
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of a viologen-based covalent organic polymer with redox-tunable polarity for dye adsorption†
Gobinda Dasa,
Tina Skorjanca,
Thirumurugan Prakasama,
Selbi Nuryyevaa,
John-Carl Olsenb and
Ali Trabolsi *a
*a
aNew York University Abu Dhabi, Saadiyat Island, P.O. Box 129188, Abu Dhabi, United Arab Emirates. E-mail: ali.trabolsi@nyu.edu
bSchool of Sciences, Indiana University Kokomo, Kokomo, IN 46904, USA
First published on 13th January 2017
Abstract
We report the efficient synthesis of a viologen-based covalent organic polymer by a microwave-assisted Menshutkin reaction. The polymer's viologen sub-units were synthesized in the 4,4′-bipyridinium dicationic state but could be reduced to radical-cationic and neutral forms. Regardless of the redox state of its viologens, the polymer was found to be thermally stable and capable of removing three dyes of differing polarities—fluorescein (FL), rhodamine B (RhB), and Nile red (NR)—from aqueous solutions. With its viologens in the dicationic state, the polymer was most efficient at removing FL, which is hydrophilic. With its viologens in the neutral state, it was most efficient at removing NR, which is hydrophobic. This strategy of using a single redox-tunable material for adsorbing multiple contaminants of different polarities represents a unique approach to water purification.
Water contamination caused by organic dyes from industrial waste is a major threat to the environment.1 Consequently, a considerable research effort has been undertaken to develop methods for capturing organic dyes.2 A wide variety of materials, including porous metal organic frameworks (MOFs),3–5 covalent organic frameworks (COFs),1,6,7 carbon-based materials,8–11 nanocomposites,12,13 and polymers,14–16 has been fabricated to facilitate their removal from waste waters. In order to have an effective material for dye-removal, the polarity of the material must be similar to that of the adsorbate molecules. It is therefore challenging to design a single material for the removal of different organic dyes because different dyes can have polarities that range from hydrophobic to hydrophilic. Many strategies have been used to adjust a host material's surface polarity.17–19 However, these methods produce adsorbents with invariant polarities that are not usually effective for adsorbing both hydrophilic and hydrophobic dyes. Furthermore, developing and scaling-up the syntheses of multiple adsorbents is not cost effective.
Viologens are a class of compounds that contain the 4,4′-bipyridinium unit and are well known for their rich redox chemistry. Viologens have three redox states that can be accessed chemically or electrochemically and can range from hydrophilic to hydrophobic.20 Viologen-based materials are therefore strong candidates for the adsorption of multiple dyes that have different polarities.21
Herein, we report the synthesis, via microwave-assisted Menshutkin reaction, of a cationic covalent organic polymer, COP++, in which bipyridinium units crosslink hexatopic cyclotriphosphazene (CTP) core moieties (Fig. 1).22,23
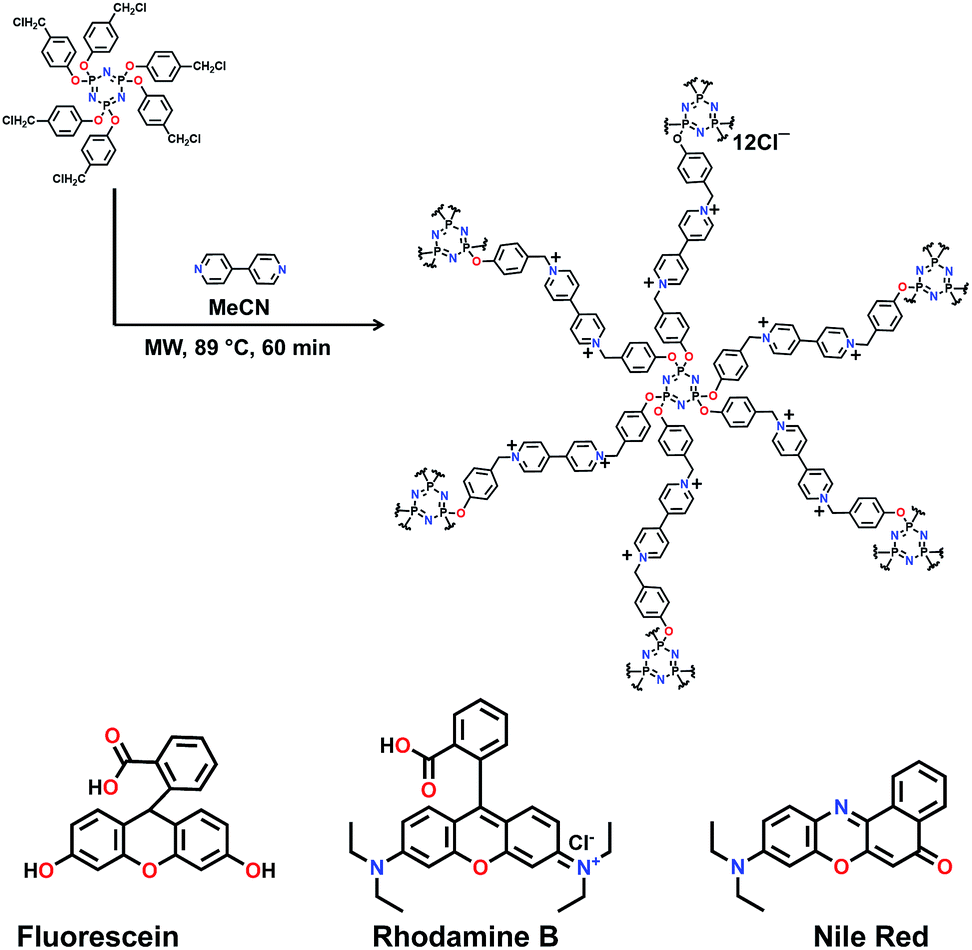 | ||
| Fig. 1 Top: Microwave-assisted Menshutkin reaction to form COP1++. Bottom: Chemical structures of the three organic dyes used in this study. | ||
Although the prepared polymer was found to be non-porous, its cationic nature and rich redox chemistry prompted us to study its interactions, in aqueous solutions, with three dyes of different polarities—fluorescein (FL), rhodamine B (RhB), and Nile red (NR). FL is hydrophilic and formally neutral, RhB is hydrophilic with a positively charged center, and NR is a hydrophobic dye.
The polarity of the polymer could be adjusted from hydrophilic, in its dicationic and radical-cationic forms (COP++ and COP1·+, respectively) to hydrophobic, in its neutral form (COP0), to match dye polarity. We accessed radical cationic and neutral forms by reaction with sodium dithionite and cobaltocene, respectively, and measured the adsorption of each dye to the polymer in each of its redox states. Consistent with its cationic nature, COP1++ exhibited fast and selective adsorption (100%) of electron-rich FL. Its adsorption of NR, on the other hand, was relatively slow and occurred to a much lesser extent (25%). In contrast, the neutral form of the polymer, COP10 adsorbed NR quickly and to a significant extent (85%). The radical-cationic polymer showed moderate to low adsorption efficiency for all three dyes.
To synthesize COP1++, a solution of 4,4′-dipyridyl (0.54 g, 3.5 mmol) in MeCN (50 mL) was added to hexakis(4-chloromethylphenoxy)cyclotriphosphazene (1.0 g, 1.02 mmol) in MeCN (50 mL). The reaction mixture was stirred under microwave irradiation of 2.45 GHz at 89 °C for 1 h and subsequently cooled to room temperature (Fig. 1 and Scheme S1†). The precipitate was filtered, washed with MeCN, H2O, acetone and n-hexane and dried under vacuum for 72 h to afford COP1++ as a yellow powder (1.56 g). The successful formation of the polymer was confirmed by Fourier transform infrared (FTIR) spectroscopy (Fig. S1†) and solid state cross polarization magic angle spinning (CP/MAS) 13C NMR (Fig. S2†). In the FTIR spectrum of COP1++, the absence of an absorption characteristic of aliphatic C–Cl stretching at 650–750 cm−1, and the presence of a strong absorption due to CH2 wagging at 1155 cm−1, confirmed the successful polymerization of hexakis(4-chloromethylphenoxy) cyclotriphosphazene and 4-4′ bipyridyl. The solid state CP/MAS 13C NMR spectra shows (Fig. S2†) a prominent peak near 65 ppm, which corresponds to the –NCH2 carbon and further confirmed polymer formation. COP1++ was not soluble in common organic solvents and was thermally more stable than the corresponding monomers (Fig. S4–S6†). Upon thermogravimetric analysis (TGA), COP1++ exhibited an initial weight loss of 5–10% below 200 °C, mainly due to solvent evaporation, and a steep weight loss at about 212 °C (Fig. S4–S6†). The broad pattern in its powder X-ray diffraction spectrum (Fig. S7†) and the roughly spherical (1 μm average diameter) particles visible in scanning electron spectroscopy (SEM) images (Fig. 2) indicate that COP1++ is amorphous. The absence of signal corresponding to nitrogen gas uptake in N2 adsorption isotherms (Fig. S8a†) measured at 77 K reveals that it is non-porous. The non-porous character of the synthesized polymer was further confirmed by measurement of its inability to adsorb CO2 (Fig. S8b†) at different temperatures.
