2004, 4, 13 CrossRef PubMed.
- M. Notarbartolo, P. Poma, D. Perri, L. Dusonchet, M. Cervello and N. D'Alessandro, Cancer Lett., 2005, 224, 53–65 CrossRef CAS PubMed.
- Y. Sadzuka, M. Nagamine, T. Toyooka, Y. Ibuki and T. Sonobe, Int. J. Pharm., 2012, 432, 42–49 CrossRef CAS PubMed.
- Y. Kwon, J. Korean Soc. Appl. Biol. Chem., 2014, 57, 273–280 CrossRef CAS.
- P. Huang, X. Qian, Y. Chen, L. Yu, H. Lin, L. Wang, Y. Zhu and J. Shi, J. Am. Chem. Soc., 2016, 139, 1275–1284 CrossRef PubMed.
- X. Liu, A. Situ, Y. Kang, K. R. Villabroza, Y. Liao, C. H. Chang, T. Donahue, A. E. Nel and H. Meng, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS PubMed.
- C. Yu, L. Qian, M. Uttamchandani, L. Li and S. Q. Yao, Angew. Chem., Int. Ed. Engl., 2015, 54, 10574–10578 CrossRef CAS PubMed.
- P. Zhang, F. Cheng, R. Zhou, J. Cao, J. Li, C. Burda, Q. Min and J. J. Zhu, Angew. Chem., Int. Ed. Engl., 2014, 53, 2371–2375 CrossRef CAS PubMed.
- K. Yang, H. Luo, M. Zeng, Y. Jiang, J. Li and X. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 17399–17407 CAS.
- L. He, H. Lai and T. Chen, Biomaterials, 2015, 51, 30–42 CrossRef CAS PubMed.
- Y. P. Chen, C. T. Chen, Y. Hung, C. M. Chou, T. P. Liu, M. R. Liang, C. T. Chen and C. Y. Mou, J. Am. Chem. Soc., 2013, 135, 1516–1523 CrossRef CAS PubMed.
- S. H. van Rijt, D. A. Bolukbas, C. Argyo, S. Datz, M. Lindner, O. Eickelberg, M. Konigshoff, T. Bein and S. Meiners, ACS Nano, 2015, 9, 2377–2389 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- J.-J. Hu, L.-H. Liu, Z.-Y. Li, R.-X. Zhuo and X.-Z. Zhang, J. Mater. Chem. B, 2016, 4, 1932–1940 RSC.
- C. Yu, L. Qian, J. Ge, J. Fu, P. Yuan, S. C. Yao and S. Q. Yao, Angew. Chem., Int. Ed. Engl., 2016, 55, 9272–9276 CrossRef CAS PubMed.
- D. W. Pack, D. Putnam and R. Langer, Biotechnol. Bioeng., 2000, 67, 217–223 CrossRef CAS PubMed.
- C. Pichon, C. Goncalves and P. Midoux, Adv. Drug Delivery Rev., 2001, 53, 75–94 CrossRef CAS PubMed.
- P. Midoux, A. Kichler, V. Boutin, J. C. Maurizot and M. Monsigny, Bioconjugate Chem., 1998, 9, 260–267 CrossRef CAS PubMed.
- S. R. Yang, H. J. Lee and J. D. Kim, J. Controlled Release, 2006, 114, 60–68 CrossRef CAS PubMed.
- I. J. Fang, I. I. Slowing, K. C. Wu, V. S. Lin and B. G. Trewyn, Chemistry, 2012, 18, 7787–7792 CrossRef CAS PubMed.
- R. O. Hynes, Nat. Med., 2002, 8, 918–921 CrossRef CAS PubMed.
- L. Pan, J. Liu, Q. He and J. Shi, Adv. Mater., 2014, 26, 6742–6748 CrossRef CAS PubMed.
- B. F. Sloane, S. Yan, I. Podgorski, B. E. Linebaugh, M. L. Cher, J. Mai, D. Cavallo-Medved, M. Sameni, J. Dosescu and K. Moin, Semin. Cancer Biol., 2005, 15, 149–157 CrossRef CAS PubMed.
- N. Li, N. Li, Q. Yi, K. Luo, C. Guo, D. Pan and Z. Gu, Biomaterials, 2014, 35, 9529–9545 CrossRef CAS PubMed.
- Y. J. Cheng, G. F. Luo, J. Y. Zhu, X. D. Xu, X. Zeng, D. B. Cheng, Y. M. Li, Y. Wu, X. Z. Zhang, R. X. Zhuo and F. He, ACS Appl. Mater. Interfaces, 2015, 7, 9078–9087 CAS.
- Y. Wang, R. Zhao, S. Wang, Z. Liu and R. Tang, Biomaterials, 2016, 75, 71–81 CrossRef CAS PubMed.
- R. Misra and S. K. Sahoo, Eur. J. Pharm. Sci., 2010, 39, 152–163 CrossRef CAS PubMed.
- R. Misra and S. K. Sahoo, Mol. Pharm., 2011, 8, 852–866 CrossRef CAS PubMed.
- J. Shen, Q. He, Y. Gao, J. Shi and Y. Li, Nanoscale, 2011, 3, 4314–4322 RSC.
- C. Lopez-Otin and L. M. Matrisian, Nat. Rev. Cancer, 2007, 7, 800–808 CrossRef CAS PubMed.
- C. Jang, J. H. Lee, A. Sahu and G. Tae, Nanoscale, 2015, 7, 18584–18594 RSC.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- H. L. Wong, R. Bendayan, A. M. Rauth, H. Y. Xue, K. Babakhanian and X. Y. Wu, J. Pharmacol. Exp. Ther., 2006, 317, 1372–1381 CrossRef CAS PubMed.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Science, 2009, 323, 1718–1722 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary table and figures. See DOI: 10.1039/c7ra01128h |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |