
A layered Na1−xNiyFe1−yO2 double oxide oxygen evolution reaction electrocatalyst for highly efficient water-splitting†
Baicheng
Weng
,
Fenghua
Xu
,
Changlei
Wang
,
Weiwei
Meng
,
Corey R.
Grice
and
Yanfa
Yan
 *
*
Department of Physics and Astronomy and Center for Photovoltaics Innovation and Commercialization, The University of Toledo, Toledo, Ohio 43606, USA. E-mail: yanfa.yan@utoledo.edu
First published on 6th December 2016
Abstract
Transition metal Ni- and Co-based oxides are potential candidates to replace expensive and scarce noble metal-based oxygen evolution reaction (OER) catalysts such as IrO2 and RuO2, which are required for efficient hydrogen production from solar water splitting and rechargeable energy storage technologies. So far, layered NiFe double hydroxide represents the best OER activity among all Ni- and Co-based oxides. Here, we report new layered Na1−xNiyFe1−yO2 double oxide OER catalysts exhibiting activity and stability surpassing those of noble metal OER catalysts including IrO2 and RuO2, and a layered NiFe double hydroxide OER catalyst. The superior catalytic properties can be ascribed to the layered structure as well as the enhanced covalency of Ni and Fe. Powered by a lead halide perovskite solar cell with a power conversion efficiency of 14.69%, a two-electrode solar water-splitting device combining a Na0.08Ni0.9Fe0.1O2 OER catalyst with a NiP hydrogen evolution reaction catalyst delivers a solar-to-hydrogen conversion efficiency of 11.22%. Our design and fabrication strategies offer insights for developing highly active electrocatalysts for water splitting and metal–air batteries.
Broader contextProducing clean and low-cost hydrogen fuels via solar-driven water splitting requires efficient oxygen evolution reaction (OER) catalysts. Currently, the most efficient OER catalysts in acidic or alkaline solutions are IrO2 and RuO2, which suffer from the disadvantages of scarcity and high cost of noble metals. Here, we demonstrate that layered Na1−xNiyFe1−yO2 double oxide electrocatalysts exhibit OER activity and stability superior to the state-of-the-art noble metal-based oxide and FeNi layered double hydroxide catalysts. The strategy for enhancing the OER activity of transition metal oxides and approach for synthesizing layered provide insights for exploiting highly efficient noble metal-free electrocatalysts for water splitting and metal–air batteries. |
Introduction
Solar-driven water splitting to produce clean and recyclable hydrogen fuels represents a desirable solution to the energy shortage and environmental pollution issues that the society is facing.1–12 Water splitting involves both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). Achieving an efficient OER is significantly more challenging than achieving an efficient HER, because OER involves multiple electron transfer and is kinetically sluggish.13–22 An efficient OER requires highly active OER catalysts, of which IrO2 and RuO2 in acidic or alkaline solutions are currently the most efficient ones. However, these OER catalysts suffer from the scarcity and high cost of noble metals, significantly limiting the production of clean and recyclable hydrogen fuels through solar-driven water splitting. Therefore, it is urgently required to develop highly active OER catalysts based on low-cost and earth-abundant elements; extensive efforts have been taken in this research direction.23–38Transition metals such as Ni- and Co-based oxides and hydroxides are promising candidates for the development of highly active, durable, and low-cost OER catalysts.26–34 For example, lithium cobalt oxide (LiCoO2), lithium nickel oxide (LiNiO2), and their analogies have been intensively studied for OER applications.36–42 LiCoO2 and LiNiO2 crystallize in the O3 phase (hexagonal R3m), in which the Li+ and Co3+/Ni3+ ions order on alternate {111} planes.36–39 It was understood that the inherent OER activity of LiCoO2 and LiNiO2 is attributed to the presence of [Co4O4]n+ and [Ni4O4]n+ cubane structural units, which provide a lower oxidation potential to Co4+ and Ni4+ and lower inter-cubane hole mobility.36–38 However, OER activities of LiCoO2 and LiNiO2 underperform that of IrO2 and RuO2.40,41 Various efforts have been made to improve their activity and stability. In general, the OER activity depends predominantly on two factors: the available active sites that can contact electrolyte1,33,34,40 and the high oxidation states of transition metal elements.43,44 It has been reported that the catalytic activity of LiCoO2 can be significantly improved by forcing Co to a higher oxidation state, which can be realized by reducing the Li content through de-lithiation or low temperature synthesis of Li1−xCoO2.41 However, de-lithiation of LiNiO2 is not successful due to the formation of Li1−xNi1+xO2, which cannot force Ni to higher charge states. Alternatively, Gupta et al. reported that Al incorporation in LiNiO2 very could improve the OER activity. They showed that Al incorporation pushed Ni to higher charge states.42 However, though pushing Co and Ni to high oxidation states has shown improved performance, the OER activities of LiCoO2 and LiNiO2 still underperform that of IrO2 and RuO2, due to the lack of high enough density of active sites for the three-dimensional crystalline structure of the O3 phase.42
Recently, the FeNi layered double hydroxide (LDH) OER catalyst has shown high activity.43–45 It was shown that the high OER activity is due to high available active sites to the electrolyte.43 Chemical exfoliation of FeNi-LDH combined with electric conducting graphene sheets resulted in low overpotential and high turnover frequency, due to the synergetic effect between the double hydroxide and graphene, which enhanced the electron transfer.44,45 In addition, ultrathin NiFe-LDH nanoplates incorporated on mildly oxidized multiwalled carbon nanotubes exhibited higher OER electrocatalytic activity and better stability than a commercial precious metal IrO2 catalyst.45 Increasing the oxidation states of Ni is a viable approach to further improve the OER activity of FeNi-LDH. Unfortunately, increasing the oxidation states of layered FeNi-LDH is very hard due to the formation of meta-phases.43–45 Thus there are few reports on study of high oxidation states of layered double hydroxide materials.
Herein, we report a unique two-step approach to combine the benefits of high chemical states and high active sites. We first synthesize NaNiyFe1−yO2 and then reduce the Na content by chemical extraction of Na, forming Na1−xNiyFe1−yO2, which is a layered double oxide consisting of [MO6] (M = Ni, Fe) octahedral layers with some residual Na atoms lying between the octahedral layers. Like NiFe-LDH, the layered Na1−xNiyFe1−yO2 double oxide structure provides more active sites than the O3 phase. The use and extraction of Na enforce Ni and Fe to high chemical states, which are confirmed by X-ray photoemission spectroscopy (XPS) measurements. As a result, the new Na1−xNiyFe1−yO2 electrocatalysts exhibit remarkable OER activity and excellent stability. For instance, Na0.08Ni0.9Fe0.1O2 reaches a 10 mA cm−2 anodic current density at a potential of 1.49 V vs. RHE as well as a low onset potential of ∼1.35 V vs. RHE and a Tafel slope of 44 mV dec−1 in alkaline solution. A water oxidation current density of ∼10 mA cm−2 can be maintained for over 70 h. The performance is superior to the state-of-the-art noble metal-based oxide catalysts, RuO2 and IrO2 as well as the NiF-LDH catalyst. Moreover, powered by a 14.69% efficiency lead halide perovskite solar cell, an unassisted two-electrode solar water-splitting device using a Na0.08Ni0.9Fe0.1O2 OER catalyst and a NiP HER catalyst is able to deliver a 11.22% solar-to-hydrogen conversion efficiency under a simulated AM 1.5G illumination. We also find that the same strategy can be applied to synthesize efficient Na1−xCo1−yFeyO2 OER catalysts. Therefore, the synthesis strategy used here provides insights for exploiting highly efficient electrocatalysts for water splitting and metal–air batteries.
Results and discussion
The powder X-ray diffraction (PXRD) patterns show that the as-synthesized NaNiyFe1−yO2via solid-reactions is highly crystalline (Fig. 1a). The NaNiyFe1−yO2 samples exhibit cubical particle morphology with sizes ranging from 300 to 700 nm. As an example, a SEM image of the as-prepared NaNi0.9Fe0.1O2 powder is shown in Fig. S1a (ESI†). The XRD patterns are in good agreement with the O3-type phase (Fig. 1b) by Rietveld fitting of the XRD patterns of NaNi0.9Fe0.1O2 and NaCo0.8Fe0.2O2 samples (Fig. S1b, ESI†). The crystal structure of NaNi1−yFeyO2 is shown in Fig. 1b. NaMO2 can have two structures: O3 type and P2 type, in which sodium ions are accommodated at octahedral and prismatic sites, respectively.46 The O3-type NaMO2 has a hexagonal lattice with a space group of R3m and consists of a cubic close-packed (ccp) oxygen array with Na cations occupying one layer, and Ni/Fe a second adjacent metal layer (Fig. 1b). Metal atoms bind with oxygen atoms forming edge-sharing [MO6] octahedra. The adjacent [NaO6] and [MO6] octahedra share O atoms. Because Na–O bonding is much weaker than M–O bonding, and Na atoms are mobile in NaMO2, the [NaO6] octahedra are not shown in Fig. 1b. It is worth noting that the [MO6] octahedra layers resemble the [M(OH)6] octahedral layers in NiFe-LDH, CoFe-LDH (Fig. S1c, ESI†).43–45 The O3-type NaMO2 and LDH are structurally similar, but chemically different. The LDH has transition metals bonded with OH with an O/H ratio of 1.22 (Fig. S1c, ESI†). On the other hand, the transitional metals in NaMO2 are bonded with O. Therefore, the transition metals are expected to exhibit higher valence states, leading to enhanced OER activity.29,43–45 | ||
| Fig. 1 Structure characterization. (a) XRD patterns of NaNiyFe1−yO2 samples (b) crystal structure of O3-type NaNiyFe1−yO2. | ||
