
Screening and validation for plasma biomarkers of nephrotoxicity based on metabolomics in male rats†
Yubo
Li
a,
Haoyue
Deng
a,
Liang
Ju
a,
Xiuxiu
Zhang
a,
Zhenzhu
Zhang
a,
Zhen
Yang
a,
Lei
Wang
a,
Zhiguo
Hou
a and
Yanjun
Zhang
*b
aTianjin State Key Laboratory of Modern Chinese Medicine, School of Traditional Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, 312 Anshan west Road, Tianjin 300193, China
bTianjin State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, 312 Anshan west Road, Tianjin 300193, China. E-mail: tianjin_tcm001@sina.com; Fax: +86-22-59596221; Tel: +86-22-59596221
First published on 5th November 2015
Abstract
Currently, drug-induced nephrotoxicity is widespread and seriously affects human health. However, the conventional indexes of renal function lack sensitivity, leading to a delay in the detection of nephrotoxicity. Therefore, we need to identify more sensitive indexes for evaluating nephrotoxicity. In this study, we used gentamicin (100 mg kg−1), etimicin (100 mg kg−1) and amphotericin B (4 mg kg−1) to establish renal injury models in rats, and we collected information using ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry in the screening stage. Thirteen nephrotoxicity metabolites were selected after multivariate statistical and integration analyses. Then, we conducted trend analysis to select 5 nephrotoxicity biomarkers [thymidine, LysoPC(16:1), LysoPC(18:4), LysoPC(20:5), and LysoPC(22:5)] whose content changed consistently at different timepoints after drug administration. To verify the sensitivity and specificity of these biomarkers for nephrotoxicity, receiver operating characteristic (ROC) and support vector machine (SVM) analyses were applied. The area under the curve of the 5 biomarkers were 0.806–0.901 at the 95% confidence interval according to the ROC analysis. We used the SVM classified model to verify these biomarkers, and the prediction rate was 95.83%. Therefore, the 5 biomarkers have strong sensitivity and high accuracy; these biomarkers are more sensitive indexes for evaluating renal function to identify nephrotoxicity and initiate prompt treatment.
1. Introduction
Currently, safety problems associated with clinical drugs are hindering their promotion.1 The kidney is easily damaged by drug-induced toxicity because it is the main excretory organ of the body. Thus, drug-induced nephrotoxicity is widespread.2,3 In recent years, serum creatinine (Scr) and blood urea nitrogen (BUN) have been commonly used as indexes for evaluating renal function. However, they are limited in their ability to detect nephrotoxicity because of their lack of sufficient sensitivity.4,5 Therefore, it is necessary to develop a sensitive and efficient method for evaluating nephrotoxicity.Metabolomics, which is an important part of systems biology, is used to investigate changes in endogenous substances when the biological system is affected by external disturbances.6–8 With research developments, metabolomics technology has been used extensively to evaluate drug toxicity. Particularly, it has promoted the study of drug-induced nephrotoxicity to gain new insights into the associated pathophysiological mechanisms.9–11 Plasma metabolomics is broadly used in human health care and drug safety evaluations because it provides a large amount of information on endogenous substances.12,13 Given its high sensitivity, extensive dynamic range and good separation ability, ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) has become one of the most versatile techniques, and is being gradually applied to various fields such as metabolomics, proteomics and traditional Chinese medicine. UPLC-MS-based metabolomics has great potential for identifying useful biomarkers for disease diagnosis (such as hepatocarcinoma and liver cirrhosis, lung cancer and pneumonia, and Alzheimer's disease and schizophrenia, etc.) and drug-induced toxicity assessment (such as cardiotoxicity, hepatotoxicity, nephrotoxicity, etc.).14–17 Metabolomics biomarkers can reveal the metabolic differences in the physiological and pathological states of organisms in a dynamic and sensitive manner.18–20
The support vector machine (SVM) is an intelligent pattern recognition technology that has been extensively used in different fields.21–23 It effectively solves the binary classification problem because it generates the optimal linear interface of two categories of substances.24 SVM provides a new direction in metabolomics and genomics data processing because of its robustness, and deals well with high-dimensional data and small sample sizes.25,26 Therefore, we utilized SVM to predict and classify the related biomarkers by feature selection and classification prediction.
In this study, we used receiver operating characteristic (ROC) and SVM to analyse plasma metabolomics data to identify biomarkers for evaluating nephrotoxicity. We used gentamicin, etimicin and amphotericin B to establish rat models of renal injury. Information on the plasma samples was collected using an UPLC quadrupole time-of-flight MS (UPLC-Q-TOF/MS) platform. After the multivariate statistical analysis, integration analysis and content analysis, we obtained nephrotoxicity biomarkers whose content changed consistently at different timepoints after drug administration. Next, we used ROC to evaluate the sensitivity and specificity of the nephrotoxicity biomarkers. Then, we predicted nephrotoxicity using these biomarkers after combining with cardiotoxicity and hepatotoxicity data by SVM. The method can provide a systematic tool for screening and validating other toxic biomarkers using metabolomics and can promote the development of metabolomics.
2. Materials and methods
2.1 Chemicals and reagents
Acetonitrile [high-pressure liquid chromatography (HPLC) grade] was purchased from Oceanpak (Gothenburg, Sweden). Formic acid (HPLC grade) was purchased from ROE (USA). Purified water was purchased from Wahaha Company (Hangzhou, China). Normal saline (NS), five nephrotoxic drugs [gentamicin (GM), etimicin (ETI), amphotericin B (AMB), thioacetamide (TAA) and cisplatin (DDP)], two cardiotoxic drugs [cyclophosphamide (CP) and 5-fluorouracil (5FU)], one hepatotoxic chemical [carbon tetrachloride (CCl4)] and one hepatotoxic drug [tetracycline (TC)] were purchased from Queensland Technology Co., Ltd (Tianjin, China) and dissolved in saline.27–362.2 Animal experiment
We purchased male Wistar rats (6 weeks old, weighing 200 ± 20 g) from Sibei Fu (Beijing) Experimental Animals Technology Co., Ltd, under license number “SCXK (Jing) 2011-0004”. The experiment was conducted at the Institute of Radiation Medicine Chinese Academy of Medical Sciences (Tianjin, China). The rats were housed in an SPF-level laboratory, and the temperature was 25 ± 1 °C. Before the experiment, the rats had free access to chow and water during the one week acclimatization period (the rats were 7 weeks old at the beginning of the experiment). This study was approved by the Animal Ethics Committee of Tianjin University of Traditional Chinese Medicine under permit number TCM-2012-078-F01. All of the experimental procedures were conducted in accordance with Chinese national legislation and local guidelines.105 rats were divided into ten groups to identify nephrotoxicity biomarkers: the NS, GM-1d, GM-3d, GM-7d, ETI-1d, ETI-2d, ETI-3d, AMB-1d, AMB-3d and AMB-7d groups. 70 rats were divided into seven groups to verify the nephrotoxicity biomarkers: the NS group, two nephrotoxicity groups (TAA and DDP), two cardiotoxicity groups (CP and 5FU), and two hepatotoxicity groups (CCl4 and TC). The groups, doses, administration routes and sampling times are shown in Table 1.36,45
| Drug | Grouping | Number | Dose | Mode of administration | Sampling time | |
|---|---|---|---|---|---|---|
| a The screening stage for nephrotoxicity biomarkers. b The validation stage for nephrotoxicity biomarkers. c Intraperitoneal injection. d Intragastric administration. e Subcutaneous injection. | ||||||
| Stage Ia | NS | NS | 15 | 5 ml kg−1 | i.p.c, single-dose | 1 day |
| GM | GM-1d | 10 | 100 mg kg−1 | i.p.c, single-dose | 1 day | |
| GM-3d | 10 | 100 mg kg−1 | i.p.c, successive administration | 3 days | ||
| GM-7d | 10 | 100 mg kg−1 | i.p.c, successive administration | 7 days | ||
| ETI | ETI-1d | 10 | 100 mg kg−1 | i.p.c, single-dose | 1 day | |
| ETI-2d | 10 | 100 mg kg−1 | i.p.c, successive administration | 2 days | ||
| ETI-3d | 10 | 100 mg kg−1 | i.p.c, successive administration | 3 days | ||
| AMB | AMB-1d | 10 | 4 mg kg−1 | i.p.c, single-dose | 1 day | |
| AMB-3d | 10 | 4 mg kg−1 | i.p.c, successive administration | 3 days | ||
| AMB-7d | 10 | 4 mg kg−1 | i.p.c, successive administration | 7 days | ||
| Stage IIb | NS | NS | 10 | 5 ml kg−1 | i.p.c, single-dose | 1 day |
| TAA | TAA | 10 | 200 mg kg−1 | i.p.c, successive administration | 6 days | |
| DDP | DDP | 10 | 6 mg kg−1 | i.p.c, successive administration | 3 days | |
| CP | CP | 10 | 200 mg kg−1 | i.p.c,successive administration | 5 days | |
| 5FU | 5FU | 10 | 125 mg kg−1 | i.g.d, single-dose | 1 day | |
| CCl4 | CCl4 | 10 | 5 mL kg−1 | i.s.e, successive administration | 2 days | |
| TC | TC | 10 | 1500 mg kg−1 | i.g.d, successive administration | 5 days | |
2.3 Sample collection
Before sample collection, all rats were fasted for twelve hours with access to water to prevent the effect of food on our final results. Blood was collected from the aorta abdominalis of each rat. Plasma was placed in a tube that had been washed with heparin sodium solution, and serum was placed in a normal tube. Then, all the rats were sacrificed, and their organs were immediately removed and stored in 10% formalin solution for pathological analysis by haematoxylin and eosin (H&E) staining. Serum and plasma were separated by centrifugation at 3500 rpm for 15 min at 4 °C. Plasma was stored at −80 °C until the metabolomics analysis. Serum was used to detect the biochemical markers.For H&E staining, the fixed tissues were embedded in paraffin wax. Then, 5 μm thick slices were cut and fixed on glass slides. The slices were deparaffinized with xylene, hydrated, stained with haematoxylin, differentiated with hydrochloric alcohol, stained with eosin and dehydrated in a graded alcohol series. Then, the slides were cleaned with xylene, and the histopathological changes were observed by light microscopy at 100× magnification.36,37
2.4 Sample pretreatment
The plasma was thawed at room temperature before processing. Then, 300 μL of acetonitrile was added to 100 μL of plasma. The mixture was vortexed for 1 min, ultrasonicated in cold water for 10 min and centrifuged at 4 °C at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. Then, the supernatant was collected for UPLC-Q-TOF/MS analysis.
000 rpm for 15 min. Then, the supernatant was collected for UPLC-Q-TOF/MS analysis.
2.5 Data acquisition
We used an UPLC-Q-TOF/MS system (Waters, USA) to acquire the metabolomics data. A 5 μL aliquot of the supernatant was injected into the ACQUITY UPLC HSS C18 column (2.1 × 100 mm, 1.7 μm; Waters, USA). The column temperature was set at 40 °C, and the flow rate was 0.3 mL min−1. The UPLC separation system included a binary solvent system with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The gradient started with 99% A followed by 0 to 0.5 min, A: 99% to 99%; 0.5 to 2 min, A: 99% to 50%; 2 to 9 min, A: 50% to 1%; 9 to 10 min, A: 1% to 1%; 10 to 10.5 min, A: 1% to 99%; and 10.5 to 12 min, A: 99% to 99%. Q-TOF/MS was performed using electrospray ionisation in positive mode. The MS parameters were as follows: drying gas flow, 10 mL min−1; auxiliary ionisation and desolvation gas, high-purity N2; desolvation temperature, 325 °C; desolvation gas flow, 600 L h−1; atomisation air pressure, 350 psi; ionisation capillary voltage, 3.5 kV; range of data acquisition, 50–1000 Da. A reference ion ([M + H]+ = 556.2771) was used to ensure accuracy during the spectral acquisition. We used quality control (QC) samples to evaluate the reliability of the data in the analysis process; the QC samples were mixed with the same amount of plasma from each sample.38 Each QC sample was detected six times to evaluate the instrument precision. Next, 6 QC samples were used to determine the method repeatability. QC samples were detected every 5 hours to test sample stability over 24 hours. Before injecting the samples, the entire system was determined to be under stable conditions.2.6 Data analysis
Information on all the plasma samples was from the UPLC-Q-TOF/MS platform. The raw data was exported using MarkerLynx V4.1 (Waters Corporation, Manchester, USA) with peak discovery, peak alignment, and filtering to determine potential discriminating variables.In the screening stage, the data was processed by multivariate statistical analysis using SIMCA-P+11.5 software (Umetrics, Sweden). In our study, principal component analysis (PCA) was used to identify the outliers in the samples, and partial least squares-discriminate analysis (PLS-DA) was used to distinguish the variables with a high contribution between the NS group and the drug treatment group at different times. The model was visualized with a score plot. The variables with a variable-importance plot (VIP) greater than 1 (VIP > 1) at different administration times were analysed using Student's t-test in SPSS 17.0 (SPSS, USA), and the variables with p < 0.05 represented potential nephrotoxicity metabolites. The potential nephrotoxicity metabolites from the three drugs at different times were processed by integration analysis to identify nephrotoxicity metabolites using Venn diagrams (http://bioinfogp.cnb.csic.es/tools/venny/index.html). The heat map was generated using Cluster software based on the relative content of each nephrotoxicity metabolite in each treatment group. Next, the change in content of nephrotoxicity metabolites at different timepoints was analysed to identify those metabolites whose content changed consistently. These metabolites were identified by MS/MS information and confirmed with HMDB (http://www.hmdb.ca/) and KEGG (http://www.genome.jp/kegg/) as nephrotoxicity biomarkers. The ROC curves of nephrotoxicity biomarkers based on the nephrotoxic drug groups were determined using the binary logistic regression model in SPSS 17.0 (SPSS, USA).
Then, we combined the data of nephrotoxic drug groups with non-nephrotoxicity (cardiotoxicity and hepatotoxicity) data to validate the nephrotoxicity biomarkers using an SVM model in MATLAB R2010a (MathWorks, USA). The peak areas of the nephrotoxicity biomarkers were the input variables, and the training set was used to build an SVM classification model with the optimal penalty parameter (c) and kernel function (g). The factor c is used to determine the characteristics of subspace-regulated learning, and g is a function for mapping from low-dimensional space to high-dimensional space.36,39 We obtained the accuracy rate of the model using the test set. Cross-validation was used to determine the confidence and experience risk ratio ranges of the model.36,40
3. Results
3.1 Histopathological examination and serum biochemical detection
We used histopathological examination to evaluate the extent of drug-induced kidney damage.41,42 The histopathological examination of the kidney in the screening stage is shown in Fig. 1. The kidneys from animals in the drug-treated groups (GM-1d, GM-3d, GM-7d, ETI-1d, ETI-2d, ETI-3d, AMB-1d, AMB-3d, and AMB-7d) were injured compared with those from animals in the NS group. The kidneys had infiltrating inflammatory cells with varying degrees of interstitial fibrosis. Additionally, the renal tubule, collecting tubule and renal pelvis showed atrophy or dilation; in some cases, the collecting tubule generated microcysts. The histopathological examination of the validation stage is shown in Fig. S1;† the organs were injured in response to drug toxicity. | ||
| Fig. 1 Histopathological examination of the kidney by H&E staining (100× magnification). | ||
Kidney damage is indicated by increased Scr and BUN contents. In our study, the levels of Scr and BUN in the drug-treated groups were compared with those in the NS group by Student's t-test (Fig. 2). The Scr and BUN levels were significantly increased (p < 0.05) only in the GM-7d, ETI-3d, and AMB-7d groups compared with the NS group. In the other groups, the two indexes were not increased significantly at the same timepoints. However, the content of the two indexes showed a temporal correlation.
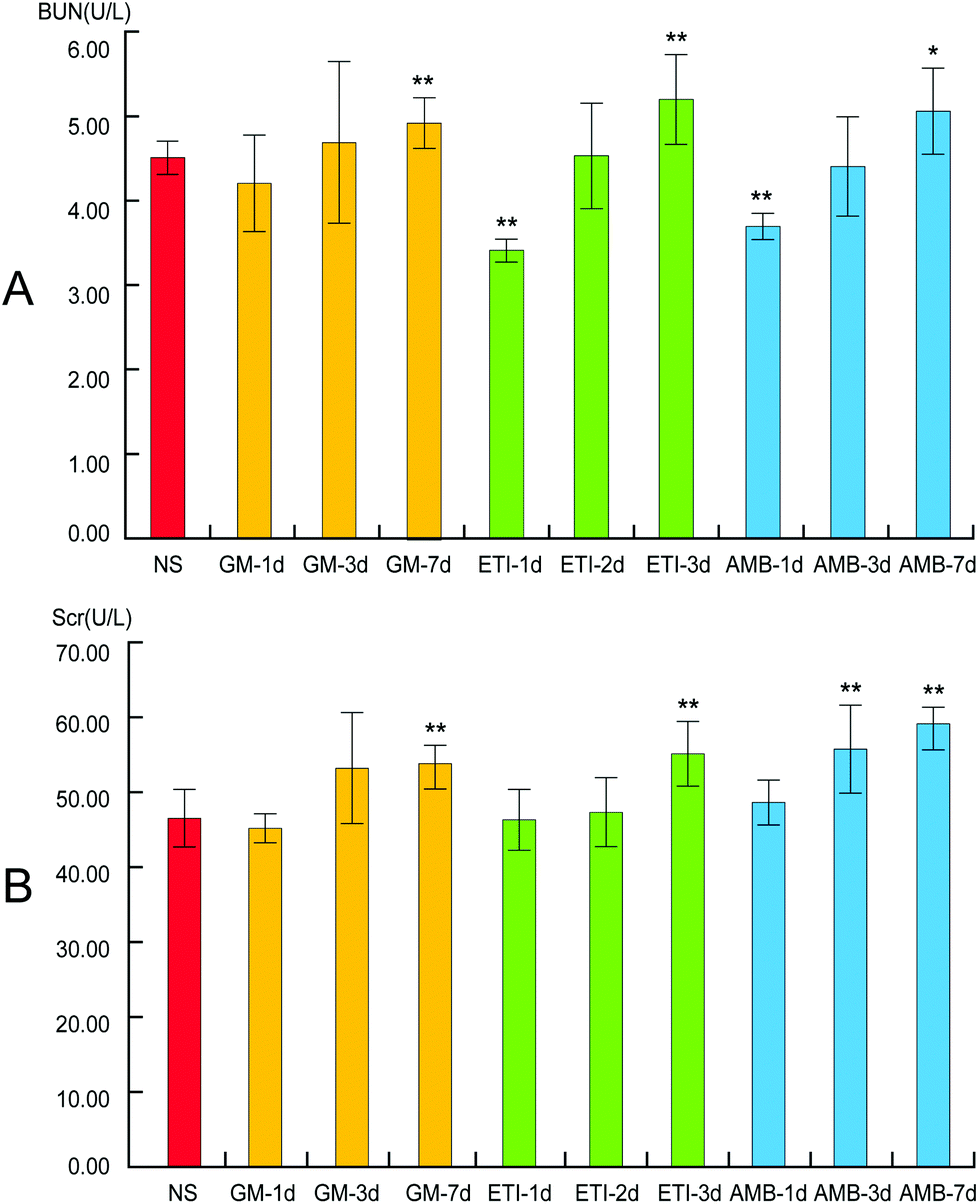 | ||
| Fig. 2 BUN and Scr levels in serum samples. (A) Changes in BUN levels. (B) Changes in Scr levels. Data are presented as the mean ± SD (*p < 0.05, **p < 0.01, compared with the NS group). | ||
3.2 Nephrotoxicity biomarker screening
The BPI chromatograms of the QC samples in positive ion mode UPLC-Q-TOF/MS are shown in Fig. S2.† The experimental results (instrument precision, method repeatability and sample stability) showed that the relative standard deviations of the peak areas and retention times of the twenty selected peaks were less than 15% (Table S1†), which indicated that the instruments and samples were stable and that the methods were reliable.We obtained the PCA and PLS-DA score plots using multivariate statistical analysis (Fig. S3 and Table S2†). Some stray samples were removed according to the PCA. We selected variables with VIP > 1 based on the PLS-DA for analysis by Student's t-test. The variables with p < 0.05 represented potential metabolites associated with each drug at different times. Then, they were processed by integration analysis to identify potential nephrotoxicity-associated metabolites of each drug (Fig. S4†). Finally, we obtained 13 nephrotoxicity metabolites, and the Venn diagram is shown in Fig. 3. The heat map of the relative content of each nephrotoxicity-associated metabolite is shown in Fig. 4. We retained 5 biomarkers whose content changed consistently in the GM, ETI and AMB groups at different times (Fig. 5). These biomarkers were identified by mass spectrometry (Fig. S5†). Detailed information regarding the 5 nephrotoxicity biomarkers is provided in Table 2.
