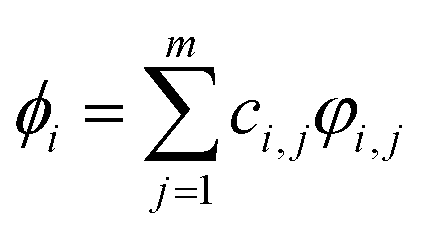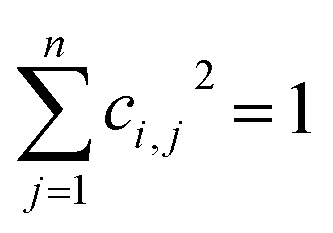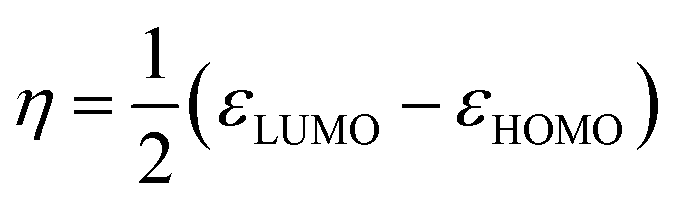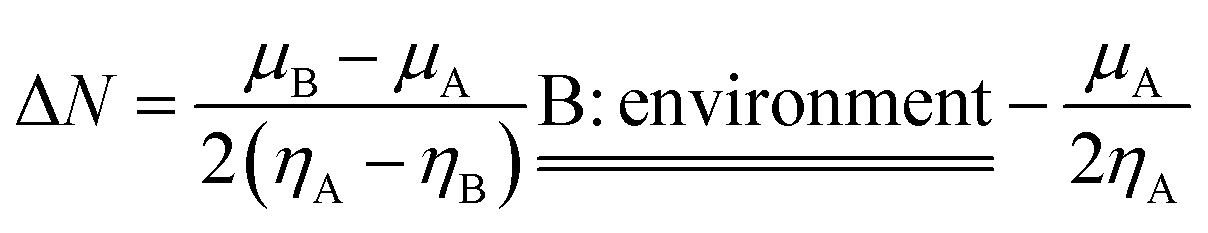DOI:
10.1039/C6RA26805F
(Paper)
RSC Adv., 2017,
7, 3586-3593
A cyclic voltammetry and PM6 semi-empirical molecular orbital method study of the capacity behaviour of an aluminum-8-hydroxyquinoline complex modified carbon paste electrode
Received
15th November 2016
, Accepted 18th December 2016
First published on 13th January 2017
Abstract
A graphite powder surface was modified by aluminum-8-hydroxyquinoline complex, and functionalised as an electron storage and transfer surface for use as one of the electrodes in an electrochemical capacitor. The electrochemical capacity behaviour of the aluminum-8-hydroxyquinoline complex modified graphite paste electrode was investigated by cyclic voltammetry experimentally and PM6 semi-empirical molecular method in MOPAC2012 software based on a theoretical model of molecular clusters. The electron-acceptor property of the 8-hydroxyquinoline-aluminum complexes improved the electrochemical capacitor performance, and resulted in a linear relation of catalytic current proportional to scan rate up to 40 V s−1, and the capacity current increases with number of scan cycles up to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Quantum chemistry calculation was based on molecular clusters composed of a piece of graphene with 38 carbon atoms and aluminum-8-hydroxyquinoline complex modifiers. The results indicate the modifications are spontaneous processes and are stable in the molecular clusters, which offers suitable LUMOs for charged electrons to be stored in, and are isolated from the π-molecular orbital sea from the graphene and parallel modifiers. Among the three aluminum-8-hydroxyquinoline complexes, the tri-8-hydroxyquinoline-aluminum complex was found to be a better modifier compared with the mono- and bi-8-hydroxyquinoline-aluminum complexes. The results accord well with the experimental data. The stable electronic properties and stability of the modifier are responsible for the good capacitor performance. These results offer a new way for quantum chemical study on electrochemical capacitor and energy storage techniques.
000 cycles. Quantum chemistry calculation was based on molecular clusters composed of a piece of graphene with 38 carbon atoms and aluminum-8-hydroxyquinoline complex modifiers. The results indicate the modifications are spontaneous processes and are stable in the molecular clusters, which offers suitable LUMOs for charged electrons to be stored in, and are isolated from the π-molecular orbital sea from the graphene and parallel modifiers. Among the three aluminum-8-hydroxyquinoline complexes, the tri-8-hydroxyquinoline-aluminum complex was found to be a better modifier compared with the mono- and bi-8-hydroxyquinoline-aluminum complexes. The results accord well with the experimental data. The stable electronic properties and stability of the modifier are responsible for the good capacitor performance. These results offer a new way for quantum chemical study on electrochemical capacitor and energy storage techniques.
Introduction
Carbon materials such as micro or nano particles,1–3 porous,4 sheets,5 and hollow particles6 with specific structures7 have been found to be the most flexible and novel electrode materials since the first super-capacitor.8 Graphite powder embedded in an organic binder is called a carbon paste electrode (CPE)9,10 and has been used for many years in electrochemical analysis and other electrochemical studies. In order to increase its functionality, CPEs have been modified by different methods, such as adsorption, chemical modification or intercalated modifiers on the surface of graphite powder,11–15 as well as the mixture of modifiers with organic binders.16–19 The theoretical or quantum chemical studies on CPEs20,21 are important for deeply understanding the electronic properties and electrochemical behaviors of CPEs composed of graphite powder with modifiers.
8-Hydroxyquinoline aluminum complexes are semiconducting materials with a unique spherical structure, high stability, film-forming property and good electronic conductivity,19 and have been widely used in the development of organic electroluminescent devices.22,23 This material commonly acts as an electron transporting host.24 Modification of a CPE with 8-hydroxyquinoline-aluminum complex may offer a home for electron storage in charging and transport to the outer circuit in discharging. This makes the electrode an electrochemical supercapacitor for energy storage. In a previous paper,25 we used the PM6 semi-empirical molecular method in MOPAC2012 software to deal with the charge transfer behavior in electrochemical catalytic oxidation of pyrogallol on an 8-hydroxyquinoline-aluminum complex modified carbon past electrode. In the present paper, the graphite powder surface was chemically modified with 8-hydroxyquinoline aluminium complexes, which not only serves as an electrochemical catalytic anode for pyrogallol oxidation, but also serves as an electrochemical capacitor with higher responding time, longer lifetime and higher stability. To understand the electron-home and capacity behavior of this modified CPE from a molecular level, a semi-empirical quantum chemical method of PM6 in MOPAC2012 software26,27 was used to investigate based on simplified molecular cluster models composed of a graphene sheet and modifiers. The calculation results were analyzed in thermodynamic and frontier orbital concepts, which gives important information for the explanation of experimental results.
