
Acute exposure of ozone induced pulmonary injury and the protective role of vitamin E through the Nrf2 pathway in Balb/c mice
Yuqing
Zhu†
ab,
Jinquan
Li†
bc,
Zhuo
Wu
b,
Yu
Lu
b,
Huihui
You
b,
Rui
Li
b,
Baizhan
Li
c,
Xu
Yang
*b and
Liju
Duan
*ad
aCollege of Public Health, Zhengzhou University, Zhengzhou, China
bSection of Environmental Biomedicine, Hubei Key Laboratory of Genetic Regulation and Integrative Biology, College of Life Science, Central China Normal University, Wuhan, China. E-mail: yangxu@mail.ccnu.edu.cn; Tel: +86-13871361954
cKey Laboratory of the Three Gorges Reservoir Region's Eco-Environment, Ministry of Education, Chongqing University, Chongqing 400045, China
dCollege of Public Health, Huazhong University of Science and Technology, Wuhan, China. E-mail: dlj@zzu.edu.cn; Tel: +86-18768869690
First published on 10th November 2015
Abstract
Ozone (O3) in the lower atmosphere is generally derived from various sources of human activity. It has become a major air pollutant in China and has been shown to adversely affect the health of humans and animals. We undertook a study to ascertain the molecular mechanism of ozone induced lung injury in mice and tried to demonstrate the protective mechanism of vitamin E. In this study, mice were exposed to clean air and three different concentrations of ozone. Oxidative stress (reactive oxygen species and malondialdehyde) and Th cytokines in the lung, serum IgE, as well as histopathological examination and the airway hyper-responsiveness (AHR) test were used to reflect inflammation and damage to the lungs of ozone-exposed mice. We then chose an effective concentration of ozone and combined treatment with vitamin E (VE) to explore the underlying mechanism of ozone-induced lung damage. The results of immunological and inflammatory biomarkers (total-immunoglobulin (Ig) E and Th cytokines) as well as histopathological examination and AHR assessment supported the notion that high doses of ozone (>0.5 ppm) could induce inflammation and lung injury in mice and that this induction was counteracted by concurrent administration of VE. The elimination of oxidative stress, the reduced Th2 responses and Ig production, and the relief of lung damage were proposed to explain the molecular mechanism of ozone induced lung injury. We also showed that VE, an antioxidant that enhanced the expression of Nrf2 and up-regulated the antioxidant genes HO-1 and NQO1, could decrease the levels of oxidative stress and alleviate ozone-induced lung injury.
Introduction
Ozone (O3) is derived from important sources of human activity, such as operating motor vehicles, and the fuel and petrochemical industries. Ozone is a secondary air pollutant generated via a series of complicated photochemical reactions involving nitrogen oxides and sunlight.1 In 2008, the U.S. Environmental Protection Agency (EPA) updated the O3 standard to 75 ppb (0.075 ppm) based on evidence that demonstrated deleterious health effects. On November 25, 2014, the EPA proposed to strengthen the standard of O3, based on extensive scientific evidence about ozone's health effects. However, today millions of Chinese are exposed to ozone levels exceeding the recommended EPA limit. In Beijing for example, the annual number of non-attainment days for 1 h O3 were 57, 67, 70, and 76, in 2003, 2004, 2010, and 2012 respectively.2 In 2013, China's Environmental Protection Ministry reported that the averaged non-attainment rates for maximum 8 h O3 in 74 cities varied between 20% and 50% during the period from May to September, and that O3 has become the second major air pollutant in many of China's cities, following PM2.5.As a strong oxidant, O3 is able to react with almost any biological tissue and induce adverse health effects in humans. When O3 is inhaled, it causes airway inflammation which can lead to constriction of the airways, increased bronchial reactivity and decreased lung function, all of which pose a significant threat to respiratory health.3 In addition, exposure to increased levels of ozone has been associated with the worsening of symptoms in patients with obstructive lung diseases such as asthma and COPD (chronic obstructive pulmonary disease),4 and increased morbidity and mortality rates in cardiovascular and respiratory patients.5,6 It has been estimated that if there was strict adherence to the established 8-hour O3 standard each year, this would result in 800 fewer premature deaths, 900![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fewer school absences and 4500 fewer hospital admissions which in turn would save an estimated $5 billion annually.7
000 fewer school absences and 4500 fewer hospital admissions which in turn would save an estimated $5 billion annually.7
However, while it is known that O3 pollution poses potential health risks, the molecular mechanisms behind ozone-induced lung resistance and inflammation are still unclear. Evidence has suggested that oxidative stress in the lung in response to ozone-induced injury contributes to inflammation.8 When O3 is inhaled, it becomes toxic by inducing peroxidation of polyunsaturated fatty acids in membrane lipids and in the lung lining fluid, giving rise to the generation of reactive oxygen species (ROS), and a mixture of lipid ozonation products.9 These products are capable of disturbing the redox balance and inducing inflammation, hence leading to lung function damage.10 Thymic stromal lymphopoietin (TSLP) is an epithelial cell-derived cytokine that has a strong influence on the polarization of dendritic cells (DCs) that drive T helper (Th) 2 cytokine production, and also plays an important role in regulating Th1/Th2 immune balance.11 In addition, ROS orchestrated Th2 responses by inducing oxidized lipids that triggered the induction of TSLP by epithelial cells mediated by toll-like receptor 4 (TLR4) and the adaptor protein TRIF.12
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor that plays an essential role in the promoter region of genes encoding antioxidant and/or detoxifying enzymes and related stress-responsive proteins.13 The small antioxidant enzymes, including NADPH, quinone oxidoreductase-1 (NQO-1), heme oxygenase-1 (HO-1) and γ-glutamate cysteine ligase (GCL), are regulated by Nrf2, responding to attenuate inflammatory damage and neutralize ROS of cells/tissues from inflammatory injuries.14 VE is a potent ROS-scavenging and chain-breaking antioxidant that may prevent the development of respiratory disease.15 Although vitamin E has previously been investigated in pulmonary disease, the lack of evidence for the protective mechanism in previous studies limits the conclusions concerning its role.
We hypothesized that O3 causes the increase of ROS in the lung, and then modulates Th-2 cell differentiation and lung inflammation, which eventually leads to lung tissue injury and lung function decline. To test our hypothesis, we first used three different exposure concentrations of ozone and analyzed ozone-induced lung injury by measuring the levels of oxidative stress and Th cytokines in the lung, the serum IgE, as well as performing a histopathological examination and an AHR (airway hyperresponsiveness) test. We next chose an appropriate exposure concentration of O3 and combined treatment with VE in order to explore the molecular mechanism of ozone-induced lung damage. Furthermore, by detecting the expression of Nrf2 and the antioxidant genes HO-1 and NQO1 in lung tissue, we tried to explore the reason why VE plays an antioxidant role and attenuates the effect of ozone-induced lung damage.
