
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilization of Amano lipase from Pseudomonas fluorescens on silk fibroin spheres: an alternative protocol for the enantioselective synthesis of halohydrins†
Irlon M. Ferreira ab,
Sergio A. Yoshiokac,
João V. Comassetode and
André L. M. Porto*a
ab,
Sergio A. Yoshiokac,
João V. Comassetode and
André L. M. Porto*a
aLaboratório de Química Orgânica e Biocatálise, Instituto de Química de São Carlos, Universidade de São Paulo, Av. João Dagnone, Ed. Química Ambiental, J. Santa Angelina, 1100, 13563-120 São Carlos, São Paulo, Brazil. E-mail: almporto@iqsc.usp.br
bUniversidade Federal do Amapá, Grupo de Biocatálise e Biotransformação em Química Orgânica, Rod. Juscelino Kubitscheck, KM 02, S/N – J. Marco Zero, 68903-419, Macapá, AP, Brazil
cLaboratório de Bioquímica e Biomateriais, Instituto de Química de São Carlos, Universidade de São Paulo, Av. Trabalhador São-Carlense, 400, 13560-970 São Carlos, SP, Brazil
dInstituto de Química, Universidade de São Paulo, Av. Professor Lineu Prestes 748, 05508-000 São Paulo, SP, Brazil
eInstituto de Ciências Ambientais, Químicas e Farmacêuticas, Universidade Federal de São Paulo, Av. Prof. Artur Riedel, 275, 09972-270 Diadema, SP, Brazil
First published on 22nd February 2017
Abstract
The search for a new, efficient, cheaper and sustainable matrix for lipase immobilization is a growing area in biotechnology. Amano lipase from Pseudomonas fluorescens was immobilized on silk fibroin spheres and used in the enzymatic kinetic resolution of halohydrins, to obtain optically active epoxides (up to 99% ee), important precursors in the synthesis of derivative antifungal azoles. This paper reinforces the versatility of silk fibroin as a support for heterogeneous catalysts.
Introduction
Enzymes are versatile biocatalysts capable of turning a wide range of reactions with high selectivity, including hydrolysis, esterification and transesterification.1–4 In many cases, the native enzymes are not ideal for commercial applications owing to their tendency to denaturate under harsh industrial conditions and separation, recovery and reutilization difficulties.5To some extent, enzyme immobilization is an effective way to overcome such limitations, since multiple fixing points of the support may limit undesirable conformational changes in the enzyme protein chains in hostile environments and the insoluble supports can be more easily recovered than the soluble not immobilized.6 Currently, a variety of supports, organics and inorganics has been reported for the immobilization of enzymes, for example, chitosan,7 cellulose,8 silica xerogel,9 or hybrid organic–inorganic,10,11 such as, zeolitic imidazolate,12 crystal nanoflower13,14 and copper hydroxysulfate nanocrystals.15
In between the biomaterials used in enzymes immobilization, the most popular is cotton fiber.16 Some studies have also been carried out on proteins like lectins17 and silk fibroin.18,19 However, it is necessary to advance in the studies of enzymatic immobilization on silk microfibroin.20,21 Silk fibroin is a class of biopolymers obtained from the Bombyx mori silk worm cocoon that consists in a protein of molecular weight between 300 and 420 kDa. Silk is a protein of a block copolymer structure dominated by large hydrophobic domains and small hydrophilic spacers. This primary sequence, upon folding into assembled silk structures, leads to organized crystalline domains (β-sheets) and less organized, more flexible domains (more hydrated).22,23
Furthermore, silk fibroin biomaterials are stable to moisture and changes in temperature and mechanically robust, due to the extensive network of physical cross-links (β-sheets) formed during the assembly process. Such biomaterial offers some important features that suggest its use as a support of enzymes due to its unique tensile strength, elasticity, good thermal stability, hygroscopicity and microbial resistance.24,25
Azole-derivative agents have been used in various treatments of anti-microbial infections. The azole portion of the molecule is essential for the high antifungal activity.26,27 The therapeutic properties of their enantiomers differ. Therefore pure enantiomers can be administered at doses half of those used for the racemic drug, which suppresses minor risks of side effects and unspecific toxicities derived from the administration of the non-active S-enantiomer.28
We recently, investigated the enzymatic kinetic resolution (EKR) of chlorohydrins using Amano AK Pseudomonas fluorescens lipase immobilized in a blend of silk fibroin–alginate. The enantioselectivity of the process was sufficient for the production of the acetates in good yields and high enantiomeric excesses.20,21
Therefore, the present paper reports on the immobilization of commercially available Amano AK Pseudomonas fluorescens lipase in a new material prepared from silk fibroin to obtain halohydrins enantio-enriched, precursors derived from azoles (Fig. 1). The main purpose of the use of biodegradable fibroin as an immobilization support was the development of an environmentally benign immobilization protocol that avoids the use of harmful chemical reagents.