Experimental and computational details
Materials and instruments
All electrochemical experiments were carried out on a CHI620b electrochemical instrument (CHI Co., Shanghai, China) with three electrodes, including a home-made platinum electrode as the counter electrode, an 8-hydroxyquinoline-aluminum complex modified carbon paste electrode as the working electrode and a KCl-saturated calomel electrode (Model 232, Shanghai, China) as the reference electrode, and all potentials reported here are with respect to this reference electrode.
Polyamide resin (Tianjin, China), epoxy resin (Wuxi, China) methyl silicone oil (Shenyang, China) and graphite powder (spectrographic grade pure) were used for the preparation of the basic carbon electrode. 8-Hydroxyquinoline (analytical grade pure) and aluminum chloride (analytical grade pure) were used for the modification of carbon electrode. Potassium chloride (analytical grade pure) was used for the preparation of the supporting electrolyte solution. All solutions were prepared with ultrapure water (18.2 MΩ, USA) for the experiment, and all experiments were carried out at room temperature.
Preparation of the basic carbon electrode
The basic carbon electrode was prepared as described in a previous work.28 Graphite powder, epoxy resin and polyamide resin were mixed into a paste, then filled into a clean glass tube about 6–7 cm in length and 5.0 mm in diameter. A piece of copper wire was inserted into the paste from other end of the tube as an electronic lead. The electrode was solidified in air for about 72 h. After the solidification, the top of the electrode was polished on sand paper, and some paste was tipped out from the top of the electrode, leaving a cavity of about 1 mm depth to hold carbon paste for the working electrode. The prepared solid carbon electrode with resistance of 10–100 Ω was used as the basic carbon paste electrode in all the experiments.
Preparation of 8-hydroxyquinoline-aluminum modified CPE
The 8-hydroxyquinoline-aluminum (AlQ) complexes modified carbon paste electrodes were prepared with different molar ratios of 1:1, 1:2 and 1:3. The molar ratios of the complexes were simply controlled by the addition of the initial reactants of aluminium chloride and 8-hydroxyquinoline in the synthesis of the three complexes. For example, an AlQ complex with 1:1 molar ratio was prepared by mixing 1.0 g of graphite powder with 0.50 mL of 3.0 mM stock solution of 8-hydroxyquinoline with 95% ethanol, and 1.50 mL of 1.0 mM AlCl3 solution with one drop of concentrated HCl stock solution. The mixture solution was heated at about 70–100 °C and stirred to rapidly evaporate solvents. After all the solvent was evaporated out, the resulting dry powder was cooled naturally at room temperature as the AlQ complex modified graphite powder. The AlQ modified graphite powder was mixed with methyl silicone oil at a mass ratio of 2:1 on a clean glass plate into a paste with a glass bar. The carbon paste was filled into the cavity of the basic carbon electrode, polished on glassy paper into a mirror-like surface, and served as the modified carbon paste electrode (modified CPE) in all of this work. After an electrochemical experiment, the paste was cleaned out from the cavity, and new paste was filled in for the next experiment. In this way the CPEs are renewable.
Molecular cluster models
All the molecules and molecular clusters were calculated in the vacuum cases, and the influence of solvent effects was not considered in the quantum chemical calculations, partly due to the calculation complexity and the organic phase is composed of methyl silicone oil and graphite powder, which reduces the influence of water molecules on the molecules and molecular clusters.
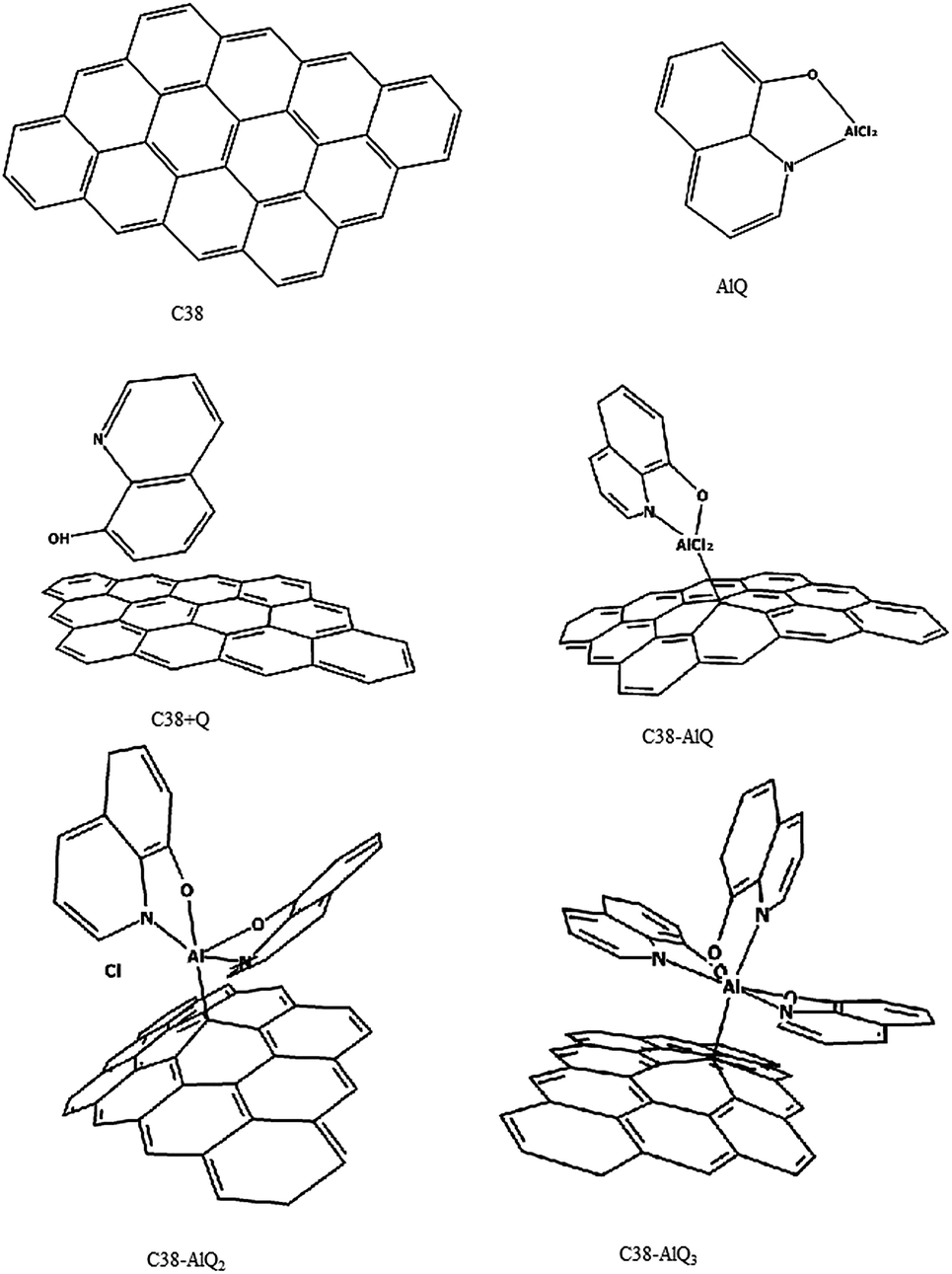
Graphite powder is a stratification of multiple layers of graphene sheets. The graphite surface exposed on the surface of graphite powder may be simply modeled with a piece of graphene sheet composed with 38 carbon carbons in match-join form of twelve six-rings of about 1.6 nm2.20,21 In this study, the molecular cluster was described as C38, as shown in Fig. 1. Molecules of mono-8-hydroxyquinoline-aluminum (AlQ) complex with two chloride ions, bi-8-hydroxyquinoline aluminum with one chloride ion and tri-8-hydroxyquinoline aluminum are given in Fig. 1 as AlQ, AlQ2 and AlQ3, respectively. The graphene sheets with AlQ, AlQ2 and AlQ3 are labelled as C38–AlQ, C38–AlQ2, and C38–AlQ3 in Fig. 1, respectively.
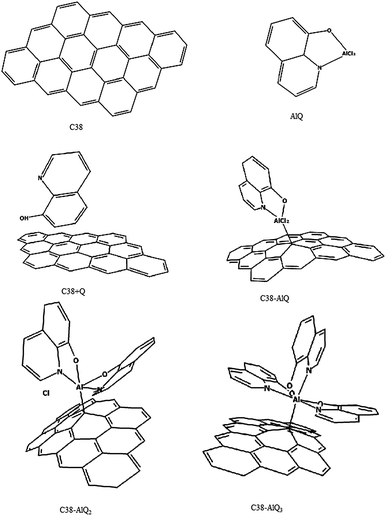 |
| | Fig. 1 Molecules or molecular clusters. C38: graphene with 38 carbon atoms; AlQ: mono-8-hydroxyquinoline aluminum with two chlorides; C38 + Q: graphene C38 with 8-hydroxyquinoline; C38–AlQ: graphene C38 modified with mono-8-hydroxyquinoline aluminum complex; C38–AlQ2: graphene C38 modified with bi-8-hydroxyquinoline aluminum complex with one chloride; C38–AlQ3: graphene C38 modified with tri-8-hydroxyquinoline aluminum complex. | |
Semi-empirical molecular orbital calculation
All quantum chemical calculations in this work were performed with semi-empirical quantum chemical methods of PM6 in MOPAC2012 software26,27 on personal computers. All molecules and molecular cluster models were designed by pcmod 7.0. These molecular models were further optimized with MOPAC2012 software before the energy and molecular orbital calculations. The calculations were stopped when the energy gradient normal drops below 0.01 kcal mol−1 in heat of formation. The heats of formation and frontier orbitals were obtained from the semi-empirical calculations, and used to analyze the stability and electron transfer properties of each molecular cluster. The semi-empirical method is not as precise a method in quantum chemical calculations as Ab initio or Gauss, but it can give out valuable information for a comparative study to a suitable reference using a personal computer in a short time. In our calculation, we chose unmodified graphene as the reference, a model of unmodified CPE, and all the modified graphenes with aluminum-8-hydroxyquinoline complexes, the modified CPE models, as the test samples. The calculation was used to reveal the differences between them, and before and after they are charged. All the molecules and molecular clusters are in vacuum conditions, and we do not consider the influence of solvent effects in the quantum chemical calculations, partly owing to the calculation complexity and because the methyl silicone oil of the organic phase is mixed with the graphite powder and reduces the influences of water molecules on the molecules and molecular clusters.
Theoretical analysis of calculation results
The heat of formation of molecules or molecular clusters can be easily obtained from the PM6 calculation in MOPAC2012 and used to evaluate the stability of molecules or molecular clusters.25 The Gibbs free energy change (ΔGr) of a reaction can be calculated with the enthalpy change (ΔHr) and entropy change (ΔSr) of the reaction, and is described as:
According to quantum chemical theory,27 the Hamiltonian of a system, Ĥi, can be described as:
| |
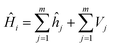 | (2) |
where
ĥj is the kinetic energy part and
Vj the potential energy part of the
jth electron in ith molecular orbital of the system:
where
ϕi is the molecular orbital and
Ei is the energy of the molecular orbital. Thus, by performing quantum chemical calculation, we can obtain the energy of the system and the frontier molecular orbital by the quantum chemical calculation. There are
n number of molecular orbitals linearly combined from
m number of valence electron orbitals of the atoms in the molecule or molecular cluster:
| |
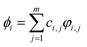 | (4) |
here
i indicates the
ith molecular orbital,
φi,j is the
jth atomic valence electron orbital of atoms in the
ith molecular orbital,
ci,j is the coefficient of the
jth atomic orbital in the
ith molecular orbital and
ci,j2 indicates the fractional probability of the
jth atomic orbital in the
ith molecular orbital, and follows the normalization principle:
| |
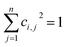 | (5) |
According to the Kohn–Sham theory, the frontier molecular orbital (FMO) composed of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) among the orbitals is very important in the description of the chemical reactive tendency of a molecule with approximations that may be useful for evaluation of electrochemical activity. Some descriptors were theoretically studied, reviewed25 and reported, such as the frontier orbital gap (ΔE), chemical potential (μ), molecular hardness (η), molecular electron affinity (ω), and charge exchange (ΔN) from A to B molecules. All of these terms can be calculated from the frontier orbital energy of the HOMO (εHOMO) and LUMO (εLUMO), and are described as follows:
| |
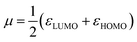 | (7) |
| |
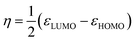 | (8) |
| |
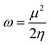 | (9) |
| |
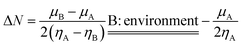 | (10) |
here Δ
N originally described the electron transfer quantity from molecule A to molecule B, but in our case we describe the electron transfer from molecule A to another electrode or output electric circuit with
μB =
ηB = 0; we call B the environment, and the new quantity may be called an exchangeable electron. In order to compare the electron transfer capacity of graphene C38, modified C38 and other molecular clusters, we used another descriptor (ΔΔ
N), which is defined as the difference of the exchangeable electrons between any molecular cluster (Δ
NMC) and graphene C38 (Δ
NC38), and may be described as:
The initial states of the modified GPEs have slightly negative values of exchangeable electrons compared with the C38 graphene, so they have some slight tendency to accept electrons from their environment.
Results and discussion
Cyclic voltammetry behavior of the modified carbon paste electrodes
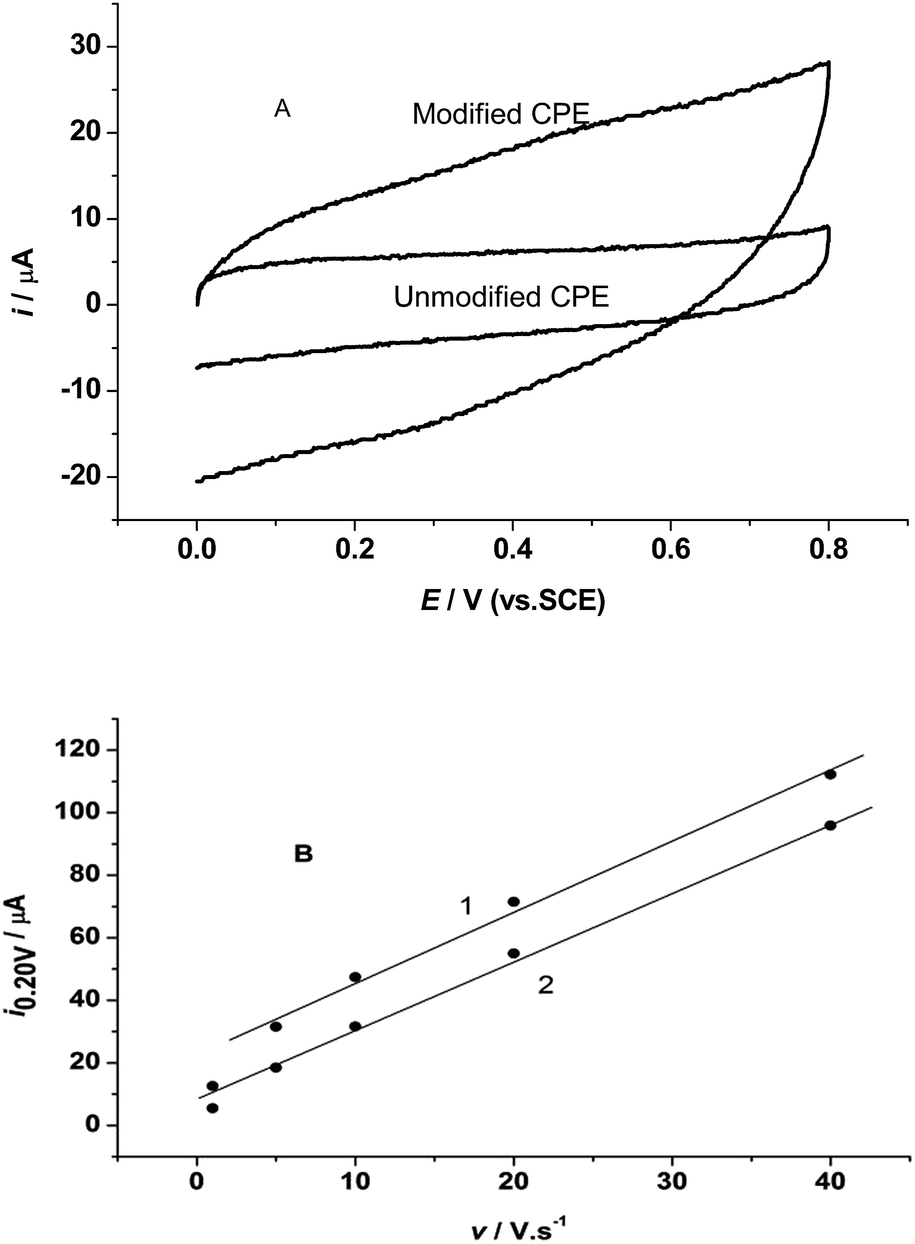
Fig. 2A shows the cyclic voltammetry (CV) curves of the unmodified and modified CPE with the tri-8-hydroxyquinoline aluminum complex in 0.5 M KCl electrolyte solution in the potential range of 0.8–0.0 V at a scan rate of 1.0 V s−1. The CV curve at the modified CPE shows a larger charging current (curve 1 in Fig. 2A) than that at the unmodified CPE (curve 2 in Fig. 2A) at the same potential range. The cathodic current on the modified CPE at 0.2 V is 2.3 times larger than that at the unmodified CPE. Both of the C linear relationships of the PEs are shown in Fig. 2B.
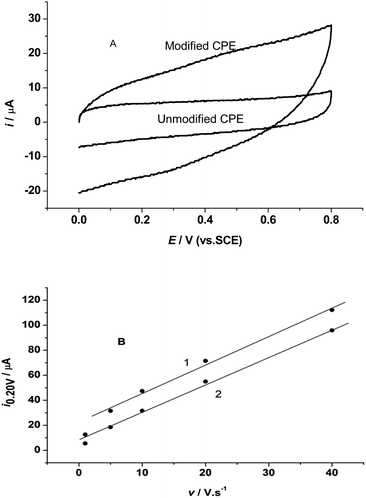 |
| | Fig. 2 (A) The cyclic voltammograms of the tri-8-hydroxyquinoline-Al complex modified (1) and unmodified (2) CPE in 0.5 M KCl solution at scan rate of 20 V s−1. (B) The linear relationships between cathodic current at 0.2 V and scan rate in CV curves. | |
The electrode responses to potential change follow the double layer capacity behavior, and both of the CPEs can be used as electrochemical capacitors. The linear relation is i0.2 V = 17.79 + 2.45v, R2 = 0.97; SD = 0.205 for the modified CPE, and i0.2 V = 6.63 + 2.28v, R2 = 0.993; SD = 0.098 for the unmodified CPE. The slope for the modified CPE is 7.5% larger than that of the unmodified CPE. This larger slope indicates that the response of the modified CPE to potential change is large than that of the unmodified CPE, and with the increase of scan rate the cathodic current must further increase faster than that of the unmodified CPE. All these results mean that the 8-hydroxyquinoline-aluminum complex can increase electrode capacity obviously.
The influence of complex ratios
The coordination ratio of the 8-hydroxyquinoline-aluminum complex is an important factor influencing the molecular structure and properties of the modified CPE. The modified CPEs with different molar ratios were set into the electrochemical cell for the cyclic voltammetric experiments. Fig. 3 shows the CV curves of the modified CPEs with different complex ratios in 0.50 M KCl electrolyte solution at a 20 V s−1 scan rate. The CV curve of the CPE modified with the 1:3 molar ratio complex (curve 1 in Fig. 3) was better than those obtained for the other ratios.
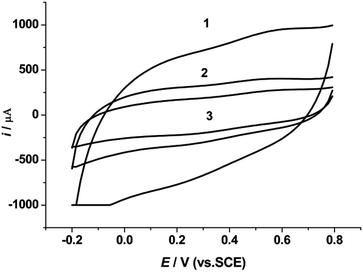 |
| | Fig. 3 Cyclic voltammograms of 8-hydroxyquinoline-aluminum complexes with different complex ratios in 0.5 M KCl electrolyte solution at a scan rate of 20 V s−1. Complex ratio: 1, 1:3; 2, 1:1; 3, 1:2. | |
The cathodic current at 0.2 V is 637.2, 303.7 and 167.8 mA for the aluminum-8-hydroxyquinoline complexes with complex ratios of 1:3, 1:1 and 1:2, respectively. The CPE of the modifier with a complex ratio of 1:3 gives the largest capacitance among the other complex ratio modifiers, which is 1.1 and 2.8 times larger than the ones with complex ratios of 1:1 and 1:2, respectively.
The influence of the modifier
The amount of the modifier (m (μM g)−1) is another factor that directly influences the capacitance of the modified CPE. Fig. 4 shows the influence of the tri-8-hydroxyquinoline-aluminum complex used for the modification of graphite by plotting the cathodic current at 0.2 V in CVs at a 20 V s−1 scan rate against the volume of modifier solution in the concentration range of 0–15.0 μM g−1. The current is about 80 mA below 3.75 μM g−1 of modifier, but when the volume of modifier was over 3.75 μM g−1, the cathodic current at 0.2 V in the CVs is nearly proportional to the amount of modifier, as described by the regression equation of:| | |
i0.3 V = 1.103 + 9.97 m (μM g)−1; R2 = 0.988, SD = 18.1
| (12) |
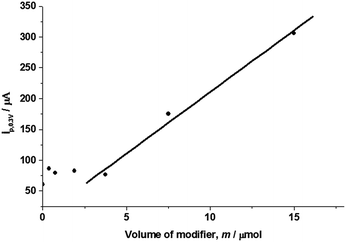 |
| | Fig. 4 The plot of cathodic current at 0.2 V against the amount of modifier of tri-8-hydroxyquinoline-Al complex in 0.5 M KCl electrolyte solution at a scan rate of 20 V s−1. | |
When the concentration of modifier solution was below 3.75 μM g−1, the graphite powder surface is partly modified with the complex, and only gives out a small increase of capacitance compared with the unmodified CPE. However, when the concentration of the modifier was over 3.75 μM g−1, the graphite powder surface was fully modified with the complexes. The modifier forms a thin film, increases the density with the increase of the volume of modifier solution and gives a linear increase of the capacitance of the CPE.
The cyclic performance and stability of the modified CPE
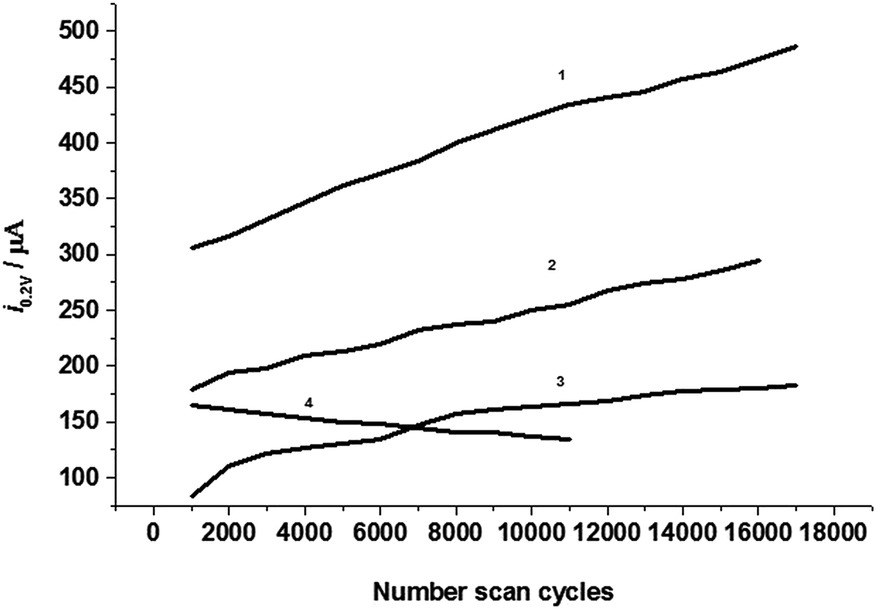
The stability or lifetime is a very important factor for the capacitor. The modification of the graphite powder surface with the electron acceptor aluminum-8-hydroxyquinoline complexes is to make a home for charged electrons and stabilize electrons in the electrode surface or the electric double layer. In this case, the electrochemical capacitor may have a long lifetime and good stability. To monitor this property, the electrode was used for performing multiple cyclic voltammetry cycles (up to 17000) continually at a scan rate of 20.0 V s−1. Fig. 5 shows the cathodic current at 0.2 V in CVs against the cycle number.
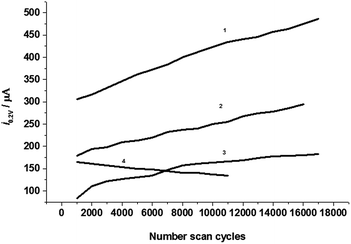 |
| | Fig. 5 The cathodic currents at 0.2 V against number of cyclic voltammetry scans in 0.5 M KCl electrolyte solution at scan rate of 20.0 V s−1 at CPEs modified by the complexes with complex ratios of (1) 1:3, (2) 1:1, (3) 1:2, and (4) 1:3 and 15 μM g−1 of modifier. | |
According to the ideal electrochemical capacitor model,1,2 the capacitance of an electrochemical capacitor would be an exponential function of the cycle number. From Fig. 5 it can be seen that curve 3 shows a nearly exponential increase, curves 1 and 2 are linearly increased, but curve 4 linearly decreased with the increase of cycles owing to the larger amount of modifier and forms a thickness modification film.
All these curves are regressed with exponential function and linear function as the following equations (from (1) to (4) in Fig. 5).
| | |
i0.2 V = 640.8 − 338.2exp[−(n − 1000)/21030], χ2 = 7.6 Å
| (13) |
| | |
i0.2 V = 781.4 − 599.0exp[−(n − 1000)/73645], χ2 = 7.6
| (14) |
| | |
i0.2 V = 193.4 − 102.8exp[−(n − 1000)/7341], χ2 = 16
| (15) |
| | |
i0.2 V = 166.3 − 0.003n, R2 = 0.988, SD = 1.2
| (16) |
If we characterize the exponential function with the specific cycle number constant, τ0 = n0, n/n0 = 1 is the cycle number that is located at the middle point of the height of the cathodic current in the curve. So the specific cycle number is 22030 for curve 1, 74645 for curve 2, and 8441 for curve 3. The specific cycle number constant may be dependent on the total capacitance of the capacitor or the efficiency and speed of the electrode charge. The modified CPE with the complex of 1:3 molar ratio has a largest capacitance and highest charging efficiently; it shows a smaller τ0 than that of CPE with a molar complex ratio of 1:2, and larger than that of the 1:1 molar ratio. This result gives further evidence that the tri-8-hydroxyquinoline-aluminum complex is the best modifier used for the electrochemical capacitor. The large amount of modifier (here over 15 μM g−1) is favorable for capacity at the initial time, but does not benefit the lifetime of the capacitor.
The thermodynamic stability of molecular clusters
The enthalpy and entropy of molecules or molecular clusters at 298 K can be easily calculated with PM6 in MOPAC2012, and used to evaluate the stability of molecules or molecular clusters. The enthalpy, entropy and Gibbs free energy of reaction for each cluster formation were also calculated according to eqn (1)–(3) and are listed in Table 1. All Gibbs free energy of cluster formation reactions show a larger negative value, which means the formation reactions are exothermal and spontaneously occur.
Table 1 The thermodynamic parameters of the molecules and molecular clusters
| Molecule/cluster |
Hf/kJ mol−1 |
Sf/kJ mol−1 K−1 |
ΔHr/kJ mol−1 |
ΔS/kJ mol−1 K−1 |
ΔGr/kJ mol−1 |
| Al(H2O)6 |
1192.48 |
0.44839 |
|
|
|
| C38 |
569.087 |
0.61107 |
|
|
|
| 8-Q |
11.4056 |
0.35569 |
|
|
|
| AlQ |
757.409 |
0.46908 |
−446.48 |
−0.3350 |
−346.65 |
| AlQ2 |
429.237 |
0.51730 |
−786.05 |
−0.64247 |
−594.60 |
| AlQ3 |
−241.96 |
0.71047 |
−1468.7 |
−0.8050 |
−1228.8 |
| C38 + Q |
494.453 |
0.81243 |
−63.229 |
−0.15434 |
−17.236 |
| C38–AlQ |
−55.421 |
0.85165 |
−1381.9 |
−0.22850 |
−1313.8 |
| C38–AlQ2 |
136.482 |
0.91549 |
−861.84 |
−0.15680 |
−815.11 |
| C38–AlQ3 |
181.146 |
1.08404 |
−1706.1 |
−0.22033 |
−1640.4 |
The initial molecular clusters are stable with lower formation energies, but after being charged with some electrons, all of them become unstable. Some of them have larger changes in formation energy. The stability of the electrochemical capacitor molecular clusters after being charged directly influences the stability and lifetime of the electrochemical capacitors. To find some evidence from the thermodynamic calculation when the molecular clusters are negatively charged with up to 10 negative electrons on the clusters, Fig. 6A shows the heat of formation of the clusters against number of electrons charged. The heat of formation of each cluster increases exponentially with the increase of the number of electrons charged to the system. They are in the order of C38 > C38–AlQ > C38–AlQ3 > C38 + Q > C38–AlQ2. The tri-8-hydroxyquinoline-Al modified CPE is in the middle of the order. The systems may keep their stabilities by justifying the distance and orientation angles of each part in the systems, for example, C38 + Q molecular cluster is stabilised by changing the distance between C38 and Q, as shown in curve 2 of Fig. 6B. The C38–AlQ and C38–AlQ2 have small and stable distances of 0.186 ± 0.0054 pm and 0.382 ± 0.645 pm from C38 to AlQ and AlQ2, respectively, due to the smaller molecular volume and strong interactions between C38 and the modifiers AlQ and AlQ2. Tri-8-hydroxyquinoline-Al has a larger but relatively stable distance of 0.919 ± 0.125 pm from C38 to AlQ3, owing to the larger molecular volume of AlQ3, and resulting in the aluminum cation not being able to easily access the surface of the graphene sheet, but it is stable through up to 10 negative charges added, like C38–AlQ.
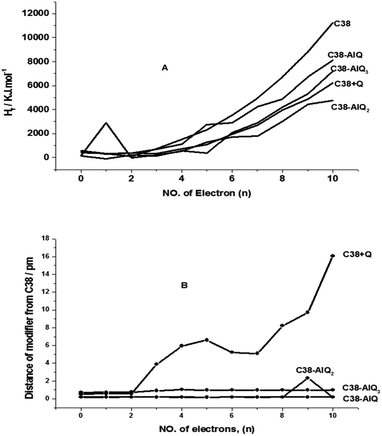 |
| | Fig. 6 (A) The heat of formation of the systems against number of electrons charged. (B) The distance between C38 and the modifier against the number of electrons charged. | |
Energy analysis of frontier orbitals
The charging electrons on the molecules and molecular clusters not only influence the stability of the systems, but also the molecular orbital structure, especially the frontier orbitals. The molecular orbitals of the molecules and molecular clusters were obtained by the semi-empirical molecular orbital method with PM6 in MOPAC2012, and all the descriptors according to eqn (9)–(14) are listed in Table 2.
Table 2 The calculated descriptors of the molecules and molecular clusters
| Molecule |
EHOMO/eV |
ELUMO/eV |
μ/eV |
η/eV |
ΔN |
ΔΔN |
| C38 |
−7.625 |
−1.555 |
4.590 |
3.037 |
0.756 |
0 |
| Q |
−8.903 |
−0.864 |
4.884 |
4.020 |
0.608 |
−0.149 |
| C38 + Q |
−9.113 |
−1.696 |
4.392 |
4.295 |
0.511 |
−0.245 |
| C38–AlQ |
−6.657 |
−1.756 |
4.273 |
3.239 |
0.660 |
−0.096 |
| C38–AlQ2 |
−6.556 |
−1.270 |
4.660 |
2.727 |
0.854 |
0.098 |
| C38–AlQ3 |
−7.551 |
−1.508 |
4.558 |
3.022 |
0.754 |
−0.002 |
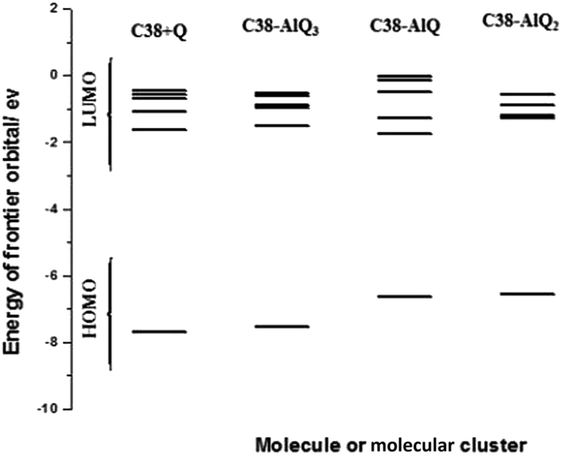
From Table 2 it can be seen that the initial states of the modified GPEs have slightly negative values of exchangeable electrons compared with the C38 graphene, so they have some slightly tendency to accept electrons from their environment substance. The energy gaps of frontier orbitals are the main factors of molecular orbital structure and are used to calculate some of the molecular descriptors. Fig. 7 shows the energy gaps against the number of electrons charged. After addition of electrons (up to 10 electrons charged) to the molecular clusters, the energy gaps are changed greatly with added electron numbers.
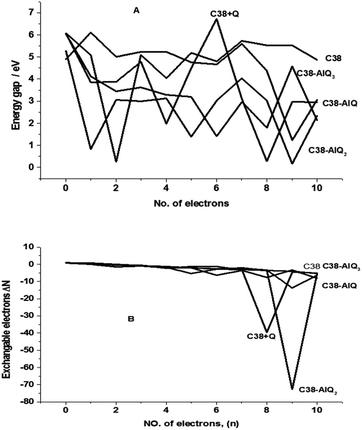 |
| | Fig. 7 The plot of orbital gap (A) and exchangeable electrons (B) against the charged electron numbers for the molecular clusters. | |
The average energy gaps are 4.74, 3.73,4.56, 2.66 and 3.33 eV for C38, C38 + Q, C38–AlQ, C38–AlQ2 and C38–AlQ3, respectively. Among the modified CPEs, the modified CPE with C38–AlQ2 has the smallest energy gap, and the next one is the modified CPE with C38–AlQ3. The smaller the gap, the easier the electron transfer from the occupied orbital to the unoccupied orbital. Fig. 7B shows the difference of exchangeable electrons of each system against charged electrons numbers.
The difference of the exchangeable electrons (ΔΔN) not changed much before the charged electrons is less than 4, but after the charged electrons it increases to over 5. However, the difference of exchangeable electrons changed greatly after addition of seven electrons, including C38 + Q; C38–AlQ and C38–AlQ2; the C38 and C38–AlQ3 modified CPEs kept at stable values, with average values of −2.24 and −2.98, respectively.
These results indicate that with the increase of the charged electron number, the average energy gap and difference of exchangeable electrons did not change greatly, which means the systems can hold the charged electrons without greatly influencing the electronic property themselves, and is also responsible for the good performance of the electrochemical capacitors.
Analysis of frontier orbital components
The components of the frontier orbitals of the LUMO and HOMO are another main factor to influence the electronic properties of the system as electron storage and electron transportation.
Fig. 8 shows the components of frontier orbitals of each molecule and molecular cluster. In the modified CPE system, all the HOMO orbitals are composed of pz atomic orbitals of the carbon atoms in C38 graphene. After the modified CPEs are charged, the charged electrons occupy the LUMOs from the molecular orbital with lower energy to the molecular orbital with higher energy. If 10 electrons are considered for the charged electrode, they will occupy 5 LUMOs of each system, which also shows the 5 LUMOs for each system in Fig. 8.
 |
| | Fig. 8 The components of the frontier orbitals of the molecular clusters. | |
In the C38 + Q molecular cluster, the C38 graphene interacts with 8-hydroxyquinoline in an approximately parallel manner, as shown in Fig. 1, so the corresponding LUMOs are composed of pz atomic orbitals of carbon atoms from C38 graphene and 8-hydroxyquinoline, and form π molecular orbitals. These kind of molecular orbitals are similar to those in the graphite case; all graphene sheets are parallel stacked together, so it is an electronic conductor, not a capacitor for electron storage. In the C38–AlQ system, the 8-hydroxyquinoline-aluminum complex stands on the surface in an approximately perpendicular manner. The third and fifth LUMOs are composed of pz atomic orbitals from C38 graphene and form π molecular orbitals, and the first two and fourth LUMOs are composed of px and py atomic orbitals from 8-hydroxyquinoline, and form another kind of molecular orbital perpendicular to the π molecular orbitals. The molecular orbitals composed of px and py atomic orbitals can be used as an energy barrier to stop the charged electrons from passing through the molecular orbital and make a reservoir to store charged electrons. In the C38–AlQ2 system, the two 8-hydroxyquinoline is slanted on the graphene surface with a small angle. Thus the second, third, and fifth LUMOs are mainly composed of pz atomic orbitals from C38 and a smaller amount of py and pz atomic orbitals from 8-quinoline, and the first and fourth LUMOs are composed of pz, py and px atomic orbitals from 8-quinoline with a smaller amount of pz atomic orbitals from C38. These mixed components make the molecular orbitals are not very suitable for electronic conductors or capacitors. In the C38–AlQ3 system, the tri-8-hydroxyquinoline aluminum complex stands on the graphene surface with one 8-hydroxyquinoline parallel to the graphene sheet and the other two are perpendicular to the sheet surface, as shown in Fig. 1. In this case, the first two LUMOs are composed of pz atomic orbitals from C38, and form π molecular orbitals, whilst the rest of the LUMOs composed of px and py atomic orbitals from 8-hydroxyquinoline form molecular orbitals that are perpendicular to the π molecular orbitals. Thus, they are good electron-holders and make this CPE a good electrochemical capacitor.
Conclusions
In the present paper, a graphite powder surface was chemically modified with 8-hydroxyquinoline aluminum complexes, and used in CPEs. The modified and unmodified CPEs show an electrochemical capacitor behavior. Among the modified CPEs, the CPE modified with tri-8-hydroxyquinoline-aluminum shows the largest capacitance. The capacitance is very stable, and increases with the increase of scan cycles up to 17000 cycles owing to the electron-acceptor property of the 8-hydroxyquinoline-aluminum complexes. The semi-empirical quantum chemical method of PM6 in MOPAC2012 software was used in quantum chemical calculations based on the molecular cluster models including graphene with 38 carbons and the 8-hydroxyquinoline aluminum complexes. The thermodynamic results indicated that the molecular clusters are stable even when charged with ten electrons. The molecular orbital analysis indicates that modification of graphene with the 8-hydroxyquinoline aluminum complexes results in the insertion of the complex frontier orbitals into the graphene frontier orbital system, which breaks its π bands into several bands, and makes some homes for electrons. This will help us to design new kinds of materials for energy storage.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 20875063), The project Foundation of the Education Department of Liaoning Province (2004-c022) and The project Foundation of Technology Bureau of Shenyang (2007GX-32).
Notes and references
- B. E. Conway, Electrochemical Supercapacitors, Kluwer Academic/Plenum Publishers, New York, 1999, vol. 1 Search PubMed.
- F. Beguin and E. Frackowiak, Supercapacitors: Materials, Systems, and Applications, Wiley-VCH, Berlin, Germany, 2013 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- I. Tanahashi, A. Yoshida and A. Nishino, Carbon, 1990, 28, 477–482 CrossRef CAS.
- S. Zhong, C. Padeste, M. Kazacos and M. Skyllas-Kazacos, J. Power Sources, 1993, 45, 29–41 CrossRef CAS.
- Y. Zhang, D. Zhang, Y. Chen, X. Sun and Y. Ma, J. Power Sources, 2012, 57, 3045–3050 Search PubMed.
- B. H. Fan, X. Mei, K. Sun and O. Yang, Appl. Phys. Lett., 2008, 93, 143103 CrossRef.
- W. J. Hong, Y. Xu, G. Lu, X. Li and G. Shi, Electrochem. Commun., 2008, 5, 1555–1558 CrossRef.
- M. M. Cordero-Rando, J. L. Hidalgo-Hidalgo de Cisneros, E. Blanco and I. Naranjo-Rodríguez, Anal. Chem., 2002, 74, 2423–2427 CrossRef CAS.
- G. Patermarakis and K. Moussoutzanis, Electrochim. Acta, 1995, 40, 699–708 CrossRef CAS.
- W. Lu, R. Hartman, L. T. Qu and L. M. Dai, J. Phys. Chem. Lett., 2011, 2, 655–660 CrossRef CAS.
- D. Cericola, P. Novak, A. Wokaun and R. Kotz, J. Power Sources, 2011, 196, 10305–10313 CrossRef CAS.
- X. Meng, M. Ionescu, M. N. Banis, Y. Zhong, H. Liu and Y. Zhang, J. Nanopart. Res., 2011, 13, 1207–1208 CrossRef CAS.
- F. Meng, X. Yan, Y. Zhu and P. Si, Nanoscale Res. Lett., 2013, 8, 179–187 CrossRef PubMed.
- J. Zang, S. Bao, C. Li, H. Bian, X. Cui and Q. Bao, J. Phys. Chem. C, 2008, 112, 14843–14847 CAS.
- M. V. Fedorov and F. A. A. Kornyshev, J. Phys. Chem. B, 2008, 112, 11868–11872 CrossRef CAS PubMed.
- J. Vatamanu, L. Cao, O. Borodin, D. Bedrov and G. D. Smith, J. Phys. Chem. Lett., 2011, 2, 2267–2280 CrossRef CAS.
- A. Meyers and M. Weck, Macromolecules, 2003, 36, 1766–1768 CrossRef CAS.
- A. N. Rudenko, F. J. Keil, M. I. Katsnelson and A. I. Lichtenstin, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075422 CrossRef.
- J. P. Reed, B. Uchoa, Y. I. Joe, Y. Gan, D. Casa and E. Fradkin, Science, 2010, 330, 805–808 CrossRef CAS PubMed.
- J. E. Knox, M. D. Halls, H. P. Hratchianz and H. B. Schlegel, Phys. Chem. Chem. Phys., 2006, 8, 1371–1377 RSC.
- P. Ratik Desai, P. Shakya, T. Kreouzis, W. P. Gillin, N. A. Morley and M. R. J. Gibbs, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 094423 CrossRef.
- C. W. Tang and S. A. Vanslylke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- Y. Zhu, A. Bao, N. Xiao, G. Su and J. Lang, RSC Adv., 2015, 11, 82853–82858 RSC.
- J. J. P. Stewart, J. Mol. Model., 2008, 149, 499–535 CrossRef PubMed.
- T. Clark, A Handbook of Computational Chemistry: A P actical Guide to Chemical Structure and Energy Calculations, Wiley, John & Sons, Incorporated, 1985 Search PubMed.
- Y. Zhu, J. Guan, L. Cao and J. Hao, Talanta, 2010, 80, 1234–1238 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
A. Baoa,
B. Naa,
G. Sua,
J. Wanga and
J. Langa
*b,
A. Baoa,
B. Naa,
G. Sua,
J. Wanga and
J. Langa