Materials and methods
The experimental protocols were approved by the Institutional Animal Care and Use Committee of Central China Normal University on March 1, 2012 (Ratification ID: CCNU-IACUC-2012-011).Animals
Male Balb/c mice (5–6 weeks; approximately 20 g) were purchased from the Hubei Province Experimental Animal Center (Wuhan, China). They were maintained in pathogen-free cages at 24 °C–26 °C and 55%–75% humidity with a 12 h light-dark cycle. Mice were kept 5 mice per cage and provided ad libitum access to a commercial diet (Hubei Province Experimental Animal Center) and filtered water. The cages we used were independent ventilation cages (IVC), each cage being equipped with separate air inlet and outlet systems.Main reagents and kits
Ozone was generated using a KTB portable ozone generator (Guangzhou, China) from ambient air with an ozone calibrator source. All other chemicals used were of analytical grade. The mouse enzyme-linked immunosorbent assay (ELISA) kits for measuring total IgE, IL-4, interferon (IFN)-γ, IL-5 and TSLP were from eBioscience (San Diego, CA, USA). To determine the total protein, a modified BCA protein assay kit was purchased from Sangon Biotech (Shanghai, China).Experimental design and animal exposure
To determine the effects of ozone exposure on mice, the experiment included 6 groups (n = 5): (A) control group, (B) VE (100 mg VE per kg) control group (intraperitoneal injection), (C) 0.1 ppm ozone exposure group, (D) 0.5 ppm ozone exposure group, (E) 1.0 ppm ozone exposure group, (F) 1 ppm ozone exposure and VE group. Mice were exposed to clean air or ozone (0.1 ppm, 0.5 ppm, 1.0 ppm) for 3 h per day, for seven days. VE was administered as an antioxidant 3 h after exposure to ozone (1 ppm). The detailed protocols are shown in Fig. 1.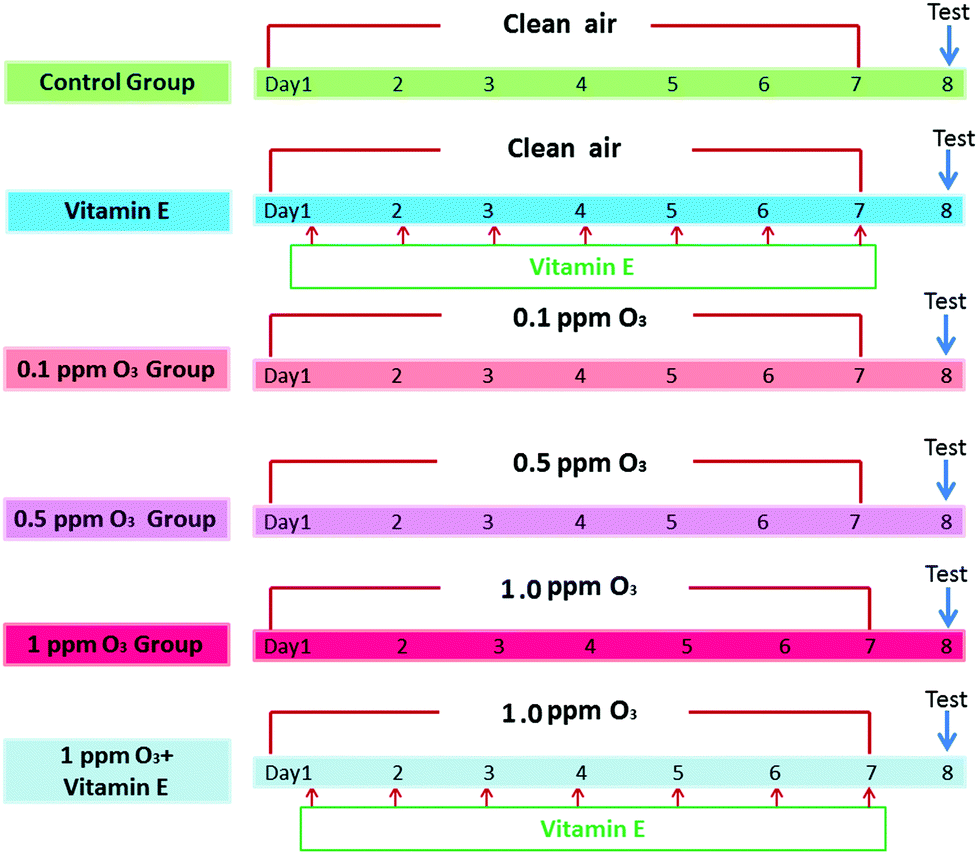 | ||
| Fig. 1 Experimental design and animal exposure. | ||
Experimental error in ascertaining the concentrations of oxidative stress-related biomarkers as well as Th cytokines and IgE concentrations due to the influence of MCH used in AHR assessments was avoided by conducting two rounds of this experiment. In the first round, after 7 days of ozone exposure, the 30 mice were used to measure Th cytokine concentrations and oxidative stress-related biomarkers in lung homogenates and serum IgE production. In the second round, another 30 mice were treated with the same protocol and then used directly for AHR tests and lung histological assays.
First round of testing
The aims of the first round were to ascertain: (i) whether ozone exposure affects serum IgE production and lung inflammation in mice; (ii) whether ozone exposure can cause oxidative stress and increase the TSLP level in the mouse lung; and (iii) whether there is any antioxidant effect of VE on these biomarkers.Quantitative analyses of total serum IgE
In the first round of experiments, 24 h after final exposure, the mice were anaesthetized with pentobarbital sodium (100 mg per kg bw, intraperitoneally). Serum samples were then collected from the heart blood by centrifugation (3000 rpm for 10 min at room temperature). Serum concentration of total-IgE was measured using the ELISA kits according to the manufacturer's instructions.Tissue sample preparation and ELISA
After serum collection, the mice were killed by cervical dislocation and the whole lung was removed using medical scissors and rinsed in ice-cold phosphate-buffered saline (PBS). Lung tissue was homogenized in a glass homogenizer on ice using 10 mL per g of ice-cold PBS at pH 7.5 to produce a 10% tissue homogenate. Then, the homogenate was centrifuged at 10000 rpm for 10 min at 4 °C and the supernatant was collected for later assessment of levels of lung cytokines (IFN-γ, IL-4 and IL-5) and TSLP using the ELISA kits according to the manufacturer's instructions.
Measurement of GSH and MDA content
The GSH test kits were provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All the operations were performed according to the manufacturer's instructions. The MDA concentration in the lung tissue homogenate was measured using a previously described procedure.16 The protein concentration was determined using the modified BCA protein assay kit (Sangon Biotech, China).Second round of testing
The aims of the second round were to ascertain: (i) whether ozone exposure causes histological lung damage in mice; (ii) whether ozone exposure can change AHR and (iii) whether there is any protective effect of VE in lung histological assays and the AHR test.Measurement of AHR
In the second round of experiments, 24 h after the final exposure, the other 30 mice were tested for AHR using the AniRes2005 Lung Function System (Bestlab ver2.0; Beijing, China) according to a previously described procedure.17Lung histological assay
After testing for AHR, the left lung was removed for preparation of histopathology slices. Samples were fixed in 10% formalin solution for 24 h at room temperature and cut into 5 μm slices of the distal pieces for H&E staining. The section was observed using the DM4000B microscope (Leica Microsystems GmbH, Wetzlar, Germany). The numbers of inflammatory cells in each sample were counted using Image-Pro Plus software (Image-Pro Plus 6.0, Media Cybernetics). The histological scores were assessed by a pathologist who knew nothing of the origin of the sample.Immunohistochemistry for IL-13, Nrf2, HO-1, NQO-1 and mast cell tryptase
The sections of lung tissue were deparaffinized, rehydrated and subjected to antigen retrieval, then incubated with 0.3% hydrogen peroxide and blocked by appropriate normal serum. Immunohistochemical detection of IL-13, Nrf2, HO-1, NQO-1 and mast cell tryptase were performed using primary antibodies anti-IL-13 (1:100, Boster, Wuhan, China), anti-Nrf2 (1:100, Proteintech, Chicago, USA), anti-HO-1 (1:50, Proteintech, Chicago, USA), anti-NQO-1 (1:200, Proteintech, Chicago, USA) and anti-mast cell tryptase (1:50, Abcam, MA, USA), respectively. After incubation in primary antibodies, the sections were sequentially incubated in an appropriate biotinylated immunoglobulin and avidin–biotin peroxidase complex. The reaction product was visualized using hydrogen peroxide (3%) and diaminobenzidine tetrahydrochloride (DAB, 5 mg/10 ml) (Sigma-Aldrich, St. Louis, MO, USA) as the chromogen. The immunohistochemical control was obtained by omitting the primary antibody. All sections were finally counterstained with hematoxylin, dehydrated and mounted in DPX (Sigma-Aldrich).