 | ||
| Fig. 2 Scanning electron microscopy images of the viologen-linked covalent organic polymer in different redox states: COP1++ cationic species (a and b); COP1·+ radical species (c and d); and COP10 neutral species (e and f). The cationic form exhibits single-sphere morphology, while the radical and the neutral polymers are more aggregated. | ||
Sodium dithionite (Na2S2O4) and cobaltocene were used to reduce COP1++ in an oxygen-free environment to afford the radical-cationic COP1·+, and the neutral COP10 forms, respectively. COP1++ (30 mg) and an excess of Na2S2O4 or cobaltocene were added to degassed H2O or CH3CN under a nitrogen atmosphere, and the mixture was shaken for one minute. Upon addition of Na2S2O4 or cobaltocene, the color of the solid powder instantly changed from yellow to dark blue or brown, respectively. With the aid of centrifugation (4000 rpm, 15 rpm), the solid was then filtered and washed three times with degassed H2O under N2 atmosphere. The solid-state EPR spectrum of the powder recovered after reduction with Na2S2O4 reveals (Fig. S9†) a sharp, well-defined peak that reflects the presence of unpaired electrons and therefore the formation of the radical-cationic form of the polymer, COP1·+. The solid state UV-Vis spectrum of COP1·+ displays (Fig. S10†) a broad band at ∼605 nm, that is also indicative of radicals. The lack of signal in the EPR spectrum of the powder isolated after reduction with cobaltocene (Fig. S9c†) indicates formation of the neutral, diamagnetic form of the polymer. SEM images of the reduced forms of the polymer confirm their structural integrity, as no changes in the size of their particles and only slight changes in the morphology of their particles are apparent (Fig. 2c–f). TGA analysis show that all three forms, COP1++, COP1·+ and COP10, are thermally stable up to 400 °C (Fig. S2–S4†). Remarkably, COP1·+ was found to be stable for 10–15 days under ambient conditions. The enhanced stability of the radical cationic state within the polymer, relative to its more fleeting existence within the monomer, can be explained by steric protection as well as radical–radical and other stabilizing interactions that can occur within the polymeric structure.
FTIR spectra of the reduced forms of the polymer reveal (Fig. S1†) pronounced intensity changes compared to the FTIR spectrum of COP1++. Such changes may be the result of the electron transfer and reflect significant perturbations of bond polarities.24 In contrast, the frequencies of most absorptions are not drastically different than those of the corresponding absorptions in the spectrum of COP1++. Thus, the structural deformations that are caused by reduction are probably largely delocalized over many bonds.
The thermal and hydrolytic stability of our material and our having the means to effectively control its polarity by chemical reduction prompted us to exam its ability to adsorb and remove three organic dyes having different polarities from aqueous solutions. Of the dyes used, FL and RhB are hydrophilic and NR is hydrophobic. In addition, RhB is a positively charged molecule with a Cl− counter anion. About 30 mg of COP1++ was added to 10 mL of each of the following dye solutions: 1 × 10−4 M aqueous FL, 1 × 10−4 M aqueous RhB and 2 × 10−4 M NR in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:methanol mixture (Fig. 3). Absorbance was monitored by UV-Vis spectrophotometry prior to and at 10 minutes intervals after polymer addition. Bold orange lines in Fig. 3 correspond to the initial and final absorbance measurements of each dye solution. The data indicate that FL is readily adsorbed by both COP1++ and COP1·+ (Fig. 3a and b). Strong hydrophilic–hydrophilic interactions between FL and COP1++ result in a complete disappearance of the dye's absorption peak. COP1++ removed ∼99% of the FL in only 4 min (Fig. 4), which represents one of the fastest adsorption rates reported for organic polymers.25 COP1·+ and COP10 removed ∼40% and ∼11% of the FL, respectively, after 60 minutes (Fig. 4), which indicates that FL has less affinity for the more hydrophobic, reduced forms of the polymer.