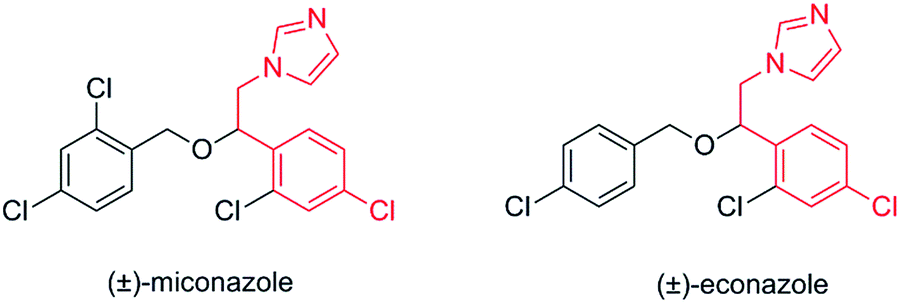 | ||
| Fig. 1 Structures of antifungal agents (±)-miconazole and (±)-econazole. | ||
Results and discussion
Initially a screening with different lipases was performed to determine the most efficient for the kinetic resolution of halohydrin. Four free lipases were used as the catalysts. The results are summarized in Table 1.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Lipasea | X | Time (h) | cb (%) | eepc (2) | eesc (1) | Ed |
| a General conditions: substrate (0.10 mmol), lipase (20 mg), vinyl acetate (0.35 mol, 30 mg), n-hexane (1 mL), 32 °C, 130 rpm.b Conversion: c = ees/(ees + eep).c ee: enantiomeric excess.d E = ln[eep(1 − ees)]/(eep + ees)/ln[eep(1 + ees)]/(eep + ees). Nc: no conversion. | |||||||
| 1 | Pseudomonas fluorescens | Br | 24 | 35 | 96 | 52 | 82 |
| 2 | Br | 48 | 48 | 92 | 88 | 70 | |
| 3 | Cl | 24 | 28 | 98 | 38 | 144 | |
| 4 | Cl | 48 | 35 | 98 | 53 | 168 | |
| 5 | Candida cylindracea | Br | 24 | 23 | 54 | 16 | 3.9 |
| 6 | Br | 48 | 36 | 48 | 27 | 3.7 | |
| 7 | Cl | 24 | 14 | 53 | 9 | 3.5 | |
| 8 | Cl | 48 | 17 | 73 | 15 | 7.4 | |
| 13 | Rhizopus niveus | Br | 24 | Nc | — | — | — |
| 14 | Br | 48 | 0.5 | 75 | 0.4 | 7 | |
| 15 | Cl | 24 | Nc | — | — | — | |
| 16 | Cl | 48 | 2.7 | 17 | 0.5 | 1.5 | |
| 17 | Aspergillus niger | Br | 24 | 4 | 12 | 0.5 | 1.3 |
| 18 | Br | 48 | 10 | 12 | 1 | 1.4 | |
| 19 | Cl | 24 | 3 | 14 | 0.5 | 7 | |
| 20 | Cl | 48 | 18 | 51 | 11 | 3.4 | |
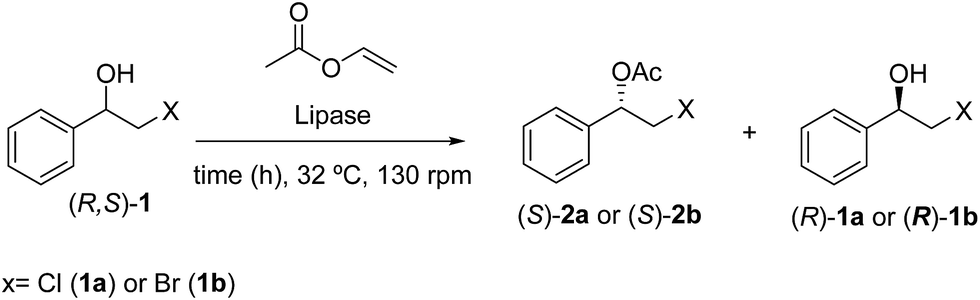
As shown in Table 1, Candida cylindracea lipase exhibited a low conversions (c = 17–36%) and selectivity of 73–48% eep for (S)-2a and (S)-2b, respectively, at 48 h reaction time (entries 6 and 8, Table 1). Rhizopus niveus lipase showed no activity for the transesterification reaction of (R,S)-1a and (R,S)-1b. The best result for a free lipase was obtained with P. fluorescens at 32 °C, 130 rpm for 48 h, yielding (S)-2a and (S)-2b with excellent optical purities (98 and 92% eep; respectively) and good conversions (c = 35 and 48%; respectively) entries 4 and 2, Table 1.
After screening with different lipases was decided immobilize lipase from P. fluorescens on silk fibroin spheres and to test the behaviour of the immobilized enzyme in reaction with (R,S)-1a (Table 2) (see Experimental).
| Entry | Mass of the silk fibroin spheres/P. fluorescens lipasea (mg) | Time (h) | cb (%) | eepc (%) (S)-2a | eesc (%) (R)-1a | Ed |
|---|---|---|---|---|---|---|
| a General conditions: Pseudomonas fluorescens lipase immobilized on silk fibroin spheres, substrate (0.12 mmol, 20 mg), vinyl acetate 25 μL (0.25 mmol), n-hexane (1 mL), 32 °C, 130 rpm.b Conversion: c = ees/(ees + eep).c ee: enantiomeric excess.d E = ln[eep(1 − ees)]/(eep + ees)/ln[eep(1 + ees)]/(eep + ees). | ||||||
| 1 | 50 | 24 | 6 | 99 | 10 | >200 |
| 2 | 100 | 24 | 10 | 99 | 11 | >200 |
| 3 | 150 | 24 | 12 | 99 | 14 | >200 |
| 4 | 150 | 48 | 26 | 99 | 34 | >200 |
| 5 | 150 | 72 | 39 | 98 | 62 | 187 |
The enzymatic resolution for (R,S)-1a in reactions performed with different amounts of the immobilized lipase for 24 h led to low conversions (6%, 10% and 12% respectively), however, with excellent enantioselectivity (E > 200) in both cases (entries 1–3, Table 2). When the reaction carried out with 150 mg of immobilized lipase remained for 48 h in 130 rpm (entry 4, Table 2), the conversion was 26% and 99% eep with excellent enantioselectivity (E = >200) for (S)-2a. When the reaction time was increased to 72 h, the conversion increased to 39% (entry 5, Table 2), the enantioselectivity (E = 187) and enantiomeric excess (98% eep) for (S)-2a. The comparison of the enantioselectivity of the free lipase with the immobilized form under similar reaction conditions demonstrates the lower enantioselectivity of the lipase in the native form. These facts suggest that immobilization of P. fluorescens lipase on silk fibroin spheres promotes changes on the enzyme conformation, leading to more effective binding of one of the enantiomers to the active site of the lipase with favorable formation of the acyl-enzyme intermediate.
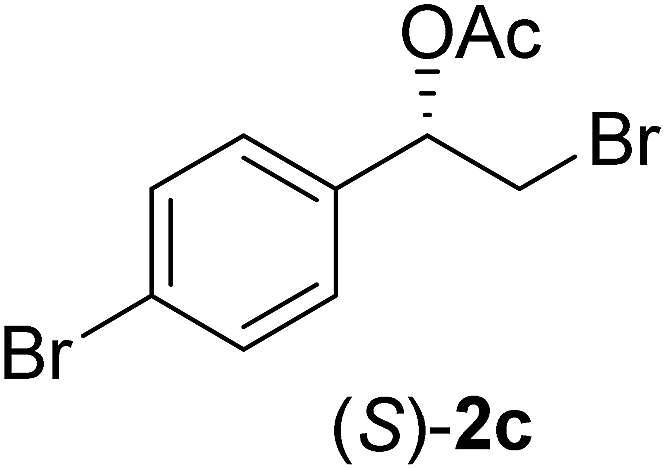
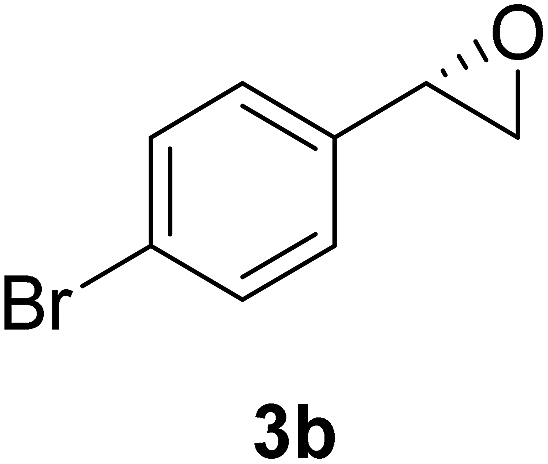
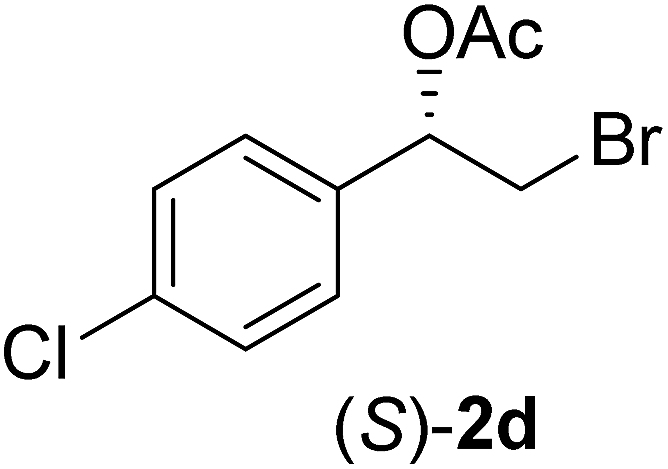
In order to examine the scope and limitations of the process, the protocol was realized on scale larger and extended to compounds (R,S)-1a–e, with different substituent groups in the aromatic ring. The enzymatic resolution of (R,S)-2-bromo-1-(4-bromophenyl)ethanol (1c) with P. fluorescens lipase immobilized on silk fibroin spheres for 96 h, led to (S)-acetate 2c in 35% conversion and 99% eep (entry 3, Table 3). For compound (R,S)-1d under similar conditions, (S)-acetate-2d was achieved with 24% conversion and 99% eep (entry 4, Table 3), whereas compound (R,S)-1e was recovered unchanged after 120 h at 32 °C and 130 rpm, what suggests the protocol is sensitive to steric effects since 1e presents a chlorine atom at the ortho position relative to the organic chain bearing the secondary alcohol.
| Entry | Substrate | Time (h) | cb (%) | eepc (%) (S)-2 | eesc (%) (R)-1 | Ed |
|---|---|---|---|---|---|---|
| a Determined by chiral GC-FID analysis.b Conversion: c = ees/(ees + eep).c ee: enantiomeric excess.d E = ln[eep(1 − ees)]/(eep + ees)/ln[eep(1 + ees)]/(eep + ees). Nc: no conversion. | ||||||
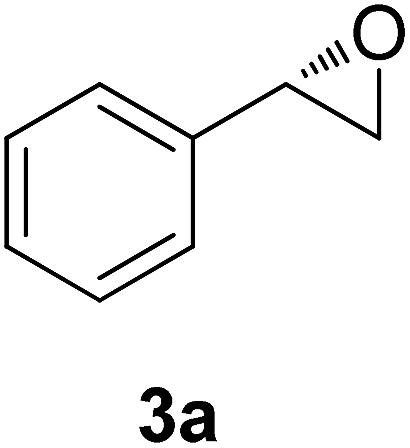
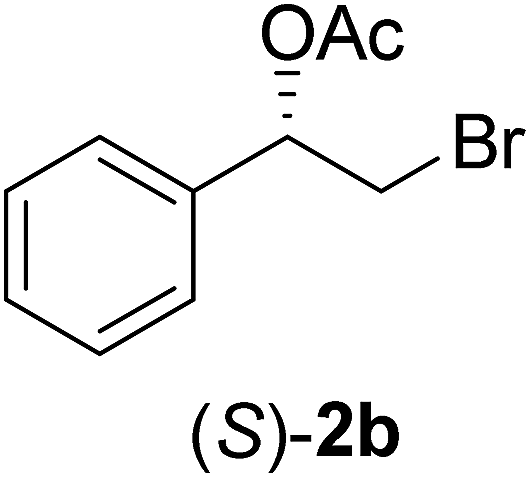
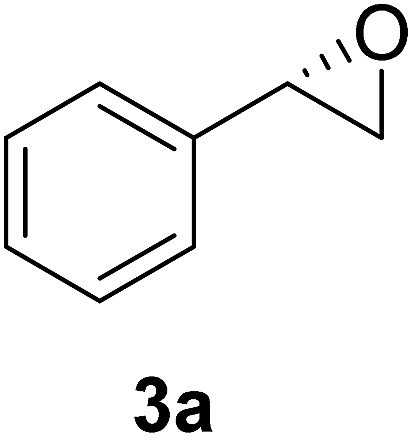
| 1 | 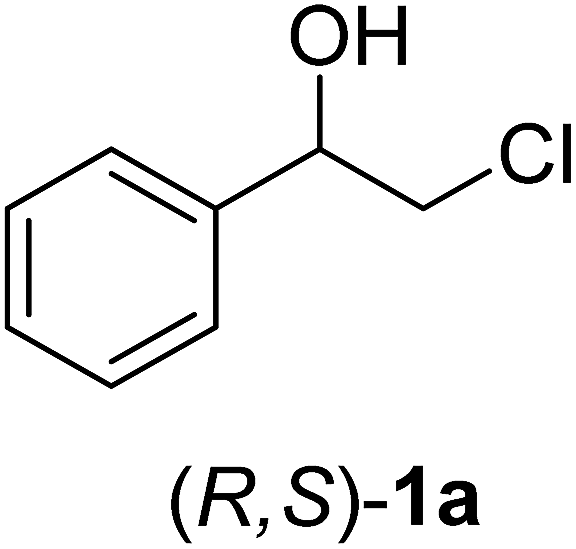 |
72 | 39 | 98 | 62 | 187 |
| 2 | 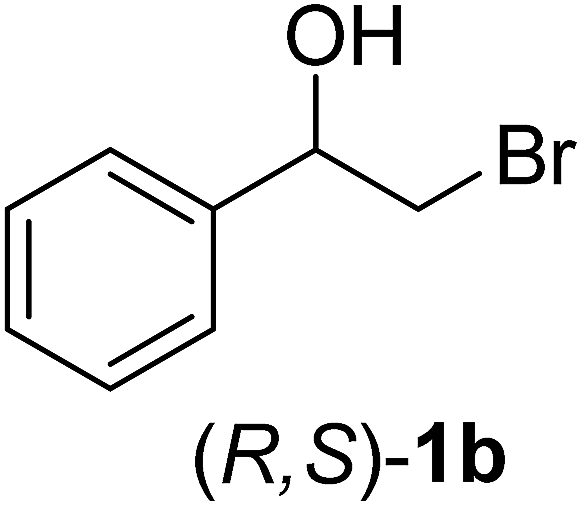 |
96 | 19 | 98 | 23 | 120 |
| 3 | 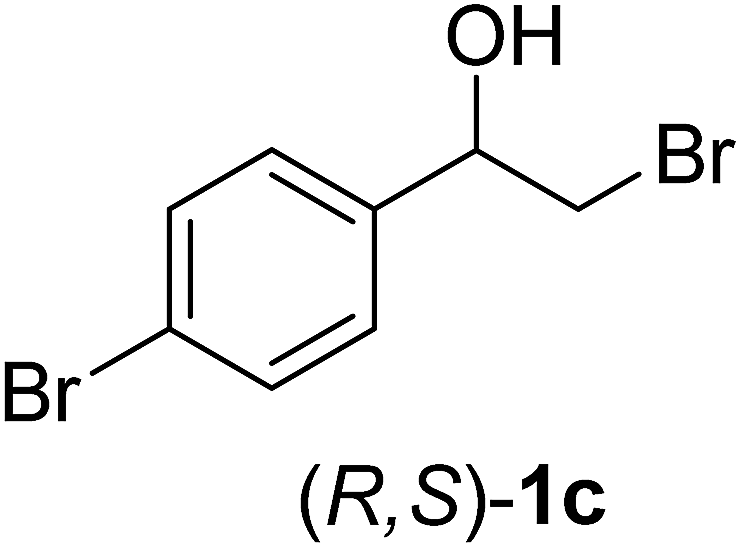 |
96 | 35 | 99 | 55 | >200 |
| 4 |  |
96 | 24 | 99 | 32 | >200 |
| 5 | 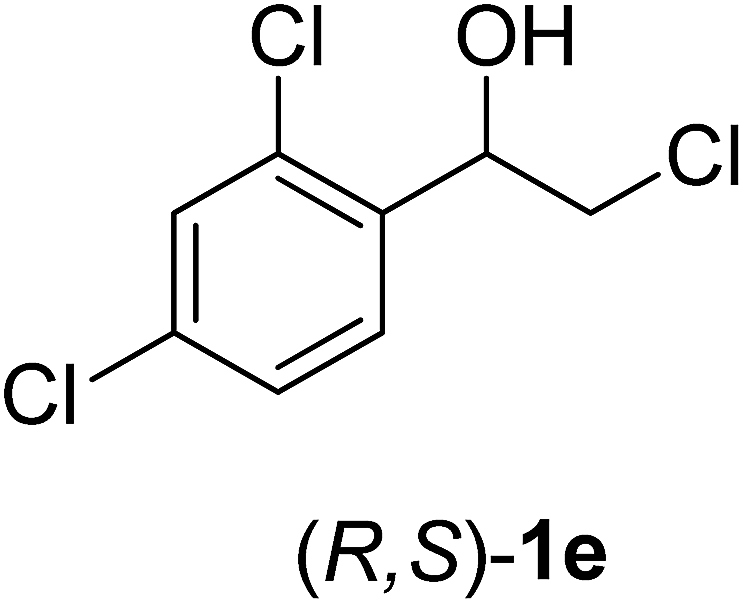 |
120 | Nc | (—) | (—) | (—) |
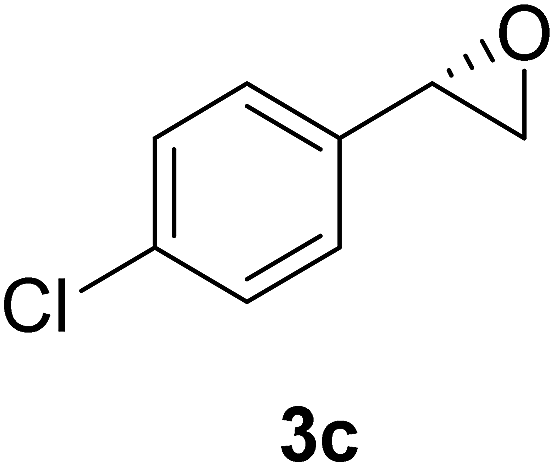
The chlorohydrin acetates (S)-2a–d (Table 3) were reacted with LiOH in ethanol for 1 h so as to yield the corresponding enantiomerically pure epoxides (S)-3a–c (Table 4). This approach provided styrene oxides of high eep.
The recycling of enzymes is important in biocatalytic processes, therefore the reutilization of the immobilized P. fluorescens lipase on silk fibroin spheres was tested. The results (Fig. 2) show that the enantiomeric excesses values did not significantly decrease after 5 consecutive operations at 32 °C, 130 rpm and 96 h reaction time, which indicates that the fibroin–lipase couple, can retain high enantioselectivity and catalytic performance in the recycling process. However, the conversion of (R,S)-1a to (S)-2a declined over successive reuses. The second cycle yielded approximately 22% of the alcohol; in the third cycle the conversion suffered a slight decrease, converted 21% of the starting (R,S)-1a in the acetate, whereas in the fourth cycle, 12% conversion was observed and in the fifth cycle a conversion of only 3% was achieved.
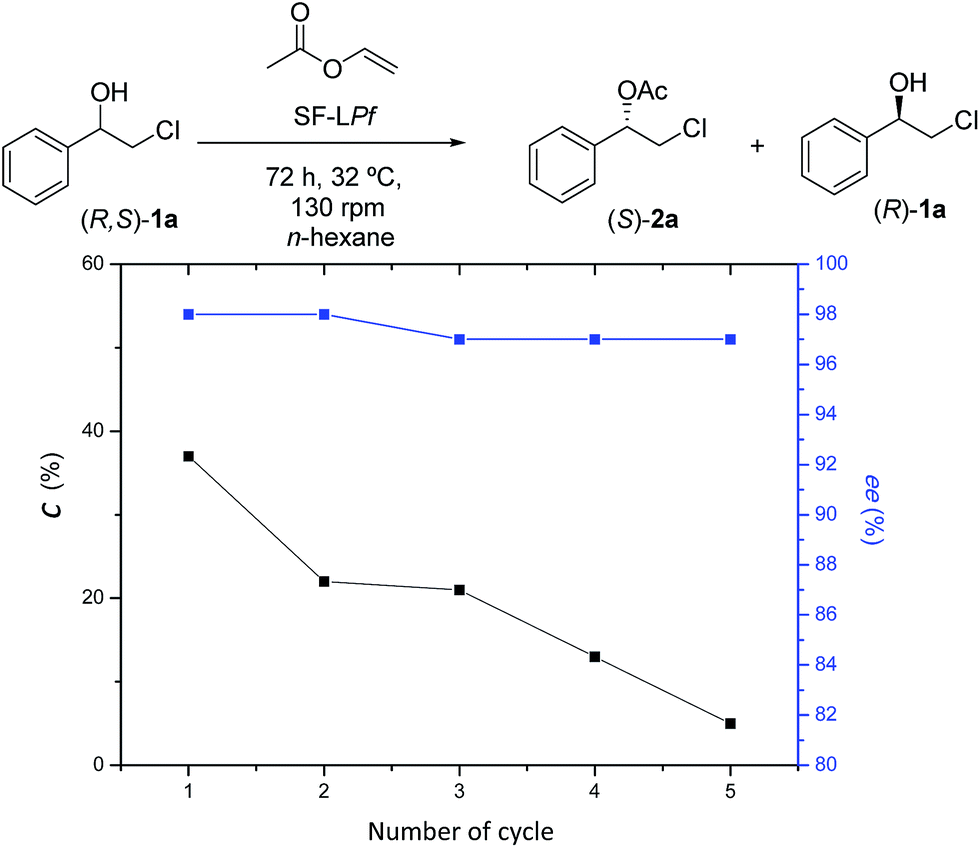 | ||
| Fig. 2 Reuse of FS-LPf in the EKR of (R,S)-1a. Conversion (■); enantiomeric excess of the (S)-2a (*). | ||
This decrease in the conversion of (R,S)-1a to (S)-2a may be due to the detachment of the enzyme from the support, since both are linked only by mechanical and physical adsorption.
The thermograms of the samples, lipase on fibroin spheres and free lipase, showed similar trends (ESI†). The initial weight loss of the samples observed around 100–110 °C is due to the loss of moisture, physically adsorbed water molecules seem to be eliminated. In the thermogram (A), with increasing the temperature, the weight residue declined at around 330 °C, probably due to the breakdown of side chain groups of amino acid residues related to silk fibroin. Similar type of improved stability was observed by Moraes et al.29 for the silk fibroin. The thermogram (B) is related to the fibroin supported lipase by adsorption, besides the peak at 330 °C the thermogram showed mass decrease also at 285 °C, suggesting alteration in the properties of the silk fibroin spheres. It is noteworthy that well oriented fibers typically have peak thermal decomposition acim to 300 °C, in contrast, silk fibroin with sheet-β not oriented tend to have next thermal decomposition at 290 °C.29
The fibroin microspheres showed irregular shapes. We guess that the unevenness on the surface of the microsphere can be due to presences of proteins β-sheet structures. The fibroin fiber presents hydrophobic groups which interact with the hydrophobic groups of the lipase of P. fluorescens. In this way it was not able to morphological difference by microscopy the immobilized of the not immobilized enzyme (Fig. 3).
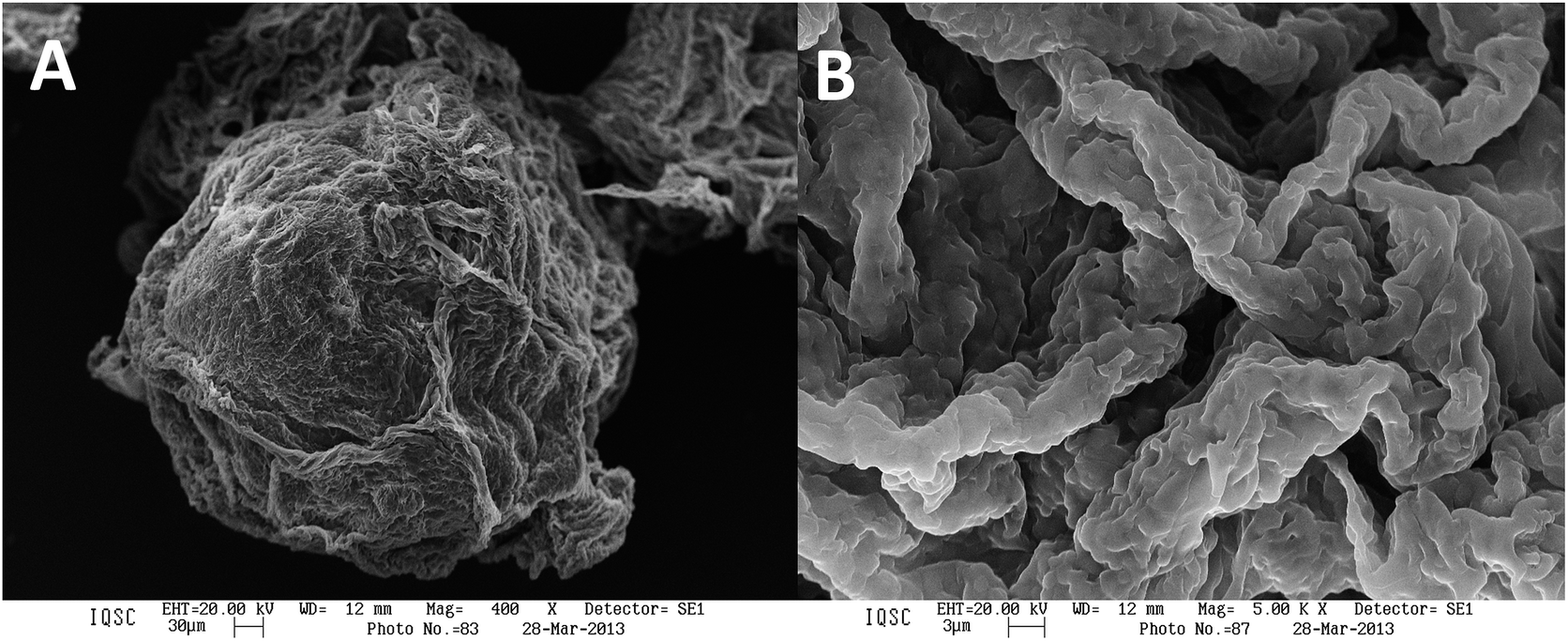 | ||
| Fig. 3 Scanning electron micrographs of silk fibroin sphere with lipase from P. fluorescens at (A) 400× and (B) 5000×. | ||
The absolute configuration of the acetates (2a–d) was assigned as S configuration by comparison with optical rotation values described in the literature. Consequently, it was possible to observe that the esterification preference is in agreement with the predictions of the Kazlauskas rule.30
Chiral β-imidazoles alcohols are important intermediates in organic synthesis and are present in several bioactive molecules. In this connection we decided to synthesize a chiral analogue derivative of antifungal agents containing an imidazole rest (Scheme 1) using a chiral epoxide prepared as described earlier (see Table 4), to show the synthetic potential of the protocol described in this paper.
