
Minimal architecture zinc–bromine battery for low cost electrochemical energy storage†
Shaurjo
Biswas
 a,
Aoi
Senju
b,
Robert
Mohr
a,
Thomas
Hodson
a,
Nivetha
Karthikeyan
a,
Kevin W.
Knehr
a,
Andrew G.
Hsieh
a,
Xiaofang
Yang
b,
Bruce E.
Koel
b and
Daniel A.
Steingart‡
*ab
a,
Aoi
Senju
b,
Robert
Mohr
a,
Thomas
Hodson
a,
Nivetha
Karthikeyan
a,
Kevin W.
Knehr
a,
Andrew G.
Hsieh
a,
Xiaofang
Yang
b,
Bruce E.
Koel
b and
Daniel A.
Steingart‡
*ab
aDepartment of Mechanical and Aerospace Engineering & the Andlinger Center for Energy and The Environment, Princeton University, NJ, 08544, USA
bDepartment of Chemical and Biological Engineering, Princeton University, NJ 08544, USA
First published on 29th November 2016
Abstract
We demonstrate a minimal-architecture zinc–bromine battery that eliminates the expensive components in traditional systems. The result is a single-chamber, membrane-free design that operates stably with >90% coulombic and >60% energy efficiencies for over 1000 cycles. It can achieve nearly 9 W h L−1 with a cost of <$100 per kWh at-scale.
Broader contextAddressing climate change by integrating renewable energy sources and enhancing efficiencies of existing non-renewable energy processes is a major global concern. To meet this challenge, low-cost grid-scale electrochemical energy storage (EES) systems are being researched extensively.1 While redox flow, lead acid, zinc alkaline and lithium ion batteries have been commercialized for stationary applications at some scale in recent years,2 research efforts are largely focused on novel, low cost and/or more efficient electrode/electrolyte active materials.3–9 However, most systems still employ traditional cell designs with expensive passive components including separators, pumps, and reactors. Unfortunately, this approach results in the balance-of-plant costs far outweighing the cost of electrochemically active materials. Multiple technoeconomic analyses10–12 put the target cost of energy storage at <$100 per kWh and power at <$600 per kW. Additional targets for sustainable EES systems are >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with <80% capacity fade, and energy efficiencies of >60%. Based on the current trajectory of mainstream battery research, these targets will be difficult to reach. Here, we demonstrate a holistic approach when considering energy storage materials and system design. We report a reconsideration of the Zn–Br2 system, and present a design that eliminates many of the expensive balance-of-plant components in traditional systems by exploiting the physical properties of the bromine and zinc materials. The result is an energy storage system that, if scaled, could be a dramatic improvement in cost per unit energy and calendar and cycle lifetime over currently-reported systems for the tradeoff of increased self discharge. 000 cycles with <80% capacity fade, and energy efficiencies of >60%. Based on the current trajectory of mainstream battery research, these targets will be difficult to reach. Here, we demonstrate a holistic approach when considering energy storage materials and system design. We report a reconsideration of the Zn–Br2 system, and present a design that eliminates many of the expensive balance-of-plant components in traditional systems by exploiting the physical properties of the bromine and zinc materials. The result is an energy storage system that, if scaled, could be a dramatic improvement in cost per unit energy and calendar and cycle lifetime over currently-reported systems for the tradeoff of increased self discharge.
|
Considerable research and development efforts in EES aim to reduce costs by an order of magnitude (per unit energy stored) while also providing order-of-magnitude longer calendar and cycle lives, a particular challenge as these are typically engineering goals which are diametrically opposed. These efforts have largely focused on developing lower-cost electrode materials while still using standard cell designs that require traditional “balance-of-plant materials”.1–9 However, an unexpected and undesired fallout from combining inexpensive electrode materials with traditional passive components is that the balance-of-plant cost outweighs the active materials cost by a wide margin. In addition to a lower cost of energy ($ per kWh), longer lifetime as well as minimal maintenance and operating costs are paramount for the success of energy storage systems. Vanadium redox flow batteries (V-RFB) and zinc bromine RFB (ZnBr–RFB) have been shown to perform with less than 20% capacity fade for 10
000 cycles,13 however no single battery type has met the cost, lifetime, and performance targets required for successful EES implementation.
Based on this techno-economic understanding, we take a more holistic approach when considering EES system design, by asking: (a) are expensive passive materials necessary to protect the inexpensive electrochemically active materials? (b) is it existentially necessary to prevent the negative and positive electrode materials from reacting with each other? We use this conceptual framework to re-design the ZnBr–RFB and take advantage of the physical properties of the electrochemically active materials.
Here, we report on a simple and scalable, low-cost, membrane-free, single-chamber, minimal architecture zinc–bromine secondary battery (MA-ZBB) design, with no forced convection, that utilizes the physical properties of liquid bromine, a porous carbon foam electrode, and also allows zinc dendrites to form freely. We demonstrate the local containment of Br2 in a carbon foam electrode, and discuss a color tracking and feedback monitoring scheme to actively control the reactive species transport and improve cell efficiencies. The MA-ZBB system has a maximum specific capacity of 76 mA h g−1 and energy density >135 W h kg−1 (normalized to the mass of ZnBr2 in the electrolyte) or >40 W h L−1 of total system volume. Including passives, each cell has a projected cost of ∼$94 per kWh, with coulombic and energy efficiencies of up to 95% and 75%, respectively, for over 1000 cycles. We present a techno-economic argument that because of the exceptionally low cost and long cycle life of the battery, many attributes that are typically flaws (e.g., self-discharge, much-lower-than-unity round trip efficiency, to name a few) can be accepted. The cost, lifetime, and performance characteristics of this engineered-to-sufficient battery are attractive for grid-scale energy storage applications.
We chose the well-studied aqueous Zn–Br2 system as a candidate system because of its high open circuit potential (1.82 V), theoretical energy (>400 W h kg−1) and demonstrated power (>100 mW cm−2) densities.14–17 Practical implementations of bromine-bearing flow systems in general, and Zn–Br2 systems in particular, are challenged by: (i) corrosive elemental bromine liquid, Br2(l), generated at the positive electrode during charging, which can diffuse through the electrolyte and react with Zn deposited at the negative electrode (crossover), leading to self-discharge and/or degrade and swell typical membranes, (ii) repeated electroplating and dissolution of zinc leads to dendrite formation, which can form a conductive bridge between the electrodes (shorting), and (iii) the low miscibility (∼2.8 vol%) and tendency of Br2(l) to stratify in aqueous solutions, which results in non-uniform concentration distributions.
Standard Zn–Br2 flow-cell designs alleviate these limitations with bromine-complexing agents to improve Br2(l) solubility,18–21 separation membranes to prevent crossover and short circuits,22 and flowing electrolyte to force bromine convection and improve the limiting current density for Zn deposition.23 These approaches are implemented at the expense of cell resistance, system efficiency, increased system size and complexity, and increased capital costs. Considering only the active materials (ZnBr2 salt), and the carbon electrodes, the cost of energy is just ∼$8 per kWh.24 At the system level, however, other costs for complexing agents, separation membranes, protective coatings, hardware for flow systems, control systems, plus the safety checks and redundancies required due to the use of corrosive Br2(l), must all be considered. Thus, despite the low cost of reactants, full systems can cost over $200 per kWh.24,25
This dramatic difference between the cost of active materials and the total system raised the following question: can one exploit the natural tendency of Zn to form dendrites, Br2 to stratify, and Zn and Br2 to react with one another to design a cost-efficient Zn–Br2 battery? If such a cell could be made to work, the costly and calendar cycle life limiting components could be eliminated and more resources could be dedicated to the electrolyte and bromine-resistant electrodes and packaging. Carbon is largely stable in bromine13,26,27 and suitable as a current collector,17,28,29 while fluorinated polymers (e.g. teflon, PVDF) are also stable in bromine and can serve as packaging materials.§ Additionally, eliminating each pump improves the system's energy efficiency by 5%.30 Thus, if a minimal architecture ZnBr2 battery can achieve a reversible capacity of 70 mA h g−1, energy density of 130 W h kg−1 ZnBr2 in solution, and perform at over 60% energy efficiency for hundreds of cycles, then grid-relevant cost targets could be hit.
In this work, we present such a battery design. This Zn–Br2 battery comprises a materials system that has been studied for over 100 years.31 The objective of this work was to demonstrate that a battery with extremely low cost and long cycle life, with adequate performance, for grid scale energy storage applications assuming scale up of the design basis. Fig. 1a shows the MA-ZBB design schematic; Fig. 1b and c show images of the cell in its discharged and charged states, respectively. The cell consists of a clear glass reactor; a carbon cloth negative current collector (Zn plating/dissolution); a 2 M ZnBr2(aq) electrolyte; and a porous, hydrophobic, carbon ‘foam’ positive current collector (bromine generation/decomposition). Here, the carbon foam electrode (CFE, Fig. 1d) is composed of graphite and carbon black. Fig. 1e shows a micrograph of its cross-section.
 | ||
| Fig. 1 MA-ZBB cell (a) design schematic. Photographs of the realized, 5 mL cell in the (b) discharged and (c) charged states show the distinct colors of the ZnBr2(aq) electrolyte (clear), dissolved Br2(aq) (yellow), and Br2(l) (red). As the MA-ZBB cell is membrane-free, zinc dendrites are able to grow towards the positive electrode, but recombine with the Br2(l) to reform Zn2+ and Br− ions. (d) Photograph and (e) cross-sectional SEM image of the CFE. | ||
The cell is assembled in a completely discharged state; the electrolyte is clear and no metallic Zn is present. As the cell is charged, bromine is generated in the CFE and Zn is plated onto the carbon cloth electrode and the nonpolar Br2(l) displaces aqueous electrolyte from the pores and cavities in the hydrophobic CFE. At low levels of capacity passed, the generated Br2(l) remains within the CFE. As charging continues and the pores fill up, excess Br2(l) spills out of the foam, collecting at the bottom of the cell due to the higher density of Br2(l) (3.1 g mL−1) compared to the electrolyte (1.5 g mL−1), creating the red layer in Fig. 1c. Thus, the CFE can trap Br2(l) within it and prevent crossover up to a certain capacity passed (which is specific to the design shown here). Strategies for managing CFE ‘lossiness’ are discussed in ESI† section.
In our new design, we attempt a physical method of separating the Br2(l) from the Zn electrode, taking advantage of the high density and low water miscibility of Br2 by placing the carbon cloth electrode at the top of the cell and the porous CFE on the bottom (Fig. 1b). With this approach, if Zn dendrites form during charging and creep towards the CFE (as in Fig. 1c), Zn(s) will react with Br2(l) on the surface of the foam and dissolve back into the electrolyte as Zn2+ and Br− ions. This spontaneous process prevents short circuiting, as the Zn(s) + Br2(l) reaction (Zn corrosion) occurs faster than zinc deposition at charging currents below 50 mA. Thus, the corrosive Br2(l) loosely held on the CFE surface acts as a natural protection from shorting, and can be exploited for ‘self-maintenance’, removing the need for separation membranes or protective coatings. Most importantly, even if the cell self-discharges due to Zn corrosion, there is no irreversible damage to the cell (the reaction is Zn(s) + Br2(l) → Zn2+ + 2Br−). Hence, the life of the cell is not limited by Zn corrosion and results in low maintenance costs. Since the cell is operating in acidic condition, there is some H2(g) generated during Zn plating. Ideally, the H2(g) would react with Br2 as well and revert back to HBr(l) form. In our experiments reported here H2(g) loss is present but not significant (<0.03% of the capacity per cycle, ESI†) because the cells are operated at such low currents. When the cell is scaled up, H2(g) recapture with Br2(l) will be essential. The design modifications for such a scheme are provided in ESI† section along with the equations occurring in the cell.
As our reactor chamber is clear, we can also take advantage of the distinct colors of the ZnBr2(aq) electrolyte, Br2(l), and dissolved Br2(aq): clear, red, and yellow, respectively (Fig. 1b and c). By calibrating the solution color with Br2(l) and Br2(aq) concentrations, we can track the generation, consumption, and transport of bromine in the system in real time to optimize battery performance and prevent unwanted processes (i.e., crossover). One such example is provided in Fig. 2: a cell is cycled at 20 mA while images are collected, and solution color is tracked at various points across the cell, indicated by points 1–5 in Fig. 2a. As Br2(l) is generated during charging and the CFE volumetric capacity is reached, Br2(l) leaks into the clear ZnBr2(aq) electrolyte. The onset of bromine leakage is detected by monitoring point 1: Br2 concentration increases at ∼3400 s after the start of charging (Fig. 2b), and the amount of bromine generated and stored in the foam is calculated from the elapsed time. In this example, given the CFE design (1 cm3, 1 g) and cell operating conditions, ∼0.019 mL of Br2(l) (18.9 mA h capacity) is stored within the foam. Points 3 and 4 are monitored to detect when Br2 reaches the plated Zn(s) during charge. If Br2 diffuses beyond point 4, Zn(s) would react with it and the cell would self-discharge. To optimize performance, a feedback loop is written into the cycling control algorithm to stop the charge step at this point. In this example, Br2(l) reaches point 4 after ∼2 h of charging (Fig. 2b). We use this as the capacity cutoff for charging in some of the electrochemical experiments below. This optical visualization, tracking, and in operando feedback technique has never been used for any bromine-based electrochemical cells, to the best of our knowledge. Note that this color-concentration tracking of bromine is to facilitate the understanding of the system at a lab-scale and is not meant for large scale field application.
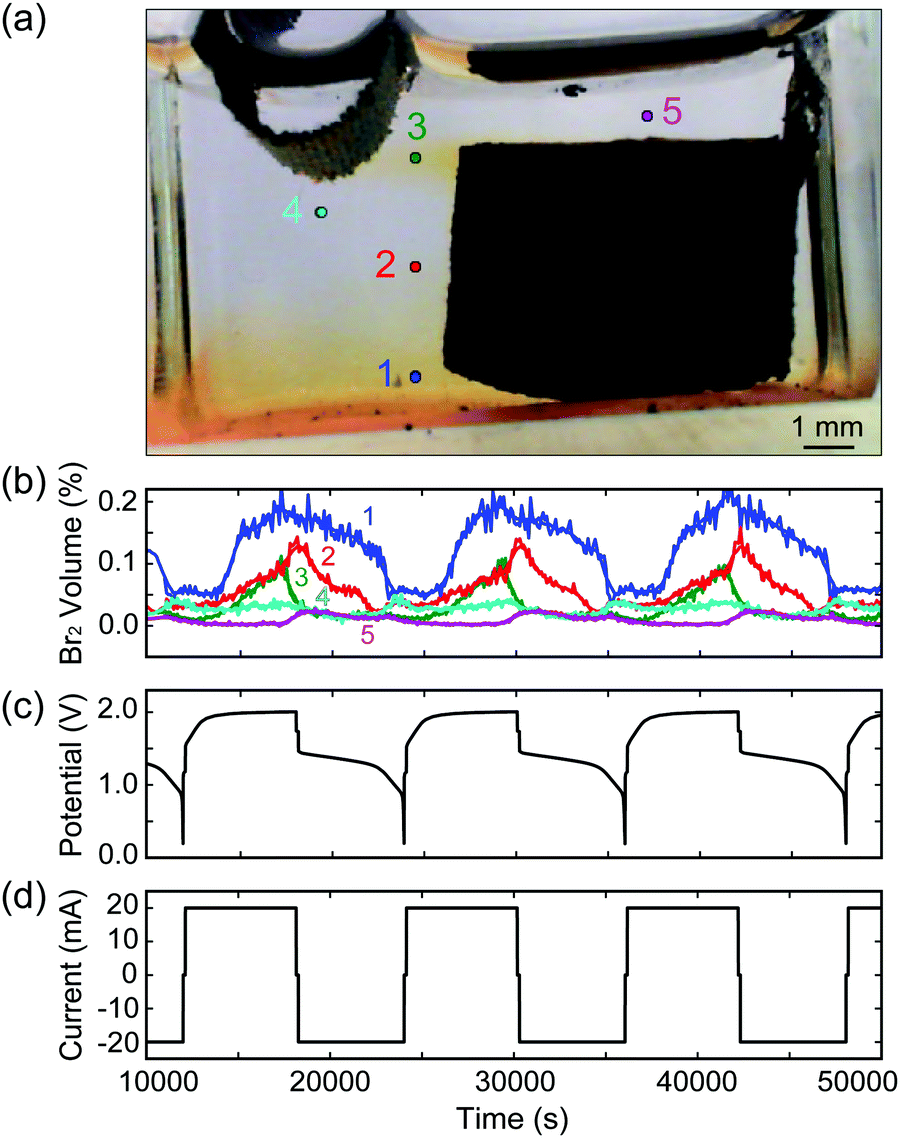 | ||
| Fig. 2 Bromine color-concentration tracking of a sample MA-ZBB cell cycling at a 20 mA constant current charge–discharge. (a) Photograph of the MA-ZBB cell; points 1–5 are the locations at which Br2 concentration is tracked, which is plotted in (b) as a function of cycle time. (c and d) show the cell potential and applied current during cycling, respectively. Note that a ∼2 h limit on the charge step is imposed based on when Br2 is detected at point 4 in order to limit crossover and self-discharge. | ||
To calibrate the color of the solution with bromine concentration, a cell with 5 mL Br2(l) (10.2% by volume) in DI water solution is placed in a closed light box illuminated with two white light sources (50 W, 6000 K color temperature) to maintain a uniform background color. To dilute the Br2(l), a 0.05 mL aliquot of 2 M ZnBr2 salt solution is added to the cell every 3 min using a calibrated syringe pump, and is stirred to obtain uniform concentration profiles. The cell is placed in a closed light box with two white light sources. Images are captured every 10 s using a Nikon D300 SLR camera with manual white light balance, aperture, shutter speed and ISO set and maintained across all experiments. The experimental setup and calibration results are provided in ESI† Fig. S4.
Optical tracking on MA-ZBB cells enables the comparison of different cells and architectures, as well as the tracking and analysis of bromine transport during operation, as depicted in Fig. 2a and b. In addition to corrosion of Zn(s) with Br2(l), Br3− and higher complexes of bromine are also responsible for self-discharge of the cell. Optically tracking the Br2(l) and Br2(aq) is performed to prevent direct corrosion as well as to be aware of the equilibrium boundary where Br3− complex is forming. Ideally, all the Br2(l) generated would be captured within the CFE, thus mitigating self-discharge, and improving cell efficiency. Note, that in our experiments, the effect of self-discharge due to Br3− and higher complexes have not been significant, as reported below.
Galvanostatic testing is performed on the MA-ZBB cell, charging at 20 mA for ∼2 h (using the color tracking control method as mentioned above) and discharging at currents ranging from 0 to 80 mA. The voltage limits set on the cell are 2.0 V and 0.2 V. Fig. 3a shows the coulombic and energy efficiencies of the cell at different discharge currents. While charging at 20 mA for 2 h utilized less than 20% of the available capacity of the cell, we chose to impose this capacity of 40 mA h to demonstrate the proof-of-concept of this MA-ZBB cell design. In the 0 mA discharge case, the cell self discharges over ∼50 h. At low discharge currents (below 20 mA in the current system), the efficiencies are limited due to self discharge, while at higher currents (>20 mA), the Br2/Br− reaction rate is limited by species transport to the positive electrode surface. The cell achieved 95% coulombic efficiency (CE) and 70% energy efficiency (EE) for 5 mA to 10 mA discharge currents, when the cell is operating far from its maximum charge capability.
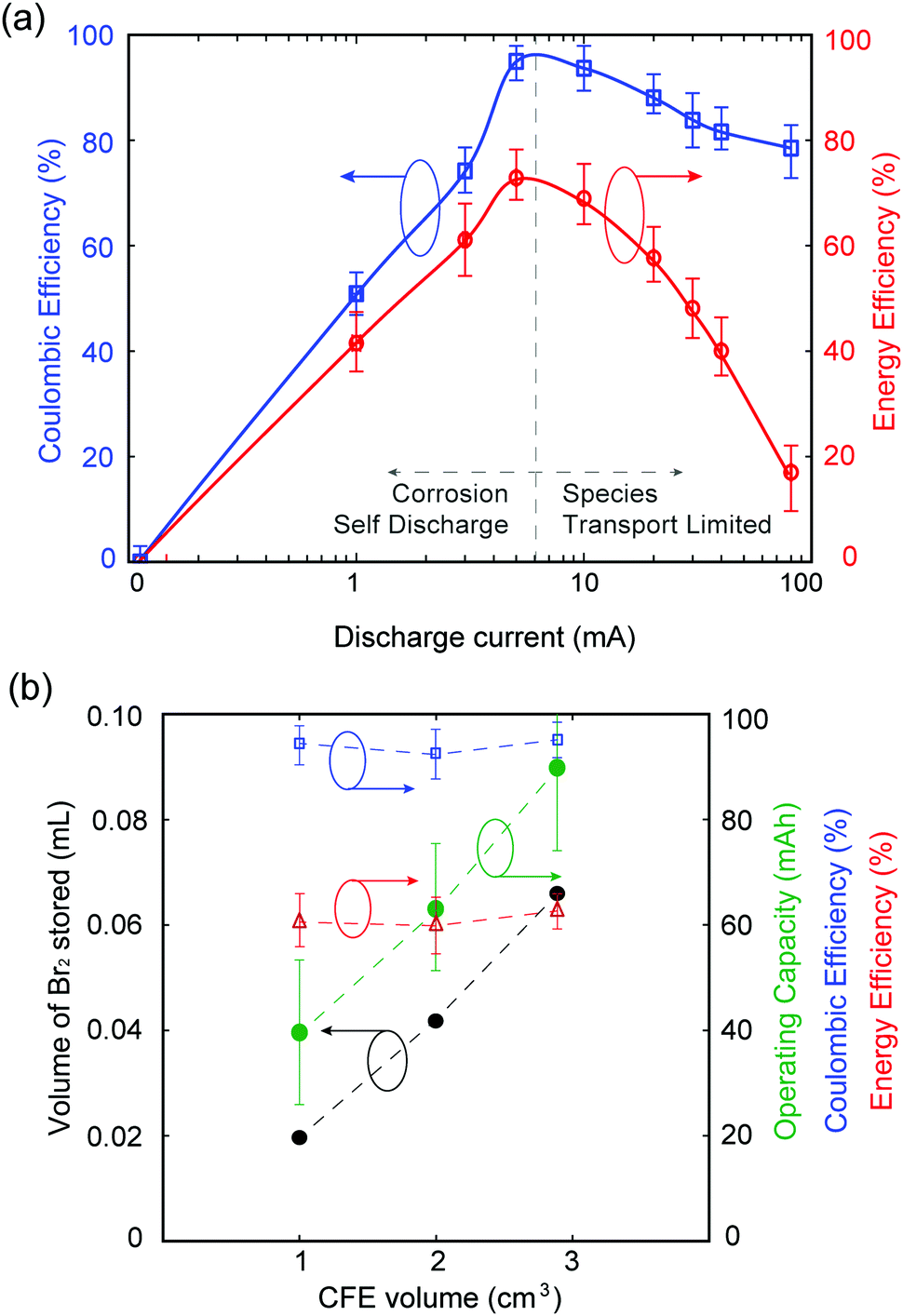 | ||
| Fig. 3 (a) Coulombic and Energy efficiencies of a cell galvanostatically charged at 20 mA for 2 h (charge capacity = 40 mA h) and discharged at 0, 1, 3, 5, 10, 20, 30, 40, and 80 mA. It is possible to achieve over 95% coulombic efficiency and 70% energy efficiency while operating at 5–10 mA. Error bars indicate range of efficiencies at each current. (b) Maximum volume of bromine stored in a CFE, and the operating capacity of a cells scale with the CFE geometric volume. CE and EE are relatively constant while the cells are subjected at 20 mA charge–discharge for 2, 3.18, and 4.51 hours using CFE with volumes of 1, 2, and 2.9 cm3, respectively. Lines are drawn as guides to the eye. | ||
Our unoptimized initial 1 cm3 CFE design has limited bromine-storing capability. The optimum MA-ZBB operating window can be broadened (i.e., higher CE and EE over a wider range of discharge currents) by decreasing the electrolyte resistance (e.g., by adding supporting salts) and by increasing CFE porosity and conductivity.
As shown in Fig. 3b, the volume of Br2(l) that can be stored in the CFE scales with the geometric volume of the foam. Three CFE of identical composition but of 1, 2, and 2.9 cm3 ± 0.07 cm3 sized are placed in 2 M ZnBr2 solution electrolyte in an identical cell setup. Upon charging at 20 mA and using the color-concentration visualization tool, 0.019, 0.042, and 0.065 mL of Br2(l) is stored in the CFEs, respectively, before it leaks out (monitoring point 1). The corresponding cells can be charged to 2, 3.18, and 4.51 h, respectively, and discharged at 20 mA current to maintain over 90% CE and 50% EE. In addition, the largest CFE with 2.9 cm3 volume operated with 90% CE and 30% EE when charged for 4.5 h at 20 mA and discharged at 50 mA. Fig. 3b shows that the maximum operating capacity for maintain reasonable battery performance scale as a function of CFE volume, without any modification to its design. Further increase in the maximum Br2(l) storage within the CFE, and thus maximum utilized cell capacity, can be obtained by modifying the CFE geometry. For example, if the CFE is a hollow cube (CFE-shell), it could potentially store larger volumes of Br2(l) within it, thus improving the operating capacity of the battery. This result is, however, beyond the scope of this paper.
In order to capture the full available capacity and attain 100% ZnBr2 capacity, yet maintain the ionic conductivity of the electrolyte solution, a 0.5 M ZnBr2 + 1.5 M ZnCl2 solution of the same volume is used in experiments described above and cycled at 20 mA charge and discharge using the 1 cm3 CFE. The charge cutoff potential is set at 2.0 V in order to prevent chlorine gas generation inadvertently (E0OCPvs. SHE = 2.2 V for ZnCl2 redox cell30). The cell charges for 3.8 h before reaching the 2.0 V voltage cutoff, and cycles with 95% CE and 60% EE. Mechanisms for Br2 complexing with Cl− in this cell and its effect on cell performance are yet to be explored.
