
Effects of probucol on cell proliferation in human ovarian cancer cells†
Lea-Yea
Chuang‡
a,
Jinn-Yuh
Guh‡
b,
Yi-Ling
Ye
c,
Ying-Ho
Lee
d and
Jau-Shyang
Huang
*d
aDepartment of Biochemistry, Kaohsiung Medical University, Kaohsiung, Taiwan
bDepartment of Internal Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
cDepartment of Biotechnology, National Formosa University, Yunlin, Taiwan
dDepartment of Biological Science and Technology, Chung Hwa University of Medical Technology, Tainan, Taiwan. E-mail: jaushyang12@hotmail.com; Fax: +886-6-2675047; Tel: +886-6-2674567-420
First published on 10th December 2015
Abstract
Probucol is considered to be an important agent in promoting anti-oxidative action and protecting against tissue injury. However, little is known about the effects of probucol on the progression of ovarian carcinoma. The aim of this study was to investigate the effects of probucol on cellular proliferation in human ovarian cancer cells (PA-1 and SKOV-3) and explore the anti-proliferative mechanism of probucol in these cells. We found that probucol decreased cell growth in PA-1 and SKOV-3 cells in a dose-dependent manner. Treatment with probucol had no effect on cytotoxicity, the percentages of Annexin V-FITC positive cells and caspase-3 activity when compared with the vehicle group. No significant differences in the protein expression of Bcl-2 and cytochrome c were observed, both of which were markers of cells undergoing apoptosis. The inhibition of cellular proliferation by probucol was caused by G1-phase arrest through regulating proteins associated with cell cycle progression, such as cyclin D1, p21Waf1/Cip1, and p27Kip1. A further study revealed that probucol strongly impaired the phosphorylation of IκBα and the nuclear translocation of NF-κB (p65). It also suppressed the activation of ERK/JNK/p38 MAPK signaling. Moreover, the NF-κB inhibitor (PDTC), the ERK inhibitor (PD98059), the JNK inhibitor (SP600125), and the p38 MAPK inhibitor (SB203580) markedly attenuated the growth of these cells. Our results indicate that probucol induces anti-proliferative effects via blocking of cell cycle progression and inactivation of NF-κB and MAPK pathways in human ovarian cancer cells.
Introduction
Probucol is a diphenolic compound with anti-hyperlipidemic, anti-oxidative, anti-diabetic, and anti-inflammatory properties that reduces tissue injury and histopathological changes.1–6 It has a long history of clinical application with established efficacy and safety profiles.2,3 Previous studies have demonstrated that probucol has diverse pharmacological properties with therapeutic effects on cardiovascular and metabolic diseases.4–8 It can also modulate the toxicity-promoting effect and can serve as a potent chemopreventive agent to suppress oxidant induced tissue injury.2,4,8 Therefore, probucol is supposed to be an excellent agent in enhancing endogenous antioxidant reserve and protecting against augmented oxidative stress.1–4Ovarian cancer is the deadliest of all gynecologic malignancies in many countries.9–11 Because this disease is nonspecific or asymptomatic at the early stage of its progression, the majority of ovarian carcinoma patients are diagnosed with advanced stage disease.10–12 For the therapy of ovarian carcinoma, cytoreductive surgery and combination chemotherapies are standard strategies.13–16 However, tumor relapse and the development of drug-resistant disease are still a knotty problem to be resolved in ovarian carcinoma treatment.
It is well documented that oxidative stress modulates cell growth or genomic stability under both physiological and pathophysiological conditions.17–19 Recent molecular and pathological evidence suggests that in progressive stages of ovarian carcinoma, the oxidative stress can contribute to the uncontrolled tumor expansion.18,19 Antioxidants, when added adjunctively, to first-line chemotherapy, may improve the efficacy of cancer therapy.19,20 More recent data showed that probucol was a potent antioxidant that can serve as a powerful chemopreventive agent to suppress oxidant induced tissue injury and carcinogenesis, in addition to being a cholesterol reducing and anti-atherogenic drug.21–23 Probucol exposure modulated iron nitrilotriacetate-dependent renal carcinogenesis and the hyperproliferative response.23 It can induce anti-angiogenesis and apoptosis in athymic nude mouse xenografted human head and neck squamous carcinoma cells.24 On the other hand, probucol was able to activate NAD(P)H:quinone reductase, one of the main detoxifying enzymes, and could then reduce chemical carcinogenesis and toxicity.25 The nanoassembly of probucol enabled novel therapeutic efficacy in the suppression of lung metastasis of breast cancer.26 Nevertheless, its potential effect on the progression of ovarian cancer has not been explored yet.
It has been reported that the nuclear factor-kappa B (NF-κB) plays an important role in the cellular redox system in various cells.27–29 In unstimulated cells, the NF-κB p50/p65 heterodimer is maintained in the cytoplasm by binding to IκBα. Upon stimulation, NF-κB dissociates from IκBα, translocates to the nucleus, activates target genes and regulates diverse cellular functions.28,29 Most of the effects of NF-κB activation on cancer cells have been linked with cancer development and poor outcomes.30,31 Cancer cells have been shown to exhibit a constitutively hyperactivated NF-κB survival signaling pathway.31 Indeed, the aberrant regulation of the NF-κB pathway is believed to be a major event contributing to malignant transformation and progression of ovarian cancer.32,33
In this study, we hypothesized that probucol, maybe, is an effective candidate for anti-ovarian cancer cells. Therefore, to examine this hypothesis, we investigated the inhibitory effects of probucol on cell proliferation in human ovarian cancer PA-1 and SKOV-3 cells and its underlying molecular mechanism for probucol-mediated cell cycle progression and NF-κB signaling. We also identified signaling molecules underlying probucol-modulated cell growth in the two cell lines. Finally, our study displayed the potential role of probucol in ovarian carcinoma chemotherapy or chemoprevention.