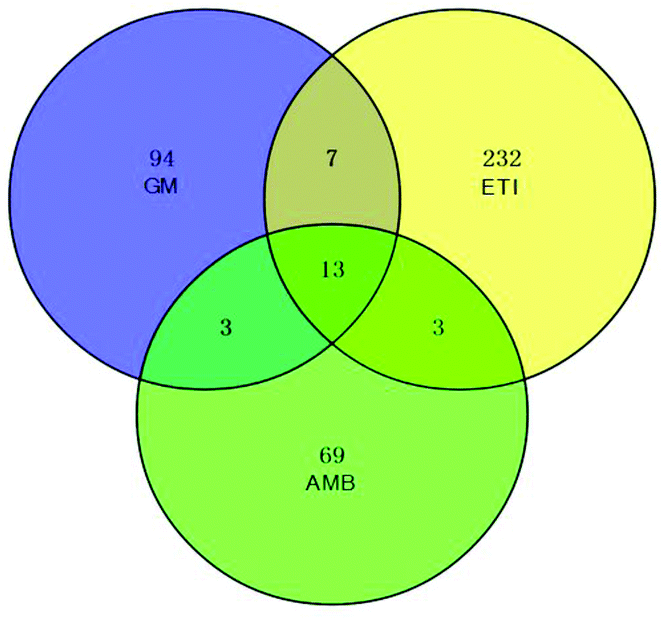 | ||
| Fig. 3 Venn diagram of the potential metabolites associated with each nephrotoxic drug (GM: 117; ETI: 255; AMB: 88) by integration analysis. Thirteen nephrotoxicity metabolites were initially obtained. | ||
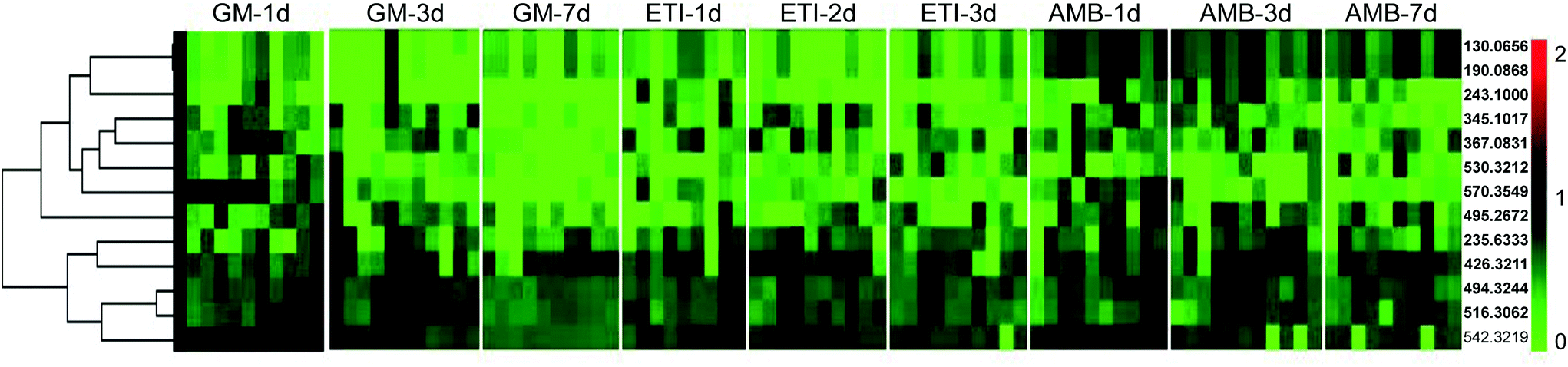 | ||
| Fig. 4 Heat map of the relative content of each nephrotoxicity-associated metabolite in all the drug-treated groups. | ||
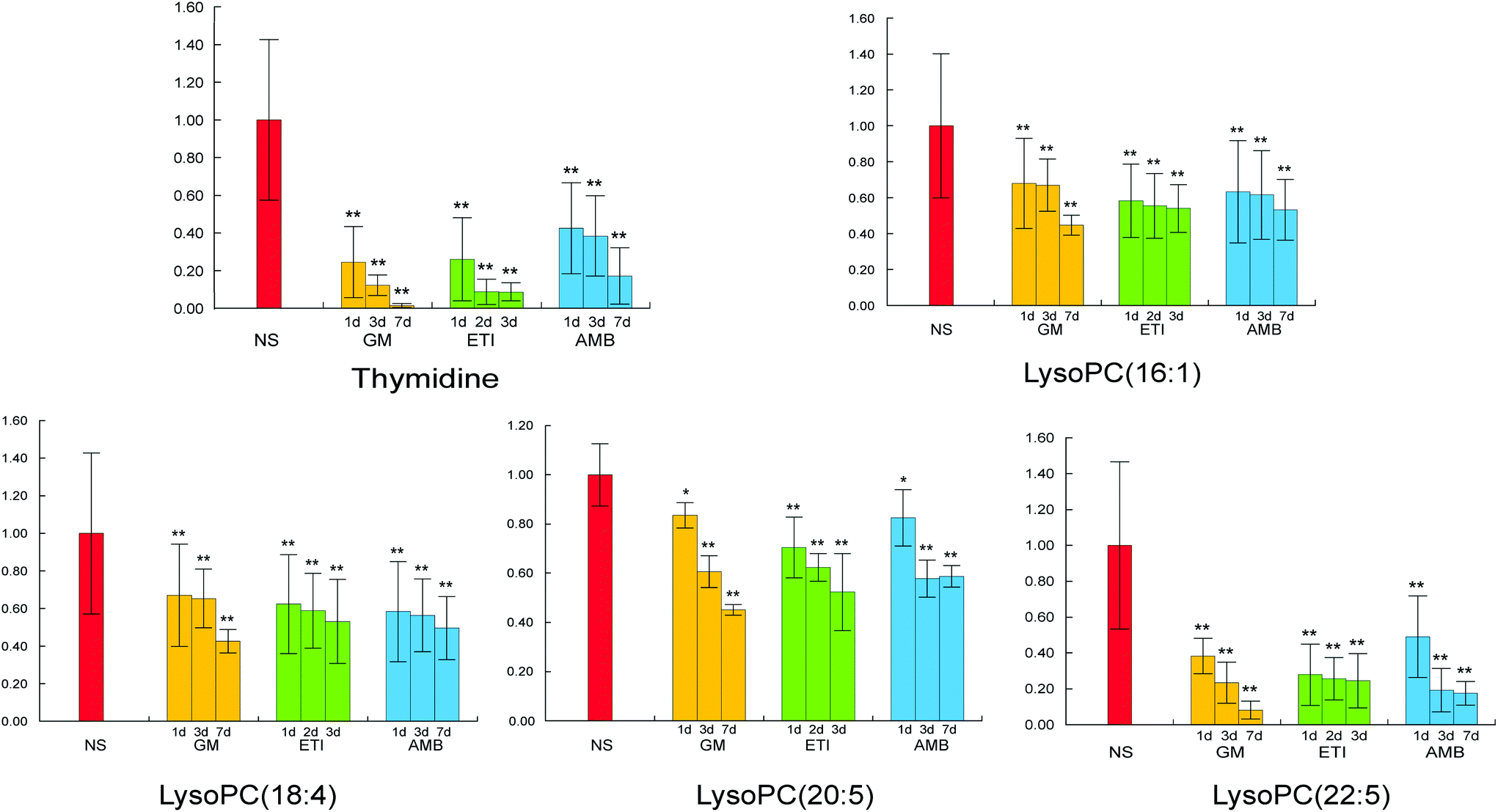 | ||
| Fig. 5 Content change of the 5 nephrotoxicity biomarkers whose content changed consistently at different timepoints after drug administration and at different administration times in the GM, ETI and AMB groups (*p < 0.05, **p < 0.01, compared with the NS group). | ||
| No. | t R (min) | Metabolites | Obsd [M + H]+ | Calcd [M + H]+ | Error (ppm) | Formula | MS/MS |
|---|---|---|---|---|---|---|---|
| 1 | 2.51 | Thymidine | 243.0987 | 243.0975 | 4.80 | C10H14N2O5 | 243.1 [M + H]+ |
| 127.1 [M + H − C5H8O3]+ | |||||||
| 2 | 5.16 | LysoPC(16:1) | 494.3244 | 494.3241 | 0.61 | C24H48NO7P | 494.3 [M + H]+ |
| 476.3 [M + H − H2O]+ | |||||||
| 184.0 [M + H − C19H34O3]+ | |||||||
| 125.0 [M + H − C22H43NO3]+ | |||||||
| 104.1 [M + H − C19H35O6P]+ | |||||||
| 3 | 5.15 | LysoPC(18:4) | 516.3062 | 516.3085 | −4.45 | C26H46NO7P | 516.3 [M + H]+ |
| 498.3 [M + H − H2O]+ | |||||||
| 184.0 [M + H − C21H32O3]+ | |||||||
| 104.1 [M + H − C21H33O6P]+ | |||||||
| 4 | 5.55 | LysoPC(20:5) | 542.3219 | 542.3241 | −4.06 | C28H48NO7P | 542.3 [M + H]+ |
| 524.3 [M + H − H2O]+ | |||||||
| 259.1 [M + H − C17H33NO2]+ | |||||||
| 184.0 [M + H − C23H34O3]+ | |||||||
| 125.0 [M + H − C26H43NO3]+ | |||||||
| 104.1 [M + H − C23H35O6P]+ | |||||||
| 5 | 6.13 | LysoPC(22:5) | 570.3549 | 570.3554 | −0.88 | C30H52NO7P | 570.4 [M + H]+ |
| 552.3 [M + H − H2O]+ | |||||||
| 184.0 [M + H − C25H38O3]+ | |||||||
| 125.0 [M + H–C28H47NO3]+ | |||||||
| 104.1 [M + H–C25H38O6P]+ |
We used ROC to evaluate the diagnostic potential of the 5 biomarkers for nephrotoxicity in the screening stage. The ROC analysis showed that the 5 nephrotoxicity biomarkers had a high accuracy for evaluating nephrotoxicity based on the area under the curve, and the sensitivity and specificity at the best cutoff points (Table 3 and Fig. 6).
 | ||
| Fig. 6 ROC curves of the 5 nephrotoxicity biomarkers [thymidine, LysoPC(16:1), LysoPC(18:4), LysoPC(20:5) and LysoPC(22:5)] at the screening stage. | ||
| Biomarkers | AUCa | Sensitivity (%) | Specificity (%) | Standard errorb | 95% CI | |
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| a The area under the curve. b Under the nonparametric assumption. | ||||||
| Thymidine | 0.901 | 0.956 | 0.714 | 0.044 | 0.815 | 0.988 |
| LysoPC(16:1) | 0.817 | 0.813 | 0.786 | 0.069 | 0.682 | 0.952 |
| LysoPC(18:4) | 0.806 | 0.846 | 0.714 | 0.064 | 0.680 | 0.933 |
| LysoPC(20:5) | 0.886 | 0.791 | 0.857 | 0.033 | 0.821 | 0.951 |
| LysoPC(22:5) | 0.830 | 0.758 | 0.786 | 0.062 | 0.710 | 0.951 |
3.3 Nephrotoxicity biomarker validation
Currently, drug-induced toxicity (nephrotoxicity, cardiotoxicity and hepatotoxicity) is widespread. When a biological system is damaged by different toxins, similar metabolic pathways, such as inflammation, may be affected.43 Thus, we verified the specificity of the 5 biomarkers by combining the data with cardiotoxicity and hepatotoxicity samples using an SVM classified model. Two-thirds of the samples in the validation stage were assigned as the training set to build the model, and the other one-third of the samples formed the test set to determine the accuracy rate of the model in the SVM classification process.36 The accuracy rate was 95.83%. The SVM model parameters in the cross-validation method are shown in Fig. 7. The data showed that the model based on the 5 nephrotoxicity biomarkers has a good predictive ability in other relevant samples.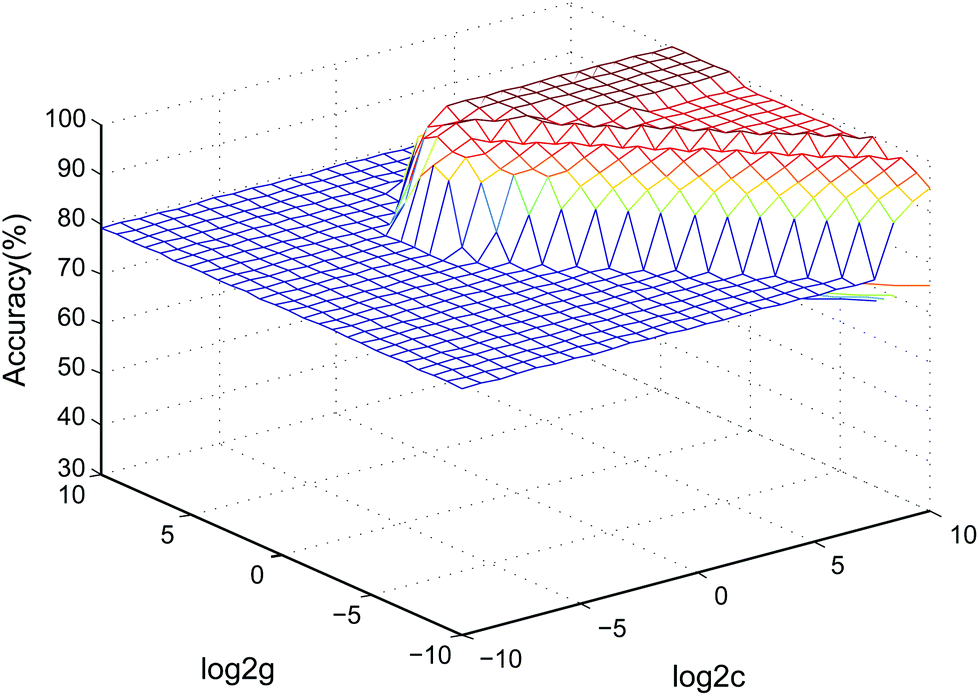 | ||
| Fig. 7 3D view of the SVM classified model of the 5 nephrotoxicity biomarkers (parameters: best c = 0.76, best g = 6.96, CV accuracy = 100%). | ||
4. Discussion
The reported studies about nephrotoxicity metabolomics biomarkers involve a variety of endogenous small molecules, compared with the combination of nephrotoxicity biomarkers [thymidine, LysoPC(16:1), LysoPC(18:4), LysoPC(20:5), and LysoPC(22:5)] that we obtained, which relate to more intricate metabolic processes in vivo and could not explain the relationship between biomarkers and nephrotoxicity on the metabolic level.44–46 However, the nephrotoxicity biomarkers we obtained can explain the mechanism of nephrotoxicity to some degree. It has been reported that the change in plasma LysoPC levels is related to drug-induced nephrotoxicity.44,45 LysoPCs belong to the class of phosphatidylcholines (PCs). PCs and LysoPCs participate in the glycerophospholipid metabolism pathway based on the KEGG database. On the one hand, PCs can produce LysoPCs by lecithin-cholesterol acyltransferase and secretory phospholipase A2.47 On the other hand, LysoPCs can produce PCs through lysophosphatidylcholine acyltransferase. In recent years, it has been reported that the mechanism of drug-induced nephrotoxicity is related to oxidative stress.48 At this time, the yield of reactive oxygen species (ROS) and reactive nitrogen species (RNS) were all excessive, and the oxidation and antioxidant systems were imbalanced, resulting in kidney tissue damage. PCs can remove peroxide in vivo. Therefore, when the kidney is damaged, PCs are required to eliminate ROS and RNS. Thus, the glycerophospholipid metabolism pathway is affected, resulting in decreased LysoPCs production, which corresponds to our experimental results. Another nephrotoxicity biomarker that we obtained is thymidine, which participates in the pyrimidine metabolism pathway. When the kidney is damaged, the content of thymidine undergoes significant changes, affecting the protein kinase C signaling pathway, thereby affecting the expression of phospholipase A2. PCs can produce LysoPCs by phospholipase A2 in the glycerophospholipid metabolism pathway, which can explain the relationship between LysoPCs and nephrotoxicity.To a certain extent, biochemical indicators reflect organ damage. However, their sensitivity and specificity are poor because they are often affected by other factors. However, histopathological analysis can reveal organ damage directly. When Scr and BUN levels are significantly increased, it is likely that the kidney has been injured by the pathological condition. Hence, we used histopathological examinations to evaluate the extent of kidney damage. From the serum biochemistry results, we ascertained that the kidneys were injured only in the GM-7d, ETI-3d, and AMB-7d groups. However, the histopathological examination revealed that the kidneys were injured in all the drug-treated groups. This finding indicates that the existing methods do not detect nephrotoxicity with adequate accuracy or sensitivity. Compared with serum biochemistry, nephrotoxicity biomarkers underwent significant changes at different timepoints after drug administration, which reveals sensitive metabolic differences in organisms. Additionally, these biomarkers can help explain the biological mechanism of drug-induced nephrotoxicity.
Currently, some studies have used single nephrotoxic drug to identify nephrotoxicity biomarkers by metabolomics. Similar metabolic processes in the body may be affected by different toxic drugs.43 Therefore, the biomarkers in the reported studies were not exclusive to nephrotoxicity. Additionally, the application of these biomarkers has not been promoted.44,45 In our study, we established nephrotoxicity models based on three nephrotoxic drugs at three different administration times to identify nephrotoxicity biomarkers. To exclude the impact of other forms of toxicity, we combined the nephrotoxicity with other toxic drugs (cardiotoxicity and hepatotoxicity) to validate our biomarkers using an SVM model. The SVM model was used to verify the accuracy of the biomarkers in predicting nephrotoxicity. The results of the SVM model showed that the best combination of biomarkers [thymidine, LysoPC(16:1), LysoPC(18:4), LysoPC(20:5), and LysoPC(22:5)] had higher sensitivity and accuracy compared with the reported studies.44,45 Therefore, this combination has potential for broad application in drug safety evaluations and drug development as well as in the clinical evaluation and prediction of drug-induced nephrotoxicity.
In our study, we established a comprehensive and systematic method for identifying nephrotoxicity biomarkers. This represents a new tool for discovering and verifying biomarkers in other areas related to metabolomics, such as drug-induced toxicity, clinical diagnostics and plant metabolomics. Furthermore, it is conducive to the development of metabolomics. To obtain specific and exclusive nephrotoxicity biomarkers, we controlled the experimental animals (male Wistar rats, 6 weeks old, weighing 200 ± 20 g) to investigate the differences in metabolism in response to different toxic drugs in this study. Considering the universal applicability of our nephrotoxicity biomarkers, we should combine them with the factors such as gender and age for verification purposes in future studies.
5. Conclusions
In this study, we performed a metabolomics study on drug-induced nephrotoxicity to identify biomarkers based on UPLC-Q-TOF/MS analysis. We initially identified 5 nephrotoxicity biomarkers whose content changed consistently at different timepoints after drug administration. Then, we used ROC and SVM to evaluate and verify the diagnostic potential of the 5 biomarkers for nephrotoxicity in different contexts. The ROC results showed that these biomarkers have a high sensitivity for nephrotoxicity. In the SVM model, the accuracy rate of these biomarkers was 95.83%, and they were specific for nephrotoxicity. Nephrotoxicity biomarkers can effectively compensate for insufficient biochemical indexes. Our study could promote the establishment of a systematic drug-induced toxicity evaluation based on metabolomics.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This project was supported by the National Basic Research Program of China (973 Program) (2011CB505300 and 2011CB505302) and the National Natural Science Foundation of China (No. 81573825). This project also was supported by the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT14R41).References
- X. Ruan, Pain Physician, 2007, 10, 357–365 Search PubMed.
- M. A. Perazella, Kidney Int., 2012, 81, 1172–1178 CrossRef CAS PubMed.
- Y. Li, Z. Y. Oo, S. Y. Chang, P. Huang, K. G. Eng, J. L. Zeng, A. J. Kaestli, B. Gopalan, K. Kandasamy, F. Tasnim and D. Zink, Toxicol. Res., 2013, 2, 352–365 RSC.
- K. B. Kim, S. Y. Um, M. W. Chung, S. C. Jung, J. S. Oh, S. H. Kim, H. S. Na, B. M. Lee and K. H. Choi, Toxicol. Appl. Pharmacol., 2010, 249, 114–126 CrossRef CAS PubMed.
- W. K. Han, V. Bailly, R. Abichandani, R. Thadhani and J. V. Bonventre, Kidney Int., 2002, 62, 237–244 CrossRef CAS.
- X. Wang, B. Yang, H. Sun and A. Zhang, Anal. Chem., 2012, 84, 428–439 CrossRef CAS.
- X. Liu, Y. Liu, Y. Qu, M. Cheng and H. Xiao, Toxicol. Res., 2015, 4, 948–955 RSC.
- Q. Huang, J. Zhang, L. Luo, X. Wang, X. Wang, A. Alamdar, S. Peng, L. Liu, M. Tian and H. Shen, Toxicol. Res., 2015, 4, 939–947 RSC.
- Y. Y. Zhao and R. C. Lint, Adv. Clin. Chem., 2014, 65, 69–89 Search PubMed.
- C. U. Niemann and N. J. Serkova, Expert Opin. Drug Metab. Toxicol., 2007, 3, 527–544 CrossRef CAS PubMed.
- Y. Y. Zhao, Clin. Chim. Acta, 2013, 422, 59–69 CrossRef CAS PubMed.
- W. Mattesa, K. Davisb, E. Fabiand, J. Greenhawc, M. Herolde, R. Loosere, W. Mellertd, S. Groetersd, H. Marxfeldd, N. Moellerf, G. Montoya-Parrad, A. Prokoudinee, B. van Ravenzwaayd, V. Straussd, T. Walke and H. Kampd, Toxicol. Lett., 2014, 230, 467–478 CrossRef PubMed.
- M. Hubalek, A. Oberguggenberger, B. Beer, V. Meraner, M. Sztankay, H. Oberacher, B. Schubert, L. Wildt, B. Seeber, J. Giesinger, G. Kemmler, B. Holzner and B. Sperner-Unterweger, Clin. Breast Cancer, 2014, 14, 291–296 CrossRef CAS PubMed.
- D. Dudzik, R. Revello, C. Barbas and J. L. Bartha, J. Proteome Res., 2015, 14, 1432–1444 CrossRef CAS PubMed.
- Y. Y. Zhao, X. L. Cheng, N. D. Vaziri, S. Liu and R. C. Lin, Clin. Biochem., 2014, 47, 16–26 CrossRef CAS PubMed.
- X. Wang, H. Sun, A. Zhang, P. Wang and Y. Han, J. Sep. Sci., 2011, 34, 3451–3459 CrossRef CAS PubMed.
- Y. Y. Zhao and R. C. Lin, Chem. – Biol. Interact., 2014, 215, 7–16 CrossRef CAS PubMed.
- C. Denkert, J. Budczies, T. Kind, W. Weichert, P. Tablack, J. Sehouli, S. Niesporek, D. Könsgen, M. Dietel and O. Fiehn, Cancer Res., 2006, 66, 10795–10804 CrossRef CAS.
- X. Zhao, F. Xu, B. Qi, S. Hao, Y. Li, Y. Li, L. Zou, C. Lu, G. Xu and L. Hou, J. Proteome Res., 2014, 13, 1101–1111 CrossRef CAS PubMed.
- C. M. Slupsky, H. Steed, T. H. Wells, K. Dabbs, A. Schepansky, V. Capstick, W. Faught and M. B. Sawyer, Clin. Cancer Res., 2010, 16, 5835–5841 CrossRef CAS PubMed.
- Y. H. Liu and Y. T. Chen, IEEE Trans. Neural Networks, 2007, 18, 178–192 CrossRef.
- K. Manivannan, P. Aggarwal, V. Devabhaktuni, A. Kumar, D. Nims and P. Bhattacharya, J. Hazard. Mater., 2012, 223–224, 94–103 CrossRef CAS PubMed.
- B. A. Johnston, B. Mwangi, K. Matthews, D. Coghill, K. Konrad and J. D. Steele, Hum. Brain Mapp., 2014, 35, 5179–5189 CrossRef PubMed.
- J. C. Alves and R. J. Poppi, Talanta, 2014, 104, 155–161 CrossRef PubMed.
- M. Hilario and A. Kalousis, Briefings Bioinf., 2008, 9, 102–118 CrossRef PubMed.
- J. X. Dong, A. Krzyzak and C. Y. Suen, IEEE Trans Pattern Anal. Mach. Intell., 2005, 27, 603–618 CrossRef PubMed.
- E. van Maarseveen, M. C. van Buul-Gast, R. Abdoellakhan, L. Gelinck, C. Neef and D. Touw, J. Antimicrob. Chemother., 2014, 69, 2581–2583 CrossRef CAS PubMed.
- B. D. Sahu, S. Tatireddy, M. Koneru, R. M. Borkar, J. M. Kumar, M. Kuncha, R. Srinivas, R. Shyam Sunder and R. Sistla, Toxicol. Appl. Pharmacol., 2014, 277, 8–20 CrossRef CAS PubMed.
- D. R. Falci, F. B. da Rosa and A. C. Pasqualotto, Mycoses, 2015, 58, 104–112 CrossRef CAS PubMed.
- N. J. Waters, C. J. Waterfield, R. D. Farrant, E. Holmes and J. K. Nicholson, Chem. Res. Toxicol., 2005, 18, 639–654 CrossRef CAS PubMed.
- Y. C. Ning, G. Y. Cai, L. Zhuo, J. J. Gao, D. Dong, S. Y. Cui, S. Z. Shi, Z. Feng, L. Zhang, X. F. Sun and X. M. Chen, Nephron Exp. Nephrol., 2013, 124, 19–27 CrossRef CAS PubMed.
- D. J. Conklin, P. Haberzettl, G. Jagatheesan, S. Baba, M. L. Merchant, R. A. Prough, J. D. Williams, S. D. Prabhu and A. Bhatnagar, Toxicol. Appl. Pharmacol., 2015, 285, 136–148 CrossRef CAS PubMed.
- C. Lestuzzi, E. Vaccher, R. Talamini, A. Lleshi, N. Meneguzzo, E. Viel, S. Scalone, L. Tartuferi, A. Buonadonna, L. Ejiofor and H. J. Schmoll, Ann. Oncol., 2014, 25, 1059–1064 CrossRef CAS PubMed.
- L. W. Weber, M. Boll and A. Stampfl, Crit. Rev. Toxicol., 2003, 33, 105–136 CrossRef CAS PubMed.
- M. Yamazaki, M. Miyake, H. Sato, N. Masutomi, N. Tsutsui, K. P. Adam, D. C. Alexander, K. A. Lawton, M. V. Milburn, J. A. Ryals, J. E. Wulff and L. Guo, Toxicol. Appl. Pharmacol., 2013, 268, 79–89 CrossRef CAS.
- Y. Li, L. Ju, Z. Hou, H. Deng, Z. Zhang, L. Wang, Z. Yang, J. Yin and Y. Zhang, J. Proteome Res., 2015, 14, 2437–2445 CrossRef CAS PubMed.
- Y. Li, X. Zhang, H. Zhou, S. Fan, Y. Wang, L. Wang, Z. Zhang, H. Deng and Y. Zhang, Anal. Methods, 2014, 6, 5909–5917 RSC.
- N. L. Kuehnbaum, J. B. Gillen, A. Kormendi, K. P. Lam, A. DiBattista, M. J. Gibala and P. Britz-McKibbin, Electrophoresis, 2015, 36, 2226–2236 CrossRef CAS PubMed.
- H. Jiang, D. Zhao, R. Zheng and X. Ma, Biomed Res. Int., 2015 DOI:10.1155/2015/781023.
- J. Luts, F. Ojeda, R. Van de Plas, B. De Moor, S. Van Huffel and J. A. Suykens, Anal. Chim. Acta, 2010, 665, 129–145 CrossRef CAS PubMed.
- M. Wagner, L. Maggiori, M. Ronot, V. Paradis, V. Vilgrain, Y. Panis and B. E. Van Beers, Eur. Radiol., 2013, 23, 2156–2164 CrossRef PubMed.
- A. Di Ieva, E. Bruner, G. Widhalm, G. Minchev, M. Tschabitscher and F. Grizzi, Sci. Rep., 2012, 2, 1–10 Search PubMed.
- B. N. M. Zordoky, A. Anwar-Mohamed, M. E. Aboutabl and A. O. S. El-Kadi, Drug Metab. Dispos., 2011, 39, 1440–1450 CrossRef CAS.
- X. Zhang, Y. Li, H. Zhou, S. Fan, Z. Zhang, L. Wang and Y. Zhang, J. Pharm. Biomed. Anal., 2014, 97, 151–156 CrossRef CAS PubMed.
- Y. Li, X. Zhang, H. Zhou, S. Fan, Y. Wang, L. Zhang, L. Ju, X. Wu, H. Wu and Y. Zhang, RSC Adv., 2014, 4, 8260–8270 RSC.
- K. J. Boudonck, M. W. Mitchell, L. Német, L. Keresztes, A. Nyska, D. Shinar and M. Rosenstock, Toxicol. Pathol., 2009, 37, 280–292 CrossRef CAS PubMed.
- U. P. Steinbrecher and P. H. Pritchard, J. Lipid Res., 1989, 30, 305–315 CAS.
- F. Yang, L. Ren, L. Zhuo, S. Ananda and L. Liu, Exp. Toxicol. Pathol., 2012, 64, 905–911 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Histopathological examination results (Fig. S1). BPI chromatograms of QC samples in the positive ion mode of UPLC-Q-TOF/MS (Fig. S2). PCA and PLS-DA score plots (Fig. S3). Potential metabolites of each drug at different times were processed by integration analysis using Venn diagrams (Fig. S4). The substance identification with mass spectrometry information (Fig. S5). The results of methodology experiments (Table S1). The parameters of PLS-DA score plots (Table S2). See DOI: 10.1039/c5tx00171d |
| This journal is © The Royal Society of Chemistry 2016 |