In a separate experiment, MA-ZBB cells are subjected to a total of 500 cycles at charge–discharge currents of 5, 50, 40, 20 mA and again 5 mA (100 cycles each), using a fixed 2 h charge step and the 1 cm3 CFE. The CE and EE are plotted against cycle life in Fig. 4a. Within the 100 cycles at each current level, the cell shows minimal loss in efficiencies. Furthermore, in the second set of 5 mA cycles, the cell operates at nearly the same efficiency as in the initial set; this indicates that there is little to no CFE degradation and minimal irreversible losses in the system during cycling. Additionally, as shown in Fig. 4b, when the cell is subjected to 20 mA charge–discharge cycles CE and EE remain relatively constant at ∼92% and ∼60%, respectively, for over 1000 cycles, further reinforcing the claim of negligible CFE degradation and irreversible chemical loss hypothesis. Preliminary XPS analysis indicates minimal degradation in the graphite of the CFE due to extended exposure to Br2(l), but a comprehensive analysis is required. Of the 8% coulombic efficiency losses, <0.03% can be attributed to hydrogen generation (see ESI†) which suggests the remaining 7.97% is caused by corrosion of the zinc electrode due to reaction with bromine and bromine complexes. Note that the corrosion of zinc causes negligible, irreversible degradation in performance as demonstrated by the relatively constant efficiencies over 1000 cycles. While it would be best not to have either loss mechanism, the bromine shuttle is more readily reversed than H2 production and is arguably a more favorable loss mechanism to contend with due to its relatively high electrochemical accessibility.
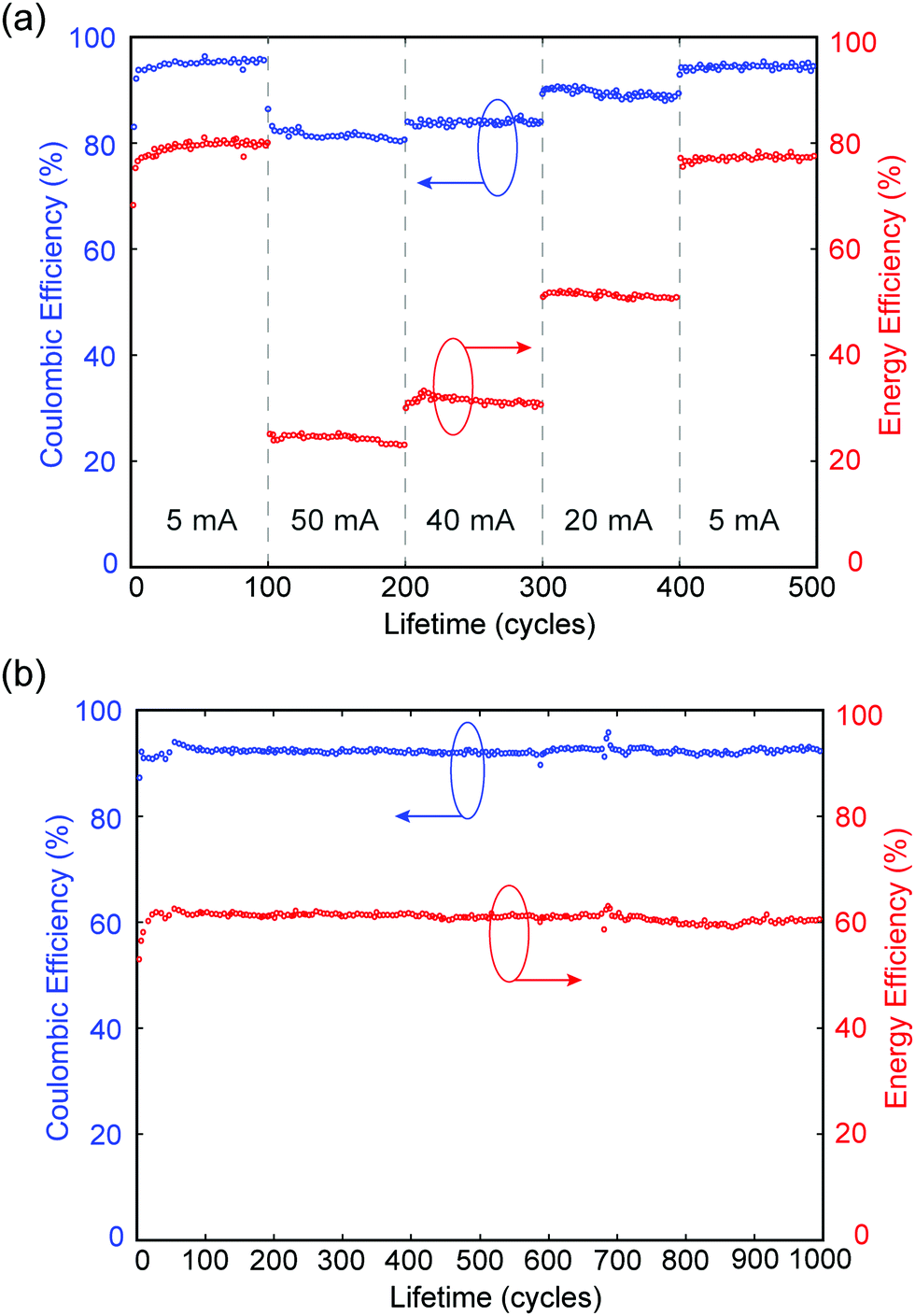 | ||
| Fig. 4 Coulombic and Energy efficiencies of: (a) a MA-ZBB cell over a total of 500 cycles (900 hours) at 5, 50, 40, 20, and 5 mA constant charge–discharge currents (2 h charge time). (b) a cell operating at 20 mA charge–discharge currents for over 1000 cycles, with negligible loss in efficiency. | ||
Finally, we estimate the cost of this lab-scale MA-ZBB cell (including passive components), based on current market prices for materials. Bill of materials cost of the 5 mL volume lab-scale cell is $0.0116 per g ZnBr2 in solution or $1.57 per L system volume. Based on the 1000 cycle cell shown in Fig. 4b, the energy density of the cell is 65.84 W h kg−1 salt or 8.89 W h L−1 system. Current cost of energy for the cell is $176.2 per kWh. Instead, if the 2.9 cm3 volume CFE is considered, the CFE cost triples, but the charge capacity increases to 144.02 W h kg−1 salt, and the system cost is reduced to $149.15 per kWh while operating at 90% CE. However, if even 80% of the full available capacity of cell (∼240 mA h, ESI†) could be utilized with innovative CFE designs like CFE-shell to trap all of the generated bromine, assuming similar 60% energy efficiency and 110% cost increase due to design complexity, this still yields a system cost of ∼$93.6 per kWh. If the full cell capacity is discharged in 2 h at 100 mA, the cost of operation would be ∼$359.7 per kW. In addition, when the design is scaled up, the cost of the most expensive component in the lab-scale cell, the glass holder, would scale as the ratio of surface area to volume. That is, if the total volume of the cell is increased by 100×, the glass cost would increase by only 20×. The glass holder can also be replaced by more inexpensive fluorocarbon plastic options when scaling up.
The ultimate figure of merit for feasibility in grid-scale energy storage system is $ per kWh over its lifetime (number of cycles) at a given energy efficiency, or the levelized cost of energy stored (LCOES). Fig. 5 shows the LCOES comparison of MA-ZBB against other battery chemistries and designs.13 Li-ion and Na–S batteries are expensive and have a life of ∼1500 cycles. Despite operating at almost 100% EE, their average LCOES is around $0.5 per kWh per cycle. Advanced lead-acid batteries have a limited cycle life, and hence has an average LCOES of $0.75 per kWh per cycle. The RFBs have an exceptionally long reported lifetime, which results in an average LCOES of less than $0.10 per kWh per cycle. However, if MA-ZBB can last for 10000 cycles, like ZnBr–RFB, the projected LCOES would be $0.017 per kWh per cycle, placing it an order of magnitude below the rest of EES available today. The full list is provided in Table S1 (ESI†).
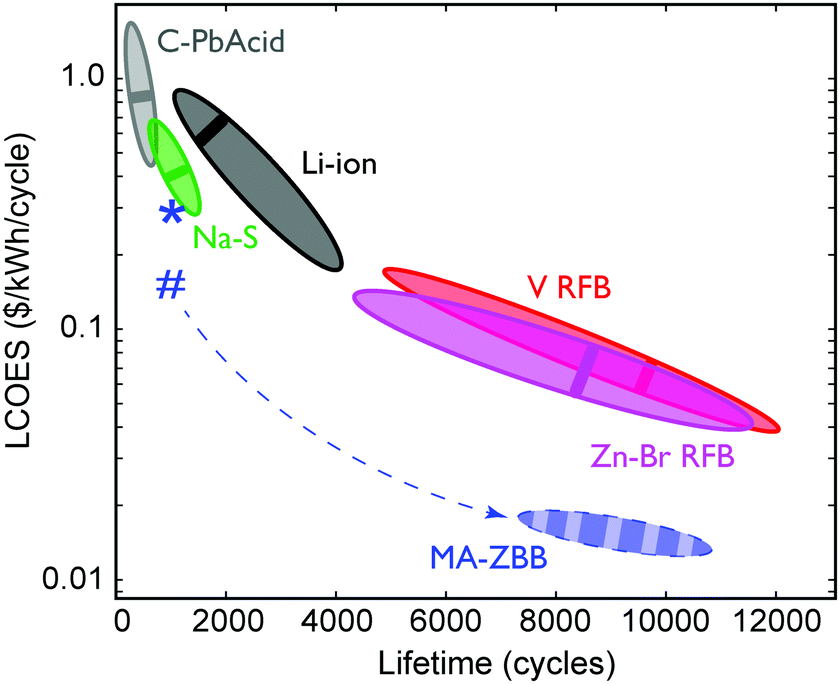 | ||
| Fig. 5 Average LCOES and its range are plotted for traditional lead acid, lithium ion, sodium sulphide, vanadium and Zn–Br RFB.13,24,25 LCOES for MA-ZBB cells currently is at $0.29 per kWh per cycle for 1000 cycles, denoted by the *. Using the 2.9 cm3 CFE and the cell cost is $0.25 per kWh per cycles, denoted by #. The cell is projected to operate for 10000+ cycles (same as V- and Zn–Br RFB) without significant loss in efficiency; the resulting LCOES could be at $0.015 per kWh, which is much lower than other traditional grid scale EES systems. The dashed arrow shows the potential learning curve for the MA-ZBB system. | ||
With this membrane-free, non-forced-flowing, minimal architecture zinc bromine battery we have achieved cell current cost $176 per kWh with over 1000 cycles and 60% energy efficiency. Our projected cost with small modifications to the CFE is $93.6 per kWh (CFE + leads: $22.03 per kWh; carbon cloth electrode: $9.82 per kWh; electrolyte: $18.71 per kWh; BOP: $43.00 per kWh). These results place our design firmly better than traditional RFB batteries. The low cost and long cycle life of MA-ZBB is achieved by eliminating the often-failing and expensive components – membrane, complexing agents, pumps and pipes – from a traditional Zn–Br2 RFB. Transitioning to activated carbon CFE to replace graphite might resolve that issue as well, as has been shown by commercially available RFB electrodes.28 And since we use similar materials to these RFBs which have been shown to cycle over 10000 cycles, we anticipate that the cell will be able to operate for over 10000 cycles without significant electrode degradation or loss in efficiencies, further reducing its lifetime cost. We further hypothesize that with economies of scale, MA-ZBB will be a compelling choice for grid-scale EES systems.
Author contributions
DS, SB, and RM conceived the design and concept. AS synthesized all the carbon foam electrodes. DS, SB, TH, and NK performed the color-concentration calibration and tracking. SB, AS, TH, and KK performed the electrochemical testing and the analysis of the results. SB, AS, and XY performed the characterization work under the supervision of BK and DS. SB, AH, KK, and DS wrote the manuscript.Author information
The authors declare no competing financial interests. NK from High Technology High School, Marlboro, NJ worked on this project as a high-school intern. AS, RM had graduated from Princeton University at the time of manuscript submission.Acknowledgements
This work was supported by the Andlinger Center for Energy and The Environment E-ffiliates Fund, and a generous gift from Israel Chemical Limited, Industrial Products. We also thank, Prof. George Scherer for his guidance on pore size analysis, and Greg Davies, Tanya Gupta, and Andrew Kim for helpful discussions.Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- T. Gupta, A. Kim, S. Phadke, S. Biswas, T. Luong, B. J. Hertzberg, M. Chamoun, K. Evans-Lutterodt and D. A. Steingart, J. Power Sources, 2016, 305, 22–29 CrossRef CAS.
- Y. Ito, M. Nyce, R. Plivelich, M. Klein, D. Steingart and S. Banerjee, J. Power Sources, 2011, 196, 2340–2345 CrossRef CAS.
- J. F. Whitacre, T. Wiley, S. Shanbhag, Y. Wenzhuo, A. Mohamed, S. E. Chun, E. Weber, D. Blackwood, E. Lynch-Bell, J. Gulakowski, C. Smith and D. Humphreys, J. Power Sources, 2012, 213, 255–264 CrossRef CAS.
- T. Ouchi, H. Kim, X. Ning and D. R. Sadoway, J. Electrochem. Soc., 2014, 161, A1898–A1904 CrossRef.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- P. W. Parfomak, CRS 7–5700 for Congress, Congressional Research Service, Washington DC, 2012, vol. R42455, pp. 37–39 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. Newman, P. G. Hoertz, C. A. Bonino and J. A. Trainham, J. Electrochem. Soc., 2012, 159, A1722–A1729 CrossRef CAS.
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef CAS.
- P. C. Butler, P. A. Eidler, P. G. Grimes, S. E. Klassen and R. C. Miles, in Handbook of Batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill, New York, NY, 2002, vol. 3, pp. 39.1–39.20 Search PubMed.
- L. Zhang, Q. Lai, J. Zhang and H. Zhang, ChemSusChem, 2012, 5, 867–869 CrossRef CAS PubMed.
- H. S. Lim, A. M. Lackner and R. C. Knechtli, J. Electrochem. Soc., 1977, 124, 1154–1157 CrossRef CAS.
- D. Aymé-Perrot, S. Walter, Z. Gabelica and S. Valange, J. Power Sources, 2008, 175, 644–650 CrossRef.
- D. J. Eustace, J. Electrochem. Soc., 1980, 127, 528–532 CrossRef CAS.
- M. Mastragostino and S. Valcher, Electrochim. Acta, 1983, 28, 501–505 CrossRef CAS.
- K. J. Cathro, K. Cedzynska, D. C. Constable and P. M. Hoobin, J. Power Sources, 1986, 18, 349–370 CrossRef CAS.
- K. J. Cathro, J. Power Sources, 1988, 23, 365–383 CrossRef CAS.
- L. Zhang, H. Zhang, Q. Lai, X. Li and Y. Cheng, J. Power Sources, 2013, 227, 41–47 CrossRef CAS.
- R. Zito Jr., US. Pat., 4482614, 1984.
- A. Larsson, Massachusetts Institute of Technology, 2009 Search PubMed.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 CAS.
- K. T. Cho, P. Ridgway, A. Z. Weber, S. Haussener, V. Battaglia and V. Srinivasan, J. Electrochem. Soc., 2012, 159, A1806–A1815 CrossRef CAS.
- W. A. Braff, M. Z. Bazant and C. R. Buie, Nat. Commun., 2013, 4, 2346 Search PubMed.
- H. Zhou, H. Zhang, P. Zhao and B. Yi, Electrochim. Acta, 2006, 51, 6304–6312 CrossRef CAS.
- D. Pletcher, Electrochemistry of Carbon Electrodes, 2015, vol. 16 Search PubMed.
- P. Leung, X. Li, C. P. de León, L. Berlouis, C. T. John Low and F. C. Walsh, RSC Adv., 2012, 2, 10125–10156 RSC.
- S. Beadley, US. Pat., 312802, 1885.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of carbon foam electrode preparation, electrode characterization, cell design, bromine concentration tracking, hydrogen generation characterization, electrochemical testing protocols, and cost analysis. See DOI: 10.1039/c6ee02782b |
| ‡ Current address: Room 213 ACEE, 86 Olden St., Princeton, NJ, 08544-5263, USA. E-mail: steingart@princeton.edu, Tel: +1 (609) 258-1257. |
| § Bromine liquid and gas is highly corrosive for most materials except for glass and certain fluorinated plastics like PVDF, PTFE, etc.: http://www.hse.gov.uk/comah/sragtech/techmeasmaterial.htm, http://www.bromaid.org/handbook/section1propertiesofbromine/section14materialsofconstruction, E. R. Booser, CRC Handbook of Lubrication and Tribology, Volume III: monitoring, materials, synthetic lubricants, and applications, CRC Press Scotia, NY, 1994, vol. 3, p. 171. |
| This journal is © The Royal Society of Chemistry 2017 |