:1 water:methanol mixture (Fig. 3). Absorbance was monitored by UV-Vis spectrophotometry prior to and at 10 minutes intervals after polymer addition. Bold orange lines in Fig. 3 correspond to the initial and final absorbance measurements of each dye solution. The data indicate that FL is readily adsorbed by both COP1++ and COP1·+ (Fig. 3a and b). Strong hydrophilic–hydrophilic interactions between FL and COP1++ result in a complete disappearance of the dye's absorption peak. COP1++ removed ∼99% of the FL in only 4 min (Fig. 4), which represents one of the fastest adsorption rates reported for organic polymers.25 COP1·+ and COP10 removed ∼40% and ∼11% of the FL, respectively, after 60 minutes (Fig. 4), which indicates that FL has less affinity for the more hydrophobic, reduced forms of the polymer.
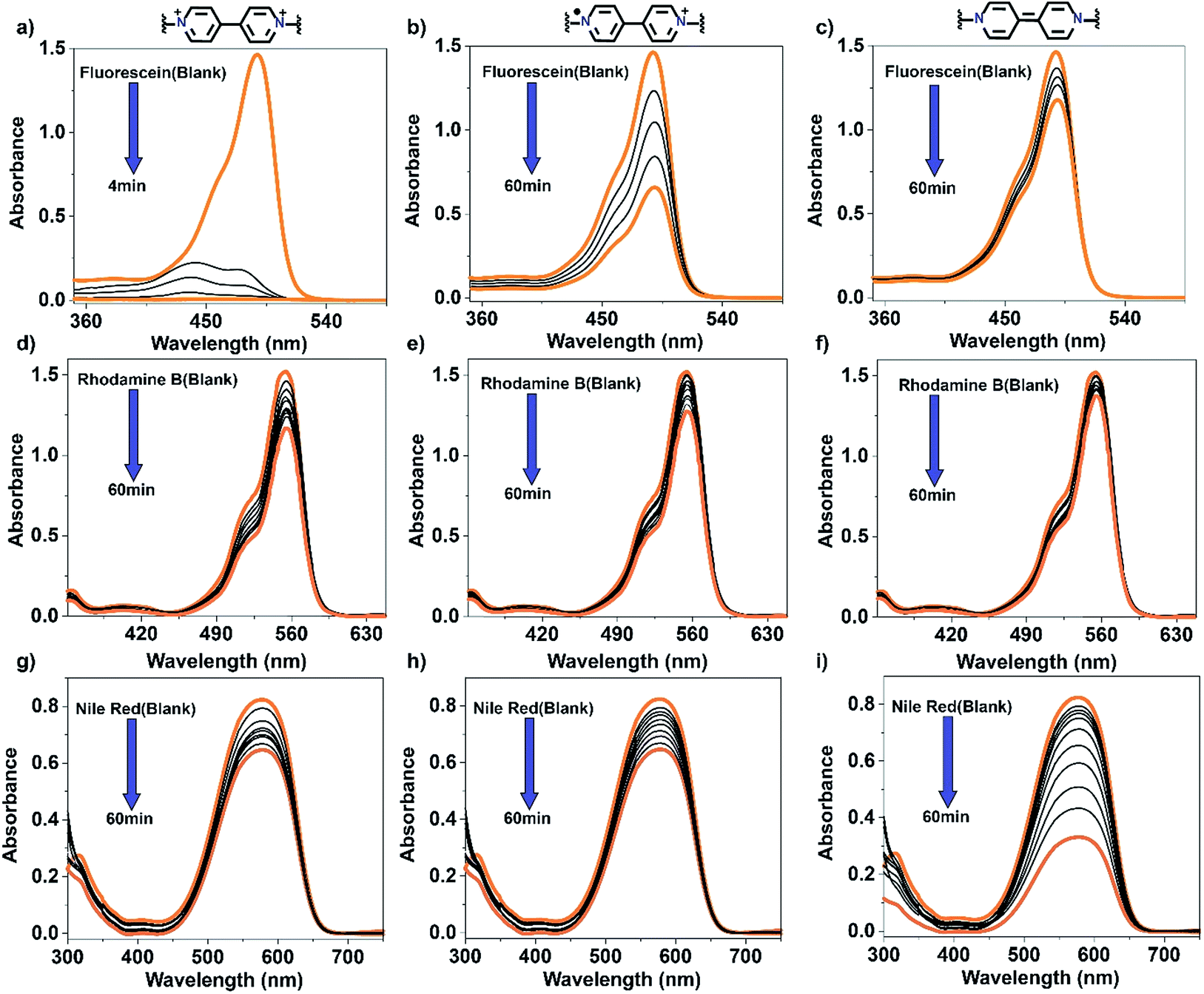 | ||
| Fig. 3 Spectroscopic monitoring of dye removal. (a–c) UV/Vis spectra of aqueous solutions of fluorescein and COP1++ (a), COP1·+ (b), or COP10 (c). (d–f) UV/Vis spectra of aqueous solutions of rhodamine B and COP1++ (d), COP1·+ (e), and COP10 (f). (g–i) UV/Vis spectra of 1:1 water:methanol solutions of Nile red and COP1++ (g), COP1·+ (h), and COP10 (i). | ||
 | ||
| Fig. 4 Adsorption capacities of the cationic, radical-cationic and neutral forms of COP1 for fluorescein, rhodamine B, and Nile Red. Adsorption time was one hour in all cases. | ||
COP1++ removed the hydrophilic RhB less efficiently than FL. A slow but steady decrease of RhB's absorption intensity in the UV-Vis measurements indicated ∼25% removal after one hour (Fig. 3d). This lower efficiency may be explained by the electrostatic repulsion between the positively charged polymer and the positively charged dye. COP1·+ was slightly less efficient at removing RhB than COP1++, adsorbing ∼20% of the dye after one hour (Fig. 3e and 4). Consistent with its increasing hydrophobic character, COP10 was the least efficient, removing a barely detectible amount of RhB after an hour (Fig. 3f and 4).
COP1++ removed a moderate amount (25%) of NR (Fig. 3g and 4) after one hour, reflecting a weak interaction between this hydrophobic dye and the positively charged polymer. COP1·+ removed slightly less NR (Fig. 3h). In contrast, COP10 removed 85% of the NR within one hour, as a result of increased hydrophobic interactions (Fig. 3i and 4).