Statistical analyses
Data are presented as mean ± SEM. Statistical graphs were generated using GraphPad Prism 5.0 (San Diego, CA, USA). A one-way ANOVA combined with Fisher's protected t-test was used to determine the significance of differences between the groups. p < 0.05 was considered significant and p < 0.01 was considered extremely significant. Data analyses were carried out using SPSS ver13 (SPSS, Chicago, IL, USA).Results
Effect of ozone exposure on the serum levels of IgE and degranulation of mast cells
To evaluate the effect of ozone exposure on serum, we measured total-IgE in the serum of mice. Fig. 2 shows the total serum IgE data after 7 days of ozone exposure. There were three important findings: (i) exposure to VE and 0.1 ppm O3 did not cause changes in total serum IgE and degranulation of mast cells; (ii) the total IgE levels for the 0.5 ppm O3, 1.0 ppm O3 and 1.0 ppm O3 + VE exposure groups were significantly increased compared to those of the control group (Fig. 2A, p < 0.01); furthermore, 1.0 ppm O3 exposure groups significantly increased the degree of mast cell degranulation (Fig. 2B and C, p < 0.01); (iii) compared with the 1.0 ppm O3 group, the 1.0 ppm O3 + VE group showed a decreased level of total IgE and mast cell degranulation (Fig. 2, p < 0.01).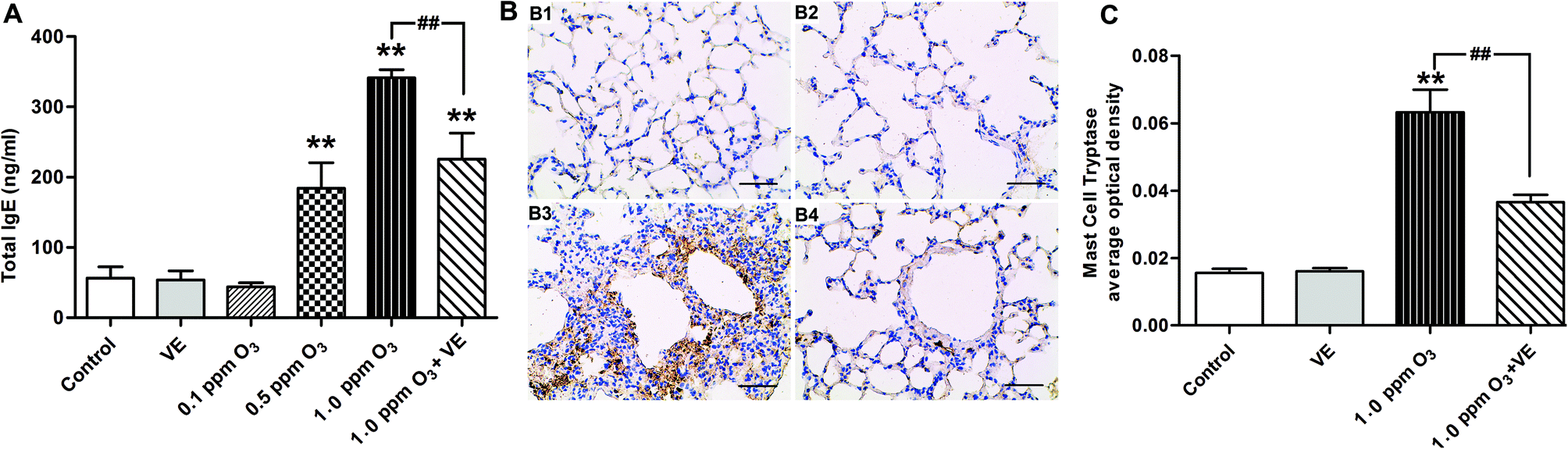 | ||
| Fig. 2 Total serum IgE levels and immunohistochemistry with tryptase. (A) Total IgE concentrations (ng ml−1). **: p < 0.01, compared with the control group; ##: p < 0.01, compared with 1.0 ppm O3 exposure group. (B) Immunohistochemistry with tryptase in lung tissue. B1–B4 represent different exposure groups (control, VE, 1.0 ppm O3, 1.0 ppm O3 + VE), scale bars = 50 μm. (C) The scores of degranulation were calculated from the expression levels of tryptase. ** p < 0.01, compared with control group; ## p < 0.01, compared 1.0 ppm O3 group with 1.0 ppm O3 + VE group. | ||
Effect of ozone exposure on the levels of Th cytokines in the lung
The levels of the Th1 cytokine IFN-γ, the Th2 cytokines IL-4, IL-5 and IL-13 were assessed in lung tissue samples (Fig. 3A–C, and 4). Administration of VE did not result in significant changes in IFN-γ, IL-4, IL-5 and IL-13 levels. Exposure to O3 (0.1 ppm O3, 0.5 ppm O3, 1.0 ppm O3 and 1.0 ppm O3 + VE groups) led to rapid drops in the levels of IFN-γ compared with the control group (Fig. 3A, p < 0.01). The protein levels of IL-4, IL-5 and IL-13 in the 1.0 ppm O3 group were significantly greater than those in the control group (Fig. 3B, C and 4, p < 0.01). Administration of VE combined with 1.0 ppm O3 exposure led to a significant decrease in the levels of IL-4, IL-5 and IL-13 compared with the 1.0 ppm O3 group (Fig. 3B, p < 0.05; Fig. 3C and 4, p < 0.01).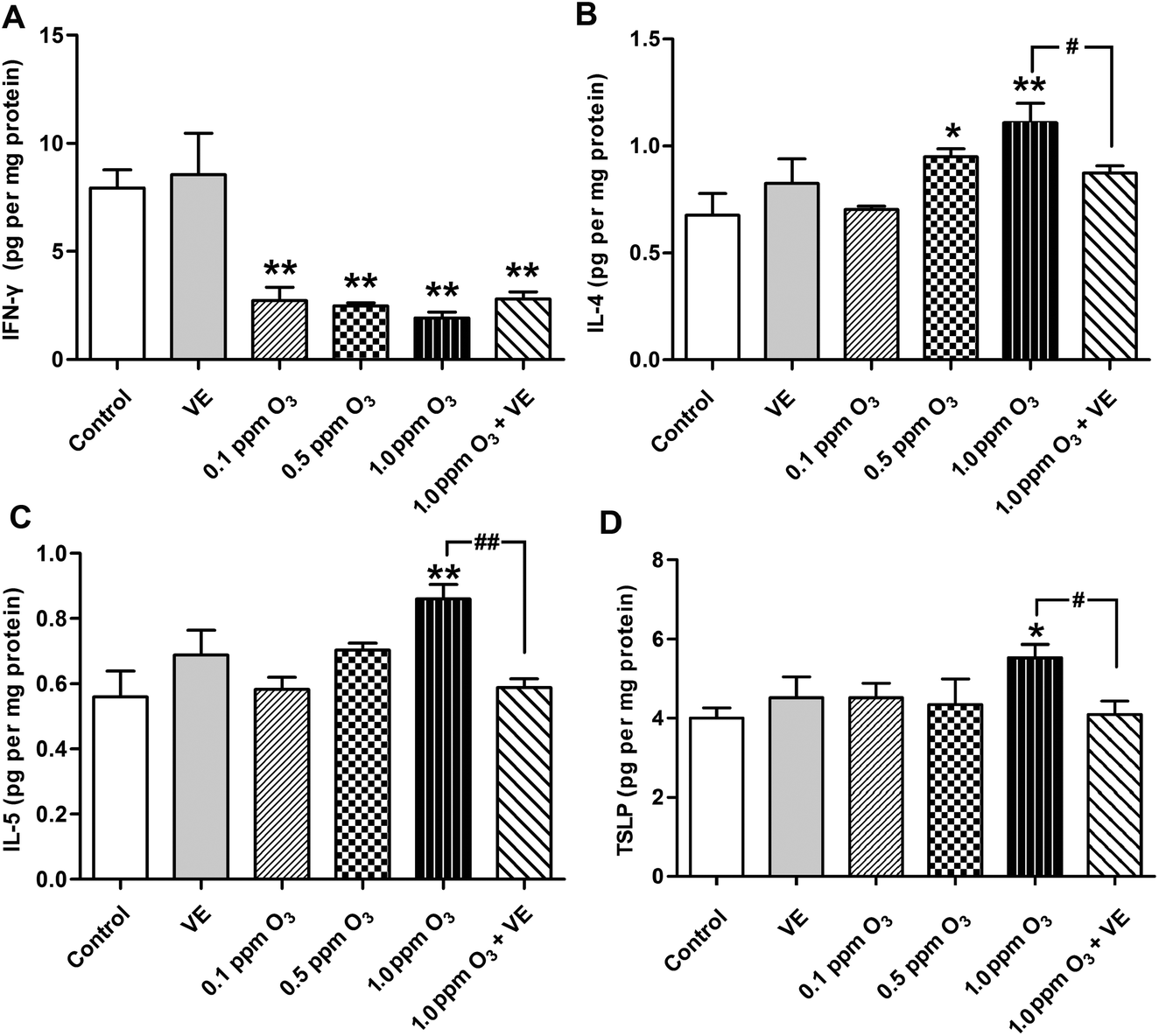 | ||
| Fig. 3 Effects of ozone on cytokine concentrations in the lung. (A) Levels of IFN-γ. (B) Levels of IL-4. (C) Levels of IL-5. (D) Levels of TSLP. *: p < 0.05, **: p < 0.01, compared with the control group; #: p < 0.05, ##: p < 0.01, compared with 1.0 ppm O3 exposure group. | ||
 | ||
| Fig. 4 Immunohistochemical analyses of IL-13 in the lung. Panel: (A1) Control group, (A2) VE (100 mg VE per kg) protected group (ip), (A3) 0.1 ppm ozone exposure group, (A4) 0.5 ppm ozone exposure group, (A5) 1.0 ppm ozone exposure group, (A6) 1.0 ppm ozone exposure and VE group. Scale bars = 50 μm. (B) Analyses of IL-13 expression levels according to the average optical density. Animal groups (in all panels): n = 3 mice per group. **: p < 0.01, compared with the control group; ##: p < 0.01, compared with 1.0 ppm O3 exposure group. | ||
Given that TSLP is an important cytokine produced by epithelial cells and has been linked to ROS and inflammation, we measured TSLP expression in the lung after ozone exposure (Fig. 3D). The VE and low ozone exposure groups (0.1 and 0.5 ppm) did not show changes in TSLP levels compared with the control group. The TSLP levels in the 1.0 ppm O3 group were significantly higher than those of the control group (p < 0.05). The 1.0 ppm O3 exposure plus VE group showed a strong alleviating effect on the results of TSLP (p < 0.01).
Ozone effects on oxidative stress in mice lungs and antioxidation of VE
To evaluate the level of oxidative stress after ozone exposure, we measured GSH and MDA in the lung. In the 1.0 ppm O3 group, the levels of GSH and MDA were significantly increased compared to those of the control group (Fig. 5A, p < 0.05; Fig. 5B, p < 0.01); compared with the 1.0 ppm O3 group, the 1 ppm O3 + VE group showed significantly decreased levels of GSH and MDA (Fig. 5, p < 0.01).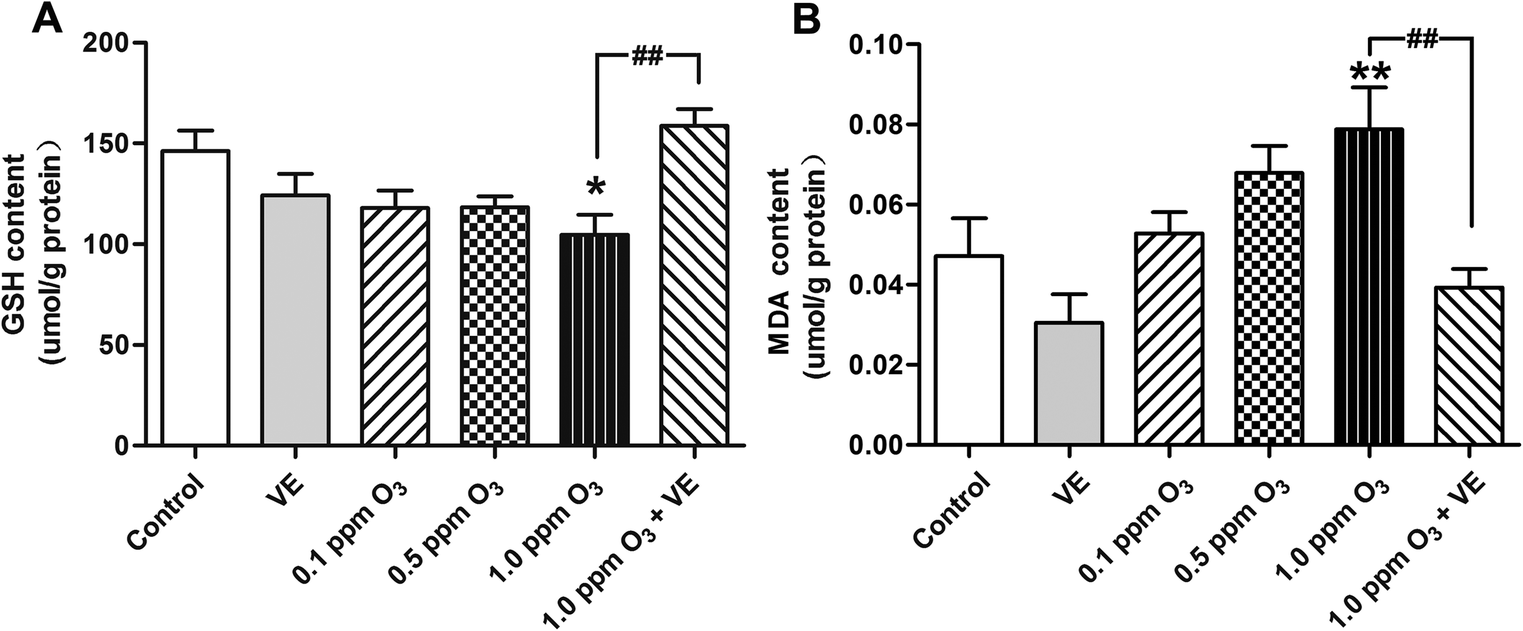 | ||
| Fig. 5 Ozone effects on oxidative stress and the antioxidant effect of VE. Concentrations of GSH (A) and MDA (B) in the lung. *: p < 0.05; **: p < 0.01, compared with the control group; ##: p < 0.01, compared with 1.0 ppm O3 exposure group. | ||
Effect of ozone exposure on histological changes
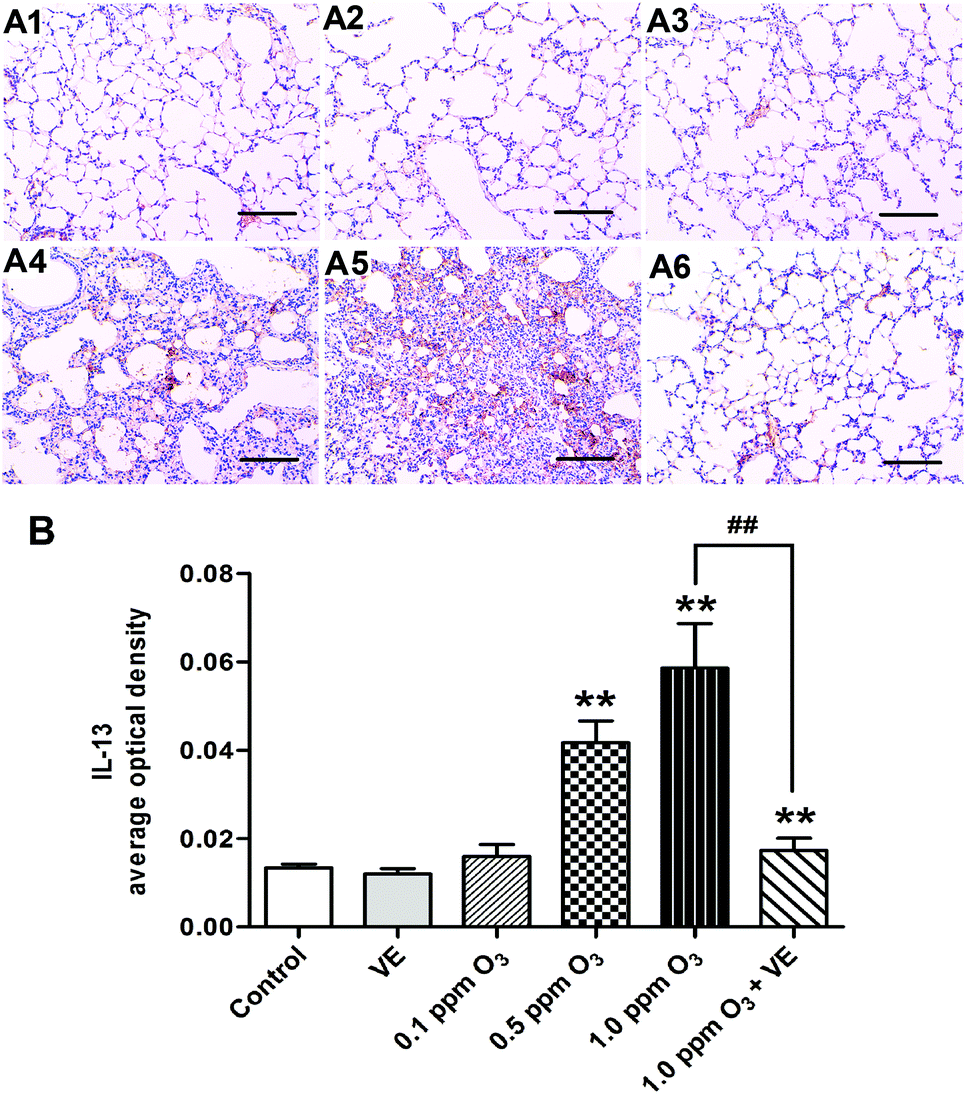
To evaluate the histological changes, we carried out hematoxylin and eosin (H&E) staining 24 h after the final O3 exposure (Fig. 6). Exposure to VE and 0.1 ppm O3 did not lead to significant pathologic alterations. The 0.5 and 1.0 ppm O3 groups caused inflammatory cell infiltration and bronchial remodeling (Fig. 6A4 and A5; 6B, p < 0.05, p < 0.01). In addition, as the concentration of O3 increased, the aggravation effect was stronger. Furthermore, the 1.0 ppm O3 exposure plus VE group showed significant attenuation of these adverse effects (Fig. 6A6 and B, p < 0.01).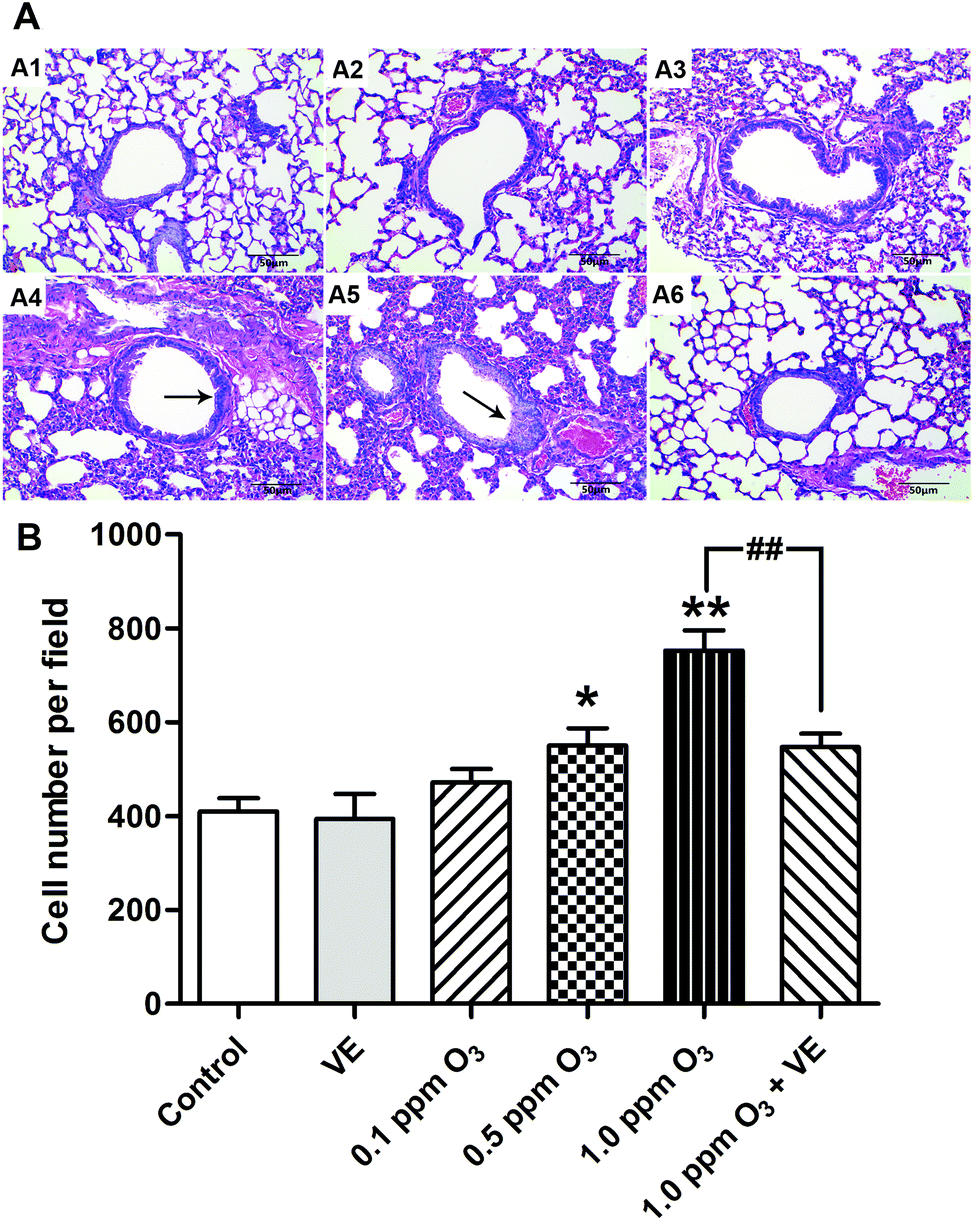 | ||
| Fig. 6 Effects of ozone treatment visualised as histopathological lung changes. (A) H&E staining. (B) Number of inflammatory cells infiltrating. Panel: (A1) Control group, (A2) VE (100 mg VE per kg) protected group (ip), (A3) 0.1 ppm ozone exposure group, (A4) 0.5 ppm ozone exposure group, (A5) 1.0 ppm ozone exposure group, (A6) 1 ppm ozone exposure and VE group. Black arrow: bronchial remodeling. *: p < 0.05; **: p < 0.01, compared with the control group; ##: p < 0.01, compared with 1.0 ppm O3 exposure group. | ||
Effects of ozone exposure on AHR in mice and the ameliorating effects of VE
Ozone treatment followed by a methacholine (MCH) challenge assay produced an increase in the R-areas of the respiratory resistances (Re and Ri, respectively) and a decrease in dynamic lung compliance (Cldyn) (Fig. 7A–C), which supported the validity of this mouse model. Continuous upward shifts of the Ri and Re curves and downward shifts of the Cldyn curves were detected as the O3 concentration increased from 0.1 to 1.0 ppm, suggesting that O3 (especially 1.0 ppm O3) adversely affected both large and small airways of the lung. Treatment with VE dramatically reduced Ri and Re values, and increased Cldyn in the 1.0 ppm O3 group.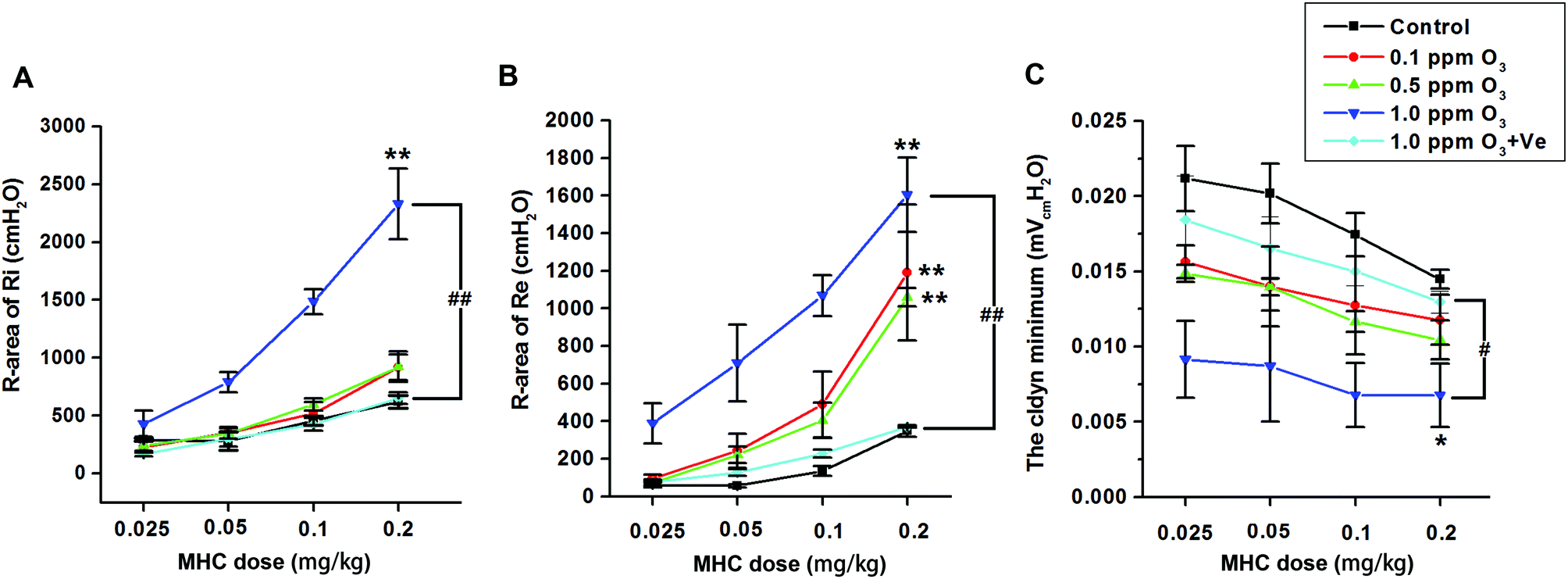 | ||
| Fig. 7 Effects of ozone treatment on AHR in mice. (A–C), Ri, Re, and Cldyn values, respectively, of control, 0.1 ppm O3, 0.5 ppm O3, 1.0 ppm O3 groups, 1.0 ppm O3 + VE groups; *: p < 0.05; **: p < 0.01, compared with the control group; #: p < 0.05, ##: p < 0.01, compared with 1.0 ppm O3 exposure group. | ||
Vitamin E up-regulating the expression of Nrf2 and antioxidant genes HO-1 and NQO1
To investigate the protective mechanism of VE in ozone induced airway inflammation, the expression of Nrf2 and the antioxidant genes HO-1 and NQO1 were detected using immunohistochemistry (Fig. 8). It was observed that the expression of the transcription factor Nrf2 was enhanced in the 1.0 ppm O3 group when VE was applied (Fig. 8A and D). The up-regulation of antioxidant genes HO-1 (Fig. 8B and E) and NQO1 (Fig. 8C and F) is shown, by comparing the 1.0 ppm O3 + VE group with the 1.0 ppm O3 group.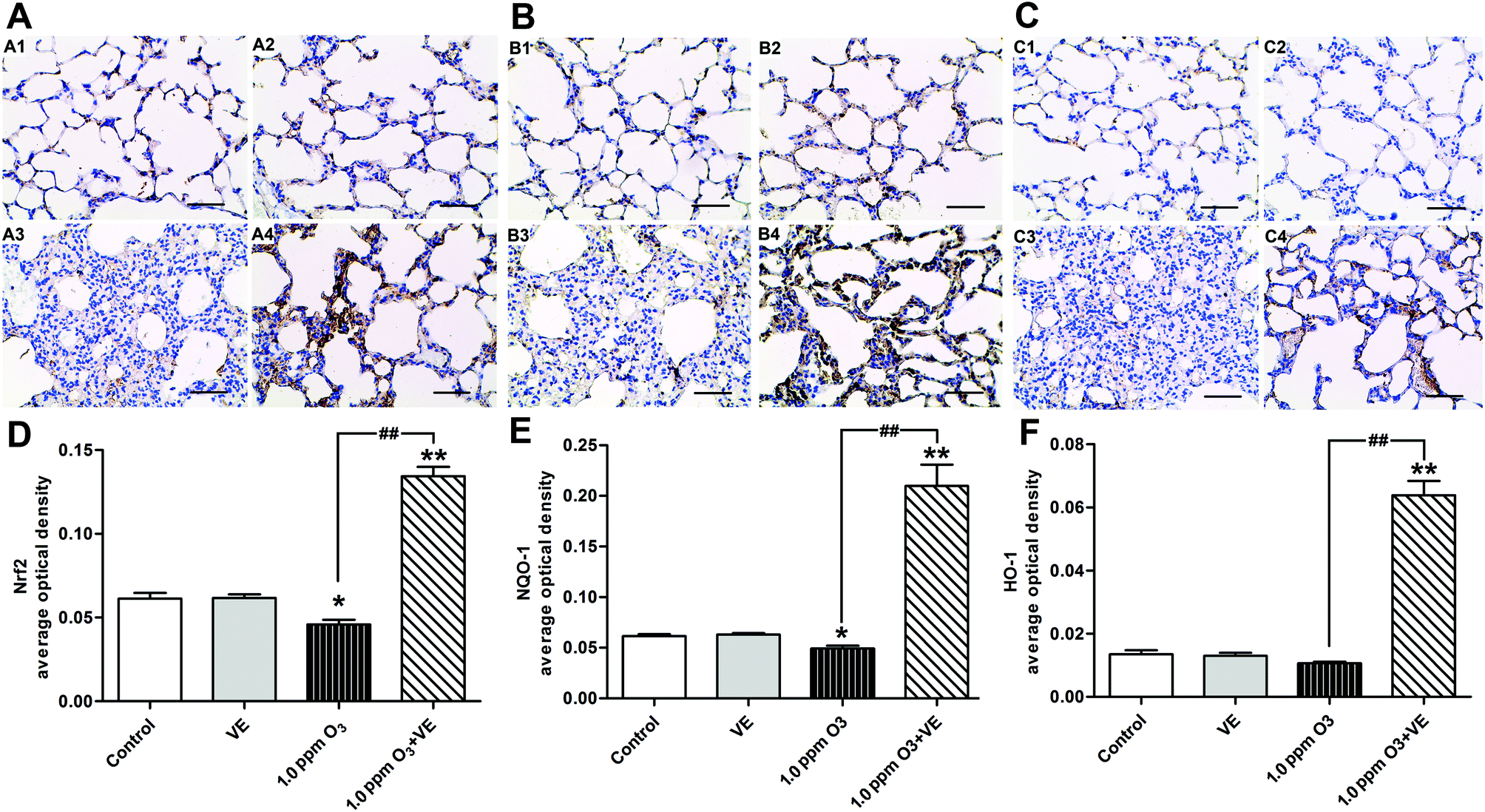 | ||
| Fig. 8 Vitamin E up-regulating the expression of Nrf2 and antioxidant genes NQO1 and HO-1. (A–C), Immunohistochemistry with Nrf2, NQO1 and HO-1 in lung tissue, respectively. Panel: (A1, B1 and C1) Control group, (A2, B2 and C2) VE (100 mg VE per kg) protected group (ip), (A3, B3 and C3) 1.0 ppm ozone exposure group, (A4, B4 and C4) 1.0 ppm ozone exposure and VE group. Scale bars = 50 μm. (D–F), The optical density of immunohistochemistry for Nrf2, NQO1 and HO-1, respectively. * p < 0.05, ** p < 0.01, compared with control group; ## p < 0.01, compared 1.0 ppm O3 group with 1.0 ppm O3 + VE group. | ||
Discussion
This study shows that high doses of ozone (>0.5 ppm) can induce inflammation and injury in the lungs of mice. This lung inflammation was concomitant with production of IgE and increased Th2 cytokines such as IL-4, IL-5 and IL-13. Also, a histopathological examination and an AHR test showed that there was lung damage in mice after high doses of ozone exposure. Furthermore, combining treatment with VE, we demonstrated that oxidative stress is an important mechanism behind ozone-induced lung injury, and activating the Nrf2 pathway is probably a potential protective mechanism of VE (Fig. 9).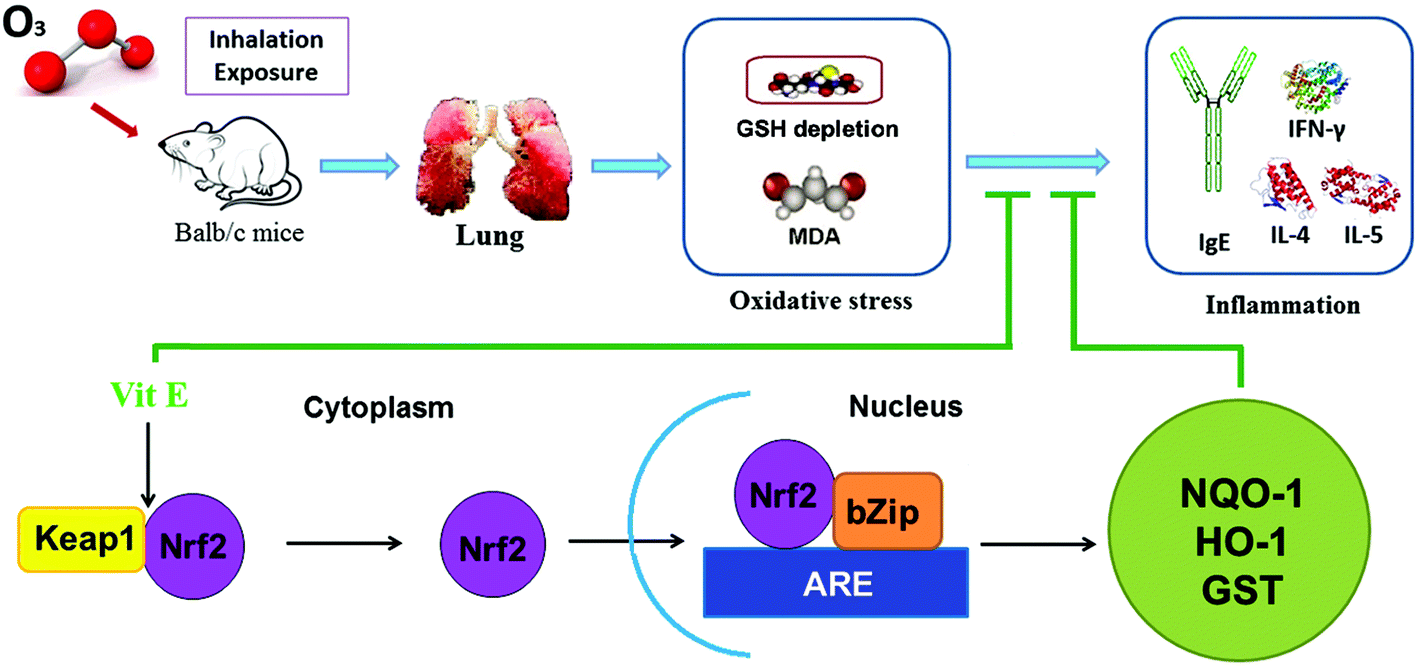 | ||
| Fig. 9 The mechanisms of ozone-induced lung injury and protective effect of VE. | ||
With increasing ozone exposure levels being experienced, the potential toxicity of this chemical is receiving increasing attention. In this study, we have investigated the ozone exposure mouse model in detail. In previous studies, mice were largely exposed to extremely high doses of ozone (usually 2.0 ppm for 3 h),18–21 which were beyond our normal environmental exposure levels. In contrast to other experiments, our study exposed the mice to three different ozone concentrations (0.1 ppm, 0.5 ppm and 1.0 ppm) for 7 days based on the actual environmental levels so as to simulate real environmental exposure.
Numerous studies have shown the connection between ozone exposure and lung inflammation.21–24 However, the precise mechanisms involved in ozone-induced inflammation remain elusive. We measured the levels of IFN-γ, IL-4, IL-5 and IL-13 in lung tissue and IgE in serum to show the levels of inflammation in this mouse model after ozone exposure. IFN-γ is a key biomarker for Th1, and IL-4 IL-5 and IL-13 represent the Th2 immune response and up-regulates IgE production to fight extracellular organisms.25,26 In this study, the O3 exposure groups displayed a decrease in IFN-γ (p < 0.01) and an increase in IL-4, IL-5 and IL-13 levels in comparison with the control group (Fig. 3B, C and 4, p < 0.01). These data suggest that the Th1/Th2 balance was broken and that the Th2 response had a dominant role in the high dose ozone (>0.5 ppm) exposure mice.
It is worth mentioning that airway remodeling and AHR, the consequence of airway inflammation, are well-established features of lung injury.27,28 H&E staining can be used to examine the pathological features of airway inflammation and structural alteration, including leucocyte infiltration in the surrounding peribronchiolar areas, epithelial folding and thickened subepithelial cell layers. In this study, airway remodeling was clearly observed in the 0.5 and 1 ppm ozone exposure groups. Mouse AHR was evaluated by measuring inspiratory resistance (Ri), expiratory resistance (Re) and dynamic lung compliance (Cldyn) using methacholine (MCH) challenge tests. The R-areas of respiratory resistances (Ri and Re) and a change in Cldyn represent variation of the large airways and the state of the small airways, respectively.29 Upward shifts of Ri (Fig. 7A) and Re (Fig. 7B) curves and downward shifts of Cldyn curves (Fig. 7C) were seen as the ozone concentration increased from 0.1 to 1.0 ppm (especially in 1 ppm), suggesting that high doses of ozone adversely affects the large and small airways of the lung.
Oxidative stress is the result of an imbalance in the production of antioxidants and free radicals. It is thought to play an important part in the pathogenesis of various types of lung inflammation.30 Ozone is a highly reactive oxidant that induces lung injury and impairs pulmonary function.1 ROS are generated by oxidative stress and have an important role in the redox-dependent regulation of signal transduction processes. However, high-levels of ROS are associated with damage to a wide variety of cellular constituents and can induce apoptosis or necrosis, which may ultimately result in pathological changes and lead to organ dysfunction or cancers.31,32 One primary result of increasing ROS is lipid peroxidation. Malondialdehyde (MDA) is a metabolite of the lipid peroxidation of membranes and usually signifies ROS damage to lipids.
Ozone can induce toxicity via a ROS-dependent pathway.33 Therefore, we hypothesized that ozone could generate oxidative stress to induce inflammation and damage to lung tissue and consequently exacerbate AHR. To verify this hypothesis, we first measured the levels of GSH and MDA in the lung after ozone exposure. Exposure to 1.0 ppm ozone led to the depletion of GSH and increases in the level of MDA indicating that oxidative stress had occurred (Fig. 4). We then administered VE, an antioxidative reagent, to relieve ozone-caused oxidative stress in mice in the 1.0 ppm ozone exposure group. Interestingly, the combined administration of VE with 1.0 ppm ozone not only caused an increase of GSH and reduction in the levels of MDA, but also antagonized inflammation effects and lung injury compared with the 1.0 ppm ozone only exposure group. Based on these results, we speculated that the adverse effects of ozone on inflammation and damage to the lung could account (at least in part) for the increased oxidative stress.
TSLP is a novel IL-7 like cytokine, which can activate dendritic cells (DCs) to induce Th2 inflammatory responses.34 When TSLP-DCs are used to stimulate naive allogeneic CD4+ T cells in vitro, they induce a unique type of Th2 cell that produces the classical Th2 cytokines IL-4, IL-5, and IL-13 and large amounts of TNF, but little or no IL-10.35 This study demonstrated that the growth trend of TSLP was coincident with the IL-4 and IL-5 cytokine data, which suggested that the activation of TSLP promoted the polarization of Th2 cells. And with TSLP increasing, the IFN-γ decreased sharply, which demonstrated that the activation of TSLP inhibited the polarization of Th1 cells. Previous studies showed that ROS orchestrated Th2 responses by inducing oxidized lipids that triggered the induction of TSLP by epithelial cells.12 In this study, the level of TSLP leading to Th2 immune responses and lung injury was shown to be counteracted by the concurrent administration of vitamin E. These findings are important as they suggest a potential role of TSLP signaling in ozone-induced oxidative stress and inflammatory responses to lung tissue injury. Meanwhile, other ways of measuring TSLP expression (i.e. western blot and PCR) and the in vivo model (i.e. TSLP knock-out mouse) should be used for the sake of further determining the role of TSLP in ozone-induced lung tissue injury. Therefore, more studies are needed for understanding the molecular mechanism of ozone-induced pulmonary injury in order to effectively prevent related health problems in the future.
The transcription factor Nrf2 is a powerful redox sensor to oxidative stress and an essential element in the regulation of many antioxidant genes.36,37 Here we propose that VE could activate the Nrf2 pathway after ozone exposure. This promoted the up-regulation of antioxidant genes NQO1 and HO-1, which inhibited the accumulation of ROS. These results demonstrate why VE plays an antioxidant role and attenuates the adverse effects of ozone. The results in the present study agree well with those reported in an investigation that Nrf2 expression in human asthmatics can be rescued by vitamin E in vivo.38 In this experiment we suppose that the expression of Nrf2 is not increased after VE alone treatment due to the redox balance in vivo. Once the redox balance is broken by environmental pollutants, such as ozone, administration of VE can exhibit antioxidation by activating the Nrf2 pathway.
Conclusion
By detecting oxidative stress and using vitamin E, we demonstrated that oxidative stress might be a mechanism of ozone-induced inflammation and lung injury, and activating the Nrf2 pathway is probably a potential protective mechanism. This result could help provide effective prevention strategies against ozone induced injury of the respiratory system.Acknowledgements
This work was funded by the National Natural Science Foundation of China (81302402) and the National Natural Science Foundation of China-Key Program (51136002).References
- N. Uysal and R. M. Schapira, Effects of ozone on lung function and lung diseases, Curr. Opin. Pulm. Med., 2003, 9(2), 144–150 CrossRef CAS PubMed.
- W. Wei, Z. Lv, S. Cheng, L. Wang, D. Ji and Y. Zhou, et al., Characterizing ozone pollution in a petrochemical industrial area in Beijing, China: a case study using a chemical reaction model, Environ. Monit. Assess., 2015, 187(6), 4620 CrossRef PubMed.
- I. S. Mudway and F. J. Kelly, An investigation of inhaled ozone dose and the magnitude of airway inflammation in healthy adults, Am. J. Respir. Crit. Care Med., 2004, 169(10), 1089–1095 CrossRef PubMed.
- A. S. Williams, S. Y. Leung, P. Nath, N. M. Khorasani, P. Bhavsar and R. Issa, et al., Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia, J. Appl. Physiol., 2007, 103(4), 1189–1195 CrossRef CAS PubMed.
- M. L. Bell, F. Dominici and J. M. Samet, A meta-analysis of time-series studies of ozone and mortality with comparison to the national morbidity, mortality, and air pollution study, Epidemiology, 2005, 16(4), 436–445 CrossRef PubMed.
- A. Gryparis, B. Forsberg, K. Katsouyanni, A. Analitis, G. Touloumi and J. Schwartz, et al., Acute effects of ozone on mortality from the “air pollution and health: a European approach” project, Am. J. Respir. Crit. Care Med., 2004, 170(10), 1080–1087 CrossRef PubMed.
- B. J. Hubbell, A. Hallberg, D. R. McCubbin and E. Post, Health-related benefits of attaining the 8-hr ozone standard, Environ. Health Perspect., 2005, 113(1), 73–82 CrossRef CAS PubMed.
- D. L. Laskin, V. R. Sunil, C. R. Gardner and J. D. Laskin, Macrophages and tissue injury: agents of defense or destruction?, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 267–288 CrossRef CAS PubMed.
- I. Rahman, A. A. van Schadewijk, A. J. Crowther, P. S. Hiemstra, J. Stolk and W. MacNee, et al., 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease, Am. J. Respir. Crit. Care Med., 2002, 166(4), 490–495 CrossRef PubMed.
- M. Al-Hegelan, R. M. Tighe, C. Castillo and J. W. Hollingsworth, Ambient ozone and pulmonary innate immunity, Immunol. Res., 2011, 49(1–3), 173–191 CrossRef CAS PubMed.
- R. He and R. S. Geha, Thymic stromal lymphopoietin, Ann. N. Y. Acad. Sci., 2010, 1183, 13–24 CrossRef CAS PubMed.
- H. Tang, W. Cao, S. P. Kasturi, R. Ravindran, H. I. Nakaya and K. Kundu, et al., The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling, Nat. Immunol., 2010, 11(7), 608–617 CrossRef CAS PubMed.
- J. Kim, Y. N. Cha and Y. J. Surh, A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders, Mutat. Res., 2010, 690(1–2), 12–23 CrossRef CAS PubMed.
- X. L. Chen and C. Kunsch, Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases, Curr. Pharm. Des., 2004, 10(8), 879–891 CrossRef CAS PubMed.
- G. Devereux, S. W. Turner, L. C. Craig, G. McNeill, S. Martindale and P. J. Harbour, et al., Low maternal vitamin E intake during pregnancy is associated with asthma in 5-year-old children, Am. J. Respir. Crit. Care Med., 2006, 174(5), 499–507 CrossRef CAS PubMed.
- J. Li, L. Li, H. Chen, Q. Chang, X. Liu and Y. Wu, et al., Application of vitamin E to antagonize SWCNTs-induced exacerbation of allergic asthma, Sci. Rep., 2014, 4, 4275 Search PubMed.
- J. Guo, B. Han, L. Qin, B. Li, H. You and J. Yang, et al., Pulmonary toxicity and adjuvant effect of di-(2-exylhexyl) phthalate in ovalbumin-immunized BALB/c mice, PLoS One, 2012, 7(6), e39008 CAS.
- C. H. Wiegman, F. Li, C.J Clarke, E. Jazrawi, P. Kirkham and P. J. Barnes, et al., A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model, Clin. Sci., 2014, 126(6), 425–440 CrossRef CAS PubMed.
- S. Hulo, H. Tiesset, S. Lancel, J. L. Edme, B. Viollet and A. Sobaszek, et al., AMP-activated protein kinase deficiency reduces ozone-induced lung injury and oxidative stress in mice, Respir. Res., 2011, 12, 64 CrossRef CAS PubMed.
- J. Bornholdt, M. Dybdahl, U. Vogel, M. Hansen, S. Loft and H. Wallin, Inhalation of ozone induces DNA strand breaks and inflammation in mice, Mutat. Res., 2002, 520(1–2), 63–71 CAS.
- A. S. Williams, P. Nath, S. Y. Leung, N. Khorasani, A. N. McKenzie and I. M. Adcock, et al., Modulation of ozone-induced airway hyperresponsiveness and inflammation by interleukin-13, Eur. Respir. J., 2008, 32(3), 571–578 CrossRef CAS PubMed.
- A. J. Connor, J. D. Laskin and D. L. Laskin, Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4, Exp. Mol. Pathol., 2012, 92(2), 229–235 CrossRef CAS PubMed.
- M. Arjomandi, H. Wong, A. Donde, J. Frelinger, S. Dalton and W. Ching, et al., Exposure to medium and high ambient levels of ozone causes adverse systemic inflammatory and cardiac autonomic effects, Am. J. Physiol., 2015, 308(12), H1499–H1509 CrossRef CAS PubMed.
- P. Zhang, F. Li, C. H. Wiegman, M. Zhang, Y. Hong and J. Gong, et al., Inhibitory effect of hydrogen sulfide on ozone-induced airway inflammation, oxidative stress, and bronchial hyperresponsiveness, Am. J. Respir. Cell Mol. Biol., 2015, 52(1), 129–137 CrossRef PubMed.
- J. F. Regal, Immunologic effector mechanisms in animal models of occupational asthma, J. Immunotoxicol., 2004, 1(1), 25–37 CrossRef CAS PubMed.
- H. J. Park, C. M. Lee, I. D. Jung, J. S. Lee, Y. I. Jeong and J. H. Chang, et al., Quercetin regulates Th1/Th2 balance in a murine model of asthma, Int. Immunopharmacol., 2009, 9(3), 261–267 CrossRef CAS PubMed.
- D. W. Cockcroft and B. E. Davis, Mechanisms of airway hyperresponsiveness, J. Allergy Clin. Immunol., 2006, 118(3), 551–559 CrossRef CAS PubMed ; quiz 60-1.
- G. Lezmi, P. Gosset, A. Deschildre, R. Abou-Taam, B. Mahut and N. Beydon, et al., Airway Remodeling in Preschool Children with Severe Recurrent Wheeze, Am. J. Respir. Crit. Care Med., 2015, 192(2), 164–171 CrossRef PubMed.
- Y. Qiao, B. Li, G. Yang, H. Yao, J. Yang and D. Liu, et al., Irritant and adjuvant effects of gaseous formaldehyde on the ovalbumin-induced hyperresponsiveness and inflammation in a rat model, Inhalation Toxicol., 2009, 21(14), 1200–1207 CrossRef CAS PubMed.
- T. Finkel, Signal transduction by reactive oxygen species, J. Cell Biol., 2011, 194(1), 7–15 CrossRef CAS PubMed.
- J. Fang, T. Seki and H. Maeda, Therapeutic strategies by modulating oxygen stress in cancer and inflammation, Adv. Drug Delivery Rev., 2009, 61(4), 290–302 CrossRef CAS PubMed.
- L. Khandrika, B. Kumar, S. Koul, P. Maroni and H. K. Koul, Oxidative stress in prostate cancer, Cancer Lett., 2009, 282(2), 125–136 CrossRef CAS PubMed.
- V. R. Sunil, K. Patel-Vayas, J. Shen, J. D. Laskin and D. L. Laskin, Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress, Toxicol. Appl. Pharmacol., 2012, 263(2), 195–202 CrossRef CAS PubMed.
- J. E. Sims, D. E. Williams, P. J. Morrissey, K. Garka, D. Foxworthe and V. Price, et al., Molecular cloning and biological characterization of a novel murine lymphoid growth factor, J. Exp. Med., 2000, 192(5), 671–680 CrossRef CAS PubMed.
- V. Soumelis, P. A. Reche, H. Kanzler, W. Yuan, G. Edward and B. Homey, et al., Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP, Nat. Immunol., 2002, 3(7), 673–680 CAS.
- R. Hu, C.L Saw, R. Yu and A. N. Kong, Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory, Antioxid. Redox Signaling, 2010, 13(11), 1679–1698 CrossRef CAS PubMed.
- M. Kobayashi and M. Yamamoto, Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation, Antioxid. Redox Signaling, 2005, 7(3–4), 385–394 CrossRef CAS PubMed.
- R. Dworski, W. Han, T. S. Blackwell, A. Hoskins and M. L. Freeman, Vitamin E prevents NRF2 suppression by allergens in asthmatic alveolar macrophages in vivo, Free Radical Biol. Med., 2011, 51(2), 516–521 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |