
Evaluation of de novo-designed coiled coils as off-the-shelf components for protein assembly†
Ajitha S.
Cristie-David
a,
Aaron
Sciore
a,
Somayesadat
Badieyan
a,
Joseph D.
Escheweiler
 a,
Philipp
Koldewey
b,
James C. A.
Bardwell
bcd,
Brandon T.
Ruotolo
a and
E. Neil G.
Marsh
*ac
a,
Philipp
Koldewey
b,
James C. A.
Bardwell
bcd,
Brandon T.
Ruotolo
a and
E. Neil G.
Marsh
*ac
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: nmarsh@umich.edu
bDepartment of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA
cDepartment of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA
dHoward Hughes Medical Institute, USA
First published on 30th March 2017
Abstract
Coiled-coil domains are attractive modular components for assembling individual protein subunits into higher order structures because they can be designed de novo with well-defined oligomerization states, topologies, and dissociation energies. However, the utility of coiled-coil designs as plug-and-play components for synthetic biology applications depends critically on them robustly maintaining their oligomerization states when fused to larger proteins of interest. Here, we investigate the ability of a series of well-characterized de novo-designed parallel coiled coils, with oligomerization states ranging from dimer to pentamer, to mediate the oligomerization of a model monomeric protein, green fluorescent protein (GFP). Six coiled-coil GFP fusion proteins were initially constructed and their oligomerization states investigated using size exclusion chromatography, analytical ultracentrifugation, and native mass spectrometry. Somewhat surprisingly, only two of these initial designs adopted their intended oligomerization states. However, with minor refinements, the intended oligomerization states of two of the four other constructs could be achieved. Parameters found to influence the oligomerization state of the GFP fusions included the number of heptad repeats and the length of the linker sequence separating GFP from the coiled coil. These results demonstrate that even for stable, well-designed coiled coils, the oligomerization state is subject to unanticipated changes when connected to larger protein components. Therefore, although coiled coils can be successfully used as components in protein designs their ability to achieve the desired oligomerization state requires experimental verification.
Design, System, ApplicationCoiled coils are short, helical protein sequences that can serve as oligomerization units to mediate the assembly of larger protein complexes. Their potential utility as components for protein engineering hinges on the fact that, based on a simple set of principles, they can be designed de novo to adopt structures with well-defined oligomerization states and tunable interaction strengths. This makes them attractive components for connecting together larger proteins to form new biological systems with new functions. However, for their utility as modular components to be fully realized, the coiled coils must reliably maintain their oligomerization states independently of the proteins to which they are attached. To test this, we examined a series of previously well-characterized coiled-coil designs to determine whether they maintained their oligomerization states when fused to a model monomeric protein. We found that only two of the six designs evaluated initially behaved as intended. After optimization, a further two designs adopted the correct oligomerization state. Our results demonstrate a limitation of coiled coils as plug-and-play components due to context-dependent changes in their structures. To successfully apply coiled coils in the construction of large-scale protein assemblies, it will likely be necessary to both carefully validate and optimize the coiled-coil interaction. |
Introduction
α-Helical coiled-coil domains represent one of the simplest and best understood protein–protein interactions. They occur widely in natural proteins, comprising up to 5% of all protein residues,1 and were among the first protein structures to be designed de novo.2,3 Simple rules, based on a repeating canonical heptad of amino acid residues, apparently govern the interactions between the α-helices of coiled-coil domains. The use of these rules has allowed the design of a wide range of small α-helical bundle proteins whose structures comprise between 2 and 7 α-helices.4–8 The helices may be designed to adopt either parallel or antiparallel topologies9 and may be identical (homooligomeric) or complementary (heterooligomeric) in sequence.10,11Further design iterations have produced coiled coils that self-assemble in response to changes in ionic strength,12 pH,13–15 or the presence of metal ions.16–18 This has led to the use of designed coiled coils to create, for example, metalloenzyme mimics,19 pH-responsive hydrogels,20,21 two-dimensional protein lattices,22,23 and three-dimensional protein nanoparticles.24 These nanoparticles have subsequently been adapted for the polyvalent display of viral epitopes to create a novel vaccine.25,26
In nature, coiled-coil interactions often mediate the assembly of larger protein domains. Well-studied examples include the dimerization of many transcription factors27 and the assembly of multi-enzyme complexes such as pyruvate dehydrogenase28 and polyketide synthases.29 Coiled coils are also important in the interaction of proteins with membranes.30–33 The ability of coiled coils to mediate the controlled assembly of large-scale protein structures has attracted the interest of synthetic biologists. Thus, in our laboratory, we have used coiled coils to design self-assembling protein cages. For example, by genetically fusing a complementary pair of designed anti-parallel heterodimeric coiled coils to a small trimeric protein, we were able to create a highly flexible but heterogeneous set of self-assembling cage-like structures.34,35 More recently, we used a parallel homo-tetrameric coiled coil to mediate the assembly of another trimeric protein into a well-defined octahedral cage.36
The potential for coiled coils to function as simple off-the-shelf connectors or recognition elements in synthetic biology has motivated efforts to curate “basis sets” of coiled coils (both homo-oligomeric and hetero-oligomeric systems) that possess well-defined oligomerization states, topologies, and interaction energies.37–40 Clearly, for these coiled coils to be useful synthetic components, it is essential that they maintain their intended oligomerization states when attached to other protein domains. However, so far, the properties of these coiled coils have been almost exclusively investigated in isolation, leaving open the possibility that they may significantly differ in their behavior when genetically fused to much larger, natural proteins. Therefore, we decided it was important to investigate how reliably such coiled coils may function as components for protein assembly.
The goal of this study was to determine how reliably coiled coil designs maintain their designed oligomerization state when fused to a larger protein domain. In other words, to what extent does the context in which the coiled coil is introduced influence the oligomerization state of the coiled coil? To answer this question, we have examined the ability of a series of well-characterized coiled coils to mediate the oligomerization of a model monomeric protein, green fluorescent protein (GFP). Our results clearly show that optimization and experimental verification is necessary if coiled coils are to be successfully used as components for protein assembly.
Results and discussion
Selection of coiled coils for evaluation
For our studies, we selected a series of crystallographically characterized four-heptad parallel coiled coils designed by Woolfson and co-workers.37,38,41 These coiled coils span the range of oligomeric states from dimer to pentamer, making them potentially useful for the assembly of a wide variety of protein structures. Four of the coiled coils were based on the well-established canonical heptad repeat sequence (XAAYKZE)4 in which the oligomerization state is primarily determined by the identity of the hydrophobic residues X and Y at the canonical ‘a’ and ‘d’ positions. The dimeric coiled coil (Protein Data Bank (PDB) ID 4DZM) employs Ile at the ‘a’ and Leu at the ‘d’ positions, with the exception of Asn at the third ‘a’ position. Two trimeric coiled coils were investigated: one (PDB ID 4DZL) employs Ile at the ‘a’ and ‘d’ positions, whereas the other (PDB ID 4DZN) employs Ile at the ‘a’ and Leu at the ‘d’ positions. The tetrameric coiled coil (PDB ID 3R4A) employs Leu at the ‘a’ and Ile at the ‘d’ positions. The residue ‘Z’ at the solvent-exposed ‘f’ position is Gln, Trp or Lys depending upon the coiled coil. The larger size of the pentameric coiled coil (PDB ID 4PN8) results in a solvent-exposed channel at the center of the helical bundle. In this case, the hydrophobic interactions between the helices occur at the ‘a’, ‘d’, and ‘g’ positions, which are occupied by Leu, Ile and Ile respectively. The sequences and oligomerization states of these coiled coils are summarized in Table 1.Fusion of coiled coils with GFP
We chose GFP as a model protein domain with which to test the coiled coils ability to mediate protein oligomerization. In addition to GFP being a very well characterized, stable, monomeric protein, its fluorescent property makes it easy to assess whether the addition of the coiled coil domain may have interfered with protein folding. We note that the GFP variant encoded in the PMCSG18 vector we used in these studies has a known tendency to dimerize at high concentrations, but at the lower concentrations used in these studies no dimerization was discernable.To construct fusion proteins in which the coiled-coil sequence was added to either the N or C terminus of GFP we used the standard molecular biology techniques described in the experimental section. A glycine-rich spacer sequence was introduced between the GFP and coiled-coil domains to alleviate steric constraints that might cause either domain to misfold (Fig. 1). Based on modeling that assumed a hydrodynamic radius for GFP of 2.4 nm, we initially set the length of this spacer to be 6 Gly residues, which is sufficient to span a distance of ∼1.8 nm. This should allow the coiled coil domain to oligomerize without introducing steric clashes between the appended GFP domains. (As discussed later, in some designs, the length of the glycine spacer was increased or the coiled-coil sequence modified from the initial design). A description of each construct studied is given in Table 2. The fusion proteins were over-expressed in Escherichia coli BL21 and purified by Ni-NTA affinity chromatography in good yields without difficulty. All constructs exhibited the characteristic fluorescent green color of GFP, indicating that the proteins were correctly folded.
 | ||
| Fig. 1 Diagram showing topology of the GFP coiled-coil fusion proteins for coiled coils fused to the C terminus of GFP (A) and to the N terminus of GFP (B). | ||
| Construct number | Position of coiled coila | Spacer lengthb | Coiled-coil sequencec | Intended oligomerization stated | ||||
|---|---|---|---|---|---|---|---|---|
| a Position of the coiled coil relative to GFP. b Number of Gly residues separating the GFP and coiled-coil domains. c Constructs 9 and 10 contain coiled coils with 5 repeating heptads. d Based on the crystal structure of the isolated coiled coil. | ||||||||
| 1 | C terminus | 6 Gly | IAALKQE | IAALKQE | IAANKQE | IAALKQE | Dimer | |
| 2 | C terminus | 6 Gly | IAALKQE | IAALKQE | IAALKQE | IAALKQE | Trimer | |
| 3 | C terminus | 6 Gly | IAAIKQE | IAAIKQE | IAAIKQE | IAAIKQE | Trimer | |
| 4 | C terminus | 6 Gly | LAAIKQE | LAAIKQE | LAAIKQE | LAAIKQE | Tetramer | |
| 5 | C terminus | 6 Gly | KIEQILQ | KIEKILQ | KIEWILQ | KIEQILQ | Pentamer | |
| 6 | C terminus | 9 Gly | KIEQILQ | KIEKILQ | KIEWILQ | KIEQILQ | Pentamer | |
| 7 | C terminus | 9 Gly | KIEQILQ | KIEKILQ | KIEQILQ | KIEQILQ | Pentamer | |
| 8 | N terminus | 6 Gly | IAAIKQE | IAAIKQE | IAAIKQE | IAAIKQE | Trimer | |
| 9 | N terminus | 6 Gly | IAAIKQE | IAAIKQE | IAAIKQE | IAAIKQE | AAIKQEI | Trimer |
| 10 | C terminus | 6 Gly | LAAIKQE | LAAIKQE | LAAIKQE | LAAIKQE | LAAIKQE | Tetramer |
| 11 | C terminus | 9 Gly | LAAIKQE | LAAIKQE | LAAIKQE | LAAIKQE | Tetramer | |
Evaluation of GFP-coiled-coil oligomerization states
To determine whether the oligomerization state of the original coiled coil was maintained in the GFP fusion constructs, we examined the oligomerization states of the various GFP constructs using three complementary techniques: size exclusion chromatography (SEC), sedimentation velocity analytical ultracentrifugation (AUC), and non-denaturing native mass spectrometry (native MS). Analytical ultracentrifugation allows the molecular mass of macromolecules in solution, as well as their size distribution, to be accurately determined. SEC does not directly measure molecular weights, but is a simple and rapid solution phase method that can report on sample homogeneity and provide an estimate of oligomerization state. In native MS, samples are ionized under very mild conditions so that non-covalent protein–protein interactions are maintained; native MS can thus provide high-resolution measurements of protein masses that allow their oligomerization states to be deduced, with the caveat that these gas phase measurements may be biased by differences in the ease with which various oligomeric species can be ionized.Characterization of monomeric wild-type GFP (as a control) and fusion proteins 1 (dimer) and 3 (trimer) by these techniques yielded consistent data indicating that each construct was fairly homogeneous and adopted its intended oligomerization state (Fig. 2A, B, and D; Table 3). However, fusion proteins 2 (expected trimer) and 4 (expected tetramer) (Fig. 2C and E) appeared to be a dimer and a trimer, respectively, by each of the techniques used to assess their oligomerization states, even though the coiled-coil domains had been demonstrated by crystallography to be trimeric and tetrameric, respectively. This result may be less surprising for fusion protein 2, as there are several structures of coiled-coil dimers in which the hydrophobic interior is packed with Ile at ‘a’ and Leu at ‘d’ positions. However, it was quite unexpected for fusion protein 4, for which the majority of synthetic coiled-coil structures with Leu at ‘a’ and Ile at ‘d’ positions are tetrameric.