The electrochemical activities of the NaNiyFe1−yO2 were evaluated using a 1.0 M KOH electrolyte. The results of LiNiO2 and NaNiO2 are also shown for comparison. LiNiO2 exhibits an onset potential of 1.57 V vs. RHE (Fig. 2a). The potential required to generate a current density of 10 mA cm−2 is about 1.63 V, and the corresponding Tafel slope is 60 mV dec−1 (Fig. S2, ESI†). The activity of LiNiO2 is comparable with the results reported in the literature.36–38 The NaNiO2 catalyst shows an onset potential of 1.46 V vs. RHE, and reaches 10 mA cm−2 at a potential of 1.56 V vs. RHE. The corresponding Tafel slope is 60 mV dec−1. Apparently, the NaNiO2 OER catalyst performs significantly better than the LiNiO2 catalyst. More strikingly, upon Fe incorporation, all NaNiyFe1−yO2 catalysts exhibit remarkably improved OER activities (Fig. 2a) compared to the NaNiO2 catalysts. With the increase of y value, the onset potentials and potentials at 10 mA cm−2 first gradually decrease to a minimum, and then increase. The catalyst shows the highest activity at y = 0.9: the NaNi0.9Fe0.1O2 catalyst exhibits an onset potential of 1.40 V vs. RHE. The potential for generating a current density of 10 mA cm−2 is about 1.52 V vs. RHE, and the corresponding Tafel slope is 44 mV dec−1 (Fig. S2, ESI†). NaNi0.8Fe0.2O2 and NaNi0.75Fe0.25O2 exhibit the same onset potential of 1.43 V vs. RHE, but their potentials at 10 mA cm−2 are 1.55 V and 1.56 V vs. RHE, respectively, and their corresponding Tafel slopes are 52 and 60 mV dec−1, respectively. The effect of the y value on activity has a similar trend to that in NiyFe1−y-LDH and/or perovskite OER catalysts (BaSrCoyFe1−yO3),43 indicating that the content of Fe would affect the e-filling level of Ni, which closes to unity at y = 0.9. To further comprehend the catalytic activity of NaNiyFe1−yO2, the relative electrochemically active surface areas (ECSA) of the samples are identified by their double-layer capacitance (Cdl) (Fig. S3, ESI†), which is determined from cyclic voltammetry measurements. Then the current densities are normalized by the ECSA.46 As shown in Table S1 (ESI†), NaNi0.9Fe0.1O2 shows the highest normalized current density, which stands for the number of active sites per relative surface area, among all NaNiyFe1−yO2 samples and it is more than 2.8 times higher than that of RuO2. To the best of our knowledge, the OER performance of NaNi0.9Fe0.1O2 is the highest among all previously reported OER catalysts coated on glassy carbon substrates (Table S2, ESI†).
 | ||
| Fig. 2 OER polarization curves of NaNiyFe1−yO2, NaNiO2, LiNiO2, and RuO2 in 1.0 M KOH solution with various y values. (a) LSV curves of Pt, RuO2, and NaNiyFe1−yO2 with y = 0.75, 0.80, 0.90 and 1.00 respectively. (b) Durability test for the NaNi0.9Fe0.1O2 and RuO2 samples at 10 mA cm−2 in 1.0 M KOH solution. | ||
The OER performance of NaNiyFe1−yO2 was also compared with that of Pt and RuO2. For Pt and RuO2 catalysts, the potential required to generate a current density of 10 mA cm−2 is about 1.78 V and 1.55 V vs. RHE, respectively, and the corresponding Tafel slopes are 246 and 73 mV dec−1, respectively. These values are considerably higher than that of NaNiyFe1−yO2 with y = 0.80 and 0.90, confirming the superior electrochemical OER performance of NaNi0.9Fe0.1O2 as compared to the-state-of-the-art noble metal catalyst. The stability of the NaNi0.9Fe0.1O2 catalyst was evaluated by chronocoulometry measurements (Fig. 2b). Under a galvanostatic current density of 10 mA cm−2, a constant operating potential of 1.52 V vs. RHE is maintained for over 70 h, whereas the RuO2 catalyst exhibits a continuous current loss during the test. These results suggest that NaNi0.9Fe0.1O2 is an efficient and stable electrocatalyst for OER. Moreover, the bulk and surface compositions of the sample were determined using inductively coupled plasma mass spectroscopy (ICP-MS) and energy-dispersive X-ray spectroscopy (EDS), respectively. The ICP-MS shows a slight decrease (0.2%) in the Na content after the OER test (Table S3, ESI†). The EDS measurements (Fig. S4, ESI†) of the sample before the stability test show an even Na distribution between the surface and the bulk, while the measurements of the sample after the stability test reveal a slight Na deficiency at the surface (Table S4, ESI†). The alkaline metal extraction during the OER test may be due to their strong ionic nature, which is common in alkaline metal-containing materials.36–42
The activity improvements observed in the NaNiyFe1−yO2 catalysts can be understood by their crystal structure and the chemical states of Ni and Fe. For example, the Na–O and Ni–O bond lengths in NaNi0.9Fe0.1O2 are 2.408(3) Å and 1.971(2) Å, respectively, which are longer than the Li–O and Ni–O bond lengths in LiNiO2 (2.0946(3) Å) and (1.9220(2) Å).47–49 As a result, the unit cell volume of NaNi0.9Fe0.1O2 is 1.26 times larger than that of LiNiO2. The volume expansion of [MO6] units is caused by the lower electronegativity of Na and Fe compared to their counterparts, Li and Ni. The unit cell volume of NaNiyFe1−yO2 decreases with the increase of the y value due to the smaller ionic radius and higher electronegativity of Ni compared to Fe. Thus, the corresponding peaks in XRD patterns shift to higher angles with the increase of y (Fig. S5, ESI†). It was proposed that a lower electronegativity of introduced metals can push up the valence state of Ni and increase the Ni–oxygen covalency.35,43 Therefore, the lower electronegativity of Na over Li and Fe over Ni leads to enhanced Ni–oxygen covalency, which is beneficial for charge transfer and hence favors OER catalytic activity.46 This is consistent with our results that NaNiyFe1−yO2 catalysts perform much better than the LiNiO2 catalysts.
The above explanations are supported by the XPS results. Fig. 3 shows the XPS spectra of Ni 2p and Fe 2p obtained from LiNiO2, NaNiO2, and NaNi0.9Fe0.1O2 samples. The peaks at 856.1 and 873.3 eV in Fig. 3a are assigned to Ni 2p3/2 and Ni 2p1/2, respectively,50 and they can be fitted with the two chemical states of Ni3+ and Ni2+, consistent with the reported literature.42 The Ni2+ chemical states may be caused by non-stoichiometric Li1−xNi1+xO2 impurities in the alternate (111) planes of the cubic close-packed oxygen sublattices of LiNiO2.42 The satellite peaks in the range of 860 to 890 eV for Ni 2p can be ascribed to Ni3+ in NiOOH, which is produced by reacting with the absorbed water from the ambient air.50 For NaNiO2, the peak positions of Ni 2p3/2 and Ni 2p1/2 shift to 857.1 and 874.1 eV, respectively. Such a position shift indicates an increase in the amount of Ni3+, further confirming that the presence of Na forces Ni to its higher oxidation state. Upon Fe incorporation, the peak positions shift to even higher binding energies. The peak positions of Ni 2p3/2 and Ni 2p1/2 in the NaNi0.9Fe0.1O2 shift to 857.6 and 876.1 eV, indicating the mixed chemical state of both Ni3+ and Ni4+.50 Furthermore, the peaks at 711.7 eV and 725.0 eV (Fig. 3b), respectively, are the characteristic signatures of Fe3+ and Fe4+.51 Higher chemical oxidation states would favor the absorption of active species and charge transfer in the rate-determining steps of OER,35,43 which explains the superior activity of NaNi0.9Fe0.1O2 as compared to NaNiO2 and LiNiO2. The ascending chemical oxidation state order of Ni in LiNiO2, NaNiO2 and NaNi0.9Fe0.1O2 samples is in good accordance with the ascending OER performance order of these samples. It is already well known that the Fe4+ chemical state is also highly favorable for OER catalysts,52 even though only few studies on OER catalysts with the Fe4+ chemical state have been reported due to complicated synthesis conditions (i.e. CaCu3Fe4O12).35 Therefore, the electrocatalytic activity improvements of NaNiyFe1−yO2 are ascribed to the combination of high chemical states of Ni and Fe.