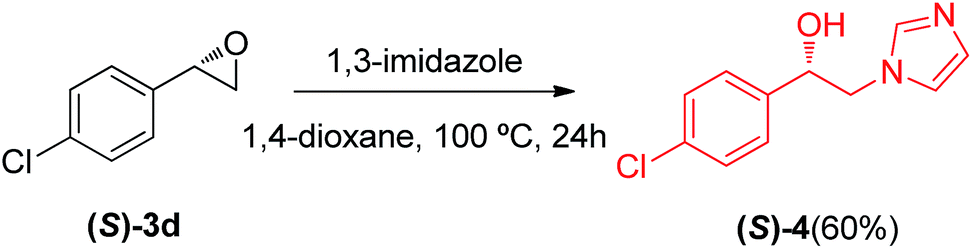 | ||
| Scheme 1 Synthesis of a chiral analogue derivative of an antifungal agent. | ||
Relative activities for lipase immobilized from P. fluorescens on silk fibroin spheres were calculated as the ratio of the activity of free lipase. The immobilized lipase showed relative activity of 82%. Losses of relative activity may be associated with the drying process of the resulting of immobilized lipase (lyophilization). More researches are needed to study the improvement of immobilization efficiency in future.
Conclusion
In conclusion, it was developed a methodology to immobilize Pseudomonas fluorescens lipase on silk fibroin spheres, used to promote the enzymatic kinetic resolution of halohydrins, precursor of chiral epoxides used in the synthesis of an analogue of antifungal agents in enantiomerically pure form. The methodology is eco-friendly and efficient in the acylation of halohydrins. The lipase from P. flourescens displays good activities and high selectivity when immobilized in silk fibroin spheres, and reinforces the versatility of silk fibroin as support for heterogeneous catalysis since it is easily removed from the reaction mixture by simple filtration. In all cases, the enantioselective enzymatic transesterification provided the expected acetates in high enantiomeric excess (99%).Experimental section
Materials
All the enzymes, lipases from Pseudomonas fluorescens (≥20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg), Candida cylindracea (4.9 units per mg), Rhizopus niveus (4.49 units per mg) and Aspergillus niger (184 units per mg) were purchased from Sigma-Aldrich Chemical Company and stored at 4 °C. Reagents (vinyl acetate, α-halobenzophenones, sodium borohydride, acetic anhydride, pyridine, LiOH and imidazole) and solvents (ethyl acetate, n-hexane, methanol, DMF, isopropanol and chloroform) were purchased from Sigma-Aldrich (USA) and Synth (Brazil). CaCO3 and CaCl2 were purchased from Synth-Brazil. The reactions were monitored by TLC with aluminum plates precoated with 60 F254 silica gel, eluting with n-hexane and ethyl acetate, and visualized by spraying with phosphomolybdic acid. The cocoon of the silkworm – Bombyx mori was provided by sericultor Helio Hatshugai, SP-Brazil.
000 units per mg), Candida cylindracea (4.9 units per mg), Rhizopus niveus (4.49 units per mg) and Aspergillus niger (184 units per mg) were purchased from Sigma-Aldrich Chemical Company and stored at 4 °C. Reagents (vinyl acetate, α-halobenzophenones, sodium borohydride, acetic anhydride, pyridine, LiOH and imidazole) and solvents (ethyl acetate, n-hexane, methanol, DMF, isopropanol and chloroform) were purchased from Sigma-Aldrich (USA) and Synth (Brazil). CaCO3 and CaCl2 were purchased from Synth-Brazil. The reactions were monitored by TLC with aluminum plates precoated with 60 F254 silica gel, eluting with n-hexane and ethyl acetate, and visualized by spraying with phosphomolybdic acid. The cocoon of the silkworm – Bombyx mori was provided by sericultor Helio Hatshugai, SP-Brazil.
Instrumentation
1H NMR and 13C NMR spectra were recorded on an Agilent Technologies 500/54 or Agilent Technologies 400/54 Premium Shielded spectrometer using CDCl3 or CD3OD as solvent and TMS as the internal standard. The chemical shifts are given in ppm and the coupling constants (J) in Hz. The data are reported as follows: chemical shift (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet) and integrated intensity. The 13C NMR chemical shifts are reported in ppm relative to the CDCl3 signal. FTIR spectra were recorded on a SHIMADZU IRAffinity-1 spectrometer samples were prepared as thin films on KBr disks (solid samples) or liquid film (liquid samples) in the 4000–400 cm−1 region. Enzymatic kinetic resolutions were carried out in a Tecnal TE-421 orbital shaker and enzymatic reactions were analyzed in a Shimadzu GC 2010 gas chromatographer equipped with an AOC 20i auto injector, a flame ionization detector (FID), and a Varian chiral column CP-Chiralsil-DEX (β-cyclodextrin) (25 m × 0.25 mm × 0.39 mm).The chromatographic conditions employed were: carrier nitrogen gas at 69.2 kPa, split ratio = 1:10, injection volume = 1.0 μL, injector temperature = 250 °C, detector temperature = 250 °C, oven temperature program initially at 120 °C for 2 min and increased at a rate of 2 °C min−1 until 165 °C for 8 min, total time of analysis = 32.5 min. The retention times for the (R,S)-alcohols obtained were: 1a, (R) = 20.5 min and (S) = 19.8 min; 1b, (R) = 13.8 min and (S) = 13.5 min; 1c (R) = 28 min and (S) = 27.5 min; 1d, (R) = 28.5 min and (S) = 27.9 min; the retention times for the (R,S)-acetates were: 2a, (R) = 16.8 min and (S) = 16.1 min; 2b, (R) = 10.7 min and (S) = 10.1 min; 2c, (R) = 18.3 min and (S) = 18.6 min; 2d, (R) = 23 min and (S) = 22.5 min. SEM images were obtained under a scanning electron microscope (Seron Technology S-4800) and DTG analysis was carried out with sample mass of silk fibroin or immobilized P. fluorescens lipase ca. 5.0 mg, in an alumina crucible, under a 50 mL min−1 flow rate dynamic nitrogen atmosphere and heating rates of 10 °C min−1 from room temperature up to 600 °C, in a TA Instrument Q-600.
Determination of activity relative
Ester hydrolysis activity was selected for the determination and assessment of immobilized P. fluorescens lipase of activity, performed with some modifications according to the method of Ye et al. (2005)31 Were mixed in a 10 mL Falcon tube, 0.5 mL of ethanol containing 14.4 mM p-NPP and 0.5 mL of phosphate buffer solution (0.1 M, pH 7.0). The reaction was started by addition of 2 mg free lipase or 20 mg immobilized lipase preparation. The mixture was incubated at 32 °C in a heating block under 400 rpm. After 5 min, the reaction was terminated by adding 0.5 mL of Na2HCO3 (0.5 M) followed by centrifuging for 10 min at 6000 rpm. The supernatant of 0.5 mL was diluted 10-fold with deionized water, the p-NP liberated was extracted by the aqueous alkaline phase, and the extraction was detected at 410 nm against a blank without enzyme using a UV-vis spectrophotometer against a blank without enzyme. Molar extinction coefficient of 12.444 mmol cm−1 for p-nitrophenol (p-NP), determined from the absorbance of standard solutions of p-NP in the reaction medium, was used.General procedure for the synthesis of (R,S)-2-halo-1-(phenyl)ethanols (1a–e)
Were added to a round-bottomed flask (100 mL), prior to small portions of 2-chloro-1-phenylethanone (2.5 mmol, 0.498 g), methanol (25 mL), followed by the addition of NaBH4 (3 mmol, 0.114 mg). The mixture was stirred for 30 min at 0 °C and for 1.5 h at room temperature. Methanol was evaporated under reduced pressure and HCl (1 mL, 10%) was added to the residue. The mixture was then extracted with ethyl acetate (3 × 20 mL) and the organic phase was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by gel silica column chromatography with a mixture of n-hexane and ethyl acetate (7:3) as eluent. The same procedure was employed for other (R,S)-2-halo-1-(phenyl)ethanols (1b–e). All spectral data of compounds 1a–e were in agreement with those described in the literature (ESI†).
General procedure for the preparation of (R,S)-2-halo-1-(phenyl)ethanol acetates (2a–e)
Acetic anhydride (0.6 mmol, 0.06 g, 56.3 μL) and pyridine (2 mmol, 0.16 g, 161.5 μL) were sequentially added to a round-bottom flask containing the appropriate (R,S)-2-halo-1-(phenyl)ethanols (0.5 mmol). The mixture was stirred overnight at room temperature. The reactions were quenched by the addition of 10% HCl (1 mL) and the acetate product was extracted with ethyl acetate (3 × 20 mL). The combined organic phases were dried over Na2SO4 and the organic solvent was evaporated under reduced pressure. The residue was then purified by silica gel column chromatography using n-hexane and ethyl acetate (9:1) as eluent.
Enzymatic kinetic resolution of (R,S)-2-halo-1-(phenyl)ethanol (1a–b)
Appropriate alcohol (R,S)-1a or (R,S)-1b (0.12 mmol), vinyl acetate (0.35 mol, 0.030 mg) and 5 mg of the appropriate lipase, n-hexane (500 μL) were added to Eppendorf® (2 mL) and stirred at 32 °C under 130 rpm. The reaction was monitored by TLC and the enantiomeric excesses (ee) of the alcohols [(S)-1a–b] and acetates [(R)-2a–b] were determined by GC-FID analysis in chiral column (Table 1). After the completion of the reaction, the mixture was filtered and the solvent was evaporated under reduced pressure and the residue was purified by silica gel column flash chromatography eluted with n-hexane and ethyl acetate (9:1).
Preparation of silk fibroin spheres
The silkworm cocoon (3.0 g) was transferred to 500 mL of a 2% Na2CO3 solution, preheated at 100 °C and stirred at this temperature for 30 min. The solid material was separated by filtration and then washed with distilled water (3 × 1000 mL). Finally, the fiber was placed in an oven to dry (70 °C) for 24 h. The fibroin was shredded and pinked in a Wiley mill (SOLAB SL31 – Embrapa – Brazil). After this step, 50 mL of a ternary solution of H2O:EtOH:CaCl2 (8:2:1 molar proportion) were used to solubilize the fibroin, which was transferred to a cellulose dialysis tube with an exclusion limit of 16 kDa and dialyzed in water for 3 d at room temperature. The dialysis water was changed every 24 hours. The fibroin solution was stored at 10 °C.4 After the fibroin solution was centrifuged (6000 rpm for 10 min) to remove impurities and larger particles. The concentration of the SF solution was adjusted to 5% (w/w) with distilled water, the determination of the final concentration was performed by the gravimetric method. The fibroin solution was transferred to a pressurized spray bottle glass. This solution was sprayed in liquid nitrogen for formation of the SF spheres. After evaporated the excess of liquid nitrogen and the spheres were lyophilized for 24 h. SF spheres has passed in sieve (6 to 13 mesh) to increase the degree of standardization.
Lipase immobilization
The 1.0 g of spheres of fibroin, 10 mL of water and 100 mg of P. fluorescens lipase were added to flask (50 mL) and gently shook for 60 min. The material containing the immobilized lipase was lyophilized (Edwards, Model: Freeze Dryer Modulyo) for 12 h. The immobilized fibroin lipase spheres were stored at 4 °C before use.Enzymatic kinetic resolution of (R,S)-1a with P. fluorescens lipase immobilized on fibroin spheres
The (R,S)-2-chloro-1-(phenyl)ethanol (0.12 mmol, 20 mg) (1a) and vinyl acetate (0.35 mol, 0.030 mg) were added to a flask vial (2 mL). n-Hexane (1 mL) was added in different flasks containing 50, 100 or 150 mg of immobilized lipase P. fluorescens on silk fibroin spheres. Each reaction mixture was stirred at 32 °C under 130 rpm (Table 2) and monitored by GC-FID. The reaction was filtered and extracted with EtOAc (3 × 15 mL), the organic layers were combined, dried over Na2SO4, the solvents were removed by distillation under reduced pressure and the residue was purified by silica gel flash column chromatography eluting with n-hexane and ethyl acetate (7:3).
Scale-up of the enzymatic kinetic resolution of (R,S)-1a by P. fluorescens lipase immobilized on fibroin spheres
(R,S)-2-Chloro-1-phenylethanol (0.36 mmol, 60 mg) (1a) and vinyl acetate (0.75 mol, 0.060 mg), 450 mg of P. fluorescens lipase immobilized on fibroin spheres and n-hexane (5 mL) were added to a vial (30 mL). The mixture was stirred at 32 °C under 130 rpm and monitored by TLC. It was then filtered and extracted with EtOAc (3 × 15 mL). The organic layers were combined and dried over Na2SO4. The solvent was removed by distillation under reduced pressure and the residue was purified by column flash chromatography with n-hexane and ethyl acetate (7:3) prior to the next synthetic step. The same procedure was employed for other (R,S)-2-halo-1-(phenyl)ethanols (1b–e). The yields of the acetates are showed in Table 3.
General procedure for the formation of epoxides (3a–c)
EtOH (1 mL), H2O (500 μL) and LiOH (26 mg, 1.1 mmol) were successively added to a solution of (S)-acetate 2a (22 mg, 0.10 mmol). The mixture was stirred at room temperature for 1 h. EtOH was removed by distillation at reduced pressure, and a solution of NaHCO3 (3 mL, 6 equiv.) and a saturated solution of NaCl (5 mL) were added. The organic phase was extracted with diethyl ether (3 × 30 mL) and the organic phases were combined and dried over Na2SO4.34 The solvent was removed by distillation under reduced pressure and purified by column flash chromatography with a mixture of n-hexane and ethyl acetate (9:1), affording (S)-3a as a colorless oil (quantitative). The same procedure was employed for the other (S)-acetate (2b–d). The yields of the epoxides (S)-3a–c are showed in Table 4.
:EtOAc (5:9.5), yield 60% (52 mg) of (S)-4 as a yellowish solid.17Absolute configuration
The optical rotations of purified alcohols 1a–d, acetates 2a–d and epoxides 3a–c were measured in CHCl3 with a JASCO P2000 polarimeter equipped with a Na-lamp 589 nm in 1 dm cuvette. The absolute configurations of alcohols 1a–d, acetates 2a–d and epoxides 3a–c were determined by comparing the specific rotations measured with those reported in the literature (ESI†).Recycling of the biocatalyst
Following the procedure described previously (section Experimental), the catalyst was recovered, washed with n-hexane (3 × 1 mL) and reused. The process was repeated for four times (the results are shown in Fig. 2).Acknowledgements
I. M. F. acknowledge Conselho Nacional de Desenvolvimento Científico e Tecnológico o (CNPq Proc. 558062/2009-1) for fellowship. A. L. M. Porto and J. V. Comasseto acknowledge to CNPq (Proc. 558062/2009-1) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Proc. 2009/50688-3) for the financial support provided to this research.Notes and references
- (a) H. Ferraz, G. Bianco, C. Teixeira, L. H. Andrade and A. L. M. Porto, Tetrahedron: Asymmetry, 2007, 18, 1070 CrossRef CAS; (b) S. S. Ribeiro, I. M. Ferreira, J. P. F. Lima, B. A. Sousa, R. C. Carmona, A. A. D. Santos and A. L. M. Porto, J. Braz. Chem. Soc., 2015, 26, 1344 CAS.
- (a) R. Ferrarini, J. V. Comasseto and A. Dos Santos, Tetrahedron: Asymmetry, 2009, 20, 2043 CrossRef CAS; (b) W. G. Birolli, M. I. Ferreira, N. Alvarenga, D. A. Santos, I. L. De Matos, J. V. Comasseto and A. L. M. Porto, Biotechnol. Adv., 2015, 33, 510 CrossRef PubMed.
- I. G. Rosset, M. Tavares, E. Assaf and A. L. M. Porto, Appl. Catal., A, 2011, 392, 136 CrossRef CAS.
- I. G. Rosset, M. Cavalheiro, E. Assaf and A. L. M. Porto, Catal. Lett., 2013, 143, 863 CrossRef CAS.
- A. Schmid, J. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2010, 409, 258 CrossRef PubMed.
- J. Wang, Y. Zheng and A. Wang, Ind. Crops Prod., 2012, 40, 178 CrossRef CAS.
- Y. Liu, S. Jia, Q. Wu, J. Ran, W. Zhang and S. Wu, Catal. Commun., 2011, 12, 717 CrossRef CAS.
- P. Lu and Y. Hsieh, J. Membr. Sci., 2009, 330, 288 CrossRef CAS.
- N. Kharrat, Y. Ben Ali, S. Marzouk, Y. Gargouri and M. Karra-Chaabouni, Process Biochem., 2011, 46, 1083 CrossRef CAS.
- X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077 CAS.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408 RSC.
- X. Wu, C. Yang, J. Ge and Z. Liu, Nanoscale, 2015, 7, 18883 RSC.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- J. Ge, J. Lei and R. N. Zare, Nat. Nano, 2012, 7, 428 CrossRef CAS PubMed.
- Z. Li, Y. Ding, S. Li, Y. Jiang, Z. Liu and J. Ge, Nanoscale, 2016, 8, 17440 RSC.
- J. D. An, D. A. Patterson, S. McNeil and M. M. Hossain, Biotechnol. Prog., 2014, 30, 806 CrossRef CAS PubMed.
- M. Saleemuddin and Q. Husain, Enzyme Microb. Technol., 1991, 13, 290 CrossRef CAS PubMed.
- Y. Zhang, Biotechnol. Adv., 1998, 16, 961 CrossRef CAS.
- Y. Zhang, Biotechnol. Adv., 2002, 20, 91 CrossRef CAS PubMed.
- I. M. Ferreira, R. Nishimura, A. B. Souza, G. Clososki, S. A. Yoshioka and A. L. M. Porto, Tetrahedron Lett., 2014, 55, 5062 CrossRef CAS.
- I. M. Ferreira, L. S. Ganseli, I. G. Rosset, S. A. Yoshioka and A. L. M. Porto, Catal. Lett., 2017, 147, 269 CrossRef CAS.
- E. Bini, D. Knight and D. Kaplan, J. Mol. Biol., 2004, 335, 27 CrossRef CAS PubMed.
- C. Foo, E. Bini, J. Huang, S. Lee and D. Kaplan, Appl. Phys. A: Mater. Sci. Process., 2006, 82, 193 CrossRef CAS.
- M. Wang, H. Jin, D. Kaplan and G. Rutledge, Macromolecules, 2004, 37, 6856 CrossRef CAS.
- L. Meli, J. Miao, J. Dordick and R. Linhardt, Green Chem., 2010, 12, 1883 RSC.
- S. Rostom, H. Ashour, H. El Razik, A. Abd El Fattah and N. El-Din, Bioorg. Med. Chem., 2009, 17, 2410 CrossRef CAS PubMed.
- F. Chevreuil, A. Landreau, D. Seraphin, G. Larcher, J. Bouchara and P. Richomme, J. Enzyme Inhib. Med. Chem., 2008, 23, 617 CrossRef CAS PubMed.
- J. Mangas-Sanchez, E. Busto, V. Gotor-Fernandez, F. Malpartida and V. Gotor, J. Org. Chem., 2011, 76, 2115 CrossRef CAS PubMed.
- (a) M. A. Moraes, G. M. Nogueira, R. F. Weska and M. M. Beppu, Polymers, 2010, 2, 719 CrossRef; (b) R. Weska, W. Vieira, G. Nogueira and M. Beppu, Mater. Res. Ibero Am. J., 2009, 12, 233 CAS.
- R. J. Kazlauskas and Q. Jing, Chirality, 2008, 70, 724 Search PubMed.
- P. Ye, Z.-K. Xu, Z.-G. Wang, J. Wu, H.-T. Deng and P. Seta, J. Mol. Catal. B: Enzym., 2005, 32, 115 CrossRef CAS.
- D. Basavaiah, U. Das and S. Roy, J. Chem. Sci., 2009, 121, 1003 CrossRef CAS.
- J. Zhang, J. Wang, Z. Qiu and Y. Wang, Tetrahedron, 2011, 67, 6859 CrossRef CAS.
- O. Pamies and J. Backvall, J. Org. Chem., 2002, 67, 9006 CrossRef CAS PubMed.
- G. Roman, J. Z. Vlahakis, D. Vukomanovic, K. Nakatsu and W. A. Szarek, ChemMedChem, 2010, 5, 1541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR spectra, GC chiral chromatography. See DOI: 10.1039/c7ra00083a |
| This journal is © The Royal Society of Chemistry 2017 |