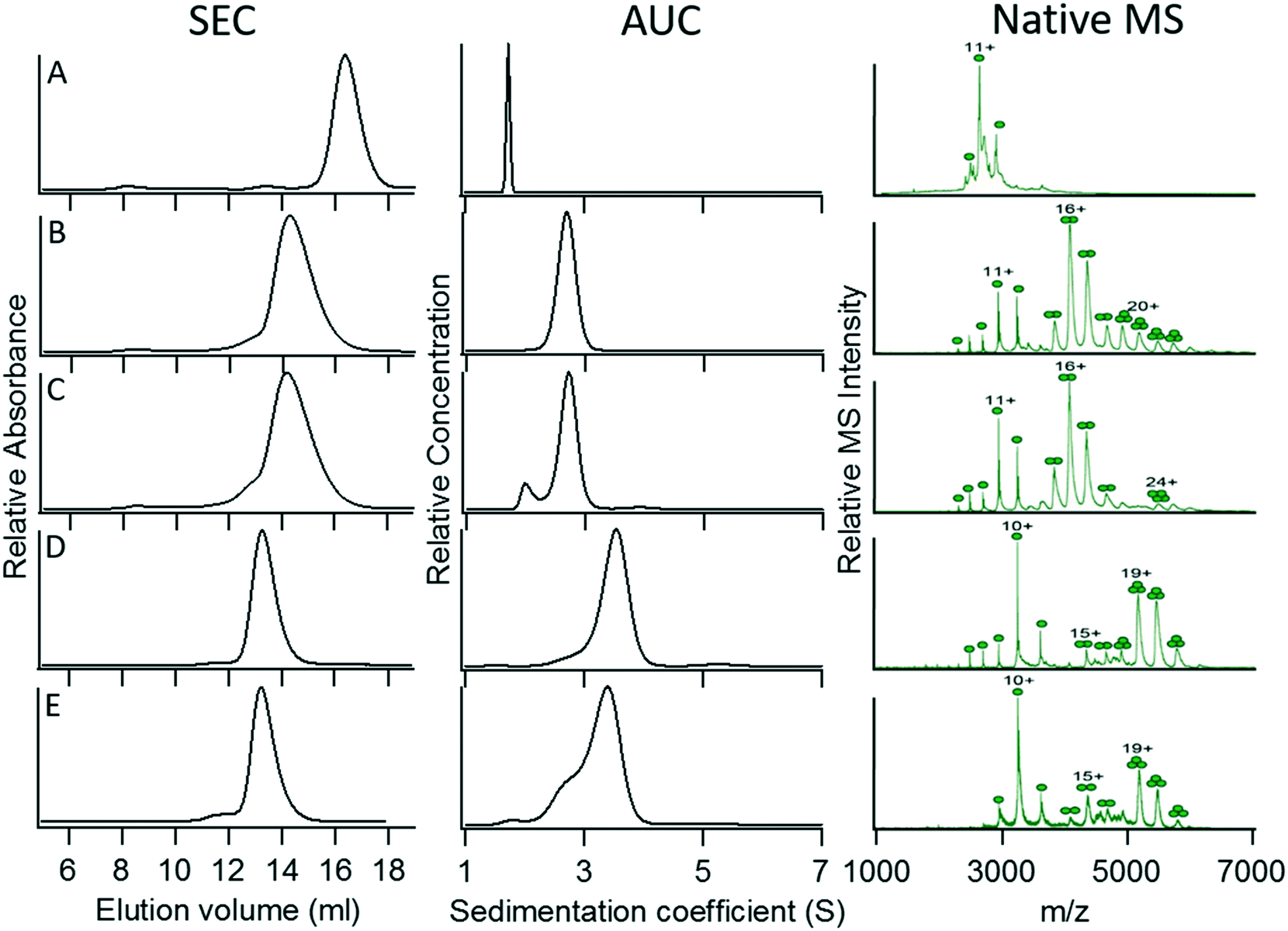 | ||
| Fig. 2 Characterization of the oligomerization state of C-terminal GFP constructs 1–4. (A) Wild-type GFP. (B) Construct 1 (intended dimer). (C) Construct 2 (intended trimer). (D) Construct 3 (intended trimer). (E) Construct 4 (intended tetramer). For details of the constructs, refer to Table 2. | ||
| GFP fusion construct | Elution volume (mL) | Sedimentation coefficient, (S)a | Frictional ratio (f/f0)b | M r (kDa) calc. from s and f/f0 | M r (Da) native MSc | Coiled-coil oligomerization stated | Observed oligomerization statee |
|---|---|---|---|---|---|---|---|
| a In samples for which more than one species is present, the sedimentation coefficient of the major species is reported. b Average frictional coefficient measured over all sedimenting species. c The molecular masses determined by native MS include varying numbers of non-specifically bound ions that derive from the buffer. Therefore, the molecular masses are not exact multiples of the protein Mr derived from the sequence. d Oligomerization state determined from the crystal structure. e Consensus from the three experimental methods used to examine the oligomerization state of the fusion proteins. f The presence of aggregated protein in the sample prevented the frictional ratio from being reliably determined from the data. | |||||||
| WT GFP | 16.3 | 1.73 | 1.31 | 30.3 ± 0.4 | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 053 ± 5 053 ± 5 |
Monomer | Monomer |
| 1 | 14.3 | 2.68 | 1.4 | 65.0 ± 4.4 | 64940 ± 80 |
Dimer | Dimer |
| 2 | 14.1 | 2.71 | 1.37 | 63.7 ± 0.6 | 65130 ± 60 |
Trimer | Dimer |
| 3 | 13.3 | 3.53 | 1.43 | 101.6 ± 5.9 | 97743 ± 30 |
Trimer | Trimer |
| 4 | 13.2 | 3.39 | 1.39 | 91.4 ± 6.8 | 98000 ± 120 |
Tetramer | Trimer |
| 5 | 8.2 | N/D | N/D | N/D | N/D | Pentamer | Aggregate |
| 6 | 11.9, 10.7, 8.1 | 4.70 | N/Df | N/D | 166300 ± 80 |
Pentamer | Pentamer-aggregate |
| 7 | 12.1 | 4.65 | 1.39 | 144.3 ± 30 | 153500 ± 160 |
Pentamer | Pentamer |
| 8 | 15.5 | 1.76 | 1.99 | 31.7 ± 0.5 | 32550 ± 6 |
Trimer | Monomer |
| 9 | 12.7 | 3.29 | 1.52 | 96.9 ± 14.1 | 104530 ± 43 |
Trimer | Trimer |
| 10 | 13.3 | 3.44 | 1.42 | 97.2 ± 9.0 | 98400 ± 120 |
Tetramer | Trimer |
| 11 | 13.4 | 3.36 | 1.41 | 88.0 ± 0.9 | 100260 ± 70 |
Tetramer | Trimer |
These results demonstrate that the addition of a large protein domain to the coiled coil has the potential to alter its oligomerization state. This observation is consistent with the previously described lability of some parallel coiled-coil designs in which the oligomerization state is sensitive to fairly subtle changes in hydrophobic core packing.6 Interestingly, in both cases in which the oligomerization state departs from what is expected, the fusion protein adopts a lower oligomerization state. This suggests that unfavorable steric interactions between the GFP domains (despite the introduction of a flexible spacer sequence) may cause reorganization of the coiled-coil structure.
We next examined the properties of a GFP fusion protein containing a pentameric coiled coil, construct 5. Although the protein was expressed as soluble, fluorescent protein (indicating that the GFP domain was correctly folded), SEC of 5 yielded a single peak in the void volume (Fig. 3A). This suggested that the protein was forming large aggregates that could not be further characterized by AUC or native MS. In this case, it seemed that a six-Gly spacer might be too short to permit the simultaneous proper folding of the GFP domain and the pentameric coiled-coil domain. We therefore increased the length of the spacer to nine Gly residues, resulting in construct 6. This construct showed significantly less tendency to aggregate, but characterization by SEC indicated that it still formed a mixture of oligomeric species (Fig. 3B). Characterization of 6 by AUC allowed the sedimentation coefficient for the major species to be determined as 4.7 S; however, the presence of aggregated proteins prevented the frictional ratio from being reliably determined so that the molecular weight could not be calculated. Characterization of 6 by native MS provided good evidence for the formation of the intended pentameric species, although this technique would not detect high molecular weight aggregates. Further mutation of a Trp residue at a solvent-exposed ‘f’ position (initially introduced to facilitate spectrophotometric quantification of the synthetic coiled coil) to a more hydrophilic Asn residue (construct 7) resulted in a more monodisperse protein species that appeared to adopt the intended pentameric oligomerization state as judged by SEC, AUC, and native MS (Fig. 3C). Although the ‘f’ position of the heptad repeat is solvent-exposed and therefore does not influence inter-helix interactions, this observation indicates that interactions between exterior residues of the coiled-coil domain and the appended protein domain (in this case GFP) may need to be considered and optimized for the protein to assemble correctly.
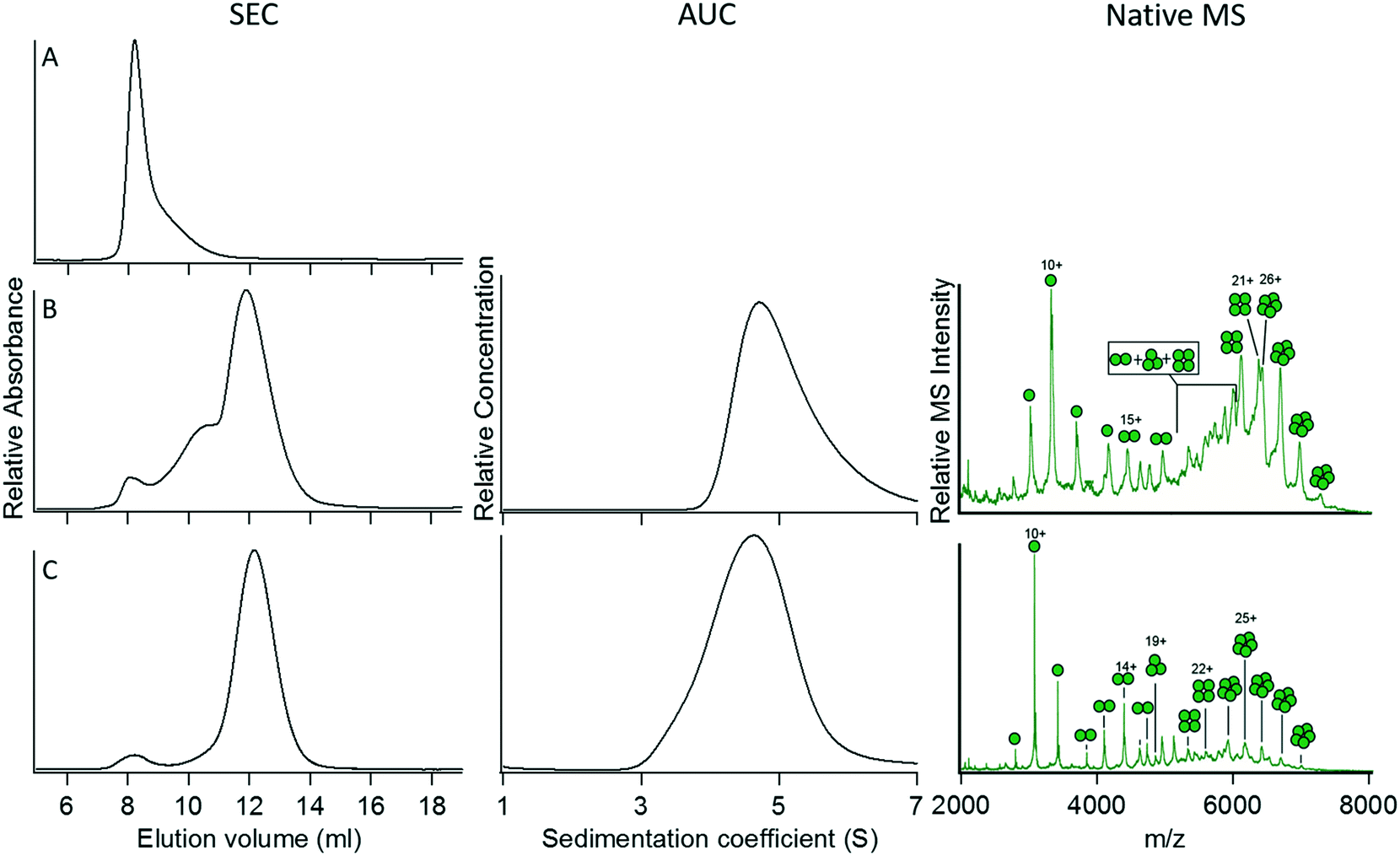 | ||
| Fig. 3 Characterization and optimization of GFP fused with the pentameric coiled coil 4PN8. (A) Construct 5, for which aggregation prevented its characterization by AUC or native MS. (B) Construct 6. (C) Construct 7. For details of the constructs, refer to Table 2. | ||
Transposition of oligomerization domain to N terminus of GFP
Construct 3 successfully mediated the trimerization of GFP when fused to its C-terminus. We therefore extended our studies on this coiled-coil domain to investigate whether it would function similarly when placed at the N terminus of GFP. The coiled-coil sequence from 4DZL (containing Ile at ‘a’ and ‘d’) was genetically fused to the N-terminus of GFP separated by the same six-Gly spacer sequence to give construct 8.Surprisingly, although construct 8 was expressed as a soluble, well-folded protein, it failed to oligomerize at all (Fig. 4A). Its elution volume (determined by SEC), sedimentation coefficient, and native mass spectrum all indicated that the protein was exclusively monomeric. In this case, it seemed unlikely that steric crowding between the GFP domains would explain the protein's failure to oligomerize. However, it is possible that placing the coiled coil domain at the N-terminus may have resulted in unintended interactions between the coiled coil and the N-terminal 6-His sequence used to purify the protein that interfered with the coiled coil's ability to oligomerize. In particular, electrostatic interactions between the (partially) positively charged 6-His sequence and negatively charged glutamate residues in the coiled coil domain could potentially disrupt the coiled coil structure.
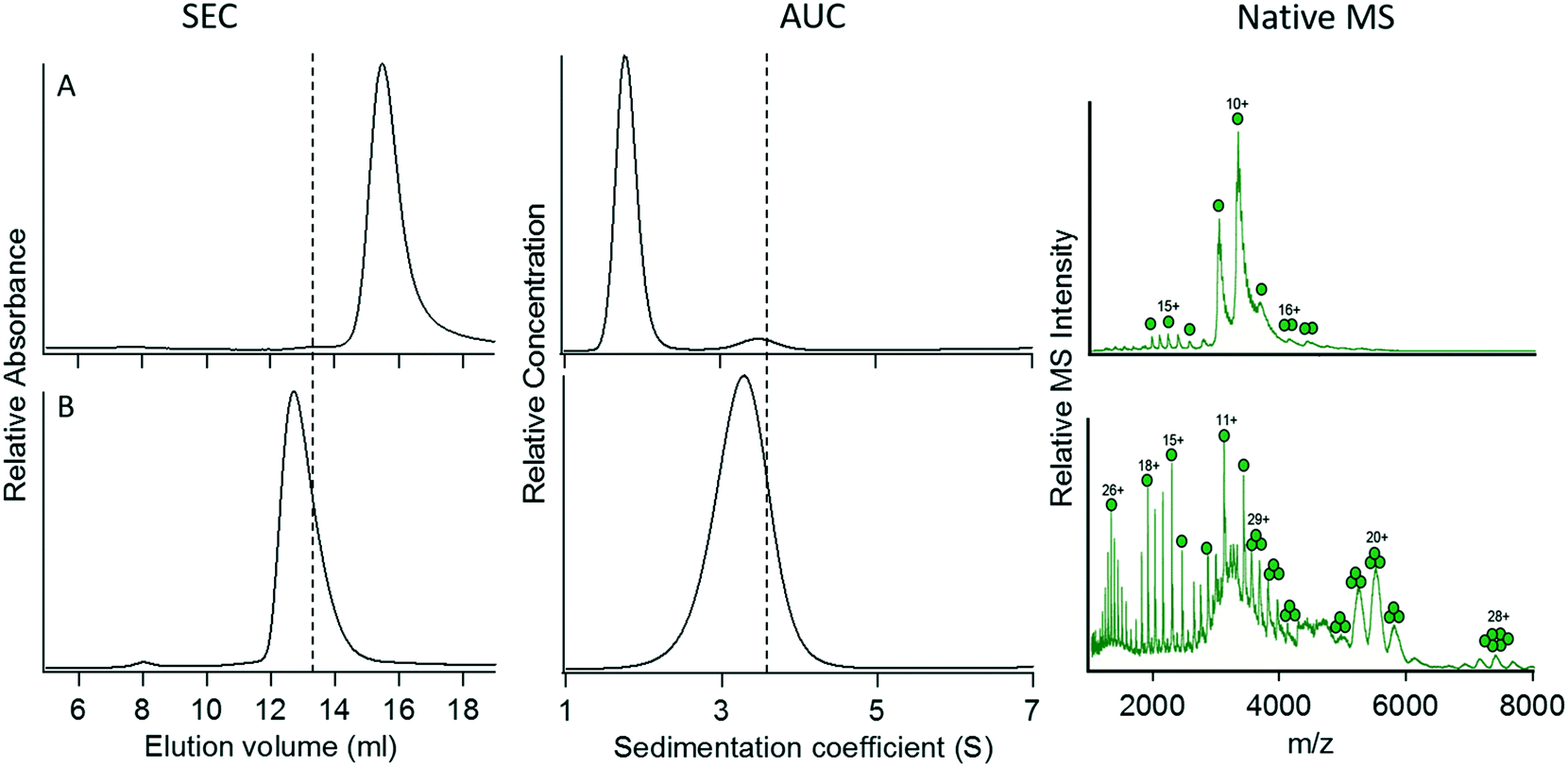 | ||
| Fig. 4 Characterization of fusion proteins in which the trimeric coiled coil 4DZL was linked to the N terminus of GFP. (A) Construct 8. (B) Construct 9. The vertical dotted lines denote the elution volume and sedimentation coefficient measured for the C-terminal fusion protein, 3. For details of the constructs, refer to Table 2. | ||
Therefore, we investigated the effect of strengthening the coiled-coil interaction by increasing the number of heptad repeats in the coiled-coil domain from 4 to 5, resulting in construct 9. The addition of the extra heptad appeared to restore the original trimeric oligomerization state of the coiled-coil domain. This suggests that increasing the strength of the coiled coil interaction was sufficient to overcome the unintended interactions between the coiled coil and either the His-tag or GFP that initially prevented the fusion protein from trimerizing.
Attempts to construct a tetrameric GFP fusion protein
Based on what we learned from optimizing the trimeric and pentameric coiled-coil designs, we attempted to construct a tetrameric fusion protein of GFP by either increasing the length of the spacer sequence between GFP and the coiled-coil domain or increasing the strength of the coiled-coil interaction. In construct 10, we increased the number of Gly residues in the spacer from 6 to 9, and in construct 11, we increased the number of heptad repeat units from 4 to 5. Both these constructs expressed as soluble, folded protein and could be purified to homogeneity. However, analysis of their oligomerization states by SEC, AUC, and native MS each indicated that 10 and 11 remained trimeric (Fig. 5). Why these GFP fusion proteins appear to adopt a trimeric rather than a tetrameric oligomerization state is unclear.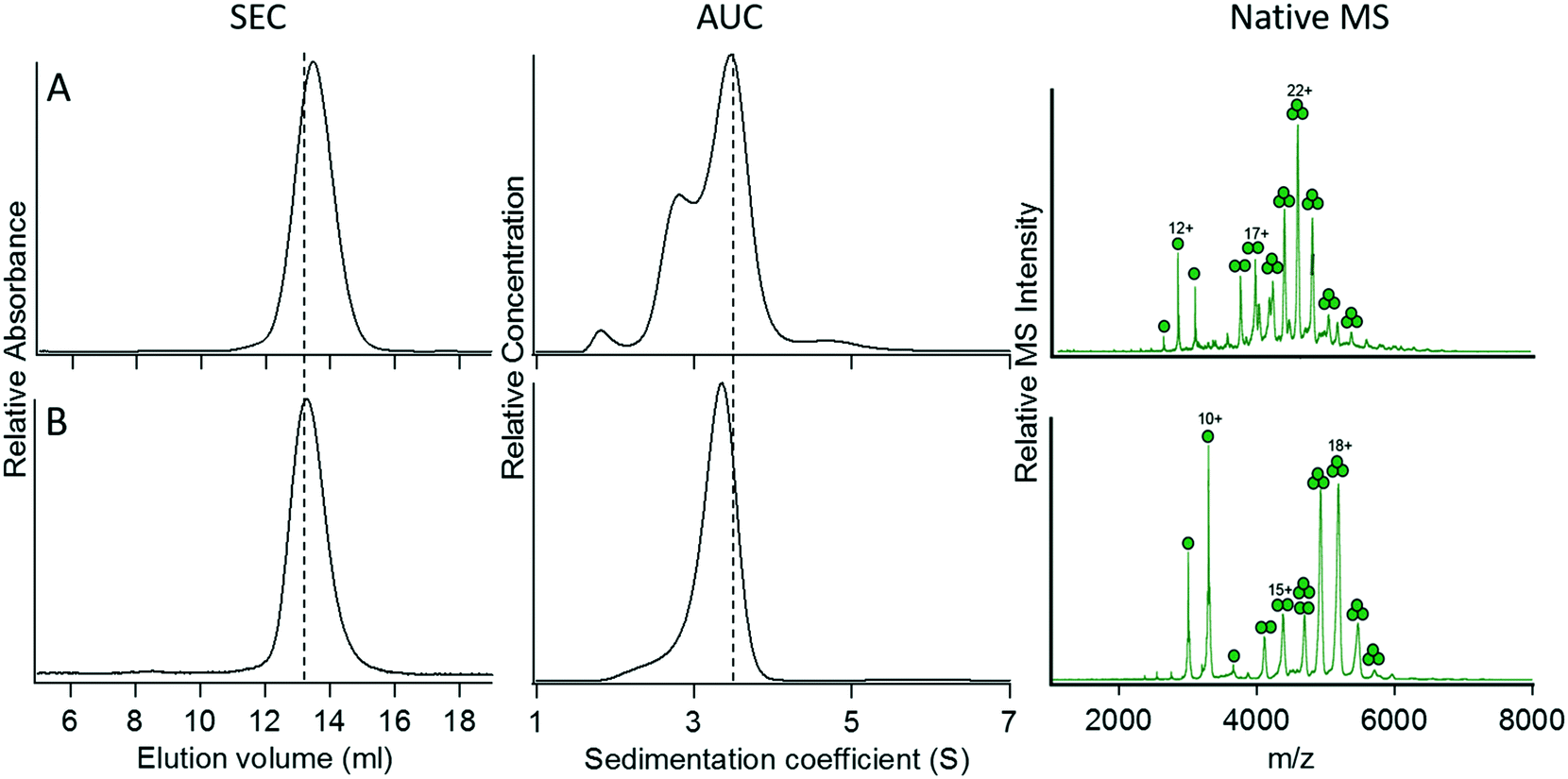 | ||
| Fig. 5 Characterization of modified versions of construct 4. (A) Construct 10, heptad strength increased to 5 repeats. (B) Construct 11, spacer length increased to 9 Gly residues. The vertical dotted lines denote the elution volume and sedimentation coefficient measured for the C-terminal fusion protein, 4. For details of the constructs, refer to Table 2. | ||
Overall, of the five crystallographically characterized coiled coils we examined, only two constructs, 1 and 3, retained their oligomerization state when fused to GFP, which served here as a model protein domain. Although they formed discrete, monodisperse assemblies, the oligomerization states of both constructs 2 and 4, which formed dimers and trimers respectively, were lower than expected based on the trimeric and tetrameric crystal structures of their respective synthetic coiled-coil domains. Interestingly, we recently employed the same tetrameric coiled-coil motif with Leu at ‘a’ and Ile at ‘d’ positions to successfully assemble an octahedral protein cage;36 in that case the coiled coil functioned as a tetramer, as intended. In contrast, the GFP fusion protein with the pentameric coiled coil, as initially constructed, formed high molecular weight aggregates, suggesting that the protein was misfolded. Lastly, the trimeric coiled coil, which was well-behaved when fused to the C terminus of GFP, failed to mediate oligomerization of GFP when transferred to the N terminus, possibly as a result of interactions between the coiled coil domain and the N-terminal His-tag.
These results illustrate both the limitations and, in some cases, advantages of coiled-coil domains as plug-and-play adaptors for the assembly of proteins. As pointed out early on by Betz and DeGrado,42 among others,6,43,44 the lability of parallel coiled coils derives from the fact that their oligomerization states are primarily specified by hydrophobic packing interactions contributed by the ‘a’ and ‘d’ sidechains. The stabilizing interactions at the interfaces of the α-helices arise from complementary electrostatic interactions between residues and ‘b’ and ‘e’ and ‘c’ and ‘g’ positions and are essentially the same for all oligomerization states between dimer and tetramer. In contrast, the interactions at the interfaces of the α-helices in anti-parallel coiled coils are different for each oligomerization state, rendering this topology inherently more robust. It seems that in several of the cases we studied, the addition of the relatively large GFP domain was sufficient to perturb the delicate balance of hydrophobic core interactions specifying the oligomerization state. Set against this, in the case of construct 9 the modularity of the coiled-coil design could be exploited to achieve the desired oligomerization state simply by adding a further heptad repeat to the coiled coil.
Our primary focus in this study was to evaluate the ability of coiled coils to act as simple components with which to assemble larger, more complex protein subunits. However, it should be noted that the choice of protein might also influence how the coiled coils behave; i.e. the protein may not be an entirely neutral component in the assembly process. In this case, for example, the latent tendency of GFP to dimerize at high concentrations could potentially influence the assembly process when individual GFP subunits are brought into close proximity by the coiled coil domain (although we observed no direct evidence for this occurring). It is possible that if had we chosen a different protein, we may have found this set of coiled coils to have been more, or less, successful as oligomerization domains.
Conclusions
Although numerous parallel coiled-coil domains have been designed as simple and robust protein oligomerization domains, our results demonstrate that their structures can be subject to unexpected changes in oligomerization state when coupled to larger protein domains. Thus, of the six fusion proteins we initially constructed (constructs 1–5 and 8), only two behaved as expected (constructs 1 and 3), which formed dimeric and trimeric assemblies. The relatively low success rate in mediating the desired self-assembly of a simple, monomeric protein comprising only a single domain should serve as a cautionary note for those considering coiled coils for use in synthetic biology applications. It appears that context effects are likely to play a greater role in modulating the structures of coiled coils than has previously been appreciated. Encouragingly, by varying the strength of the coiled coil and the length of the space between the two protein domains (parameters that might intuitively be expected to affect the ability of the coiled-coil domain to oligomerize), we were able to achieve the intended oligomerization states of two of the fusion proteins (constructs 7 and 9), representing pentameric and trimeric assemblies. These observations suggest that by screening a fairly sparse matrix of spacer lengths and coiled coil strengths, it should be possible to identify conditions for successful protein assembly in many cases.Experimental
Construction of genes encoding fusion proteins
Codon-optimized genes encoding the various coiled-coil designs and Gly spacer units were commercially synthesized and introduced into the expression vector pMCSG18 that encodes GFP as a reporter gene, either 3′ or 5′ to the GFP gene. The complete sequence of each of the fusion proteins is given in the ESI.†Protein expression and purification
Expression constructs were transformed into E. coli BL21(DE3) cells. Cells were grown in 2xYT medium with 100 mg L−1 ampicillin at 37 °C. At an OD600 of 0.8, the temperature was reduced to 18 °C, and at an OD600 of 1.0, protein expression was induced by addition of 0.1 mM IPTG. Cells were grown for a further 18 h and harvested by centrifugation.All purification steps were performed on ice or at 4 °C. Cell pellets were resuspended in 50 mM HEPES buffer, pH 7.5, containing 1 M urea, 300 mM NaCl, 25 mM imidazole, 5% glycerol, SigmaFAST protease inhibitor, and 1 mg mL−1 lysozyme, then lysed by sonication. The lysate was clarified by centrifugation at 48000g for 30 min and injected onto a HisTrap Ni-NTA column, washed with several volumes of the same buffer, and eluted with 50 mM HEPES buffer, pH 7.5, containing 300 mM NaCl, 500 mM imidazole, and 5% glycerol. Fractions containing GFP were pooled and dialyzed against 25 mM HEPES buffer, pH 7.5, containing 100 mM NaCl and 2 mM EDTA. The protein was then concentrated by ultrafiltration on a PM30 membrane and further purified by SEC on a Superdex 200 300/10 column equilibrated in the same buffer. Fractions containing proteins of the desired oligomerization state were pooled and further concentrated for analysis.
Size exclusion chromatography
100 μL of protein sample at a concentration of 1 mg mL−1 was injected onto a Superdex 200 300/10 column equilibrated at 4 °C in dialysis buffer described above, and proteins were eluted at 0.4 mL min−1.Analytical ultracentrifugation
Sedimentation velocity analysis was performed as described previously34,36 using a Beckman Proteome Lab XL-I analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) equipped with an AN60TI rotor. Samples were dialyzed against 25 mM HEPES buffer, pH 7.5, containing 100 mM NaCl and 1 mM EDTA. The hydrodynamic behavior of the various proteins was analyzed at a protein concentration of ∼0.2 mg mL−1 (A280 = 0.2). Samples were loaded into pre-cooled cells containing standard sector-shaped 2-channel Epon centerpieces with 1.2 cm path-length (Beckman Coulter, Indianapolis, IN) and allowed to equilibrate at 6 °C for 2 h in the non-spinning rotor prior to sedimentation. Proteins were sedimented at 40000 rpm and the sedimentation of the protein constructs was monitored continuously at a wavelength of 280 nm. Sedimentation velocity data was analyzed with the program SEDFIT (version 15.01b).45 Sedimentation distribution plots were generated using the continuous c(s) distribution model, with a confidence level for the ME (maximum entropy) regularization of 0.7. Molecular weights (Mr) including their standard deviation were calculated by integration of a c(M) continuous distribution. For that, the ME (maximum entropy) regularization was set to 0.5. Buffer density and viscosity were calculated using SEDNTERP (http://sednterp.unh.edu).
Native mass spectrometry
Samples prepared for mass spectrometry were purified as described above and concentrated to 40 μL. The minimum concentration of protein required for analysis is 1 μM, with higher concentrations preferred. Samples were then loaded into gold plated needles prepared in house as previously described.46 Nano-electrospray-ion-mobility-TOF mass spectrometry was performed using a Synapt G2 Traveling-Wave instrument (Waters Corp, Manchester, U.K.). Ions were generated by applying a voltage of 1.5 kV between the needle and the instrument source, with further voltage drops aiding in acceleration and desolvation as ions passed through the skimmer region of the instrument. The quadrupole region was set to RF-only mode for collection of complete mass spectra, and in some cases was tuned to isolate selected peaks for MS/MS analysis. A range of collision energies was tested for enhanced transmission and desolvation of the ions, and in some cases dissociation of the ion into its component subunits. The base values for collision energies were 20–50 V; however, energies up to 150 V were utilized for dissociation experiments. The IMS region of the instrument was operated at 4 mBar of nitrogen, with wave heights and wave velocities of 15 V and 150 m s−1, respectively. The instrument time of flight mass analyzer was operated in sensitivity mode, and mass spectra were collected from 1000 to 15000 m/z. Data analysis was performed using the manufacturer-provided Masslynx software.
Acknowledgements
This research was supported by grants from the Army Research Office ARO W911NF-11-1-0251 and W911NF-16-1-0147 to E. N. G. M.References
- E. Wolf, P. S. Kim and B. Berger, Protein Sci., 1997, 6, 1179–1189 CrossRef CAS PubMed.
- B. Apostolovic, M. Danial and H.-A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 RSC.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS PubMed.
- J. Liu, W. Yong, Y. Deng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16156–16161 CrossRef CAS PubMed.
- J. Liu, Q. Zheng, Y. Deng, C.-S. Cheng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15457–15462 CrossRef CAS PubMed.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CAS.
- N. R. Zaccai, B. Chi, A. R. Thomson, A. L. Boyle, G. J. Bartlett, M. Bruning, N. Linden, R. B. Sessions, P. J. Booth, R. L. Brady and D. N. Woolfson, Nat. Chem. Biol., 2011, 7, 935–941 CrossRef CAS PubMed.
- B. C. Buer, J. L. Meagher, J. A. Stuckey and E. N. G. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4810–4815 CrossRef CAS PubMed.
- M. G. Oakley and P. S. Kim, Biochemistry, 1998, 37, 12603–12610 CrossRef CAS PubMed.
- A. Kashiwada, H. Hiroaki, D. Kohda, M. Nango and T. Tanaka, J. Am. Chem. Soc., 2000, 122, 212–215 CrossRef CAS.
- E. N. G. Marsh and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5150–5154 CrossRef CAS PubMed.
- P. Burkhard, M. Meier and A. Lustig, Protein Sci., 2000, 9, 2294–2301 CrossRef CAS PubMed.
- N. E. Zhou, C. M. Kay and R. S. Hodges, J. Mol. Biol., 1994, 237, 500–512 CrossRef CAS PubMed.
- J. Yang, C. Xu, C. Wang and J. Kopeček, Biomacromolecules, 2006, 7, 1187–1195 CrossRef CAS PubMed.
- Y. Yu, O. D. Monera, R. S. Hodges and P. L. Privalov, Biophys. Chem., 1996, 59, 299–314 CrossRef CAS PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. O'Halloran, J. E. Penner-Hahn, W. F. DeGrado and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 6195–6196 CrossRef CAS.
- X. Li, K. Suzuki, K. Kanaori, K. Tajima, A. Kashiwada, H. Hiroaki, D. Kohda and T. Tanaka, Protein Sci., 2000, 9, 1327–1333 CrossRef CAS PubMed.
- K. H. Lee, C. Cabello, L. Hemmingsen, E. N. G. Marsh and V. L. Pecoraro, Angew. Chem., Int. Ed., 2006, 45, 2864–2868 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef CAS PubMed.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- C. J. Lanci, C. M. MacDermaid, S.-G. Kang, R. Acharya, B. North, X. Yang, X. J. Qiu, W. F. DeGrado and J. G. Saven, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7304–7309 CrossRef CAS PubMed.
- S. Raman, G. Machaidze, A. Lustig, U. Aebi and P. Burkhard, Nanomed.: Nanotechnol., Biol. Med., 2006, 2, 95–102 CrossRef CAS PubMed.
- S. A. Kaba, C. Brando, Q. Guo, C. Mittelholzer, S. Raman, D. Tropel, U. Aebi, P. Burkhard and D. E. Lanar, J. Immunol., 2009, 183, 7268–7277 CrossRef CAS PubMed.
- Y. Yang, P. Ringler, S. A. Müller and P. Burkhard, J. Struct. Biol., 2012, 177, 168–176 CrossRef CAS PubMed.
- M. Miller, Curr. Protein Pept. Sci., 2009, 10, 244–269 CrossRef CAS PubMed.
- H. I. Jung, A. Cooper and R. N. Perham, Eur. J. Biochem., 2003, 270, 4488–4496 CrossRef CAS PubMed.
- T. J. Buchholz, T. W. Geders, F. L. Bartley, K. A. Reynolds, J. L. Smith and D. H. Sherman, ACS Chem. Biol., 2009, 4, 41–52 CrossRef CAS PubMed.
- N. B. Last, D. E. Schlamadinger and A. D. Miranker, Protein Sci., 2013, 22, 870–882 CrossRef CAS PubMed.
- E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250 CrossRef CAS PubMed.
- A. A. Polyansky, A. O. Chugunov, A. A. Vassilevski, E. V. Grishin and R. G. Efremov, Curr. Protein Pept. Sci., 2012, 13, 644–657 CrossRef CAS PubMed.
- B. C. Buer, J. Chugh, H. M. Al-Hashimi and E. N. G. Marsh, Biochemistry, 2010, 49, 5760–5765 CrossRef CAS PubMed.
- D. P. Patterson, M. Su, T. M. Franzmann, A. Sciore, G. Skiniotis and E. N. G. Marsh, Protein Sci., 2014, 23, 190–199 CrossRef CAS PubMed.
- D. P. Patterson, A. M. Desai, M. M. B. Holl and E. N. G. Marsh, RSC Adv., 2011, 1, 1004–1012 RSC.
- A. Sciore, M. Su, P. Koldewey, J. D. Eschweiler, K. A. Diffley, B. M. Linhares, B. T. Ruotolo, J. C. A. Bardwell, G. Skiniotis and E. N. G. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8681–8686 CrossRef CAS PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- F. Thomas, A. L. Boyle, A. J. Burton and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 5161–5166 CrossRef CAS PubMed.
- T. S. Chen and A. E. Keating, Protein Sci., 2012, 21, 949–963 CrossRef CAS PubMed.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- S. F. Betz and W. F. DeGrado, Biochemistry, 1996, 35, 6955–6962 CrossRef CAS PubMed.
- B. C. Buer, R. de la Salud-Bea, H. M. A. Hashimi and E. N. G. Marsh, Biochemistry, 2009, 48, 10810–10817 CrossRef CAS PubMed.
- L. M. Gottler, R. de la Salud-Bea and E. N. G. Marsh, Biochemistry, 2008, 47, 4484–4490 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete protein sequences of each of the fusion proteins listed in Table 2. See DOI: 10.1039/c7me00012j |
| This journal is © The Royal Society of Chemistry 2017 |