Materials and methods
Reagents
Anti-ERK1/2, -JNK, -p38 MAPK, -p21Waf1/Cip1, -p27Kip1, -Bcl-2, -c-Myc, -cytochrome c, -cdk4, -cyclin D1, -PCNA, -p65, and -IκBα antibodies were purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA. Anti-phospho-IκBα, -phospho-ERK1/2, -phospho-JNK, -phospho-p38 MAPK, and histone H3 antibodies were obtained from Abcam, Cambridge, MA. The HRP-conjugated goat anti-rabbit or anti-mouse secondary antibody, streptavidin-peroxidase, and the enhanced chemiluminescence kit were obtained from Amersham Corp, Arlington Heights, IL. The lactate dehydrogenase (LDH)-cytotoxicity assay kit was purchased from BioVision, Mountain View, CA. PD98059, SP600125, and SB203580 were purchased from Calbiochem, La Jolla, CA. N,N′-methylenebisacrylamide, acrylamide, SDS, ammonium persulfate, Temed, and Tween 20 were purchased from Bio-Rad Laboratories, Hercules, CA. FBS, DMEM, antibiotics, molecular weight standards, trypsin-EDTA, trypan blue dye, and all medium additives were obtained from Life Technologies, Gaithersburg, MD. Probucol, staurosporine, dimethyl sulfoxide (DMSO), pyrrolidine dithiocarbamate (PDTC), propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay kit, Annexin V-FITC apoptosis detection kit, caspase-3 activity assay kit, and anti-β-actin antibodies, and all other chemicals were purchased from Sigma-Aldrich Chemical, St. Louis, MO.Culture conditions
The human ovarian cancer cell lines (PA-1 and SKOV-3) were obtained from the American Type Culture Collection (Manassas, VA, USA). These cells were grown in culture flasks (Nunclon, Denmark) and maintained in DMEM supplemented with 100 i.u. per ml penicillin, 100 μg per ml streptomycin and 10% FBS in a humidified 5% CO2 incubator at 37 °C. In this study, cells were exposed to serum-free (0.1% FBS) DMEM supplemented with probucol or probucol's vehicle (0.05% DMSO) for 4 h prior to timed exposure to DMEM containing 10% FBS. For cell number analysis, cells (1.2 × 105 cells per well) were seeded in 6-well (9.6 cm2 per well) culture plates (Nunclon) and grown in the added test agents. The subconfluent cells were harvested by using 0.25% trypsin-EDTA. Cells were resuspended in equal volumes of the medium and trypan blue (0.05% solution) and counted using a haemocytometer. Trypan blue dye exclusion was used to assess the cell viability. Each experimental data point represents the mean of duplicate wells from three independent experiments.Cell viability assay
The cell viability (MTT) assay was performed to evaluate the proliferation of PA-1 and SKOV-3 cells. Cells were plated and incubated for 24 h in a 96-well plate. Then probucol (5, 10, 50, 100, 500 μM) and vehicle (0.05% DMSO) were added to the wells. Cells with no added treatment were used as control. After treatment, 10 μl of sterile MTT dye was added to each well, and the cells were incubated for 6 h at 37 °C. Then 100 μl of acidic isopropanol (0.04 M HCl in isopropanol) were added and thoroughly mixed. Spectrometric absorbance at 595 nm (for formazan dye) was measured with the absorbance at 655 nm for reference. According to the cell viability assay and cell number analysis, the lower and non-toxic concentration of probucol (100 μM) is used in subsequent experiments.Cytotoxicity assay
The LDH content was determined by using an LDH-cytotoxicity assay kit, which is based on a coupled enzymatic reaction that results in the conversion of a tetrazolium salt into a red formazan product. The increase in the amount of formazan produced in a culture supernatant directly correlates with the increase in the number of lysed cells. The formazan was quantified spectrophotometrically by measuring its absorbance at 490 nm. A group of wells were treated with 1% Triton X-100 solution for maximum LDH release. The mean of the background value was subtracted from all other values. Cytotoxicity in experimental samples was expressed as a percentage of the LDH release of 10% FBS-treated cells (control).Caspase-3 activity assay
Caspase-3 activity was measured using a colorimetric activity kit as per the manufacturer's instructions. This assay measures the hydrolysis of acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) by caspase-3 that results in the release of p-nitroanilide (pNA). Absorbances were measured at 405 nm and the results were calculated using a standard curve derived from known concentrations of pNA.Western blotting
One day before treatment, 1.5 × 106 cells were seeded in T-75 flasks. Cell growth medium was replaced 24 h after seeding, followed by the addition of test compounds after medium change. For protein analysis, total cell lysates were harvested and lysed in 1× SDS-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (0.375 M Tris-HCl, pH 6.8, 30% glycerol, 10% SDS, 2% β-mercaptoethanol, 0.1% bromophenol blue), and heated for 3 min at 95 °C. Samples were resolved by 10% SDS-PAGE, and then transferred to Protran membranes (0.45 μm, Schieicher & Schuell, Keene, NH, USA). The membranes were blocked with a blocking solution and subsequently probed with primary antibodies. The membrane was incubated in 4000× diluted HRP-conjugated goat anti-rabbit or anti-mouse secondary antibody. The protein bands were detected using the enhanced chemiluminescence (ECL) system. For the NF-κB and ERK/JNK/p38 MAPK activation assays, proteins were resolved by SDS-PAGE and transferred to Protran membranes. The membranes were probed with anti-phospho-IκBα (1 μg ml−1), anti-phospho-ERK1/2 (1 μg ml−1), anti-phospho-JNK (1 μg ml−1), anti-phospho-p38 MAPK (1 μg ml−1), anti-IκBα (1 μg ml−1), anti-ERK1/2 (0.75 μg ml−1), anti-JNK (1 μg ml−1), and anti-p38 MAPK (0.75 μg ml−1) antibodies. Immunoreactive proteins were detected with the ECL system as described above. The intensity of Western blot bands was quantified by densitometric analysis. Results were expressed as the ratio of the intensity of the protein of interest to that of β-actin or the indicated protein from the same sample.Assay of cytochrome c release
To obtain cytosolic fractions, cells were harvested and washed once in cold PBS, and then incubated with mitochondria isolation buffer (300 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 20 mM HEPES, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 1 μg per ml leupeptin, 10 μg per ml pepstatin and 10 μg per ml aprotinin) on ice for 30 min. Then, cells were disrupted by 20 passages through a 26-gauge needle. The disrupted cells were centrifuged at 750g for 10 min at 4 °C. The resulting supernatant was then twice centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C, and the supernatant was collected as the cytosolic fraction. For the assay of cytochrome c release, the cytosolic fraction was electrophoresed on 12% SDS-PAGE, and Western blot was then performed using an anti-cytochrome c antibody.
000g for 20 min at 4 °C, and the supernatant was collected as the cytosolic fraction. For the assay of cytochrome c release, the cytosolic fraction was electrophoresed on 12% SDS-PAGE, and Western blot was then performed using an anti-cytochrome c antibody.
Cell cycle analysis and Annexin V staining assay
The relative cell size and DNA content were assessed by flow-cytometry analysis. At various time points, cells were harvested and fixed with 100% ethanol and then placed at −20 °C overnight. After fixation, cells were centrifuged and washed once with PBS containing 1% bovine serum albumin. For staining with DNA dye, cells were resuspended in 0.5 to 1 ml of PI solution containing RNase and incubated at 37 °C for 30 min, followed by overnight incubation at 4 °C. For Annexin V staining, 1 × 104 cells were collected and stained with Annexin V-FITC according to the manufacturer's instruction. The PI and Annexin V stained cells were collected and analyzed using BD flow-cytometry systems. The percentage of Annexin V-FITC positive cells was considered as the percentage of apoptotic cells.Statistical analysis
Analysis and graphing of data were performed with Prism 3.0 (GraphPad Software, San Diego, CA). Data are expressed as means ± SEM. Statistical analysis was performed by ANOVA for multiple group comparison and by Student's t-test for direct comparison of two groups. P values < 0.05 were considered significant.Results and discussion
Effect of probucol on proliferation in human ovarian cancer cells
To determine whether probucol modulated cellular proliferation in human ovarian cancer cell lines (PA-1 and SKOV-3), cells were seeded in culture plates in equal numbers and left to grow to 30 to 40% confluence in a medium containing 10% FBS. Cells were washed twice with PBS buffer and maintained in medium containing 0.1% FBS for 48 h. The growth-arrested cells were then treated with probucol or probucol's vehicle (0.05% DMSO) in the presence of 10% FBS for 3 d. As shown in Fig. 1, raising the ambient probucol concentration (5, 10, 50, 100, 500 μM) causes a dose-dependent decrease in cell viability (Fig. 1a) and cell number (Fig. 1b) when compared with control (10% FBS) or probucol's vehicle in PA-1 and SKOV-3 cells. The high concentrations (100 μM and 500 μM) of probucol cause a significant decrease in cellular mitogenesis when compared with control or vehicle. In addition, the effects of probucol and the cytotoxic-inducer staurosporine on the membrane integrity and cytotoxicity of these cells were also clarified through evaluating the release of LDH into the experimental medium. We found that staurosporine (0.1 μM) and probucol (500 μM) promoted the LDH release when compared with control (Fig. 1c). According to the cell viability assay and cell number analysis, the lower and non-toxic concentration of probucol (100 μM) was used in subsequent experiments.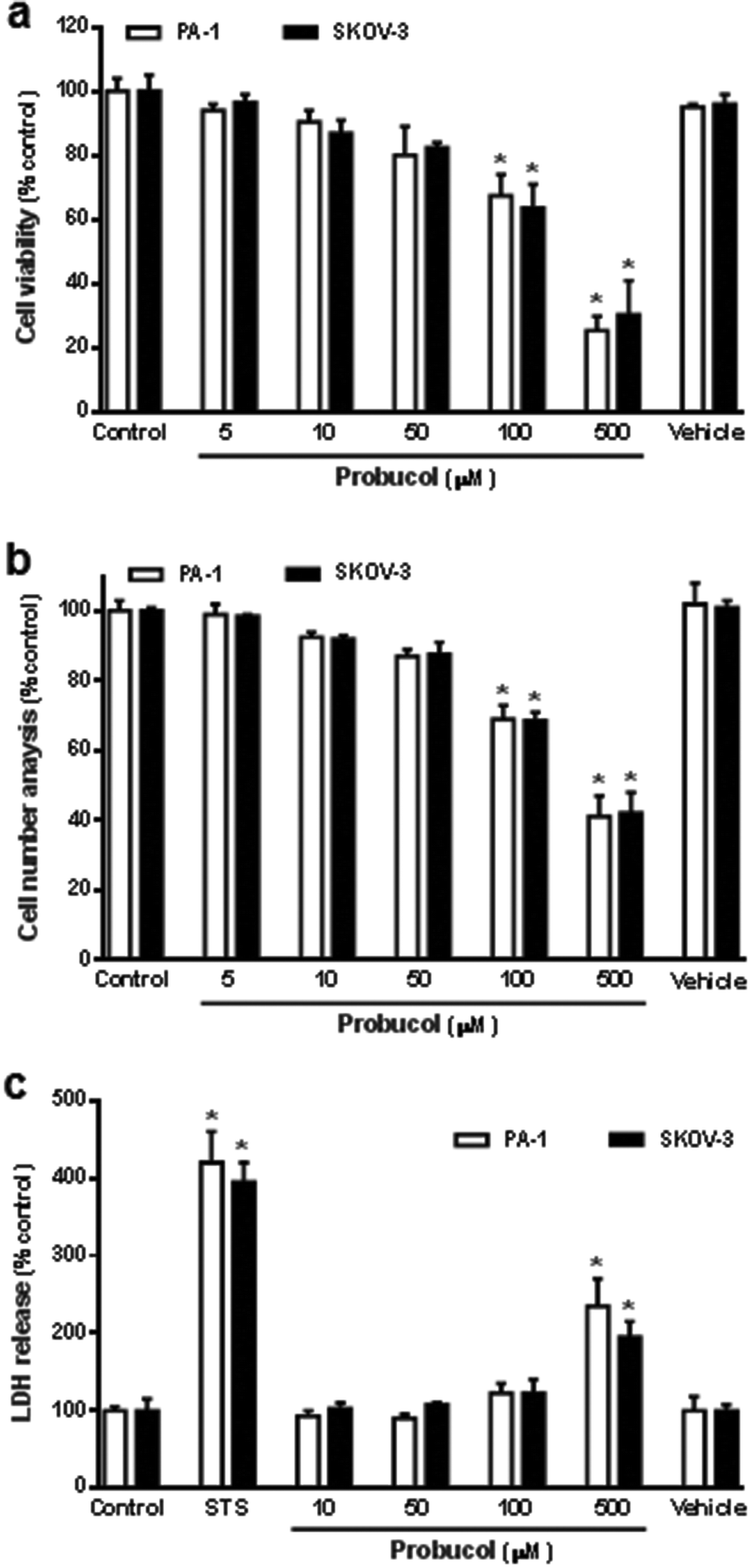 | ||
| Fig. 1 Effects of different concentrations of probucol on cellular mitogenesis and cytotoxicity in human ovarian cancer cell lines (PA-1 and SKOV-3). Serum-deprived cells were treated with 10% FBS (control), probucol (5, 10, 50, 100, and 500 μM), or probucol's vehicle (0.05% DMSO) for 3 d. Assayed for cell viability (a), cell numbers (b), and cytotoxicity (c) are described under “Materials and methods”. 0.1 μM staurosporine (STS) was used as a cytotoxic-inducer control. Results were expressed as the mean ± SEM (n = 6). *p < 0.05 versus control. | ||
Effect of probucol on apoptosis in human ovarian cancer cells
In order to assess whether probucol inhibited cellular proliferation through an apoptotic mechanism, we analyzed in detail, the changes in expression of the anti-apoptotic protein Bcl-2 and the apoptogenic protein cytochrome c and caspase-3. As shown in Fig. 2a and b, probucol (100 μM) treatments had no obvious effect on Bcl-2 and cytosolic cytochrome c expression as compared with control or vehicle in PA-1 and SKOV-3 cells. However, the pronounced decrease in Bcl-2 and increase in cytosolic cytochrome c were detected after the apoptotic-inducer staurosporine (0.1 μM) treatment. Furthermore, no obvious differences were observed regarding the percentages of Annexin V-FITC positive cells (Fig. 2c) and the caspase-3 activation (Fig. 2d) between probucol (100 μM) and control cultures in these cells. However, staurosporine did markedly increase the number of apoptotic cells and caspase-3 activity.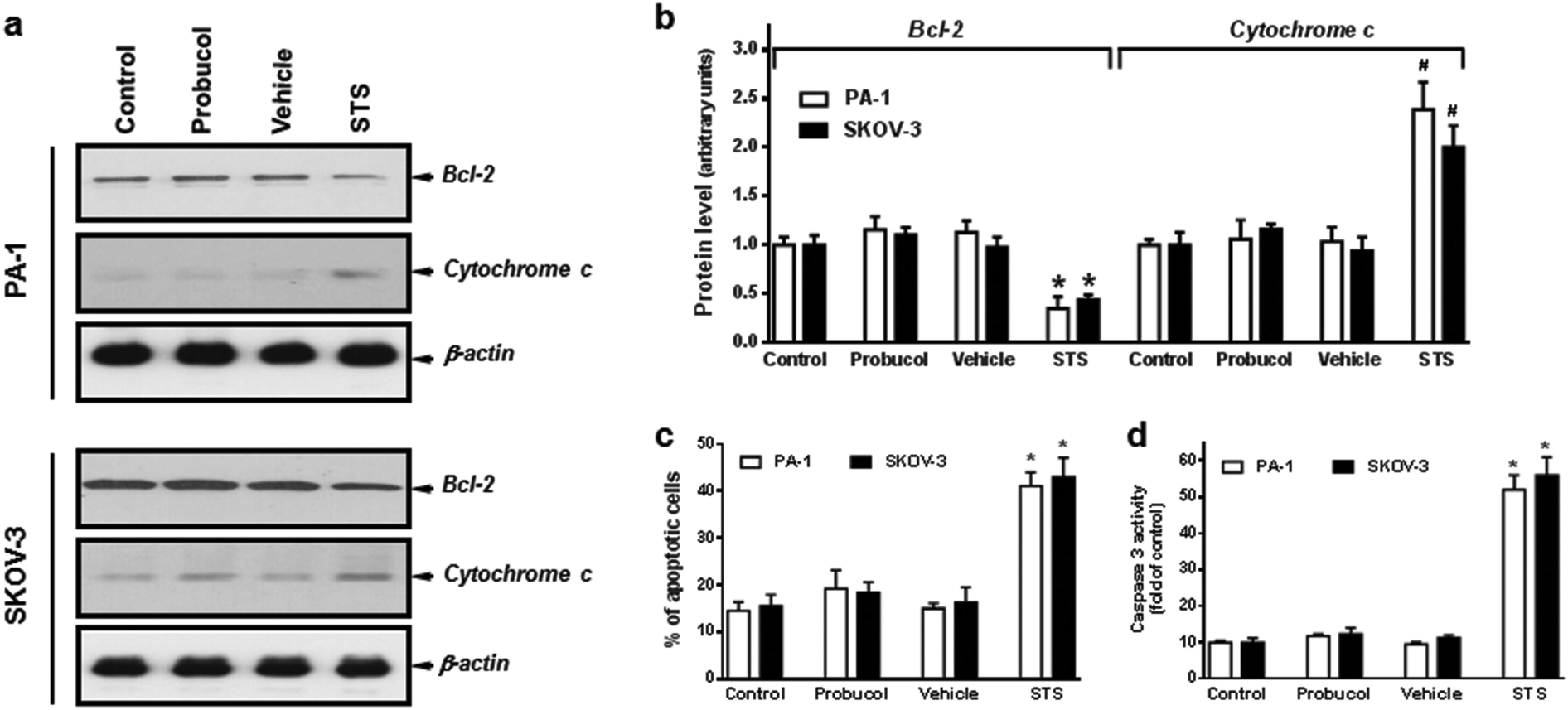 | ||
| Fig. 2 Effects of probucol on apoptosis regulatory molecules and caspase-3 activity in PA-1 and SKOV-3 cells. Total cell lysates from cells treated with 10% FBS (control), probucol (100 μM), vehicle, or staurosporine (0.1 μM) for 3 d were subjected to Western blot analysis for Bcl-2 and cytochrome c (a), Annexin V-FITC staining (c) and caspase-3 activity assay (d). Assayed for cytochrome c release in the cytosolic fraction is described under “Materials and methods”. Staurosporine (STS) was used as an apoptotic-inducer control. (b) Laser densitometry of the gels showed in (a) and two additional experiments. These are representative experiments, each performed at least three times in PA-1 and SKOV-3 cells. *p < 0.05 versus control. | ||
Indeed, probucol has been demonstrated to modulate cell proliferation and cytotoxicity in many tissues and organs.5–7 It regulates programmed cell death and exerts both agonistic and antagonistic effects on apoptotic signaling.24,25,34 Some lines of evidence have indicated that probucol increased the expression of anti-apoptotic protein and reduced apoptosis. It attenuates cyclophosphamide-induced oxidative apoptosis, p53 and Bax signal expression in rat cardiac tissues.35 Pretreatment with probucol significantly blocked apoptosis and increased the expression of the Bcl-2 protein in human umbilical vein endothelial cells.36 It also inhibited the lipopolysaccharide-reduced expression of the Bcl-2 protein and then suppressed apoptosis in vascular smooth muscle cells.37 Additionally, it could protect renal proximal tubular cells from methylguanidine-induced apoptosis.38 Nevertheless, in the present study, we found that probucol did not alter the cytotoxicity, the percentages of Annexin V-FITC positive cells and caspase-3 activation in human ovarian cancer cells. Moreover, it had no significant effect on Bcl-2 and cytosolic cytochrome c expression. Because probucol did not modulate apoptosis and cytotoxicity, our findings strongly suggest that it probably exerted its anti-proliferative effects by inhibiting cell cycle progression in these cells.