In order to recycle the different forms of the polymer, we conducted regeneration tests that corresponded to the cases where dye removal had been most efficient: for COP1++ removal of FL and for COP10 removal of NR. In these experiments, dye-loaded polymer samples were dried at 60 °C and then immersed in different solvents at room temperature. Dye release was monitored by UV-Vis and fluorescence spectroscopies. The release of FL from FL-loaded COP1++ was measured in a saturated aqueous solution of sodium chloride and confirmed by an increase in the intensity of FL's characteristic absorption band. Ninety percent of the FL was released within one hour (Fig. S11†). We infer that the competitive binding of chloride anions to the polymer facilitates the release of FL molecules.
NR was removed from COP10 by immersion in methanol, and the process was followed by fluorescence spectroscopy. The neutral polymer released ∼90% of the dye in one hour (Fig. S12 and S13†). After regeneration, the polymer samples were analyzed by FTIR (Fig. S14†) and SEM (Fig. S15†), and their spherical morphologies were found to be intact, confirming their structural robustness.
In summary, microwave radiation was used to efficiently prepare, by Menshutkin coupling, a thermally stable cationic polymer, in which 4,4′-bipyridinium units link hexatopic phosphazene core moieties. The redox responsive viologen units of the polymer allowed access to radical cationic and neutral states. Switching the viologens' redox states allowed the polymer's polarity to be adjusted from the hydrophilic dicationic form to hydrophobic neutral one. The three forms of the polymer were tested for their ability to remove three organic dyes from aqueous solutions. The cationic polymer was highly selective and efficient for the adsorption of fluorescein, which is a hydrophilic dye; showed moderate uptake of rhodamine B, which is positively charged and hydrophilic; and demonstrated poor uptake for Nile red, which is hydrophobic. The radical cationic form of the polymer displayed poor uptake and selectivity for all three dyes. The neutral form was highly selective for Nile red as compared to fluorescein and rhodamine B. Thus, this versatile polymer with its easily adjustable redox states is a promising material for the removal of dyes of different polarities from waste water.
Acknowledgements
The research described here was sponsored by New York University Abu Dhabi (NYUAD), UAE. G. D., T. S., T. P., S. N. and A. T. thank NYUAD for its generous support of the research program at NYUAD. The research was carried out by using the Core Technology Platform resources at NYUAD. The authors thank Professor Kyriaki Polychronopoulou for the CO2 gas uptake measurements.Notes and references
- S.-B. Yu, H. Lyu, J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Polym. Chem., 2016, 7, 3392–3397 RSC.
- A. K. Gupta, R. R. Naregalkar, V. D. Vaidya and M. Gupta, Nanomedicine, 2007, 2, 23–39 CrossRef CAS PubMed.
- X. Zhao, S. Liu, Z. Tang, H. Niu, Y. Cai, W. Meng, F. Wu and J. P. Giesy, Sci. Rep., 2015, 5, 11849 CrossRef PubMed.
- S. Duan, J.-X. Li, X. Liu, Y. Wang, S. Zeng, D. Shao and T. Hayat, ACS Sustainable Chem. Eng., 2016, 4, 3368–3378 CrossRef CAS.
- Y.-C. He, J. Yang, W.-Q. Kan, H.-M. Zhang, Y.-Y. Liu and J.-F. Ma, J. Mater. Chem. A, 2015, 3, 1675–1681 CAS.
- W. Zhang, F. Liang, C. Li, L.-G. Qiu, Y.-P. Yuan, F.-M. Peng, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 186, 984–990 CrossRef CAS PubMed.
- T. Wang, K. Kailasam, P. Xiao, G. Chen, L. Chen, L. Wang, J. Li and J. Zhu, Microporous Mesoporous Mater., 2014, 187, 63–70 CrossRef CAS.
- Z. Zhang and J. Kong, J. Hazard. Mater., 2011, 193, 325–329 CrossRef CAS PubMed.
- J. N. Tiwari, K. Mahesh, N. H. Le, K. C. Kemp, R. Timilsina, R. N. Tiwari and K. S. Kim, Carbon, 2013, 56, 173–182 CrossRef CAS.
- B. Chen, Q. Ma, C. Tan, T. Lim, L. Huang and H. Zhang, Small, 2015, 11, 3319–3336 CrossRef CAS PubMed.
- X. Liu, L. Yan, W. Yin, L. Zhou, G. Tian, J. Shi, Z. Yang, D. Xiao, Z. Gu and Y. Zhao, J. Mater. Chem. A, 2014, 2, 12296–12303 CAS.
- P. Hu, L. Han and S. Dong, ACS Appl. Mater. Interfaces, 2013, 6, 500–506 Search PubMed.
- A. K. Sarkar, A. Saha, A. Tarafder, A. B. Panda and S. Pal, ACS Sustainable Chem. Eng., 2016, 4, 1679–1688 CrossRef CAS.
- P. Kuhn, K. Krüger, A. Thomas and M. Antonietti, Chem. Commun., 2008, 5815–5817 RSC.
- Z. Wang, J. Guo, J. Ma and L. Shao, J. Mater. Chem. A, 2015, 3, 19960–19968 CAS.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2015, 529, 190–194 CrossRef PubMed.
- Z. Gan, A. Zhao, M. Zhang, W. Tao, H. Guo, Q. Gao, R. Mao and E. Liu, Dalton Trans., 2013, 42, 8597–8605 RSC.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2012, 116, 6873–6878 CAS.
- L. Xu, Y. Luo, L. Sun, S. Pu, M. Fang, R.-X. Yuan and H.-B. Du, Dalton Trans., 2016, 45, 8614–8621 RSC.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- G.-Q. Gao and A.-W. Xu, New J. Chem., 2014, 38, 4661–4665 RSC.
- K. Wadhwa, S. Nuryyeva, A. C. Fahrenbach, M. Elhabiri, C. P. Iglesias and A. Trabolsi, J. Mat. Chemi. C, 2013, 1, 2302–2307 RSC.
- G. Das, T. Prakasam, S. Nuryyeva, D. Suk Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, M. Jouiad, F. Ravaux and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 CAS.
- O. Poizat, C. Sourisseau and J. Corset, J. Mol. Struct., 1986, 143, 203–206 CrossRef CAS.
- S. Pirillo, L. Cornaglia, M. L. Ferreira and E. H. Rueda, Spectrochim. Acta, Part A, 2008, 71, 636–643 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26332a |
| This journal is © The Royal Society of Chemistry 2017 |