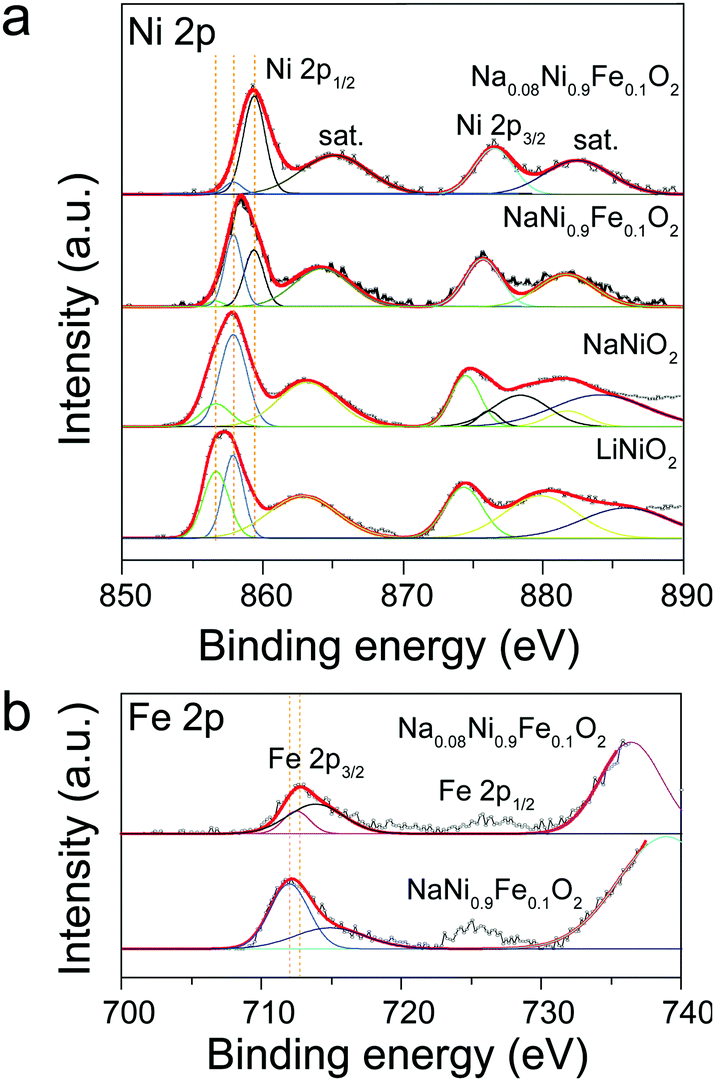 | ||
| Fig. 3 XPS spectra obtained from LiNiO2, NaNiO2, NaNi0.9Fe0.1O2 and Na0.08Ni0.9Fe0.1O2 samples. (a) Ni 2p, (b) Fe 2p states. | ||
As an analogy of NaNiyFe1−yO2, NaCoyFe1−yO2 samples were also studied. NaCoyFe1−yO2 samples also crystallize in the O3-type crystal structure (Fig. S6, ESI†). Among all synthesized catalysts in this family of alloys, NaCo0.8Fe0.2O2 exhibits the best performance (Fig. S6b, ESI†). Even though the activity is not as good as that of NaNi0.9Fe0.1O2, it still outperforms noble metal electrocatalysts including RuO2 (Table S2, ESI†).
Given that the active sites of NaNiyFe1−yO2 materials are the layered [MO6] octahedra, partial extraction of Na cations could expose more [MO6] octahedra active sites to the electrolyte, which is beneficial for activity improvement. In sodium-ion batteries, de-intercalation of Na gradually converts NaMO2 from the O3-type to the P3-type, because gliding of [MO6] layers results in the formation of more symmetric structures. The electrochemical performance (Fig. 4) of Na de-intercalated NaNi0.9Fe0.1O2 samples was evaluated. ICP-MS measurements suggest that the samples with 12, 24, 48 and 72 h Na-extraction have compositions of Na0.41Ni0.9Fe0.1O2, Na0.24Ni0.9Fe0.1O2, Na0.08Ni0.9Fe0.1O2 and Na0.01Ni0.9Fe0.1O2, respectively. With the increase of Na-extraction duration, the performance improves and reaches the highest activity at 48 h (Na0.08Ni0.9Fe0.1O2), but further increase of Na-extraction time leads to decreased activity. For Na0.41Ni0.9Fe0.1O2 and Na0.24Ni0.9Fe0.1O2 samples, the potentials at 10 mA cm−2 decrease to 1.50 V and 1.49 V vs. RHE, respectively (Fig. 4a), which are lower than that of NaNi0.9Fe0.1O2 (1.52 V vs. RHE). The corresponding Tafel slopes are 60 and 43 mV dec−1, respectively (Fig. 4b). Na0.08Ni0.9Fe0.1O2 shows the best performance with an onset potential of 1.40 V vs. RHE and a Tafel slope of 40 mV dec−1, outperforming the best noble metal catalysts, LiMO2 analogies, and most of other reported catalysts on glassy carbon substrates (Table S2, ESI†). The SEM images reveal that the morphology of Na0.08Ni0.9Fe0.1O2 changes to layered flakes from the cubes before Na extraction (Fig. S7, ESI†). The XRD pattern obtained from the Na0.08Ni0.9Fe0.1O2 sample shows only two peaks at 12.0° and 24.1° (Fig. 4c), resembling the XRD pattern of (001)-oriented NiFe-LDH,44,45 further confirming the layered structure. XRD simulations indicate that the structure of Na0.08Ni0.9Fe0.1O2 can be considered as the structure of NaNi0.9Fe0.1O2 in the absence of Na atoms (Fig. S8b, ESI†). After Na extraction, the unit cell has an expansion of 0.2 nm along the [001] direction, possibly due to the weaker van der Waals interaction between two adjacent [MO6] octahedral layers. To further verify the structure of Na de-intercalated NaNi0.9Fe0.1O2, Raman spectroscopy was conducted (Fig. S9, ESI†). Two featured peaks centered around 450 and 650 cm−1 are ascribed to Ni–O and Fe–O bonds.53,54 The broadness of the peaks may be caused by the structural asymmetry. The similarity between NaNi0.9Fe0.1O2 and Na0.08Ni0.9Fe0.1O2 reveals that the [MO6] layers are maintained after 48 h Na-extraction. The improved activity after Na de-intercalation is attributed to the exposure of more active sites to the electrolyte. However, excessive extraction of Na will not allow a stable double oxide structure to persist, due to the relatively high chemical oxidation state of Ni and Fe. The stability of Na0.08Ni0.9Fe0.1O2 was evaluated by chronocoulometry measurements (Fig. 4d). Under a galvanostatic current density of 10 mA cm−2, the Na0.08Ni0.9Fe0.1O2 catalyst exhibits a nearly constant operating potential of 1.50 V vs. RHE for more than 40 h, demonstrating an excellent stability of the layered oxide material. Further evaluation through ICP-MS (Table S5, ESI†), EDS (Table S5, ESI†), XRD (Fig. S10, ESI†), Raman spectra (Fig. S9, ESI†), and SEM (Fig. S11, ESI†) confirm that the structure, chemical state, and composition of a Na0.08Ni0.9Fe0.1O2 sample after the stability test exhibit negligible change, demonstrating that the Na0.08Ni0.9Fe0.1O2 sample is very stable under operation. Na0.08Ni0.9Fe0.1O2 may experience further Na extraction during OER testing. While the ICP-MS measurements show only 0.08% decrease in the Na content, EDS analysis did not exhibit Na content variation due to the sensitivity limit. Therefore, Na0.08Ni0.9Fe0.1O2 demonstrates a higher electrocatalytic activity and stability than NaNi0.9Fe0.1O2. In addition, the EDS and ICP-MS did not detect K in our catalysts after activity tests in the KOH electrolyte. Therefore, the electrolyte should not affect the stability of NaNi0.9Fe0.1O2.
 | ||
| Fig. 4 Electrochemical and structural characterization of NaNi0.9Fe0.1O2 samples. (a) LSV curves of NaNi0.9Fe0.1O2 samples for various Na extraction times. (b) Corresponding Tafel plots of (a). (c) XRD patterns of NaNi0.9Fe0.1O2 samples for various Na extraction times. (d) Durability test for the Na0.08Ni0.9Fe0.1O2 sample (48 h of Na extraction) at 10 mA cm−2 in 1.0 M KOH solution. | ||
The Na0.08Ni0.9Fe0.1O2 sample exhibits higher chemical states of Ni and Fe compared to NaNi0.9Fe0.1O2 (Fig. 2). The corresponding XPS peaks of Ni 2p3/2, Ni 2p1/2, Fe 2p3/2 and Fe 2p1/2 obtained from the sample Na0.08Ni0.9Fe0.1O2 are 858.7 and 880.5 eV, 712.6 eV and 725.7 eV, respectively. Therefore, the increased OER activity of Na0.08Ni0.9Fe0.1O2 is due to the higher covalency of Ni. Electrocatalysts often show broad redox waves, and the valence state always shuttles between oxidation states and resting states.55,56 The slight variation in oxidation states will strongly affect the absorbance of oxidative oxygen species O22−/O−. In particular, in the rate-determining steps of OER, the higher chemical states could promote the absorption capacity, thus resulting in higher activities.35,43 Electrochemical impedance spectroscopy (EIS) was carried out to evaluate the charge transfer during oxygen evolution (Fig. S12, ESI†). The semicircular arc diameter is related to charge transfer resistance (Rct), and smaller diameter means smaller Rct. and therefore, improved charge transfer in the rate-determining steps of OER.40 The semicircular arc diameter of Na0.08Ni0.9Fe0.1O2 in the EIS plot is much smaller than that of RuO2, indicating that Na0.08Ni0.9Fe0.1O2 has very high electron generation and fast charge transfer for OER. These results further confirm that Na0.08Ni0.9Fe0.1O2 is an excellent OER catalyst with high activity and superior stability.
Given the excellent activity and stability, Na0.08Ni0.9Fe0.1O2 and NaNi0.9Fe0.1O2 OER catalysts coated on Ni foam were integrated with NiP nanoparticle HER catalysts coated on Ni foam to form a two-electrode water-splitting device. NiP was chosen as an alternative to a noble metal HER catalyst. The HER catalytic activities of NiP were also evaluated in 1.0 M KOH solution (Fig. S13a and b, ESI†). NiP has the lowest onset potential of around −100 mV vs. RHE. The cathodic current density reaches up to −10 mA cm−2 at a potential of −150 mV vs. RHE. Furthermore, the stability test manifests that NiP can still maintain stable potentials at −10 mA cm−2 for over 6 h (Fig. S13b, ESI†). Thus, it has been demonstrated that the NiP is a superior HER electrocatalyst exhibiting high activity and stability. The Na0.08Ni0.9Fe0.1O2 and NaNi0.9Fe0.1O2 catalysts coated on nickel foam show even higher performance. They reach 10 mA cm−2 current density at a potential of 1.39 and 1.41 V (vs. RHE) respectively (Fig. S13, ESI†). A water electrolysis polarization curve obtained using the combination of RuO2(+) with Pt(−) coating on Ni foam exhibits an onset potential difference of 1.40 V, and achieves 10 mA cm−2 current densities at a potential difference of 1.62 V (Fig. 5a). The IrO2(+) and Pt(−) electrode combination shows inferior activities, with an onset potential difference of 1.50 V and a potential difference of 1.74 V at 10 mA cm−2. The NaNi0.9Fe0.1O2 and NiP electrode combination shows a slightly higher performance than the RuO2(+) and Pt(−) electrode combination – with the same onset potential but a slightly lower potential of 1.61 V at 10 mA cm−2. Strikingly, the Na0.08Ni0.9Fe0.1O2 and NiP electrode combination shows much higher activities – an onset potential difference of 1.4 V and a potential difference of 1.54 V at 10 mA cm−2. The long-term stability test is further conducted by full water splitting at 1.60 V applied potential difference. The RuO2(+) and Pt(–) electrode combination exhibits a gradual decay from 9.5 to 7.9 mA cm−2 in 5 h. In contrast, the current density of the Na0.08Ni0.9Fe0.1O2 and NiP electrode combination is stable above 15.5 mA cm−2 for over 12 h of continuous testing (Fig. 5b). Gas chromatography measurements confirm a high faradaic efficiency of H2 and O2 at the predicted ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S14, ESI†).
:1 (Fig. S14, ESI†).
 | ||
| Fig. 5 (a) Overall water-splitting characteristics of different catalyst electrodes in a two-electrode configuration. (b) Durability test for the Na0.08Ni0.9Fe0.1O2 sample in a two-electrode configuration at 1.60 V vs. RHE in 1.0 M KOH solution, compared with noble metal eletrodes. (c) J–V curves of the perovskite tandem cell under simulated AM 1.5G 100 mW cm−2 illumination. (d) Current density–time curve of solar-driven water splitting under chopped simulated AM 1.5G 100 mW cm−2 illumination. Inset image in (d) is the schematic of the solar-driven water splitting device using the Na0.08Ni0.9Fe0.1O2 electrocatalyst and perovskite solar cells. | ||
The Na0.08Ni0.9Fe0.1O2 and NiP electrode combination was also tested in an unassisted solar-driven water splitting device. A series-connected lead halide perovskite solar cell, which is composed of two perovskite sub-cells with an open circuit potential (Voc) of 1.10 V each, was used to power the system. The series-connected perovskite solar cell exhibits a Voc of 2.16 V, a short circuit current density (Jsc) of 9.96 mA cm−2, and a power conversion efficiency of 14.69% when illuminated under 100 mW cm−2 AM 1.5G solar irradiation (Fig. 5c). The Na0.08Ni0.9Fe0.1O2 and NiP two-electrode system delivered a current density of 9.12 mA cm−2 and was stable for up to 2 h (Fig. 5d). Further lifetime testing was limited by the stability of the lead halide perovskite cell. A corresponding solar-to-hydrogen conversion efficiency of ∼11.22% was obtained from the polarization curve. The current density response under chopped simulated sunlight illumination during the stability test indicates good reversibility of the electrodes and relatively rapid response to changes in photocurrent.
Conclusions
New noble metal-free layered Na1−xNiyFe1−yO2 oxide electrocatalysts have been successfully developed for efficient water splitting. Owing to Na extraction, enhanced charge states of Ni and Fe, and a layered microstructure, Na1−xNiyFe1−yO2 catalysts exhibit excellent OER activity. Among these catalysts, Na0.08Ni0.9Fe0.1O2 exhibits the highest OER activity, higher than LiMO2, noble metal electocatalysts (e.g. RuO2, IrO2 and Pt) and most of reported OER catalysts including the NiFe or CoFe layered double hydroxides. When integrated with a 14.69% efficient perovskite solar cell, the Na0.08Ni0.9Fe0.1O2 and NiP two-electrode system delivered a solar-to-hydrogen efficiency of 11.22% under an AM 1.5G simulated illumination. Due to the scalable synthetic method, wide material availability, and high catalytic activity and stability, the Na1−xNiyFe1−yO2 OER catalysts are attractive candidates for substituting noble metal-based catalysts toward highly efficient and low-cost electrolysis or solar-driven water splitting.Acknowledgements
This paper presents results from an NSF project (award number CBET-1433401) competitively-selected under the solicitation “NSF 14–15: NSF/DOE Partnership on Advanced Frontiers in Renewable Hydrogen Fuel Production via Solar Water Splitting Technologies”, which was co-sponsored by the National Science Foundation, Division of Chemical, Bioengineering, Environmental, and Transport Systems (CBET), and the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office. The work was also partially supported by the National Science Foundation under contract no. CHE-1230246 and DMR-1534686. This research used the resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- B. Kong, J. Tang, Y. Y. Zhang, C. Selomulya, X. G. Gong, Y. Liu, W. Zhang, J. P. Yang, W. S. Wang, X. T. Sun, Y. F. Wang, G. F. Zheng and D. Y. Zhao, J. Am. Chem. Soc., 2015, 137, 4260 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- W. J. Yin, H. Tang, S. H. Wei, M. M. Al-Jassim, J. Turner and Y. Yan, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045106 CrossRef.
- A. P. Goodey, S. M. Eichfeld, K. K. Lew, J. M. Redwing and T. E. Mallouk, J. Am. Chem. Soc., 2007, 129, 12344 CrossRef CAS PubMed.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Science, 2010, 327, 185 CrossRef CAS PubMed.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, D. B. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, J. Am. Chem. Soc., 2011, 133, 1216 CrossRef CAS PubMed.
- Y. W. Chen, J. D. Prange, S. Duhnen, Y. Park, M. Gunji, C. E. D. Chidsey, E. D. Christopher and P. C. McIntye, Nat. Mater., 2011, 10, 539 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 138, 37 CrossRef.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- A. Kudo and Y. Misek, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. D. Hemandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler Jr, Science, 2002, 297, 2243 CrossRef CAS PubMed.
- K. Maeda, T. Tanaka, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286 CrossRef CAS PubMed.
- B. C. Weng, F. H. Xu and J. G. Xu, J. Nanopart. Res., 2014, 16, 2766 CrossRef.
- B. C. Weng, F. H. Xu and J. G. Xu, Nanotechnology, 2014, 25, 455402 CrossRef PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52 CrossRef CAS PubMed.
- Z. Zhang and J. T. Yates Jr., Chem. Rev., 2012, 112, 5520 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- J. R. McKone, A. P. Pieterick, H. B. Gray and N. S. Lewis, J. Am. Chem. Soc., 2013, 135, 223 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeis, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- Z. M. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542 CrossRef CAS PubMed.
- W. D. Chemelewski, H.-C. Lee, J. F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- M. Cabán-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide, Nat. Mater., 2015, 14, 1245 CrossRef PubMed.
- X. Yang, A. Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K. W. Huang, Y. Han and L. J. Li, Nano Energy, 2015, 15, 634 CrossRef CAS.
- C. W. Tung, Y. Y. Hsu, Y. P. Shen, Y. Zheng, T. S. Chan, H. S. Sheu, Y. C. Cheng and H. M. Chen, Nat. Commun., 2015, 6, 8106 CrossRef CAS PubMed.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Muller, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 1 CrossRef.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- G. Gardner, J. Al-Sharab, N. Danilovic, Y. B. Go, K. Ayers, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2016, 9, 184 CAS.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P. C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CAS.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150 CrossRef CAS PubMed.
- A. Gupta, W. D. Chemelewski, C. B. Mullins and J. B. Goodenough, Adv. Mater., 2015, 27, 6063 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- N. Yabuuchi and S. Komaba, Adv. Mater., 2014, 15, 043501 Search PubMed.
- E. Lee, J. Lu, Y. Ren, X. Luo, X. Zhang, J. Wen, D. Miller, A. DeWahl, S. Hackney, B. Key, D. Kim, M. D. Slater and C. S. Johnson, Adv. Energy Mater., 2014, 4, 1400458 CrossRef.
- C. Zhu, D. Wen, S. Leubner, M. Oschatz, W. Liu, M. Holzschuh, F. Simon, S. Kaskel and A. Eychmüller, Chem. Commun., 2015, 51, 7851 RSC.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, K. Guruprakash, A. S. Prakash, M. L. Doublete and J. M. Tarascon, Chem. Commun., 2013, 49, 11376 RSC.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, J. Mater. Chem. A, 2015, 3, 7179 CAS.
- A. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090 CrossRef PubMed.
- L. A. Stern and X. Hu, Faraday Discuss., 2014, 176, 363 RSC.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, H. R. Winkler and A. M. Muller, J. Am. Chem. Soc., 2014, 136, 13118 CrossRef CAS PubMed.
- C. W. Tung, Y. Y. Hsu, Y. P. Shen, Y. Zheng, T. S. Chan, H. S. Sheu, Y. C. Cheng and H. M. Chen, Nat. Commun., 2015, 6, 8106 CrossRef CAS PubMed.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, characterization and supporting figures. See DOI: 10.1039/c6ee03088b |
| This journal is © The Royal Society of Chemistry 2017 |