Effects of probucol on cell cycle regulatory molecules and cell cycle progression
To gain further insight into the mechanism exerted by probucol, we next wished to determine whether probucol is responsible for inhibition of cell cycle progression in human ovarian cancer cells. Western blot analysis revealed that probucol (100 μM) cannot affect protein synthesis of cdk4 and c-Myc (Fig. 3a and b) in PA-1 and SKOV-3 cells. However, probucol clearly reduced the protein synthesis of cyclin D1 and PCNA. Interestingly, protein levels of p27Kip1 and p21Waf1/Cip1 were significantly induced by probucol. Furthermore, flow-cytometry analysis showed that the percentages of cells in the G0/G1 phase increased while the percentages of cells in the G2/M phase decreased in the probucol-treated cells compared with the control groups (Fig. 4). These results indicated that suppression of cyclin D1 and PCNA and induction of p21Waf1/Cip1 and p27Kip1 are the underlying mechanisms by probucol to promote inhibition of cell cycle progression in human ovarian cancer cells.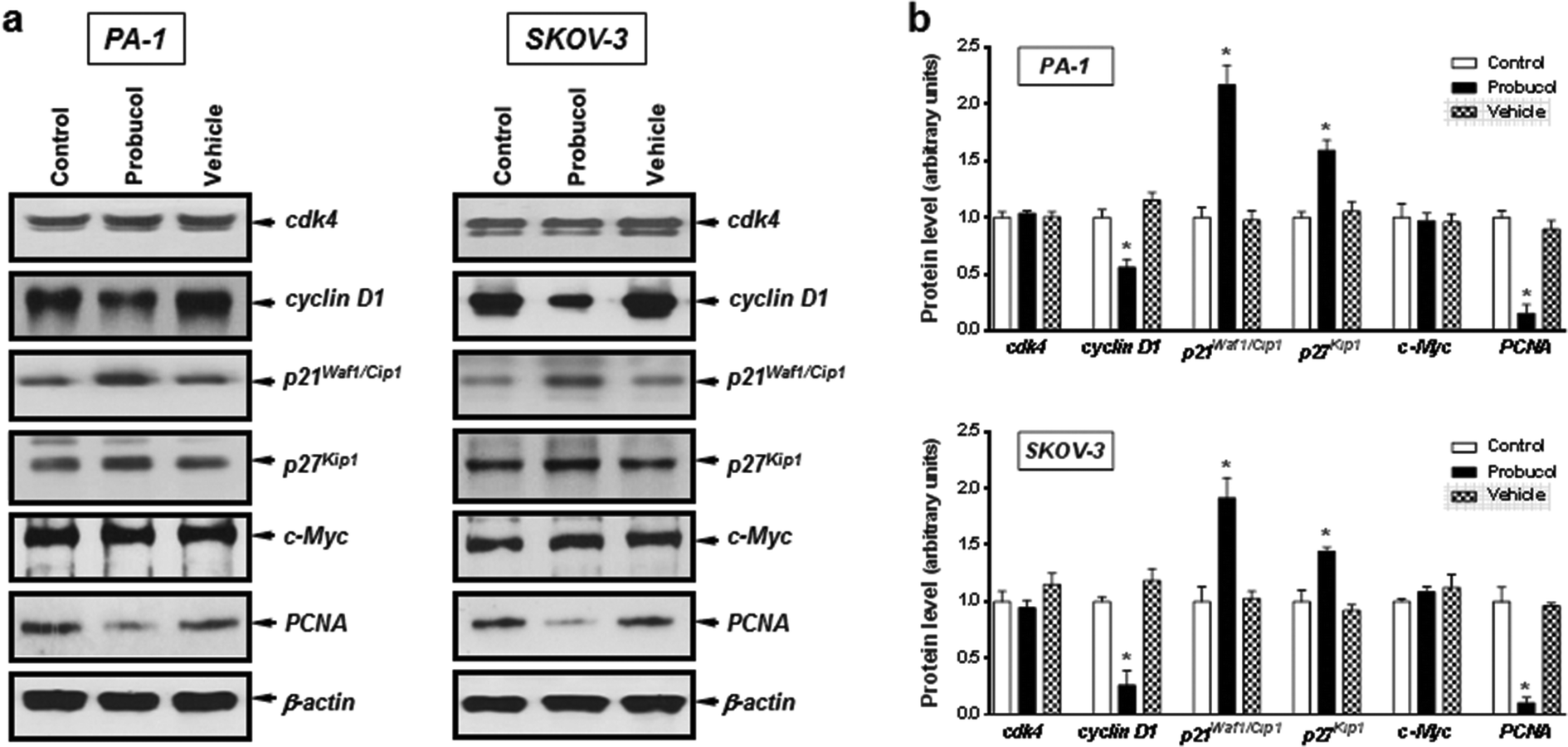 | ||
| Fig. 3 Effects of probucol on the protein levels of cdk4, cyclin D1, p21Wafi/Cip1, p27Kip1, c-Myc and PCNA in PA-1 and SKOV-3 cells. Total cell lysates from cells treated with 10% FBS (control), probucol (100 μM), or vehicle, for 12 h were subjected to Western blot analysis for cdk4, cyclin D1, p21Wafi/Cip1, p27Kip1, c-Myc and PCNA (a). This is a representative experiment independently performed three times in PA-1 and SKOV-3 cells. (b) Laser densitometry of the gels are shown in (a) and two additional experiments. *p < 0.05 versus control. | ||
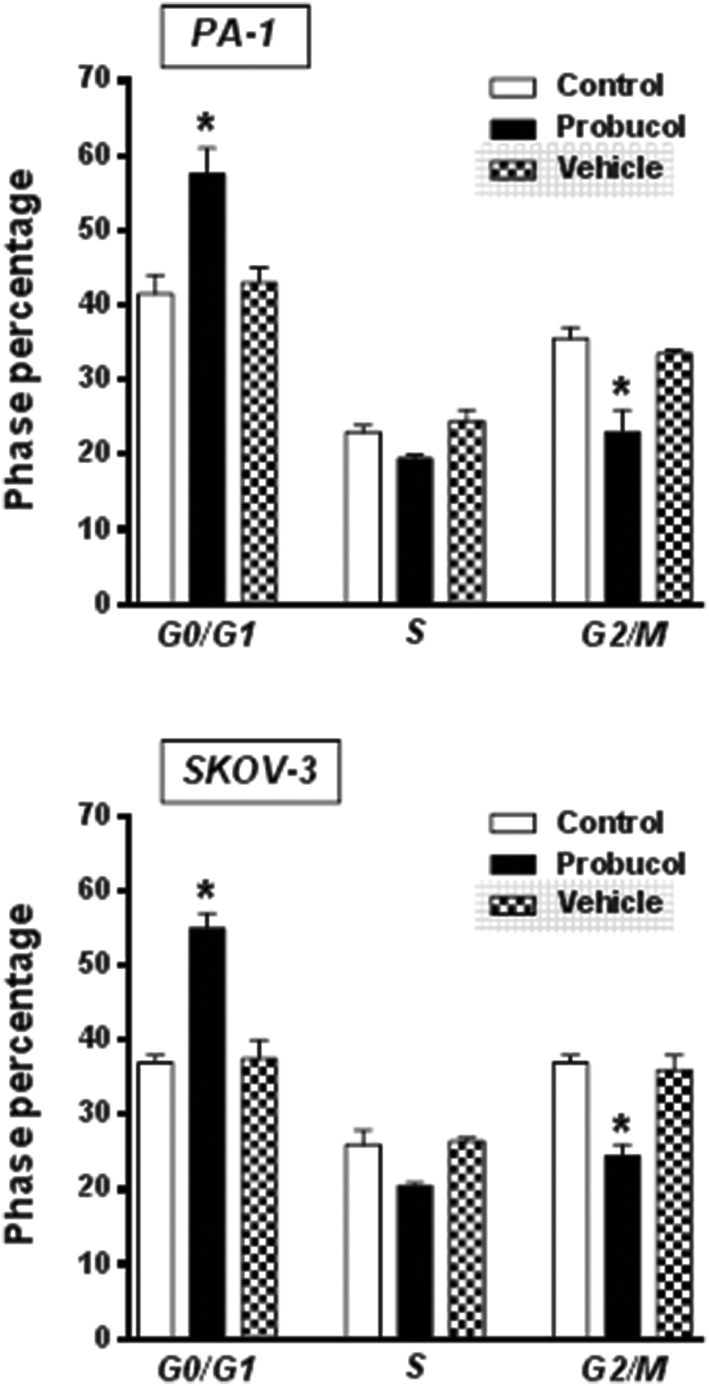 | ||
| Fig. 4 Effects of probucol on cell cycle distribution in PA-1 and SKOV-3 cells. The cell pellets from cells treated with 10% FBS (control), probucol (100 μM), or vehicle, for 24 h were fixed, stained, and subjected to BD flow-cytometry systems for cell cycle distribution. This is a representative experiment independently performed three times in PA-1 and SKOV-3 cells. *p < 0.05 versus control. | ||
Effects of probucol and some kinase inhibitors on NF-κB and ERK/JNK/p38 MAPK activation in human ovarian cancer cells
Evidence is accumulating to indicate that the NF-κB signaling pathway may be an important feature of ovarian carcinoma.32,33,39 To investigate whether probucol and the NF-κB signaling pathway played roles in ovarian cancer cell proliferation, probucol and the NF-κB inhibitor PDTC were used to pretreat PA-1 and SKOV-3 cells. We found that phospho-IκBα and nuclear p65 were strongly reduced by probucol treatment while the protein levels of IκBα and cytosolic p65 were enhanced (Fig. 5a and b). In addition, phosphorylation of IκBα was markedly attenuated by administration of PDTC (10 μM).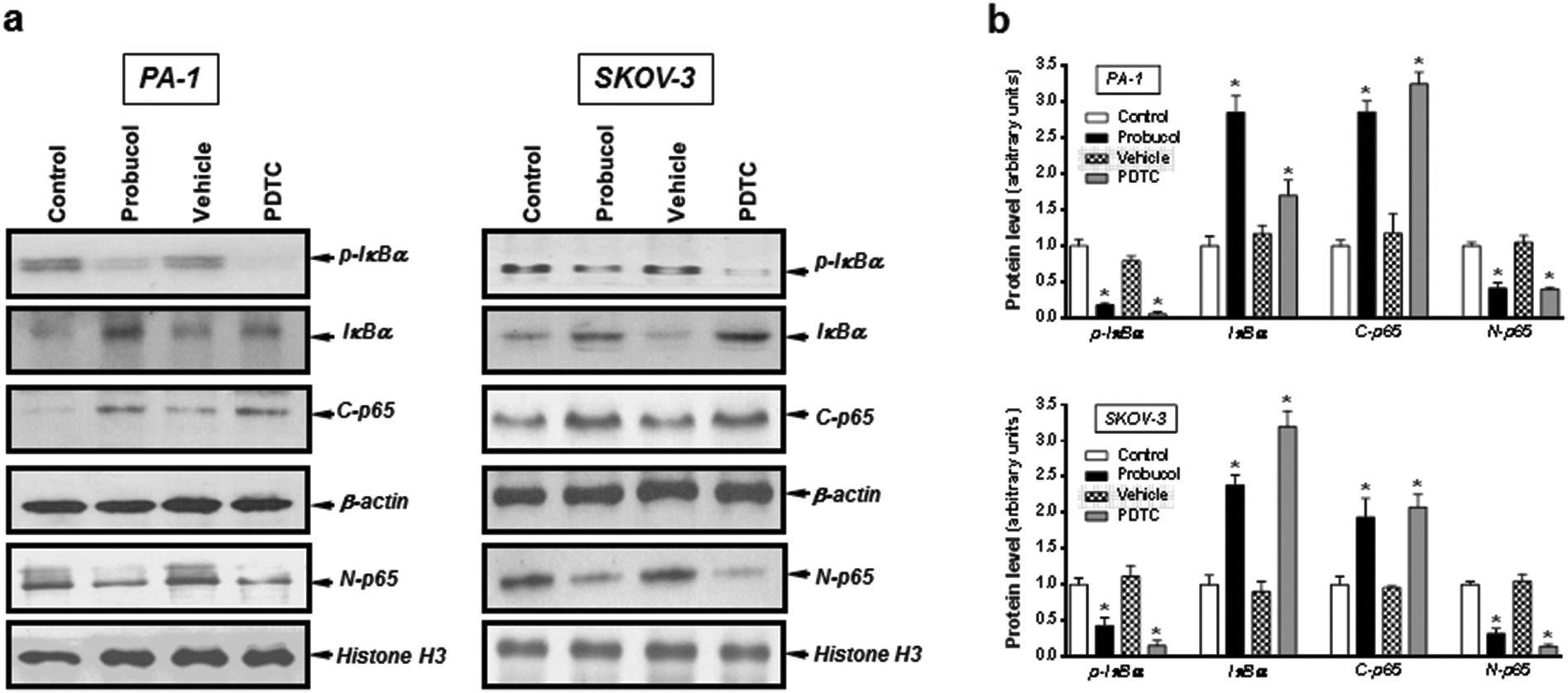 | ||
| Fig. 5 Effects of probucol on NF-κB activation in PA-1 and SKOV-3 cells. Cytosolic or nuclear protein from cells treated with 10% FBS (control), probucol (100 μM), vehicle, or PDTC (10 μM) for 12 h were subjected to Western blot analysis for phospho-IκBα (p-IκBα), IκBα, cytosolic p65 (C-p65), and nuclear p65 (N-p65) (a). Histone H3 was used as an internal control for nuclear fraction. This is a representative experiment independently performed three times in PA-1 and SKOV-3 cells. (b) Laser densitometry of the gels showed in (a) and two additional experiments. *p < 0.05 versus control. | ||
To clarify whether the ERK/JNK/p38 MAPK pathway played a role in cell growth, the ERK kinase inhibitor PD98059, the JNK kinase inhibitor SP60012, and the p38 MAPK kinase inhibitor SB203580 were used to pretreat PA-1 and SKOV-3 cells. After exposure of cultured cells to probucol and kinase inhibitors for 12 h, we found that probucol significantly reduced phospho-ERK, phospho-JNK and phospho-p38 MAPK without obviously affecting the ERK, JNK and p38 MAPK protein levels (Fig. 6a–c). Phosphorylation of ERK, JNK and p38 MAPK was significantly reduced by administration of PD98059 (10 μM), SP600125 (5 μM), and SB203580 (5 μM), respectively. On the other hand, we further examined the effects of PDTC, PD98059, SP600125, and SB203580 on cell viability and cell numbers in PA-1 and SKOV-3 cells. As depicted in Fig. 7, compared with the control groups, growth inhibition was clearly observed in these cells treated with probucol (100 μM), PDTC (10 μM), PD98059 (10 μM), SP600125 (5 μM), and SB203580 (5 μM). Additionally, growth inhibition was enhanced by probucol plus each kinase inhibitor in these cells. These results indicated that down-regulation of the NF-κB and ERK/JNK/p38 MAPK signaling pathways are important mechanisms by probucol to promote the inhibition of cellular proliferation in human ovarian cancer cells.
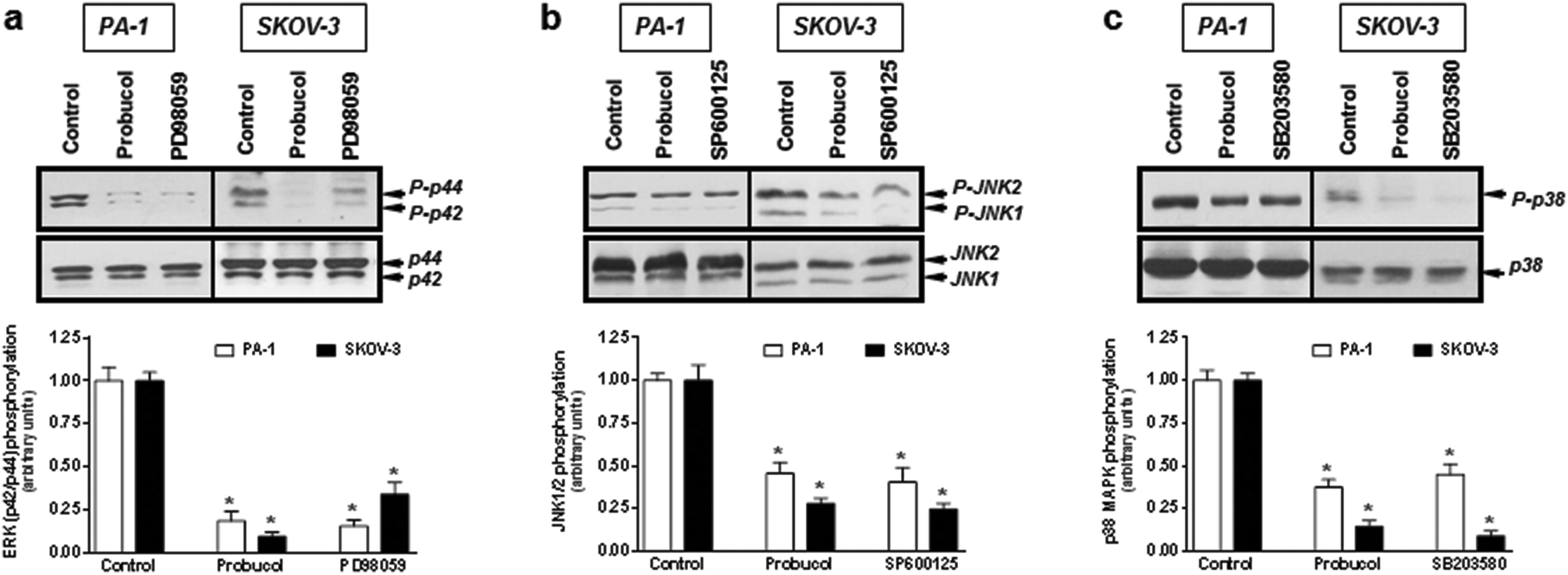 | ||
| Fig. 6 Effects of probucol on ERK/JNK/p38 MAPK activation in PA-1 and SKOV-3 cells. Total cell lysates from cells treated with probucol (100 μM), PD98059 (10 μM), SP600125 (5 μM), or SB203580 (5 μM) in the presence of 10% FBS (control) for 12 h were subjected to Western blot analysis for phospho-p42/p44 MAPK (a), phospho-JNK (b), and phospho-p38 MAPK (c) (upper panel) or proteins corresponding to the above phosphorylated proteins (lower panel). This is a representative experiment independently performed three times in PA-1 and SKOV-3 cells. *p < 0.05 versus control. | ||
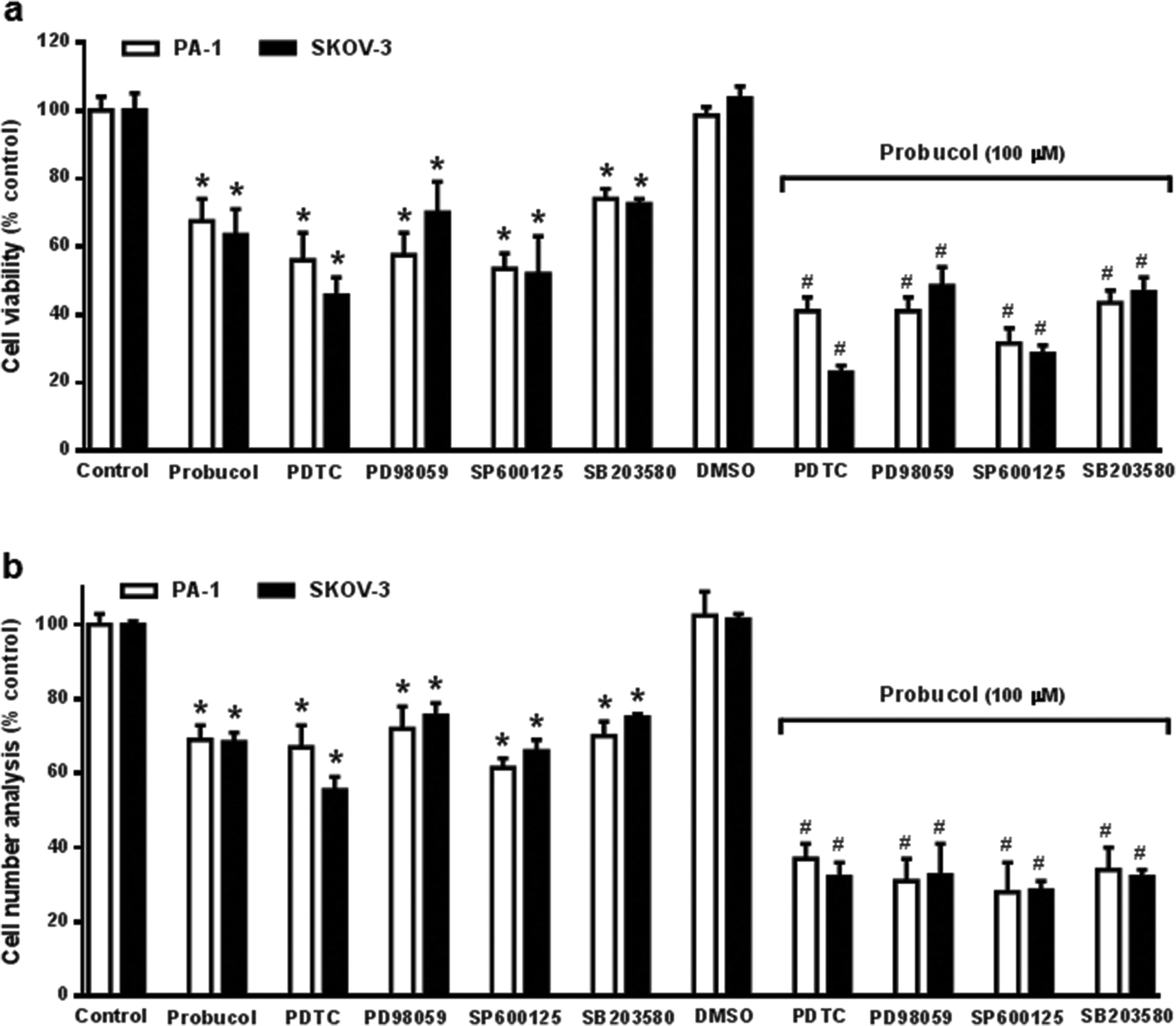 | ||
| Fig. 7 Effects of probucol, the NF-κB inhibitor, and the ERK/JNK/p38 MAPK blockade on cellular mitogenesis in PA-1 and SKOV-3 cells. Serum-deprived cells were treated with 10% FBS (control), probucol (100 μM), PDTC (10 μM), PD98059 (10 μM), SP600125 (5 μM), SB203580 (5 μM), probucol + PDTC, probucol + PD98059, probucol + SP600125, or probucol + SB203580 in the presence of 10% FBS (control) for 3 d. DMSO is the solvent used to dissolve the above kinase inhibitors. Assays for cell viability (a) and cell numbers (b) are described under “Materials and methods”. Results were expressed as the mean ± SEM (n = 6). *p < 0.05 versus control; #p < 0.05 versus probucol. | ||
To further evaluate whether NF-κB and ERK/JNK/p38 MAPK pathways mediate cellular mitogenesis of PA-1 and SKOV-3 cells, we first knockdown p65, ERK, JNK and p38 MAPK by specific siRNAs, as shown in ESI Fig. 1.† These protein levels decreased significantly in silenced cells than those in non-silencing RNA cells, confirming successfully, the presence of silenced p65, ERK, JNK and p38 MAPK in these cells. Also in these cases, knockdown of ERK, JNK and p38 MAPK strongly reduced the phosphorylation of ERK, JNK and p38 MAPK, respectively. We then investigated the effects of probucol, p65 siRNA, ERK siRNA, JNK siRNA and p38 MAPK siRNA on cellular mitogenesis in PA-1 and SKOV-3 cells. ESI Fig. 2† showed that the knockdown of p65, ERK, JNK and p38 MAPK by specific siRNAs caused a marked drop in cell viability, and the cell numbers further implicate the involvement of NF-κB and ERK/JNK/p38 MAPK pathways in growth control. Additionally, growth inhibition was enhanced by probucol plus each kinase inhibitor in these cells. Taken together, these results suggested that probucol down-regulates cellular proliferation through NF-κB and ERK/JNK/p38 MAPK pathways.
Several reports have indicated that both the NF-κB and MAPK signaling pathways play important roles in cell proliferation, differentiation, transformation, apoptosis, and the regulation of a variety of transcription factors and gene expressions.40–43 Inactivation of the NF-κB and MAPK signaling pathways are involved in anti-oxidative effects under various pathological conditions.28,42–44 Recent studies have implied that probucol may decrease NF-κB or MAPK activation and exert its unique pharmacologic actions and protective effects in many tissues and organs.45,46 In human aortic endothelial cells, probucol could suppress the activation of NF-κB and inhibit endothelial apoptosis.46 Besides, probucol reversed adriamycin-induced cardiomyopathy by inhibiting the ERK/JNK/p38 MAPK pathway via diminishing oxidative stress.47 In the current results, probucol treatment displayed a marked decrease in cell viability through inhibiting the NF-κB and ERK/JNK/p38 MAPK pathways in human ovarian cancer cells. This suggests that probucol is able to inhibit cell growth, one of the main features acquired by downregulation of NF-κB and ERK/JNK/p38 MAPK signaling, and could then reduce the proliferative potential of ovarian cancers.
Conclusions
In conclusion, the present work explains the mechanism of action of probucol that modulates proliferation and cell cycle progression in human ovarian cancer cells (Fig. 8). We found that probucol caused inhibition of cellular proliferation partly by obstructing cell cycle progression in two human ovarian cancer cell lines – PA-1 and SKOV-3. However, no apparent apoptosis was evident in any of the cell lines. The model suggested in Fig. 8 demonstrates that the ability of probucol to induce cell cycle arrest was verified by the observation that it significantly decreased protein expression of cyclin D1 and PCNA but increased the protein levels of p21Waf1/Cip1 and p27Kip1. The exposure of probucol also attenuated NF-κB and ERK/JNK/p38 MAPK activation. On the other hand, the specific inhibitors (e.g., PDTC, PD98059, SP600125, and SB203580) significantly inhibited cellular growth in these cells. Overall, this is the first report suggesting that probucol has potent inhibitory effects against human ovarian cancer cell proliferation, and NF-κB and ERK/JNK/p38 MAPK pathways may be important targets of probucol (Fig. 8). The blockade of these signaling pathways by probucol may be an effective strategy in the treatment of ovarian cancers.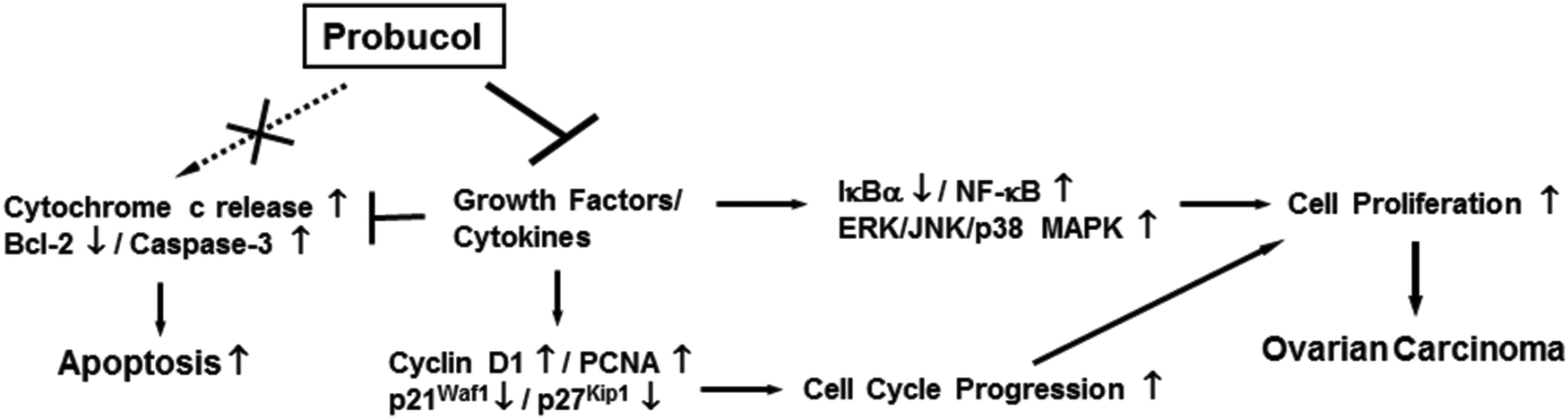 | ||
| Fig. 8 Proposed effects of probucol on cell proliferation and apoptosis in human ovarian carcinoma. The NF-κB and ERK/JNK/p38 MAPK cascades are important growth factors/cytokines-induced signaling pathways contributing to induction of cell proliferation in human ovarian cancer cells. Aberrant growth of these cells can lead to ovarian carcinoma. In the presence of probucol, the activities of NF-κB and ERK/JNK/p38 MAPK were inhibited and the suppression of cell cycle progression was partly mediated by enhancing p21Waf1/Cip1 and p27Kip1 but reducing cyclin D1 and PCNA. Nevertheless, probucol did not induce apoptosis by modulating cytochrome c release and caspase-3 activity in human ovarian cancer cells. | ||
Abbreviations
| ERK | Extracellular signal regulated kinase |
| IκBα | Inhibitory kappa B alpha |
| JNK | c-Jun N-terminal kinase |
| LDH | Lactate dehydrogenase |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-kappaB |
| PCNA | Proliferating cell nuclear antigen |
| PDTC | Pyrrolidine dithiocarbamate |
| STS | Staurosporine. |
Conflict of interest
The authors have declared no conflict of interest.Acknowledgements
This work was supported by Research Grants (NSC-102-2314-B273-001 to J.-S. Huang) from the National Science Council, Republic of China.References
- A. Adameova, Y. J. Xu, T. A. Duhamel, P. S. Tappia, L. Shan and N. S. Dhalla, Curr. Pharm. Des., 2009, 15, 3094–3107 CrossRef CAS PubMed.
