
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titania nanotube stabilized BiOCl nanoparticles in visible-light photocatalysis†
B. Buchholcza,
H. Haspel‡
a,
A. Oszkób,
A. Kukoveczac and
Z. Kónya *ad
*ad
aDepartment of Applied and Environmental Chemistry, University of Szeged, Rerrich Béla tér 1, H-6720 Szeged, Hungary. E-mail: konya@chem.u-szeged.hu; Fax: +36-62-544619; Tel: +36-62-544620
bDepartment of Physical Chemistry and Materials Science, University of Szeged, Rerrich Béla tér 1, H-6720 Szeged, Hungary
cMTA-SZTE “Lendület” Porous Nanocomposites Research Group, Rerrich Béla tér 1, H-6720 Szeged, Hungary
dMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, Rerrich Béla tér 1, H-6720 Szeged, Hungary
First published on 14th March 2017
Abstract
Photocatalysis is a green approach in environmental organic pollutant decomposition. Lately, considerable improvement in the activity of photocatalysts has been achieved with the realization of p–n heterostructures due to the lifetime extension of the photogenerated charge carriers. Herein, we report a facile synthesis approach for decorating n-type titanate nanotubes with p-type V–VI–VII compound semiconductor BiOCl nanoparticles. It is well-known that BiOX (X = Cl, Br, I) materials form nanometer-thick platelets, which can eventually assemble into micrometer size flower-like 3D structures. Here, we demonstrate that the tubular titanate support can stabilize BiOCl on its surface in the form of nanoparticles measuring a few nanometers in diameter, instead of forming the well-known bismuth-oxyhalide nanoflowers. Subsequent calcination at 400 °C transforms the pristine titanate structures into one-dimensional anatase nanotubes, along with the formation of a heterojunction at the interface of the emerging Bi2Ti2O7 and anatase phases. The resulting nanocomposite shows activity in visible-light photocatalytic test reactions.
1. Introduction
Photocatalysis is a green chemistry approach to catalysis, since the initiator of the reaction is the incident electromagnetic irradiation; mostly UV,1 but sometimes UV-VIS,2 visible,3 and near infrared.4 Photocatalytic (and photochemical) studies often use light sources with high-energy emission, whereas only about 6% of the solar radiation energy is in the UV range, while 50% of the energy arrives between 400 and 700 nm at sea level. Thus, sustainable development demands harvesting visible-light energy more effectively.Titanate nanotubes are layered (rolled-up) titanium-oxide materials [trititanates, (Hx, Na2−x)Ti3O7], with a large specific surface area of 170–250 m2 g−1 accompanied by a specific pore volume of 0.5–0.8 cm3 g−1.5 Titanates can easily be doped by various elements, e.g., nitrogen6 and decorated by different nanoparticles immobilized on the surface via simple wet impregnation techniques.7,8 Protonated trititanate nanotubes (TiONT) can be transformed into anatase TiO2 nanotubes; phase transformation can be promoted or inhibited by the exchanged interlayer ions, the supported nanoparticles and doping elements.6,7 Although n-type semiconductor TiOx materials attracted considerable attention due to their photocatalytic performance,9 their activity is limited by short charge carrier lifetime due to fast recombination. Semiconductor p–n heterojunctions can, however, facilitate electron–hole pair separation via forming an internal electric field at the interface10 and thus suppressing charge carrier recombination. This, in turn, significantly improves the photocatalytic activity of the structure.11
The V–VI–VII semiconductor bismuth oxyhalide [BiOX (X = Br, Cl, I)] is a new photocatalyst family, which has drawn considerable attention due to the promising properties of its members in organic contaminant degradation12 even when utilizing visible-light irradiation.13 BiOX is usually a p-type semiconductor with a layered structure, constructed by X–Bi–X–Bi–X sheets. These materials are capable of generating electron–hole pairs with long lifetime, which makes them potentially effective photocatalysts.14 BiOX materials can be synthesized via various methods (see, e.g., Table 4 in ref. 15), forming mainly hundreds of nanometers long and tens of nanometer thick nanosheets and nanoflakes, which eventually assemble into 3D flower-like, micron sized structures. As-prepared materials have low specific surface area, generally lower than 10 m2 g−1.16 In order to increase the accessibility of the organic contaminants by the photoinduced radicals at the surface, two options arise: find a new synthetic way to prepare BiOX17 with a larger specific surface area, or immobilize BiOX nanoparticles on the surface of a suitable support. Utilizing TiOx supports is a straightforward way to go, and numerous reports have been published in this topic as demonstrated by the review of BiOX literature as summarized in Table 1.
| Photocatalyst | Photocatalyst | Test system | Ref. |
|---|---|---|---|
| BiOI/TiO2 | 4 nm TiO2 NPs on >100 nm BiOI plates | MO under VIS | 18 |
| BiOI/TiO2 NTA | 8 nm BiOI coating on 100 nm/2 μm NTA | MO under VIS | 19 |
| BiOI/TiO2 (P25) | 24 nm TiO2 NP on 2 nm BiOI or microplates | Phenol under VIS | 20 |
| BiOCl/TiO2 hybrid | Microparticles and aggregates | Eosin Y under UV | 21 |
| BiOI/TiO2 (A) | Not shown or discussed | RhB under UV and VIS | 22 |
| BiOI/TiO2 (A) | TiO2 on 100 nm/8 nm BiOI plates | MO under VIS | 23 |
| BiOBr/TiO2 | 40 nm BiOBr flakes on TiO2 agglomerates | RhB under VIS | 24 |
| BiOCl/TiO2 NTA | >100 nm/30 nm BiOCl NS on TiO2 NTA | MO, PCP under UV | 25 |
| BiOI/titania NT | >100 nm/10 nm BiOI on 140 nm nanotubes | — | 26 |
| BiOI/TiO2/textile | >100 nm/>10 nm BiOI NS on TiO2 layer | MO under UV-VIS | 27 |
| BiOI/TiO2 NF | 1 μm/20 nm BiOI on 1 μm/>100 nm fiber | RhB under VIS | 28 |
| BiOI/TiO2 fiber | 300 nm/>10 nm BiOI on 10 μm/0.5 μm fiber | MB under VIS | 29 |
| BiOBr/TiO2/G | BiOBr core + TiO2 flakes ≥ microspheres | RhB under VIS | 30 |
| BiOCl/TiO2 NF | μm/>10 nm BiOCl on μm/>100 nm fiber | RhB under UV | 31 |
| BiOCl/TiO2 (R@A) | >100 nm anatase/rutile on 200–400 nm BiOCl | PEC water splitting | 32 |
| BiOX/TiO2 NR | 100 nm/20 nm BiOX on 100 nm ribbons | SA, RhB, MO under Sun | 33 |
| BiOBr/TiO2 NTA | 30 nm/1 nm BiOBr on 200 nm TiO2 NTA | MO under UV-VIS | 34 |
| BiOI/TiO2 NTA | 20 nm/1 nm BiOI on 200 nm TiO2 NTA | MO under UV-VIS | 35 |
| BiOI/TiO2 | Nanometer sized TiO2 NPs on BiOI flakes | BPA under VIS | 36 |
| BiOCl/porous TiO2 | ∼μm/∼nm BiOCl flakes on porous TiO2 | RhB under UV-VIS | 37 |
| BiOCl/porous TiO2 | ∼μm/∼nm BiOCl flakes on porous TiO2 | RhB under UV-VIS | 38 |
| BiOBr/a-TiO2 | ∼μm BiOBr plates on amorphous TiO2 NP | MO, phenol under VIS | 39 |
| BiOI/3D TiO2 | >100 nm BiOI flakes on 300 nm porous TiO2 | PV test as solar cell | 40 |
| BiOI/TiO2 NRA | 0.1–3 μm BiOI flakes on ∼μm rutile TiO2 rods | PEC under UV-VIS | 41 |
| BiOBr/TiO2 | 100 nm/10 nm BiOBr on ∼100 nm TiO2 tubes | RhB under VIS | 42 |
| BiOCl/TiO2 | ∼μm/200 nm BiOCl plates on >100 nm pellets | Phenol under UV-VIS | 43 |
| BiOCl/TiO2 fiber | >10 nm/>50 nm BiOCl on 200 nm TiO2 fiber | RhB under UV | 44 |
| BiOI/TiO2 NF | ∼1 μm BiOI NS on ∼μm/200 nm TiO2 fiber | MB under VIS | 45 |
| BiOBr/TiO2 | 100/10 nm BiOBr flakes on ∼μm/100 nm TiO2 | RhB under VIS | 46 |
| BiOI/TiO2 NBA | Sporadic BiOI NP on micron long nanobelts | MO under VIS | 47 |
| BiOI/TiO2 nanobelt | >10 nm BiOI flakes on micron long nanobelt | MO under VIS | 48 |
| BiOI/TiO2 nanotube | >10 nm flakes on micron long nanotubes | PEC water splitting | 49 |
| BiOI/TiO2 | >10 nm TiO2 NPs on BiOI platelets | MO under UV-VIS | 50 |
| BiOCl/TiO2 | From ∼μm flowers to 20 nm particles on flakes | RhB under VIS | 51 |
| BiOCl/TiO2−x | TiO2 NPs on 50–100 nm BiOCl nanosheets | RhB under VIS | 52 |
| BiOBr/TiO2 NB | >100 nm BiOBr NSs on 50–200 nm nanobelts | PEC and RhB under VIS | 53 |
| BiOI/TiO2 NF | >100 nm BiOI NSs on micron sized nanofibers | MO under VIS | 54 |
| BiOX/TiO2 | ∼20 nm TiO2 NPs in ∼μm BiOX microspheres | Different dyes under VIS | 15 |
However, a careful examination of the studies revealed that reports on actual BiOX nanoparticle formation are scarce. To name of few, BiOX (X = Cl, Br, I) nanoparticles with diameter between 3 and 22 nm were formed in reverse microemulsions,55 BiOCl (10–15 nm), BiOBr (20 nm) and BiOI (40–100 nm) particles by microwave irradiation56 and 2.7 nm thick, 50 nm nanosheets using PVP in mannitol solution,57 BiOI nanoparticles with diameter of several nanometers distributed on BiOCl sheets via ionic liquid assisted ultrasonic method, 5–15 nm BiOBr nanosheets in a hydrothermal synthesis,58 and BiOI quantum dots were formed on reduced graphene-oxide.59 There are also reports on the formation of few nanometer nanosheet fragments under electron beam irradiation, e.g., 50–500 nm long and 2–12 nm thick flakes in HRTEM.60 These methods generally yield smaller nanosheets instead of real nanoparticles, and required complex synthesis methods. Moreover, to the best of our knowledge, reports on BiOX nanoparticle decorated titanium-oxide heterostructures are completely absent from the literature.
Herein, we report the facile fabrication of a hierarchical heterostructure of p-type BiOCl nanoparticles and n-type one-dimensional titanium-oxide nanotubes. Porous titanate nanotubes can immobilize small diameter BiOCl nanoparticles on their surface. The evolution of the p–n heterojunctions takes place during a subsequent thermal annealing, in parallel with the formation of a Bi2Ti2O7 phase in the one-dimensional anatase support, resulting in a photocatalytically active heterostructure.
2. Experimental
2.1. Preparation of photocatalysts
Titanate nanotubes were synthesized via the hydrothermal route.61 A white suspension was formed by mixing 50 g anatase TiO2 (99.8%, Sigma-Aldrich) and 1 L 10 M NaOH (99.93%, Molar) for 1 hour, then the system was transferred into a PTFE-lined stainless steel autoclave (diameter 120 mm, height 250 mm) and kept at 130 °C for 24 hours while rotating the autoclave continuously at 3 rpm around its short axis. The resulting white precipitate was washed with 0.01 M aqueous HCl (Molar) solution to neutral pH and finally, with deionized water. The prepared TiONT (characterized by the composition HxNa2−xTi3O7 where x > 1.8) was dried in air at 60 °C for 48 hours.In the catalyst synthesis 1 g protonated titanate nanotube was suspended in 50 ml deionised water for 2 hours in an ultrasonic bath. Subsequently, BiCl3 was added to the continuously stirred suspension at an atomic ratio of Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi = 10:1 and 100:1 depending on the target composition, and stirred for 24 hours. BiOCl was formed in the following reaction:
:Bi = 10:1 and 100:1 depending on the target composition, and stirred for 24 hours. BiOCl was formed in the following reaction:
| BiCl3 + H2O → BiOCl + 2HCl | (1) |
The product was washed with distilled water to get rid of unreacted BiCl3 and dried at 80 °C for 24 hours. Subsequent heat treatment at temperatures between 300 and 900 °C with 100 °C increments was applied to a part of each sample for 1 hour. The as-prepared and BiOCl decorated samples were designated as NT (nanotubes) and BNT (BiOCl decorated nanotubes), respectively. Calcined samples were denoted as HTNT and HTBNT, corresponding to heat treated nanotubes and heat treated BiOCl/nanotube composites, respectively. Additionally, BNT and HTBNT samples were marked according to the calculated BiOCl percentage as well. Unless indicated otherwise, results are shown for HTNT and HTBNT samples prepared by annealing at 400 °C. Bulk BiOCl was also synthesized as reference. 2 g precursor salt vigorously stirred in 200 ml distilled water for 2 h. The as-prepared precipitate was washed with distilled water and dried for one day at 80 °C.
2.2. Characterization methods
The completion of the nanostructure synthesis and the morphology of the composites was confirmed by Transmission Electron Microscopy (TEM) using a FEI Tecnai G2 20 X-Twin operated at 200 kV, and by Scanning Electron Microscopy (SEM) using a Hitachi S-4700 Type II FE-SEM instrument. Elemental analysis was carried out using the Röntec QX2 energy dispersive X-ray spectrometer mounted in the SEM. Nitrogen adsorption–desorption isotherms were recorded at 77 K in a Quantachrome Nova 2200 surface area analyzer after applying a prior outgassing step to remove adsorbed contaminants. The specific surface area of the samples was calculated in the Brunauer–Emmett–Teller (BET) model, while the pore size distribution functions were determined using the Barrett–Joyner–Halenda (BJH) method. The band gap of the as-prepared TiONT and the composites was determined with an Ocean Optics USB4000 UV-VIS spectrometer using a DH-2000-BAL UV-Vis-NIR light source and a diffuse reflectance probe. Crystal phase changes were monitored by X-ray diffractometry (XRD) using a Rigaku Miniflex II unit operated with a Cu Kα source (λ = 0.1542 nm) at 30 kV and 15 mA. Diffractograms were recorded in the 10–70° 2Θ range at a 4° min−1 scan rate. X-ray photoelectron spectra were recorded in a SPECS instrument equipped with a PHOIBOS 150 MCD 9 hemispherical electron energy analyzer, using the Kα radiation of the Al anode (hν = 1486.6 eV). The X-ray gun was operated at 210 W (14 kV, 15 mA). The analyzer was operated in the FAT mode, with the pass energy set to 20 eV. Five scans were summed to get a single high-resolution spectrum. The binding energy scale was corrected by the deconvolution of the complex C 1s region, where the position of the adventitious carbon peak was fixed at 285.1 eV.2.3. Photocatalytic experiments
The photocatalytic properties of the pristine and heat-treated materials were tested by methyl orange decolorization under visible light irradiation. A 40 W Medicor Q 250 mercury-vapor lamp with a UV cut-off filter was used as light source in a batch reactor, thermostated to 25 °C by a Julabo F12 thermostat. In each experiment 10 mg sample was continuously stirred in 10 mg l−1 methyl orange aqueous solution, irradiated for 1, 2, 4 and 8 hours. Before each measurement, the solution was stirred in dark for one hour to reach the adsorption–desorption equilibrium. The change in methyl orange concentration was monitored at the wavelength of the maximum absorption of the solution (λ = 464 nm) with a Hitachi U-2001 UV-VIS spectrophotometer. Degussa P25 was used as a reference photocatalyst to compare the catalytic performance of all samples.3. Results and discussion
3.1. Particle size and morphology
The size and morphology of the pristine nanotubes and nanotubes decorated with 10% BiOCl nanoparticles are observable in the electron microscopic images in Fig. 1. The TEM investigation revealed that the pristine, elongated, protonated trititanate nanostructures have 5–6 nm inner and 10–11 nm outer diameters. The interlayer distance in the nanotube walls was found to be 0.79 nm. The average tube length was between 100 and 300 nm as shown in Fig. 1a. Fig. 1b and c depict immobilized BiOCl nanoparticles on the nanotube surface. An average particle size of 4.9 ± 0.9 nm was obtained for BiOCl, which is approx. half of the nanotubes' outer diameter, as it is seen in the inset of the magnified TEM image in Fig. 1c. SEM images in Fig. 1d and e show entangled 3D TiONT aggregates measuring a few micrometers in diameter, covered by BiOCl coating. Such large assemblies are characteristic to BiOX materials, and to BiOX/TiOx composites.18–54 | ||
| Fig. 1 TEM images of pristine titanate nanotubes (NT) (a) and nanotubes decorated with 10% BiOCl (BNT10) at low (b) and high magnifications (c). The inset graph in part (c) depicts the BiOCl particle size distribution determined from TEM images. The morphology of the BiOCl/TiONT composite is illustrated in the SEM images (d) and (e). | ||
The electron microscopic studies indicate an intimate contact between the 0D BiOCl nanoparticles and 1D NTs, therefore, it seems plausible that the supported nanoparticles could connect adjacent n-type nanotubes through p-type BiOCl heterojunction bridges, forming a quasi-continuous n/p/n-type semiconductor network.
3.2. Specific surface area and pore size distribution
The specific surface area and the porous structure of the pristine, nanoparticle decorated and modified nanotubes were characterized by nitrogen adsorption–desorption measurements. Fig. 2 shows the total isotherms and the corresponding BJH pore size distributions as an inset graph.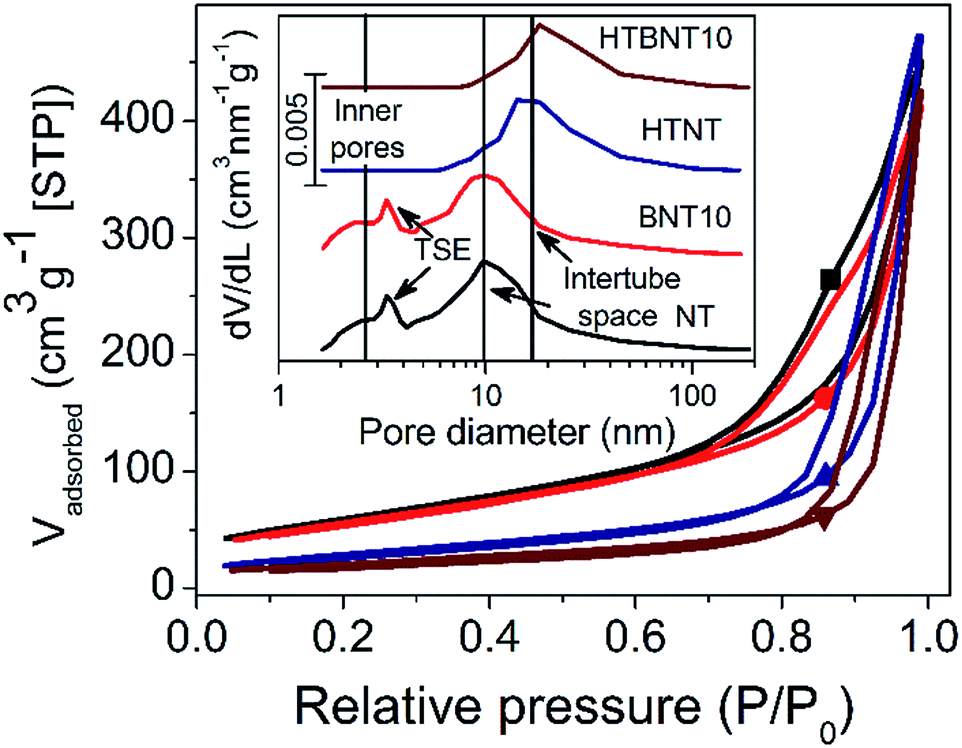 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of pristine titanate nanotubes (NT) “■”, nanotubes decorated with 10% BiOCl (BNT10) “●”, and the pristine (HTNT) “▲” and 10% BiOCl decorated TiONT after heat treatment at 400 °C (HTBNT) “▼”. The inset graph depicts the corresponding pore size distributions, indicating the inner and external pores, where “TSE” stands for the “tensile strength effect” artefact. | ||
In all cases type IV adsorption isotherms with H3 hysteresis loops were obtained. The specific surface areas of the pristine and BiOCl decorated nanotubes were found to be very similar (224 and 205 m2 g−1, respectively) as summarized in Table 2. These values compare favourably with data reported for certain high specific surface area BiOCl/TiO2 composite systems in the literature (occasionally as low as 45 (ref. 44) or 39 m2 g−1 (ref. 54)). However, it should be noted that heterostructures with somewhat higher surface areas above 100 m2 g−1 38,43 or even higher, up to around 230 m2 g−1 21 have also been described in the literature. The P25 and BiOCl exhibit relatively low specific surface area (48 and 16 m2 g−1, respectively).
| Sample | as (m2 g−1) | Vpore (cm3 g−1) |
|---|---|---|
| NT | 224 | 0.70 |
| BNT10 | 205 | 0.75 |
| HTNT | 106 | 0.64 |
| HTBNT10 | 75 | 0.68 |
| P25* | 48 | — |
| BiOCl (bulk)* | 16 | — |
The pore size distribution curves before thermal annealing are characterized by two distinct broad peaks at 2.5 and 10 nm pore diameter, and a further sharp one at 3.5 nm. The first broader peak was assigned to the inner pores and wall defects of the tubes, while the second one corresponds to the external cavities among nanotubes, i.e., the intertube space.5 The peak at 3.5 nm diameter arises from the well-known tensile strength effect (TSE) artefact that appears often in pore size distribution curves calculated by Kelvin equation based methods (like the BJH method) from the desorption branch of the isotherm. The TSE artefact is recognizable by the steep drop of the desorption branch onto the adsorption branch at p/p0 = 0.45 for nitrogen sorption,62 which is caused by the collapse of the liquid nitrogen meniscus.
It is clear that the deposition of small surface area BiOCl nanoparticles lowers the surface area of the support, and hence, that of the whole system proportionally. However, subsequent heat treatment significantly affects the specific surface area. Elevated temperature first transforms the layered tubular structures into single-layer nanotubes, and then into TiO2 nanorods. Consequently, the specific surface area drops by ∼50% and ∼64% in the case of titanate (∼106 m2) and BiOCl-decorated nanotubes (∼75 m2), respectively. The total pore volume of the samples was found to be around 0.7 cm3 g−1, which is a typical value for titanate nanotubes.5 With the intertube cavities dominating the overall porosity of the samples, the heat treatment induced nanorod formation caused only a minor decrease in this value. The specific surface area and pore size/volume of the 1% BiOCl decorated samples before and after heat treatment fit in the series in Table 1 (see data Fig. S1 and Table S1 in ESI†) well.
3.3. Crystal structure and phase transformation
It is well known that anatase TiO2 can easily transform into rutile structure at 700–1000 °C.63 Protonated trititanate nanocomposites can transform into anatase phase at around 400 °C (ref. 64 and 65) due to the influence of the supported nanoparticles. Both the resulting structure and morphology can be preserved, or on the contrary, destroyed by the complex effects of the particles.8,66 We therefore annealed the NT and BNT10 samples at 400 °C, and chose this system for further investigations as a potential candidate for the catalytic photodegradation of organic pollutants. Results of the TEM, SEM and XRD investigations are summarized in Fig. 3.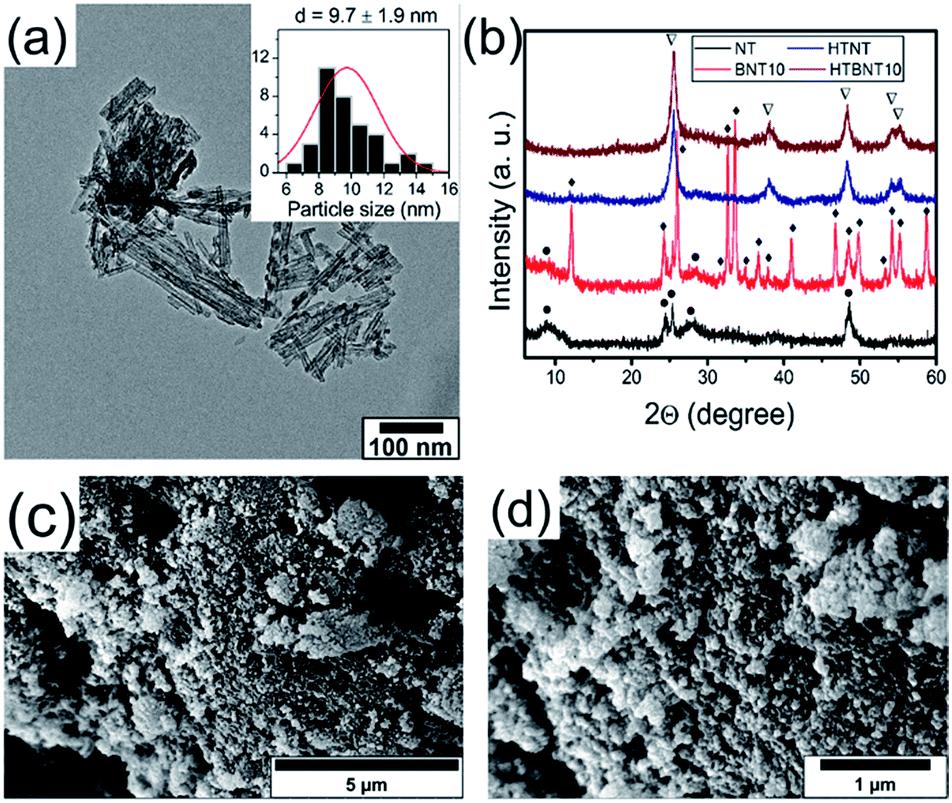 | ||
| Fig. 3 TEM image of the 10% BiOCl decorated TiONT after heat treatment at 400 °C (HTBNT10) (a). XRD patterns of pristine titanate nanotubes (NT) and nanotubes decorated with 10% BiOCl (BNT10) before and after thermal annealing (b). Symbol “•” corresponds to titanate, “∇” to anatase TiO2, “♦” to BiOCl. The morphology of the BiOCl/TiONT composite is shown in the SEM images before (c) and after (d) thermal annealing. | ||
Titanate nanotubes were converted into anatase TiO2 but preserved their morphology up to 400 °C as depicted in Fig. 3a. The 400 °C calcination shortened the nanotubes and kept their tubular morphology mostly intact, although some titanate sheets started fusing into nanorods already. Such sintered features can also be seen in Fig. 3c and d. The XRD patterns in Fig. 3b show reflections characteristic to TiONT and tetragonal BiOCl [JCPDS 00-006-0249] in the pristine samples. Features associated with bismuth-containing species' are almost completely missing from the pattern of calcined BNT10. The almost identical diffraction patterns imply nearly identical titanium-oxide structures in the heat-treated samples, even though the presence of bismuth is clearly verified by the EDS measurements presented in Fig. 5. The same phenomenon was observed in the BNT1 sample before and after annealing (see Fig. S3 in the ESI†).
The ‘disappearance’ of bismuth-containing species' could be explained by the fragmentation of the nanoparticles into even smaller, XRD-invisible pieces as it was found earlier for e.g., TiO2-supported Rh nanoparticles.67 This hypothesis was tested by attempting to sinter/recrystallize them using high temperature annealing. Pristine and BiOCl/TiONT samples were annealed at temperatures up to 900 °C for 1 h and the resulting phase transformations were observed by XRD. Results are shown in Fig. 4. Reflections at 2θ = 9.3°, 24.4°, 25.5°, 27.8° and 48.5° are characteristic to layered trititanate nanotubes,68 where the broad first reflection corresponds to the interlayer distance in the rolled-up structure. These reflections are hardly seen in Fig. 4b (non heat-treated and 200 °C curves), since intensive reflections from the well-crystallized tetragonal BiOCl overlap with the support's diffraction pattern. As a result of the heat treatment between 300 and 700 °C, both NT and BNT10 show anatase TiO2 reflections at 2θ = 25.6°, 37.1°, 38.0°, 38.6°, 48.2°, 54.0°, 55.2° (JCPDS card no. 21-1272).7 The reflections in the XRD profile of BNT10 are sharper than those in pristine nanotubes, implying a higher degree of crystallinity. In the case of BNT10 the anatase to rutile phase transformation starts at as low as 700 °C, whereas in pristine nanotubes this transition commences only at 900 °C as indicated by the rutile TiO2 reflections at 2θ = 27.6°, 36.2°, 39.3°, 41.4°, 44.3°, 54.5° and 56.8° (JCPDS card no. 21-1276) in Fig. 3b. XRD measurements on annealed, 1% BiOCl decorated titanate nanotubes (see Fig. S2 in the ESI†) further justify that the presence of BiOCl facilitates the anatase to rutile phase transformation.
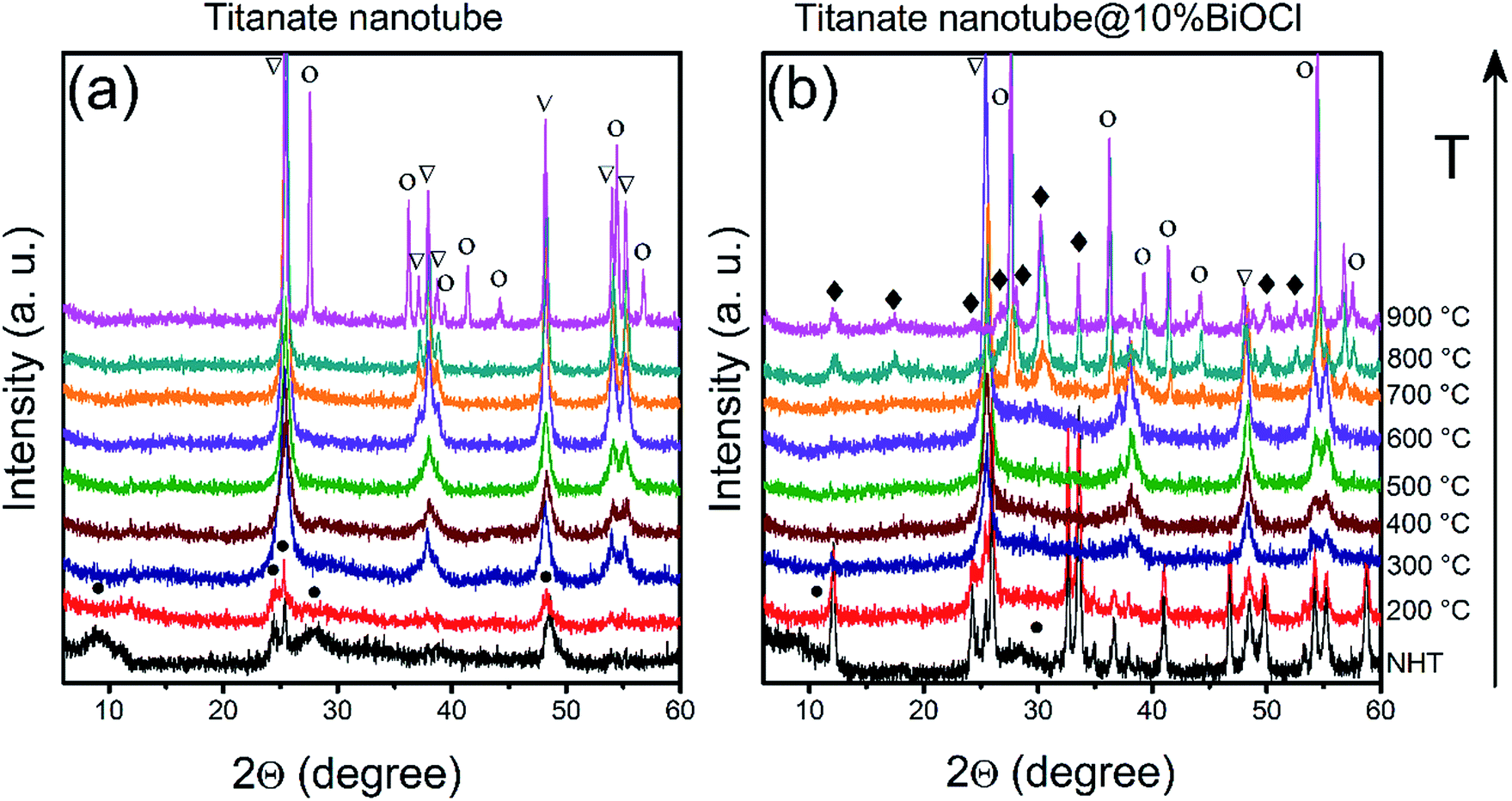 | ||
| Fig. 4 XRD patterns of pristine titanate nanotubes (a) and nanotubes decorated with 10% BiOCl (BNT10) (b) before and after thermal annealing. Symbol “•” corresponds to titanate, “∇” to anatase TiO2, “○” to rutile TiO2 and “♦” to tetragonal BiOCl. NHT: non heat-treated sample. | ||
The peaks at around 2θ = 28–30° in the pattern of BNT10 (but not in that of NT) indicate the presence of monoclinic bismuth titanium oxide (Bi2Ti4O11, JCPDS card no. 83-0673) above 700 °C. This feature helps to shed light on the underlying phase transformations, as it is well-known from the literature, that bismuth titanates with various stoichiometries (e.g., Bi2Ti2O7, Bi2Ti4O11) can be formed in the reaction of BiOCl and titania.69 At lower temperatures Bi2Ti2O7 remains amorphous (and consequently, undetectable by XRD), while above 700 °C the monoclinic Bi2Ti4O11 phase emerges. This is in accordance with our results, which means that above 200 °C, the BiOCl–TiONT system serves as a precursor for the formation of bismuth-titanate materials.
The Bi2Ti2O7 phase is known for its visible light photocatalytic activity in the form of e.g., individual microspheres70 and nanorods,71 or in TiO2-based composite structures.72,73 However, the characteristic marks of the crystalline pyrochlore-type structure of Bi2Ti2O7 cannot be identified in the XRD patterns of the studied system: the amorphous phase seems to transform directly into monoclinic Bi2Ti4O11. We assign this to a support effect, as it is widely known that phase transformation of surface demobilized nanoparticles can be largely affected by the support itself.7,8
3.4. Elemental composition
The elemental composition of the as-prepared nanotubes and the nanoparticle-nanotube systems was investigated by energy dispersive X-ray spectrometry. Characteristic spectra are depicted in Fig. 5.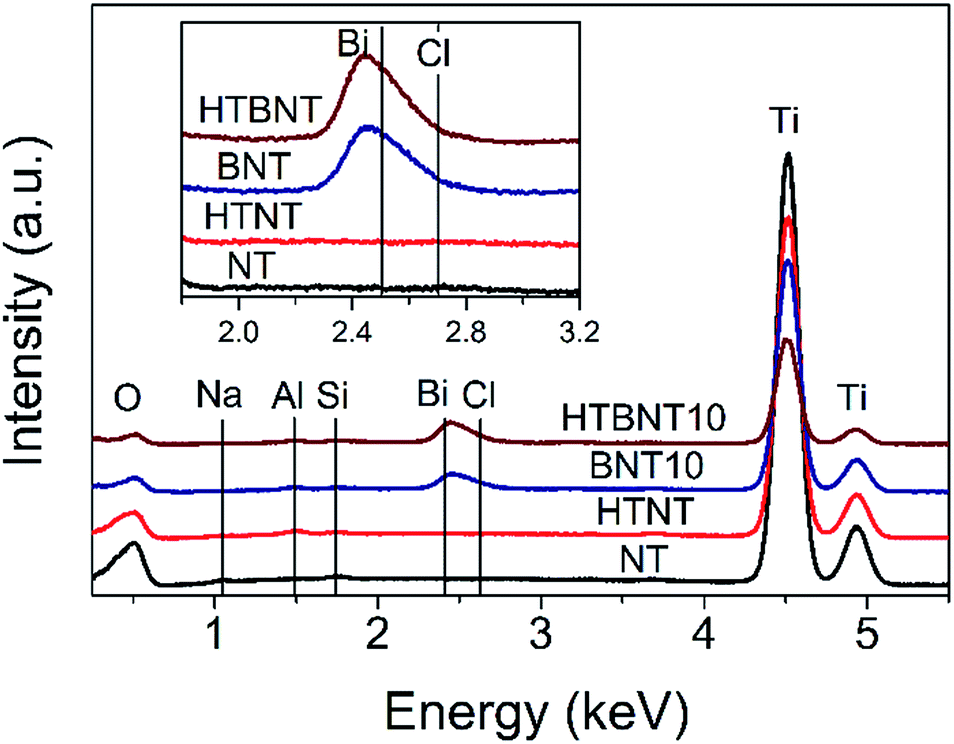 | ||
| Fig. 5 EDS spectra of pristine titanate nanotubes (NT), nanotubes decorated with 10% BiOCl (BNT10), and pristine (HTNT) and 10% BiOCl decorated TiONTs after heat treatment at 400 °C (HTBNT). | ||
All samples contained titanium (4.508 and 4.932 keV) and oxygen (0.525 keV) as expected in a titanate structure. The lack of Na at 1.041 keV confirms the success of the protonation of the as-prepared nanotubes, while the weak Al (1.487 keV) and Si (1.739 keV) signals are due to the sample holder and the detector, respectively. The characteristic peaks of Bi (2.419 keV) and Cl (2.621 keV) show up in the spectra of the BiOCl decorated samples, and are preserved even after the heat treatment.
X-ray photoelectron spectroscopy was employed to characterize the oxidation state of Bi, Ti, O and Cl in the system. Results are seen in Fig. 6 for Bi 4f (a), Ti 2p and Bi 4d (b), O 1s (c) and Cl 2p (d).
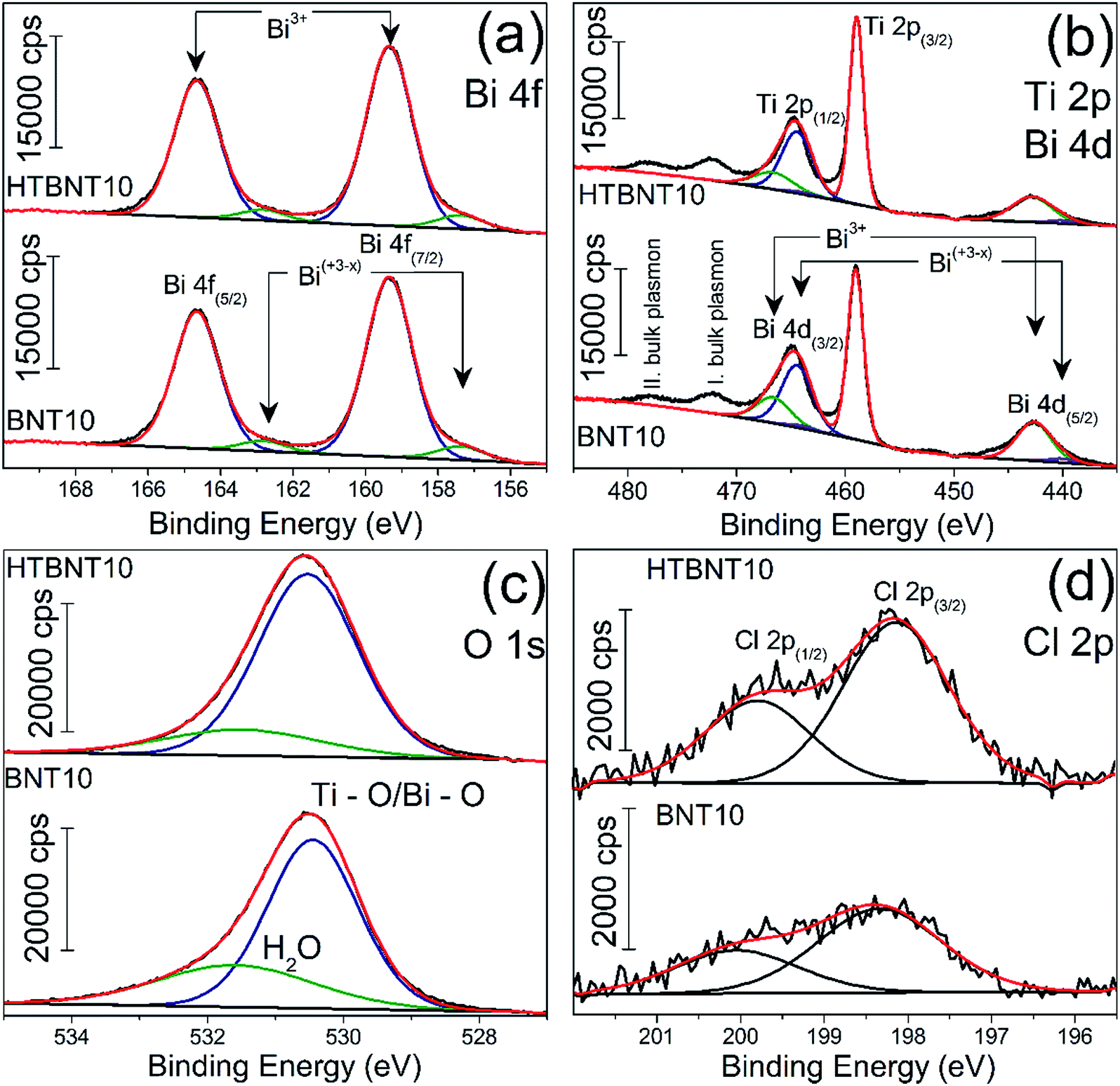 | ||
| Fig. 6 High-resolution XP spectra of the 10% BiOCl decorated TiONT before (BNT10) and after heat treatment (HTBNT10) in the Bi 4f (a) Ti 2p, Bi 4d and Ti 2p (b), O 1s (c) and Cl 2p (d) region. | ||
The bismuth spectrum features two intense peaks at 159.4 and 164.6 eV, as it was also found in BiOCl ultrathin nanosheets earlier.74
These were assigned to trivalent oxidation states of bismuth: Bi3+ 4f7/2 and Bi3+ 4f5/2, respectively. At the low binding energy side, with an energy shift of ∼2 eV, an additional spin–orbit doublet overlapped the major Bi 4f photoemission at 157.4 and 162.8 eV. Similar XP spectra were observed in a series of bismuth oxyiodides with different composition.12,14 The interpretation of those findings was based on the XPS study of Bi4Ti3O12 ferroelectric ceramics, where the authors identified bismuth in a sub-stoichiometric Bi(+3−x) state. The latter was generated due to the enhanced concentration of oxygen vacancies in the vicinity of bismuth cations in the Bi2O2 layers of the material.
Fig. 6b shows the Ti 2p photoemission spectra overlapped by the Bi 4d core level peaks at 159.43 and 164.82 eV which belong to Bi3+ 4d5/2 and Bi3+ 4d3/2, respectively.75 The non-stoichiometric Bi 4d peaks are also detectable by further deconvolution at lower binding energies at 440.11 and 463.98 eV, and we assigned them as Bi(+3−x) 4d5/2 and Bi(+3−x) 4d3/2, respectively. Ti 2p photoemission spectra show a spin–orbit doublet separated by 5.7 eV at 459 and 464.47 eV and characteristic to Ti 2p3/2 and Ti 2p1/2, respectively. We identified only Ti4+ but the titanate nanotubes might also contain Ti3+ as reported by Bavykin et al.5 Its features can be overlapped by those of Ti 4d and the detectability also depends on the type of baseline.5 The two unfitted peaks observable at higher binding energies are probably related to various plasmon effects of titanium according to the TiN XPS studies of D. Jaeger et al.76 We identified the peaks at 472.85 and 478.3 eV as the bulk plasmons of titanium. Heat treatment of the pristine samples does not cause any shift in the Ti 2p spectra in agreement with the previous findings of Pótári et al.8
We deconvoluted the asymmetric O 1s peak in Fig. 6c into two components. The peak at 530.55 eV binding energy might be characteristic to different oxides in the bulk phase. It is commonly assigned to the O2− in Ti–O or Bi–O bonds.74,77 The other component around 531.55–531.63 eV belongs to the structural and/or chemisorbed water. It can be seen that the intensity of the former decreased with the heat treatment along with a shift towards higher binding energies. This behavior is typical for titanate nanotubes during structural water loss, where the majority of the chemisorbed water was found to remain in the system.8
The peaks at binding energies 198.1 and 199.8 eV are characteristic to Cl 2p3/2 and Cl 2p1/2 of Cl−, respectively. After heat treatment at 400 °C, neither the Bi 4f and 4d, nor the Ti 2p, nor the Cl 2p spectrum changed, indicating the stability of the decorating nanoparticles up to this temperature, while a sub-stoichiometric bismuth content remained in the structure.
3.5. Optical properties and band gap of the structures
The band gap energy of a semiconductor is an important (but not exclusive) feature for the prediction of its photocatalytic activity. Lower values mean that less excitation energy is needed to generate excited electron–hole pairs, which in turn, can induce photocatalytic reactions. Our system has been built up by two different types of semiconductor materials: titanate and TiO2 nanotubes are n-type, while BiOCl nanoparticles are p-type semiconductors in most of the cases.44 However, n-type BiOCl samples were also reported,78,90 suggesting the potential effect of the synthesis method used. Furthermore, BixOyClz materials can switch between the p- and n-type characteristics with the change in the actual composition.79 As our supported BiOCl nanoparticles cannot be synthesized in freestanding form to analyse their actual conduction properties, and large body of data show that BiOX is a p-type intrinsic semiconductor, we discuss our findings in this frame. Nonetheless, it is worth noting, that due to the appropriate band edge positions, our qualitative conclusions would remain unaffected if an n-type BiOCl was considered.The optical properties and the corresponding band gaps of the pristine nanotubes, the BiOCl modified materials and their calcined counterparts were determined from diffuse reflectance UV-VIS spectra. These results are shown in Fig. 7, where absorption spectra of NT, HTNT (a), and BNT10, HTBNT10 (b) samples were compared to that of Degussa P25.
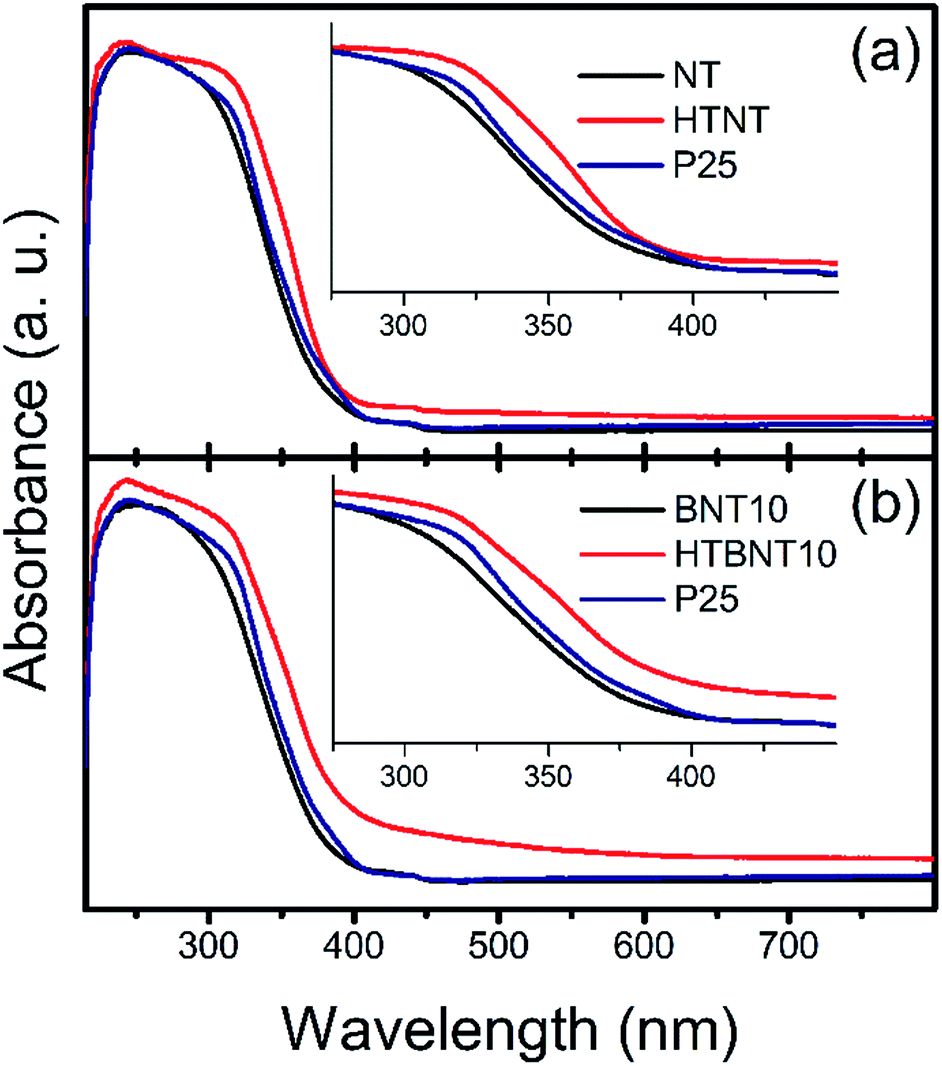 | ||
| Fig. 7 UV-VIS absorption spectra of the pristine titanate nanotubes (a), and nanotubes decorated with 10% BiOCl (b) before (NT and BNT10) and after (HTNT and HTBNT10) annealing. Results for Degussa P25 are also shown for comparison. | ||
It is seen that pristine and 10% BiOCl decorated nanotubes are characterized by lower absorbance in the whole UV-VIS region, accompanied by a rise in absorption only at a lower wavelength in the UV regime compared to P25; thus, spectra are shifted to higher photon energies. After calcination at 400 °C, however, both samples exhibit a higher overall response, which is more pronounced in the case of HTBNT10. Moreover, the absorption step has a lower slope, which implies a lower band gap, and thus, higher excitability in the visible light regime. The 1% BiOCl sample shows similar, but somewhat smaller changes in the UV-VIS regime as can be seen in Fig. S4 in the ESI.† The elevated tail or baseline at higher wavelengths in the spectra of the calcined samples (HTNT, HTBNT10) is characteristic to the formation of defect sites, e.g., oxygen vacancies in black BiOCl80 and Bi/BiOCl composite,81 Bi3+-oxygen vacancy associates in BiOCl nanosheets,57 or Ti3+, Ti2+, and oxygen vacancies in reduced BiOCl/TiO2−x heterojunctions.52
Band gap energies were calculated from both the steep part of the absorbance spectra and by means of the Kubelka–Munk (KM) plot.82 In the latter representation the [F(R∞)hν]1/2 function is plotted against hν for an indirect band gap semiconductor. Here, the value of F(R∞) is derived from the absorption coefficient (α), which can be calculated from the equation of α = A(hν − Eg)n/hν,83 where A is a constant, hν is the energy of the incident light, Eg is the band gap energy and n depends on the nature of the electron transition in the investigated system and equals to 2 in TiO2.84 Band gap energy is then calculated by the extrapolation of the linear range in the spectra to the energy axis, or alternatively, to an extended baseline of the low-energy part of the KM-plot if the elevation of the latter is considerable.85 Band gap values are summarized in Table 3. It is noteworthy that results are largely unaffected by using the extended baseline evaluation.
| Sample | Band gap energies (eV) | |
|---|---|---|
| From abs. spectra | From KM-plots | |
| NT | 3.23 | 3.02 |
| HTNT | 3.21 | 3.06 |
| BNT10 | 3.22 | 3.00 |
| HTBNT10 | 3.10 | 2.85 |
| P25 | 3.17 | 2.95 |
There is a steady ∼0.2 eV difference between the sets of values calculated directly from the spectra and from the Kubelka–Munk plots. The latter method provided lower energies, thus higher visible range absorption properties. Despite of this quantitative discrepancy, identical trends can be found in both datasets.
The band gap of the pristine nanotubes is around 3.2 eV, close to that of P25, and remains practically unchanged upon the deposition of BiOCl nanoparticles. The band gap of BiOCl samples varies between 3.5 and 3.0 eV – the actual value changes with the dominant crystal facets (e.g., Eg(110) < Eg(001)) and subsequent heat treatment. A comprehensive study on nanosized BiOX (X = Cl, Br, I) showed that particle size also has an effect on the band gap in these structures.86 In our case, thermal annealing affects the band gap only if originally semiconductor BiOCl nanoparticles decorate the titanate surfaces. In this case, Eg drops to around 2.9 eV in the HTBNT10 sample. The band gap of the Bi2Ti2O7 phase was found to be between 2.9 and 3.0 eV.73 The transition between the UV and visible regions is around 3.1 eV excitation energy (λ ∼400 nm), and decreasing band gap energy implies higher excitability in the visible regime. The values in Table 3 further confirm the qualitative findings from the UV-VIS spectra of Fig. 7. Results for the 1% BiOCl samples are seen in Fig. S5 and S6 in the ESI.† Trends derived from the UV-VIS spectrum and Kubelka–Munk plots are similar to those observed for the 10% BiOCl samples.
3.6. Photocatalytic activity
The investigated materials were subjected to photocatalytic testing. Visible light irradiation was used to decolorize and decompose an organic dye (methyl orange, MO) as a substrate. The spectrum of the applied light source with and without using a UV cut-off filter is depicted in Fig. S7.† Although the usage of dye decolorization reactions for photocatalytic activity assessment has been criticized recently,87,88 such tests are still widely used in the literature.89 Thus a summary of MO decolorization properties of BiOX/TiOx composites can be found in Table S2,† while the UV-VIS spectra of MO under visible light irradiation are shown in Fig. S8 in the ESI.† The kinetic curves (variation in MO concentration with irradiation time) are depicted in Fig. 8a in linear representation, where lines are guides to the eye. Since unsupported BiOCl nanoparticles cannot be synthesized via the used methods, and large BiOCl flakes from other methods would not be appropriate to compare our results to, we used P25 as a standard reference photocatalyst instead. The same procedure has already been used in visible-light dye decolorization experiments.90 As intensive light sources can initiate chemical processes in compounds with high absorbance, and catalysts with high adsorption capacity can also cause artifacts in photocatalytic measurements, we tested our system for different sources of error. These results are summarized in Fig. 8b.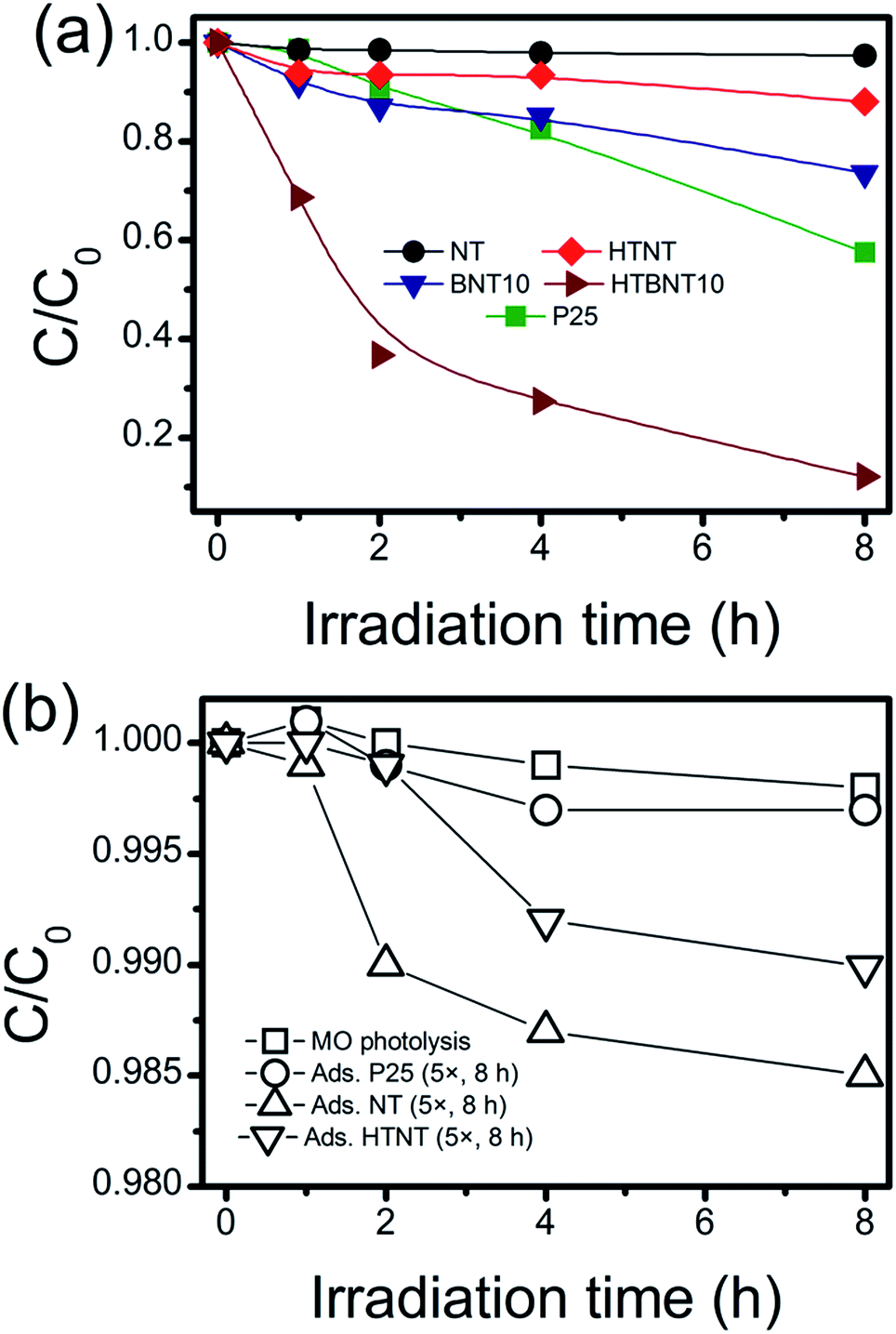 | ||
| Fig. 8 Photocatalytic decolorization curves of methyl orange by visible light irradiation in linear ([MO] vs. irradiation time) representation for NT, HTNT, BNT10 and HTBNT10 samples (a), where lines are guides to the eye, and catalytic performances are compared to that of P25. Effect of adsorption and photolysis on the decolorization of the organic dye (b). Note the higher amount of support in the adsorption experiments! | ||
MO photolysis does not happen in the used setup (Fig. 8b), and the effect of adsorption itself cannot be measured within the experimental error. Therefore, we applied 5× higher support (TiONT, HTNT, P25) concentration to rule out any potential adsorption-related issues. The high amount of high surface area nanotube caused only 1.5% MO loss in an 8 hour-span. It is worth noting that these experiments reflected the differences in the specific surface area of the supports in the amount of dye loss well. The MO consumption was less than 3% in the irradiated TiONT samples in the absence of the BiOCl particles from the titanate surface; hence, bare supports did not show any considerable activity in the dye decolorization experiments, even in a relatively long time span of 8 hours. Although a subsequent 400 °C calcination (HTNT) somewhat enhanced the photocatalytic activity of TiONT, it can still be considered low, as only about 10% of the initial MO was decomposed after eight hours of irradiation. We suggest that this enhancement could be the result of the formation of anatase from the trititanate phase.5 Deposition of BiOCl nanoparticles onto the surface of the pristine titanate nanotubes (BNT10) resulted in an even higher catalytic activity, i.e., a more than two-fold improvement compared to that of the heat treated nanotubes. Although this material cannot compete with the long-term catalytic performance of Degussa P25, is remarkable that the corresponding degradation curves run together in the first 4 hours of the irradiation. Subsequently, P25 degraded 10% more MO in the following two-hour time span. Major improvement was achieved by the calcination of the BiOCl decorated nanotubes at 400 °C. These samples were found to be more active compared to any of the investigated catalysts right from the beginning of the irradiation. They degraded more than 60% of the initial MO within the first 2 hours of irradiation (c/c0 = 0.5 at t ≈ 100 min), and finally eliminated 90% of the organic dye in 8 hours. The 1% BiOCl containing sample (BNT1) shows similar photocatalytic activity to that of the heat treated nanotubes, and a subsequent annealing does not have any further effect on this characteristic, as it is seen in Fig. S9 in the ESI.†
Photocatalytic decolorization of methyl orange can usually be described by the first-order kinetic equation of lnc/c0 = kt,88 where c is the actual and c0 is the initial concentration of the substrate, k is the apparent reaction rate constant (min−1) and t is the irradiation time. If a chemical reaction follows first-order kinetics, the plot of ln(c/c0) against irradiation time provides a linear relationship where the slope of a linear fit gives the apparent reaction rate constant k. These values for our system were summarized in Table 4.
| Sample | k (min−1) |
|---|---|
| NT | (6.94 ± 1.47) × 10−5 |
| BNT10 | (6.82 ± 0.68) × 10−4 |
| HTNT | (2.96 ± 0.53) × 10−4 |
| HTBNT10 | (4.81 ± 0.45) × 10−3 |
| P25 | (1.06 ± 0.09) × 10−3 |
| BiOCl (bulk) | (2.42 ± 0.12) × 10−3 |
We found that the pristine nanotubes (NT) have indeed the lowest rate constant among the investigated catalysts. The deposition of BiOCl nanoparticles (BNT10) improved this parameter by an order of magnitude from around 7 × 10−5 min−1 to ∼7 × 10−4 min−1. The 400 °C heat treatment on the as-prepared nanotubes (HTNT) caused a further two-fold improvement in the catalytic activity compared to that of the BiOCl decorated nanotubes (BNT10). The calcined BNT10 sample (HTBNT10) has even an order of magnitude higher rate constant. The latter sample shows ∼4.5× higher decolorization rate than the widely-known photocatalyst Degussa P25.
The re-usability of the HTBNT10 catalyst was assessed by performing five consecutive 8 hour long MO degradation tests. Results presented in Fig. S12 in the ESI† clearly indicate that the catalyst remained stable and active during this test. Even though photocatalytic performance optimization is out of the scope of this study, the observed stability of HTBNT10 confirms that the material is worth further efforts in this direction.
The promotional effect of a composite photocatalyst can be developed via various mechanisms. If both members of a heterostructure can be excited by the actual irradiation, then charge carriers are generated in either of the materials. This happens, e.g., in TiO2-based composite photocatalysts under UV irradiation.21,38,43 The generated electrons are transported from the higher energy conduction band of bismuth-halogenide to the conduction band of TiO2, while holes are removed from the lower level valence band of TiO2 to the valence band of the BiOX. However, if the excitation energy is insufficient to generate charge carriers in either material, only one-way carrier transport is possible in the structure. A review of the literature revealed the edge positions of the valence and conduction bands in BiOCl, TiONT, and TiO2. These energies are summarized in Fig. S10 in the ESI.† Average values were used in delineating the underlying mechanism. A scheme is proposed in Fig. 9 to explain the photocatalytic activity of the 10% BiOCl decorated sample before (a) and after the formation of the p–n heterojunction.
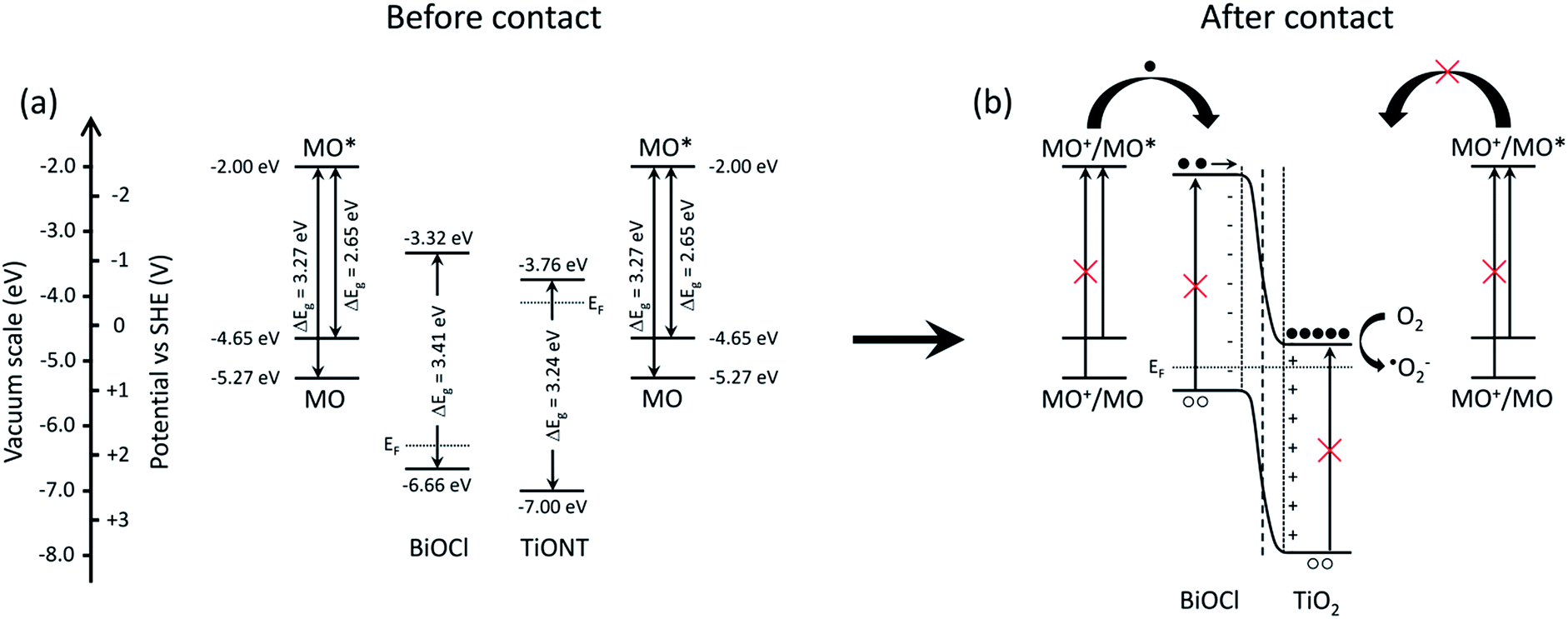 | ||
| Fig. 9 Schematics of the band structure of the BiOCl decorated titania nanotubes (a) and the suggested mechanism of charge generation and subsequent charge separation processes (b). | ||
Qualitatively, the same scheme can be applied to the heat treated BNT1 and BNT10 samples, as the band gap of Bi2Ti2O7 is very similar to that of the pristine materials, and band edges are at higher potentials compared to that of the titanium-oxides.
It is obvious from the band gaps, that methyl orange has the highest absorption cross-section in our system, while the solid cannot be effectively excited by visible light irradiation. The latter effect mainly originates from sub-band gap states introduced during thermal annealing by defect formation, and evidenced by the elevated baseline in UV-VIS spectra in Fig. 7. However, in a system, where an irradiated organic dye is present in the vicinity a metal or semiconductor support, heterogeneous electron transfer, the so-called dye sensitization takes place.91 In photoinduced heterogeneous electron transfer a light absorber is photoexcited and the support acts as an acceptor for the excited electron, leaving the absorber in an oxidized state. The dye donor molecule should be bound to the adsorbent via an appropriate anchor group. For efficient charge separation a semiconductor with a sufficiently large band gap is needed to prevent its direct excitation, while the proper band alignment is also a prerequisite.92
In an indirect dye photosensitization process the initially photoexcited dye molecule injects electrons into an empty electronic state of the conduction band of the semiconductor. The transfer is usually fast. Typically, rise times in the order of tens of femtoseconds were measured in hot electron injection from chemically anchored Ru-dye molecules into the empty electronic states of colloidal anatase TiO2, while the reverse electron transfer of the electron wave packet is practically impossible. The tunneling barrier and the anchor groups fundamentally determine the injection rate, as the adequate overlap between the vibrational states of the reactants' and the products', and the anchoring chromophore groups facilitates ultrafast electron transfer.93 The injected electrons transform the surface-adsorbed molecular oxygen to, e.g., O2−˙ and OH˙ active species. The valence band of the semiconductor does not participate in the indirect photosensitization as it does in direct photoexcitation processes.94
In BiOCl single-crystalline nanosheets, BiOCl was found to act only as an electron-transfer mediator, promoting charge separation of the injected electron and the cationic methyl orange radical.95 We suggest that the photocatalytic activity in our system is due to a similar mechanism, where photogenerated electrons in the excited dye and oxygen species play key roles. In the former study, an active species trapping experiment confirmed the vital role of electrons and oxygen. In our case, since visible light irradiation was used, only the MO molecules were excited as it is seen in Fig. 9. Interestingly, pristine TiONT and TiO2/P25 do not exhibit any photocatalytic activity, as dye sensitization cannot take place with these titanium-oxide forms. The exact reason for the lack of sensitization effect is still unclear. The deposition of BiOCl onto the supports, however, opened up a route to the sensitization process, where the annealing-induced p–n heterojunction formation further facilitates charge carrier separation. Due to the favorable band edge positions, injected electrons move to the conduction band of the titania support, where those are not able to recombine due to the lack of holes. This and the development of an inner electric field at the contact zone of a p-type BiOCl nanoparticle and an n-type titanium-oxide establishes an effective charge separation, and hence, an elongated carrier lifetime. Such lifetime extension was found in a sulphur-doped g-C3N4/BiVO4 composite visible-light photocatalyst, where the formation of the heterostructures extended the charge carrier lifetime compared to that of the pristine components.96 In summary, the photocatalytic activity of the prepared BiOCl/1D titania system stems from the following: (a) good adsorption of the model substrate on titania; (b) efficient charge injection between the dye and the solid; (c) favorable band alignment; (d) the presence of vacancies in the structure extends VIS absorption; (e) high performance heterojunctions, which improve charge separation and elongate charge carrier lifetime via the reduction of the recombination rate.
4. Conclusions
A facile synthesis approach was developed to decorate titanate nanotubes with V–VI–VII compound p-type semiconductor BiOCl nanoparticles. Despite the large body of research done on developing visible light photocatalytic systems using BiOCl lately, data on titania supported BiOCl nanoparticles is lacking. The reaction between BiCl3 and water usually produces BiOCl nanoflakes with the size of hundreds of nanometers, assembled into micron-sized 3D flowers. Here we demonstrated that titanate nanotubes can successfully stabilize smaller BiOCl nanoparticles with ∼5 nm diameter on their surfaces. The BiOCl deposition transformed the originally photocatalytically inactive titanate nanotubes into a system exhibiting photocatalytic activity comparable to that of P25 in the visible light range. Subsequent calcination at 400 °C, was found to improve the catalytic properties of the nanocomposite system further due to the formation of a Bi2Ti2O7/anatase interface and the charge separation across the evolved heterojunction.Acknowledgements
The financial support of the Hungarian Research Development and Innovation Office through grants NKFIH OTKA K 112531, K 120115 and GINOP-2.3.2-15-2016-0013 is acknowledged.References
- H. Kazuhito, I. Hiroshi and F. Akira, Jpn. J. Appl. Phys., 2005, 4, 8269 Search PubMed.
- M. Hodos, E. Horváth, H. Haspel, Á. Kukovecz, Z. Kónya and I. Kiricsi, Chem. Phys. Lett., 2004, 399, 512 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- W. Qin, D. Zhang, D. Zhao, L. Wang and K. Zheng, Chem. Commun., 2010, 46, 2304 RSC.
- D. V. Bavykin and F. C. Walsh, Titanate and titania nanotubes: synthesis, properties and applications, Royal Society of Chemistry, Cambridge, UK, 2010 Search PubMed.
- B. Buchholcz, H. Haspel, A. Kukovecz and Z. Konya, CrystEngComm, 2014, 16, 7486 RSC.
- D. Madarasz, G. Potari, A. Sapi, B. Laszlo, C. Csudai, A. Oszko, A. Kukovecz, A. Erdohelyi, Z. Kónya and J. Kiss, Phys. Chem. Chem. Phys., 2013, 15, 15917 RSC.
- G. Pótári, D. Madarász, L. Nagy, B. László, A. Sápi, A. Oszkó, A. Kukovecz, A. Erdőhelyi, Z. Kónya and J. Kiss, Langmuir, 2013, 29, 3061 CrossRef PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893 RSC.
- S. Bai, W. Jiang, Z. Li and Y. Xiong, ChemNanoMat, 2015, 1, 223 CrossRef CAS.
- W. W. Lee, C. Lu, C. Chuang, Y. Chen, J. Fu, C. Siao and C. Chen, RSC Adv., 2015, 5, 23450 RSC.
- J. Di, J. Xia, M. Ji, S. Yin, H. Li, H. Xu, Q. Zhang and H. Li, J. Mater. Chem. A, 2015, 3, 15108 CAS.
- K. Zhang, C. Liu, F. Huang, C. Zheng and W. Wang, Appl. Catal., B, 2006, 68, 125 CrossRef CAS.
- Y. I. Choi, K. H. Jeon, H. S. Kim, J. H. Lee, S. J. Park, J. E. Roh, M. M. Khan and Y. Sohn, Sep. Purif. Technol., 2016, 160, 28 CrossRef CAS.
- S. Weng, Z. Pei, Z. Zheng, J. Hu and P. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 12380 CAS.
- J. Song, Q. Fan, W. Zhu, R. Wang and Z. Dong, Mater. Lett., 2016, 165, 14 CrossRef CAS.
- X. Zhang, L. Zhang, T. Xie and D. Wang, J. Phys. Chem. C, 2009, 113, 7371 CAS.
- G. Dai, J. Yu and G. Liu, J. Phys. Chem. C, 2011, 115, 7339 CAS.
- Y. Li, J. Wang, B. Liu, L. Dang, H. Yao and Z. Li, Chem. Phys. Lett., 2011, 508, 102 CrossRef CAS.
- Z. Liu, X. Xu, J. Fang, X. Zhu and B. Li, Water, Air, Soil Pollut., 2012, 223, 2783 CrossRef CAS.
- H. Li, Y. Cui, X. Wu, L. Hua and W. Hong, Acta Phys.-Chim. Sin., 2012, 28, 1985 CAS.
- Z. Liu, X. Xu, J. Fang, X. Zhu, J. Chu and B. Li, Appl. Surf. Sci., 2012, 258, 3771 CrossRef CAS.
- X. Wei, H. Cui, S. Guo, L. Zhao and W. Li, J. Hazard. Mater., 2013, 263, 650 CrossRef CAS PubMed.
- Y. Cai, P. Wang, Y. Ye, J. Liu, Z. Tian, Y. Liu and C. Liang, RSC Adv., 2013, 3, 19064 RSC.
- W. Zhao, Z. Liu, S. Shan, W. Zhang, J. Wang, Z. Ma, J. Xu and H. Chen, Sci. Rep., 2014, 4, 4426 Search PubMed.
- D. Wu, H. Wang, C. Li, J. Xia, X. Song and W. Huang, Surf. Coat. Technol., 2014, 258, 672 CrossRef CAS.
- C. Liao, Z. Ma, G. Dong and J. Qiu, Appl. Surf. Sci., 2014, 314, 481 CrossRef CAS.
- Y. Zhang, S. Liu, Z. Xiu, Q. Lu, H. Sun and G. Liu, J. Nanopart. Res., 2014, 16, 2375 CrossRef.
- X. Wei, C. Chen, S. Guo, F. Guo, X. Li, X. Wang, H. Cui, L. Zhao and W. Li, J. Mater. Chem. A, 2014, 2, 4667 CAS.
- L. Li, M. Zhang, Y. Liu and X. Zhang, J. Colloid Interface Sci., 2014, 435, 26 CrossRef CAS PubMed.
- W. Fan, X. Yu, S. Song, H. Bai, C. Zhang, D. Yan, C. Liu, Q. Wang and W. Shi, CrystEngComm, 2014, 16, 820 RSC.
- X. Cao, Z. Lu, L. Zhu, L. Yang, L. Gu, L. Cai and J. Chen, Nanoscale, 2014, 6, 1434 RSC.
- L. Ruan, J. Liu, Q. Zhou, J. Hu, G. Xu, X. Shu and Y. Wu, New J. Chem., 2014, 38, 3022 RSC.
- J. Liu, L. Ruan, S. B. Adelojuc and Y. Wu, Dalton Trans., 2014, 43, 1706 RSC.
- Y. Chen, X. Xu, J. Fang, G. Zhou, Z. Liu, S. Wu, W. Xu, J. Chu and X. Zhu, Sci. World J., 2014, 647040 Search PubMed.
- M. Guerrero, A. Altube, E. García-Lecina, E. Rossinyol, M. Dolors Baró, E. Pellicer and J. Sort, ACS Appl. Mater. Interfaces, 2014, 6, 13994 CAS.
- D. Sun, J. Li, L. He, B. Zhao, T. Wang, R. Li, S. Yin, Z. Feng and T. Sato, CrystEngComm, 2014, 16, 7564 RSC.
- X. Wang, W. Yang, F. Li, J. Zhao, R. Liu, S. Liu and B. Li, J. Hazard. Mater., 2015, 292, 126 CrossRef CAS PubMed.
- Y. Zhang, Q. Pei, J. Liang, T. Feng, X. Zhou, H. Mao, W. Zhang, Y. Hisaeda and X. Song, Langmuir, 2015, 31, 10279 CrossRef CAS PubMed.
- L. Wang and W. A. Daoud, Appl. Surf. Sci., 2015, 324, 532 CrossRef CAS.
- C. Xue, X. Xu, G. Yang and S. Ding, RSC Adv., 2015, 5, 102228 RSC.
- F. Duo, Y. Wang, C. Fan, X. Mao, X. Zhang, Y. Wang and J. Liu, Mater. Charact., 2015, 99, 8 CrossRef CAS.
- K. Wang, C. Shao, X. Li, X. Zhang, N. Lu, F. Miao and Y. Liu, Catal. Commun., 2015, 67, 6 CrossRef CAS.
- S. Luo, C. Tang, Z. Huang, C. Liu, J. Chen and M. Fang, Ceram. Int., 2016, 42, 15780 CrossRef CAS.
- T. Xin, L. Xiangli, Y. Tao and Z. Yang, Trans. Tianjin Univ., 2016, 22, 211 CrossRef.
- Q. Teng, X. Zhou, B. Jin, J. Luo, X. Xu, H. Guan, W. Wang and F. Yang, RSC Adv., 2016, 6, 36881 RSC.
- Z. Li, M. Wang, J. Shen, Z. Zhu and Y. Liu, RSC Adv., 2016, 6, 30037 RSC.
- X. Zhang, H. Yang, B. Zhang, Y. Shen and M. Wang, Adv. Mater. Interfaces, 2016, 3, 1500273 CrossRef.
- J. Li, J. Zhong, Y. Si, S. Huang, L. Dou, M. Li, Y. Liu and J. Ding, Solid State Sci., 2016, 52, 106 CrossRef CAS.
- W. Li, Y. Tian, H. Li, C. Zhao, B. Zhang, H. Zhang, W. Geng and Q. Zhang, Appl. Catal., A, 2016, 516, 81 CrossRef CAS.
- R. Fu, X. Zeng, L. Ma, S. Gao, Q. Wang, Z. Wang, B. Huang, Y. Dai and J. Lu, J. Power Sources, 2016, 312, 12 CrossRef CAS.
- Y. Zhao, X. Huang, X. Tan, T. Yu, X. Li, L. Yang and S. Wang, Appl. Surf. Sci., 2016, 365, 209 CrossRef CAS.
- K. Wang, C. Shao, X. Li, F. Miao, N. Lu and Y. Liu, Materials, 2016, 9, 90 CrossRef.
- J. Henle, P. Simon, A. Frenzel, S. Scholz and S. Kaskel, Chem. Mater., 2007, 19, 366 CrossRef CAS.
- G. Li, F. Qin, R. Wang, S. Xiao, H. Sun and R. Chen, J. Colloid Interface Sci., 2013, 409, 43 CrossRef CAS PubMed.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411 CrossRef CAS PubMed.
- K. Li, W. W. Lee, C. Lu, Y. Dai, S. Chou, H. Chen, H. Lin and C. Chen, J. Taiwan Inst. Chem. Eng., 2014, 45, 2688 CrossRef CAS.
- Z. Liu, W. Xu, J. Fang, X. Xu, S. Wu, X. Zhu and Z. Chen, Appl. Surf. Sci., 2012, 259, 441 CrossRef CAS.
- M. Zhao, L. Dong, C. Li, L. Yu and P. Li, Chin. Phys. Lett., 2015, 32, 098101 CrossRef.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160 CrossRef CAS.
- T. Kanyó, Z. Kónya, Á. Kukovecz, F. Berger, I. Dékány and I. Kiricsi, Langmuir, 2004, 20, 1656 CrossRef.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2010, 46, 855 CrossRef.
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807 CrossRef CAS.
- S. H. Lim, J. Luo, Z. Zhong, W. Ji and J. Lin, Inorg. Chem., 2005, 44, 4124 CrossRef CAS PubMed.
- A. Rónavári, B. Buchholcz, Á. Kukovecz and Z. Kónya, J. Mol. Struct., 2013, 1044, 104 CrossRef.
- A. Berkó, I. Ulrych and K. C. Prince, J. Phys. Chem. B, 1998, 102, 3379 CrossRef.
- H. Zhang, G. R. Li, L. P. An, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2007, 111, 6143 CAS.
- T. Kidchob, L. Malfatti, D. Marongiu, S. Enzo and P. Innocenzi, J. Am. Ceram. Soc., 2010, 93, 2897 CrossRef CAS.
- Z. Bian, Y. Huo, Y. Zhang, J. Zhu, Y. Lu and H. Li, Appl. Catal., B, 2009, 91, 247 CrossRef CAS.
- L. Z. Pei, H. D. Liu, N. Lin and H. Y. Yu, J. Alloys Compd., 2015, 622, 254 CrossRef CAS.
- H. Liu, Y. Chen, G. Tian, Z. Ren, C. Tian and H. Fu, Langmuir, 2015, 31, 5962 CrossRef CAS PubMed.
- D. Zhou, H. Yang, Y. Tu, Y. Tian, Y. Cai, Z. Hu and X. Zhu, Nanoscale Res. Lett., 2016, 11, 193 CrossRef PubMed.
- Y. Wu, B. Yuan, M. Li, W. Zhang, Y. Liu and C. Li, Chem. Sci., 2015, 6, 1873 RSC.
- Č. Jovalekić, M. Pavlović, P. Osmokrović and L. Atanasoska, Appl. Phys. Lett., 1998, 72, 1051 CrossRef.
- D. Jaeger and J. Patscheider, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 523 CrossRef CAS.
- S. Kang, R. C. Pawar, Y. Pyo, V. Khare and C. S. Lee, J. Exp. Nanosci., 2016, 11, 259 CrossRef CAS.
- S. Weng, B. Chen, L. Xie, Z. Zheng and P. Liu, J. Mater. Chem. A, 2013, 1, 3068 CAS.
- Y. Myung, F. Wu, S. Banerjee, A. Stoica, H. Zhong, S. Lee, J. Fortner, L. Yang and P. Banerjee, Chem. Mater., 2015, 27, 7710 CrossRef CAS.
- L. Ye, K. Deng, F. Xu, L. Tian, T. Peng and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 82 RSC.
- Y. Yu, C. Cao, H. Liu, P. Li, F. Wei, Y. Jiang and W. Song, J. Mater. Chem. A, 2014, 2, 1677 CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 293 Search PubMed.
- R. Beranek and H. Kisch, Photochem. Photobiol. Sci., 2008, 7, 40 CAS.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042 CrossRef CAS.
- Z. Chen, H. N. Dinh and E. Miller, Photoelectrochemical Water Splitting – Standards, Experimental Methods, and Protocols, Springer, Heidelberg, 2013 Search PubMed.
- L. Chen, S. Yin, R. Huang, Y. Zhou, S. Luo and C. Au, Catal. Commun., 2012, 23, 54 CrossRef CAS.
- X. Yan, T. Ohno, K. Nishijima, R. Abe and B. Ohtani, Chem. Phys. Lett., 2006, 429, 606 CrossRef CAS.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21 CrossRef CAS.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J. Herrmann, Appl. Catal., B, 2002, 39, 75 CrossRef CAS.
- J. Hu, W. Fan, W. Ye, C. Huang and X. Qiu, Appl. Catal., B, 2014, 158–159, 182 CrossRef CAS.
- D. Chatterjee and A. Mahata, J. Photochem. Photobiol., A, 2002, 153, 199 CrossRef CAS.
- J. Nieto-Pescador, B. Abraham and L. Gundlach, J. Phys. Chem. Lett., 2014, 5, 3498 CrossRef CAS PubMed.
- T. Hannappel, B. Burfeindt, W. Storck and F. Willig, J. Phys. Chem. B, 1997, 101, 6799 CrossRef CAS.
- J. Jiang, K. Zhao, X. Xiao and L. Zhang, J. Am. Chem. Soc., 2012, 134, 4473 CrossRef CAS PubMed.
- Y. Zhang, Z. Jiang, J. Huang, L. Y. Lim, W. Li, J. Deng, D. Gong, Y. Tang, Y. Lai and Z. Chen, RSC Adv., 2015, 5, 79479 RSC.
- H. J. Kong, D. H. Won, J. Kim and S. I. Woo, Chem. Mater., 2016, 28, 1318 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28490f |
| ‡ Present address: King Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC) and Physical Sciences and Engineering Division (PSE), Thuwal, 23955-6900, Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2017 |