- S. Yamashita and Y. Matsuzawa, Atherosclerosis, 2009, 207, 16–23 CrossRef CAS PubMed.
- D. Tanous, N. Hime and R. Stocker, Redox. Rep., 2008, 13, 48–59 CrossRef CAS PubMed.
- R. Stocker, Curr. Opin. Lipidol., 2009, 20, 227–235 CrossRef CAS PubMed.
- J. W. Heinecke, J. Exp. Med., 2006, 203, 813–816 CrossRef CAS PubMed.
- S. P. Moubayed, T. M. Heinonen and J. C. Tardif, Curr. Opin. Lipidol., 2007, 18, 638–644 CrossRef CAS PubMed.
- M. T. Saleem, M. C. Chetty and S. Kavimani, Ther. Adv. Cardiovasc. Dis., 2014, 8, 4–11 CrossRef CAS PubMed.
- J. R. Walker, A. Sharma, M. Lytwyn, S. Bohonis, J. Thliveris, P. K. Singal and D. S. Jassal, J. Am. Soc. Echocardiogr., 2011, 24, 699–705 CrossRef PubMed.
- P. S. Munksgaard and J. Blaakaer, Gynecol. Oncol., 2012, 124, 164–169 CrossRef CAS PubMed.
- Z. Stanojevic, B. Djordjevic, S. B. Pajovic, J. Zivanov-Curlis and S. Najman, J. BUON., 2009, 14, 7–18 CAS.
- R. Longuespée, C. Boyon, A. Desmons, D. Vinatier, E. Leblanc, I. Farré, M. Wisztorski, K. Ly, F. D'Anjou, R. Day, I. Fournier and M. Salzet, Cancer Metastasis Rev., 2012, 31, 713–732 CrossRef PubMed.
- L. Batista, T. Gruosso and F. Mechta-Grigoriou, Int. J. Biochem. Cell Biol., 2013, 45, 1092–1098 CrossRef CAS PubMed.
- M. Fung-Kee-Fung, T. Oliver, L. Elit, A. Oza, H. W. Hirte and P. Bryson, Curr. Oncol., 2007, 14, 195–208 CrossRef CAS PubMed.
- J. Liu and U. A. Matulonis, Clin. Cancer Res., 2014, 20, 5150–5156 CrossRef CAS PubMed.
- S. Banerjee and M. Gore, Oncologist, 2009, 14, 706–716 CrossRef CAS PubMed.
- A. A. Secord, A. B. Nixon and H. I. Hurwitz, Gynecol. Oncol., 2014, 135, 349–358 CrossRef CAS PubMed.
- W. C. Burhans and N. H. Heintz, Free Radicals Biol. Med., 2009, 47, 1282–1293 CrossRef CAS PubMed.
- J. Chiu and I. W. Dawes, Trends Cell Biol., 2012, 22, 592–601 CrossRef CAS PubMed.
- M. Tertil, A. Jozkowicz and J. Dulak, Curr. Pharm. Des., 2010, 16, 3877–3894 CrossRef CAS PubMed.
- P. Rajendran, N. Nandakumar, T. Rengarajan, R. Palaniswami, E. N. Gnanadhas, U. Lakshminarasaiah, J. Gopas and I. Nishigaki, Clin. Chim. Acta, 2014, 436, 332–347 CrossRef CAS PubMed.
- E. el-Demerdash, E. D. el-Denshary, M. el-Didi, N. Al-Gharabli and A. M. Osman, Anticancer Res., 2002, 22, 977–984 CAS.
- M. Zarkovic, X. Qin, Y. Nakatsuru, S. Zhang, Y. Yamazaki, H. Oda, T. Ishikawa and T. Ishikawa, Carcinogenesis, 1995, 16, 2599–2601 CrossRef CAS PubMed.
- M. Iqbal, Y. Okazaki and S. Okada, Mol. Cell. Biochem., 2007, 304, 61–69 CrossRef CAS PubMed.
- G. Nishimura, S. Yanoma, H. Mizuno, K. Kawakami and M. Tsukuda, Jpn. J. Cancer Res., 1999, 90, 1224–1230 CrossRef CAS PubMed.
- M. Iqbal and S. Okada, Pharmacol. Toxicol., 2003, 93, 259–263 CAS.
- Z. Zhang, H. Cao, S. Jiang, Z. Liu, X. He, H. Yu and Y. Li, Small, 2014, 10, 4735–4745 CrossRef CAS PubMed.
- A. Siomek, Acta Biochim. Pol., 2011, 59, 323–331 Search PubMed.
- M. J. Morgan and Z. G. Liu, Cell Res., 2011, 21, 103–115 CrossRef CAS PubMed.
- C. Pantano, N. L. Reynaert, A. Vliet and Y. M. Janssen-Heininger, Antioxid. Redox Signalling, 2006, 8, 1791–1806 CrossRef CAS PubMed.
- H. Jing and S. Lee, Mol. Cells, 2014, 37, 189–195 CrossRef PubMed.
- T. Okamoto, T. Sanda and K. Asamitsu, Curr. Pharm. Des., 2007, 13, 447–462 CrossRef CAS PubMed.
- A. E. Drummond and P. J. Fuller, Mol. Cell. Endocrinol., 2012, 359, 85–91 CrossRef CAS PubMed.
- K. L. White, D. N. Rider, K. R. Kalli, K. L. Knutson, G. P. Jarvik and E. L. Goode, Cancer Causes Control, 2011, 22, 785–801 CrossRef PubMed.
- H. J. Oskarsson, L. Coppey, R. M. Weiss and W. G. Li, Cardiovasc. Res., 2000, 45, 679–687 CrossRef CAS PubMed.
- Y. A. Asiri, Oxid. Med. Cell. Longev., 2010, 3, 308–316 CrossRef PubMed.
- Z. Dai, D. F. Liao, D. J. Jiang, H. W. Deng and Y. J. Li, Naunyn Schmiedebergs Arch. Pharmacol., 2004, 370, 314–319 CrossRef CAS PubMed.
- J. F. Li, S. Chen, J. D. Feng, M. Y. Zhang and X. X. Liu, Exp. Mol. Pathol., 2014, 96, 250–256 CrossRef CAS PubMed.
- F. Wang, B. Yang, G. H. Ling, C. Yao and Y. S. Jiang, Ren. Fail., 2010, 32, 978–985 CrossRef CAS PubMed.
- L. Hernandez, S. C. Hsu, B. Davidson, M. J. Birrer, E. C. Kohn and C. M. Annunziata, Cancer Res., 2010, 70, 4005–4014 CrossRef CAS PubMed.
- A. C. Ledoux and N. D. Perkins, Biochem. Soc. Trans., 2014, 42, 76–81 CrossRef CAS PubMed.
- E. K. Kim and E. J. Choi, Biochim. Biophys. Acta, 2010, 1802, 396–405 CrossRef CAS PubMed.
- D. J. Erstad and J. C. Cusack Jr., Surg. Oncol. Clin. N. Am., 2013, 22, 705–746 CrossRef PubMed.
- E. F. Wagner and A. R. Nebreda, Nat. Rev. Cancer, 2009, 9, 537–549 CrossRef CAS PubMed.
- E. D. Owuor and A. N. Kong, Biochem. Pharmacol., 2002, 64, 765–770 CrossRef CAS PubMed.
- M. Zhang, J. Wang, J. H. Liu, S. J. Chen, B. Zhen, C. H. Wang, H. He and C. X. Jiang, Mol. Med. Rep., 2013, 7, 177–182 CAS.
- M. Aoki, T. Nata, R. Morishita, H. Matsushita, H. Nakagami, K. Yamamoto, K. Yamazaki, M. Nakabayashi, T. Ogihara and Y. Kaneda, Hypertension, 2001, 38, 48–55 CrossRef CAS PubMed.
- H. Lou, I. Danelisen and P. K. Singal, Am. J. Physiol. Heart Circ. Physiol., 2005, 288, 1925–1930 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tx00088b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |