
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ tuning of magnetization via topotactic lithium insertion in ordered mesoporous lithium ferrite thin films†
Christian
Reitz‡
a,
Christian
Suchomski‡
*b,
Di
Wang
ac,
Horst
Hahn
ad and
Torsten
Brezesinski
 *a
*a
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: torsten.brezesinski@kit.edu
bInstitute of Physical Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: christian.suchomski@phys.chemie.uni-giessen.de
cInstitute of Nanotechnology, Karlsruhe Nano Micro Facility, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dKIT-TUD Joint Research Laboratory Nanomaterials, Institute of Material Science, Technical University of Darmstadt, Jovanka-Bontschits-Str. 2, 64287 Darmstadt, Germany
First published on 1st August 2016
Abstract
The synthesis and characterization of cubic mesostructured lithium ferrite (α-LiFe5O8) with 20 nm diameter pores and nanocrystalline walls is reported. The material is prepared in the form of thin films by sol–gel dip-coating using a poly(isobutylene)-block-poly(ethylene oxide) amphiphilic diblock copolymer as the porogen. Electron microscopy, X-ray scattering and diffraction, time-of-flight secondary ion mass spectrometry, Raman and X-ray photoelectron spectroscopy all show that α-LiFe5O8 can be templated to produce high-quality films that are chemically and phase-pure and thermally stable to over 600 °C. Magnetometry measurements indicate ferrimagnetic behavior below 300 K, with the coercivity exhibiting a T1/2 dependence. This novel mesoporous spinel material – when used as an electrode in secondary battery cells – can reversibly store charge via topotactic Li insertion, which allows for the intriguing possibility of tuning the magnetization at room temperature in a facile and controlled manner. The general approach is simple and should be applicable to a variety of other magnetic materials that are capable of reacting electrochemically with Li to produce reduced phases.
Introduction
Synthetic ferrites such as Y3Fe5O12 (garnet), BaFe12O19 (hexaferrite) and Mn1−xZnxFe2O4 (spinel) represent a ubiquitous class of hard and soft-magnetic materials that are widely used in various applications.1,2 Herein, we focus on lithium ferrite, LiFe5O8 (referred to as LFO in the following), which has been shown to exhibit a high magnetization (2.5 μB per formula unit) and one of the highest Curie temperatures of all spinel ferrites, a narrow resonance linewidth and high electrical resistivity, making it promising for microwave and magnetic device applications.3–5 Furthermore, LFO has attracted interest as a low-cost cathode active material for rechargeable Li-ion batteries in the past.6 LFO exists in two crystal forms, the “ordered” alpha phase with space group P4332 (or enantiomorphic P4132) and the “disordered” beta phase with space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m.7,8 A phase change in the material occurs at temperatures above 730 °C, with a transformation from the alpha phase to the beta phase (order/disorder transition).9,10
m.7,8 A phase change in the material occurs at temperatures above 730 °C, with a transformation from the alpha phase to the beta phase (order/disorder transition).9,10
In recent years, it has been shown that material properties can be tailored by microstructural engineering and controlling the physical dimensions and shape to meet a specific application. However, the synthesis of single-phase and small grain size LFO has proven to be difficult in practice. This is due, in part, to both the formation of impurity phases such as α-Fe2O3 or LiFeO2 during annealing and the high volatility of lithium (or Li2O) and oxygen loss.11,12 In this work, we demonstrate a simple synthetic route to large-pore mesoporous alpha-LFO thin films with nanocrystalline walls by utilizing an evaporation-induced self-assembly (EISA) process.
The EISA process was introduced in the late 1990's and has led to the fabrication of many important phases and materials, most of them being either siliceous in nature or binary metal oxides.13–23 Despite the progress made, particularly in the past decade, the preparation of mixed-metal oxides that combine features such as high crystallinity and thermal stability as well as well-developed nanoscale porosity is very challenging. Here, we make use of the superior templating properties of a large poly(isobutylene)-block-poly(ethylene oxide) amphiphilic diblock copolymer structure-directing agent to achieve chemically and phase-pure alpha-LFO films with cubic pore symmetry.24–27 Notably, they show no signs of major structural degradation or crack formation during the thermal crystallization process and further exhibit stable magnetic properties at room temperature, unlike other templated mesostructured spinel ferrites.28–32
These key properties and the fact that LFO can reversibly take up Li in a topotactic way in a specific voltage range make the films an ideal model system for studying in situ changes in magnetization upon lithiation. Usually, the magnetic properties can only be tuned by strain mediation or electrostatic charge carrier doping, both of which requires precise interface and interphase engineering.27,33–37 The mesoporous morphology of the sol–gel derived material is beneficial as it provides short diffusion path lengths for facile Li insertion, while the film geometry paves the way to novel device structures and concepts (e.g., for future non-volatile memory and logic applications). Similar non-destructive approaches have been reported recently by Tsuchiya et al. and Dasgupta et al. for all-solid-state thin film Li-ion batteries with 10 nm-thick Fe3O4 anode layer and Li half-cells with γ-Fe2O3 electrode containing polymer binder and carbon black additive, respectively.38,39
Collectively, we show that topotactic Li insertion into LFO allows for the intriguing possibility of controlling the bulk magnetic state in a highly reversible manner and that this electrochemically mediated approach has great potential in the development of tailored magnetic nanostructures.
Experimental section
Materials
Li(OOCCH3) (99.95%), Fe(NO3)3 × 9H2O (≥99.999%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 99.95%), 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMIM-TFSI, ≥97.0%), ethanol (EtOH, ACS reagent), tetrahydrofuran (THF, ≥99.9%) and 2-methoxyethanol (99.8%) were purchased from Sigma-Aldrich. H[C(CH3)2CH2]107C6H4(OCH2CH2)150OH (PIB107-b-PEO150) was obtained from BASF SE and used as received. 700 μm-thick Si(001) wafers were purchased from SiMat.LFO film synthesis
PIB107-b-PEO150 (45 mg), Fe(NO3)3 × 9H2O (454.5 mg) and Li(OOCCH3) (14.8 mg) were dissolved in a mixed solvent of EtOH (1.5 mL), THF (0.2 mL) and 2-methoxyethanol (0.5 mL). Films were prepared by dip-coating at 25 °C and 12% relative humidity onto Si(001), indium tin oxide (In2O3/SnO2, 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 wt%, ITO)-coated Si(001) and quartz glass substrates. After drying for 5 min, the as-made films were transferred to an oven at 130 °C for 1 h and then heated to 300 °C (0.5 °C min−1), followed by aging for 12 h. Finally, they were heated to 630 °C (10 °C min−1) both to burn out the polymer structure-directing agent and to crystallize the wall structure.
:10 wt%, ITO)-coated Si(001) and quartz glass substrates. After drying for 5 min, the as-made films were transferred to an oven at 130 °C for 1 h and then heated to 300 °C (0.5 °C min−1), followed by aging for 12 h. Finally, they were heated to 630 °C (10 °C min−1) both to burn out the polymer structure-directing agent and to crystallize the wall structure.
Cell assembly
Half-cells were assembled in an argon-filled glovebox by stacking 600 μm-thick Li metal foil (China Lithium Ltd), GF/A separator (GE Healthcare Life Sciences, Whatman) and mesoporous LFO film on ITO-coated Si(001) substrate. The electrolyte used was a solution of LiTFSI (20 wt%) in EMIM-TFSI. The water content was <10 ppm, as determined by Karl Fischer titration.Instrumentation
Scanning electron microscopy (SEM) images were collected on a MERLIN from Carl Zeiss operated at 5 keV. Transmission electron microscopy (TEM) imaging was performed on an aberration corrected Titan 80-300 (FEI) equipped with an energy-dispersive X-ray spectroscopy (EDX) detector and Gatan Tridiem image filter. The microscope was operated at 300 keV in TEM mode for high-resolution TEM (HRTEM) and in nanoprobe mode for scanning TEM (STEM). X-ray diffraction (XRD) was carried out on an X'Pert PRO from PANalytical instruments (λ = 0.15418 nm) equipped with an X'Celerator RTMS detector. Grazing incidence small-angle X-ray scattering (GISAXS) measurements were conducted at the German synchrotron radiation facility HASYLAB at DESY (λ = 0.1088 nm, beam size = 22.6 × 23.3 μm2, sample/detector distance = 3787 mm) by use of a DECTRIS Pilatus 1 M area detector (981 × 1043 pixels, pixel size = 172 × 172 μm2). The angle of incidence was set to 0.2°. For data analysis, DPDAK 1.2.0 software was used.40 X-ray photoelectron spectroscopy (XPS) data were acquired on a PHI VersaProbe scanning XPS microprobe equipped with a monochromatic Al-Kα source and hemispherical electron energy analyzer. The electron takeoff angle was set to 45° and the C 1s peak at 284.8 eV was used as an energy reference to correct for charging. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) was performed on a TOF-SIMS 5 from ION-TOF GmbH. A 25 keV Bi+ primary ion beam (I = 0.06 pA, area = 100 × 100 μm2, ion dose density = 1.24 × 1012 ions per cm2) was used to generate secondary ions. For sputter etching, a beam of 1 keV O2+ (I = 254.76 nA, area = 200 × 200 μm2, ion dose density = 1.34 × 1018 ions per cm2) was used. Raman spectra were acquired on a SENTERRA dispersive Raman microscope from Bruker Optics equipped with an Olympus objective (MPLN 100×, F.N.22, N.A.0.9) and Nd:YAG laser (λ = 532 nm, P = 2 mW). An MPMS XL-5 superconducting quantum interference device (SQUID) magnetometer from Quantum Design was used for magnetic susceptibility measurements in the field range from +45 to −45 kOe. Cyclic voltammetry was performed in the potential range from 3.2 to 1.4 V with respect to Li+/Li at a scan rate of 1 mV s−1 by use of an Autolab PGSTAT302 potentiostat.Results and discussion
The films employed in this work were prepared by dip-coating onto polished substrates, including Si(001) and ITO-coated Si(001) wafers and quartz glass. Structural characterization was performed on samples heated to temperatures from 530 to 630 °C by electron microscopy and GISAXS. Fig. 1 and Fig. S1 (ESI†) show results from TEM and SEM of mesoporous LFO films heated to 630 °C. From these data, we conclude that the self-assembling does not depend on the type of substrate used. Top view SEM images show a partially ordered mesophase with open 20 nm diameter pores and 10 nm-thick walls. The films appear to be free of cracks and other structural defects, and the network of pores persists throughout the bulk, as shown by cross-sectional SEM. STEM confirms the homogeneity of composition, while HRTEM demonstrates the high crystallinity with walls composed of randomly oriented spinel grains. | ||
| Fig. 1 Electron microscopy of mesoporous LFO thin films on (a–d) Si(001) and (e) ITO-coated Si(001) substrates heated to 630 °C. (a and b) Top view SEM and (c) Both panel (c) and the inset in (c) show high-angle annular dark-field STEM images at different magnifications. (d) HRTEM image. (e) Cross-sectional SEM image collected with a sample tilt of 80°. | ||
Fig. 2 shows synchrotron-based GISAXS patterns at an angle of incident of 0.2° obtained ex situ on a mesoporous LFO film of thickness around 200 nm on ITO-coated Si(001) substrate. Line cuts along the highest intensity in qy are also shown in the figure. Oscillations and strong in-plane scattering maxima from the ITO layer and LFO, respectively, are clearly visible. The primary reasons for the lack of scattering in qz are the low film thickness and that the material is already crystalline to some degree after heating to 530 °C and the pore ordering is lower than that of other polymer-templated mesostructured spinel ferrites reported in the literature.41 However, given that Li compounds in general are rather poor sol–gel precursors (glass formers), it is quite remarkable that LFO films with both a well-defined pore structure and uniform composition can be produced by facile coassembly strategies. As seen, the full width at half maximum (FWHM) of the first order peak at qy = 0.195 nm−1 increases and the second order peak decreases in intensity with an increase in heating temperature to 630 °C. This can be attributed to distortions induced by crystallization and grain growth. Severe structural degradation is observed at temperatures equal to or above 700 °C. Also, it can be seen that the in-plane lattice contraction is negligible, which helps explain why the films are crack-free on the micrometer level. Collectively, the GISAXS data agree with the results from electron microscopy that LFO can be templated to produce thermally stable films with a cubic pore network.
 | ||
| Fig. 2 (a–c) Synchrotron-based GISAXS patterns of a mesoporous LFO thin film on ITO-coated Si(001) substrate heated to different temperatures of (a) 530 °C, (b) 580 °C and (c) 630 °C. The corresponding intensity color map is shown on the right. (d–f) Horizontal line cuts along the qy-direction indicated by the yellow arrows. | ||
The crystallization behavior was investigated by means of XRD (Fig. 3a). The onset of crystallization was found to be at 510 °C, and the reflections can be indexed to the alpha phase of LFO. The average crystallite size was calculated from the FWHM of the (220) and (311) reflections at 30.5° and 35.7° 2θ, respectively, by use of the Scherrer equation. This analysis provides values ranging from (7 ± 1) nm at 530 °C to (11 ± 1) nm at 630 °C. On the basis of XRD, the presence of second phases such as α-Fe2O3, LiFeO2 or other ferric oxides can be ruled out. These could potentially be formed in the Fe-rich material, particularly at elevated temperatures.42 Overall, this finding is exciting because phase-pure LFO can usually only be achieved by prolonged heating well above 700 °C, which, however, is detrimental to maintaining nanoscale features due to sintering effects. We believe that the mesoporous morphology characterized by very thin walls is beneficial for the control of the amorphous to crystalline transformation.
 | ||
| Fig. 3 Crystallinity and phase composition of mesoporous LFO thin films. (a) XRD patterns for different heating temperatures. The line pattern corresponds to ICSD reference card no. 35769 for alpha-LFO.7 (b) Raman spectra obtained on a bare quartz glass (gray) and mesoporous LFO film on quartz glass (magenta) heated to 630 °C. (c) TOF-SIMS depth profiling showing 7Li+ and 56Fe+. | ||
The phase composition was also investigated via Raman spectroscopy. According to group theory, alpha-LFO has forty Raman-active phonon modes (6 × A1 + 14 × E + 20 × F2), while beta-LFO possesses only five symmetry allowed first-order modes (1 × A1g + 1 × Eg + 3 × F2g).43,44 A representative non-polarized Raman spectrum obtained on a mesoporous LFO film on quartz glass substrate heated to 630 °C is shown in Fig. 3b. In total, eighteen bands are observed between 100 and 900 cm−1, the peak frequencies of which are consistent with those reported for single crystals of alpha-LFO (see Table S1, ESI†).44 The lack of Raman bands is probably due to superposition of the spectrum from the substrate as well as line broadening and decreasing intensity because of the small crystallite size (phonon confinement).45,46 Nevertheless, the data neither indicate polymorphism nor the presence of second phases, thus confirming purity of the sol–gel derived material.
TOF-SIMS was used to obtain information on the spatial distribution of Li and Fe. Depth profiling results are shown in Fig. 3c. Both the 7Li+ and 56Fe+ signals are constant, which implies that the metal ions are homogeneously distributed throughout the bulk of the films. Minor variations are observed in the vicinity of the sample/substrate interface. This is likely due, in part, to the different chemical environment, which strongly affects the ionization probability.
Although XPS is not suited to determine the atomic Li/Fe ratio because of the relatively low amount of lithium in LFO and the fact that the Li 1s and Fe 3p levels are superimposed, this technique was applied to gain insight into the oxidation state of iron and the bonding state of oxygen. Fig. S2 (ESI†) shows both a survey spectrum and detailed scans of the Fe 2p and O 1s regions. Apart from a weak Si 2p substrate peak, only Fe, Li, O and C levels are clearly visible. The Fe 2p data establish that iron is – as expected – in the oxidation state +3. The O 1s data indicate two different bonding states, corresponding to lattice oxygen and hydroxyl oxygen. The presence of OH groups is not unexpected because, after synthesis, the mesoporous films were stored at ambient conditions. Given that the wall thickness and XPS information depth are similar, the results can be considered representative of the whole sample.
In a nutshell, the data in Fig. 3 and Fig. S2 (ESI†) demonstrate that the crystallite size is on the same order as the wall thickness after heating to 630 °C and the films are single-phase with no major fluctuations in composition.
The magnetic properties of the mesoporous LFO films heated to 630 °C were studied by DC SQUID magnetometry. Fig. 4a shows zero-field-cooled (ZFC) and field-cooled (FC) curves measured at 100 Oe with the magnetic field oriented parallel to the substrate plane. The magnetization increases as the temperature is increased until a broad maximum is reached at about 310 K, which can be associated with the blocking temperature, TB. In this temperature range and above the thermal energy overcomes the magnetic coupling forces, resulting in superparamagnetic relaxation. Fig. 4b shows in-plane hysteresis curves at different temperatures. M(H) loops measured at 5 and 300 K with the magnetic field oriented parallel as well as perpendicular to the substrate plane are shown in Fig. S3 (ESI†). From the latter data, it is inferred that the films have the magnetic easy axis in the plane. This means the energy needed to reach “saturation magnetization” is higher in the off-plane direction, which can be well-explained by the shape anisotropy. The in-plane and out-of-plane hysteresis curves indicate soft-magnetic behavior below 300 K with moderate low-temperature coercivities, HC, of 360 Oe and 430 Oe, respectively.
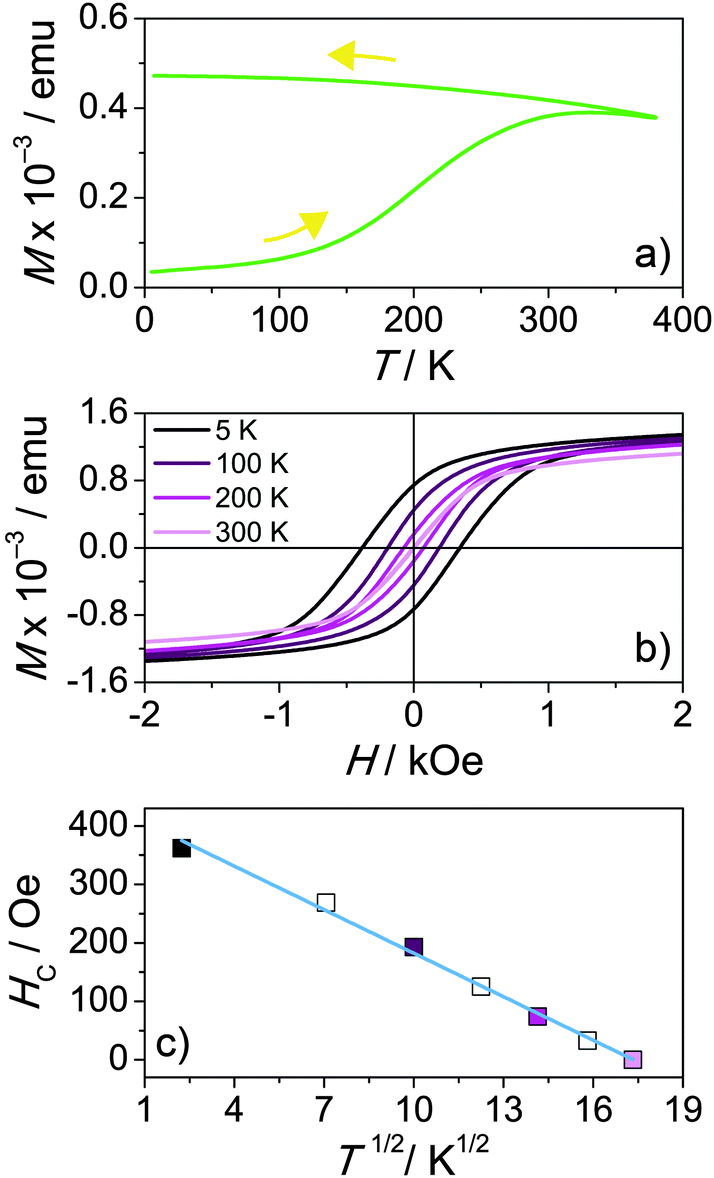 | ||
| Fig. 4 In-plane magnetic properties of mesoporous LFO thin films. (a) ZFC/FC curves measured in an applied field of 100 Oe. (b) Hysteresis curves within the field range of ±2 kOe at different temperatures of 5 K, 100 K, 200 K and 300 K. (c) Plot of HCvs. T1/2. The blue line is a linear fit to the data. | ||
Fig. 4b further shows that HC decreases with increasing temperature and exhibits a T1/2 dependence. This kind of dependence is usually observed for non-interacting single-domain particles below TB.47–49Fig. 4c presents a linear fit of the HC data against T1/2 according to the expression:

Spinel ferrites have been shown to undergo conversion reactions at low potential when used as an anode material in Li-ion batteries.51–53 These electrochemical reactions are not fully reversible and lead to large volume changes and significant hysteresis between charge and discharge, which is partly why they will likely not be used in practical (next-generation) energy storage devices, despite high theoretical specific capacities. However, in recent years, it has been shown that spinel ferrites such as ZnFe2O4 can store Li via topotactic insertion at relatively high potentials (≥1.0 V with respect to Li+/Li).54–56 This should allow for the possibility of modulating the magnetization in a facile and reversible manner, which makes them well-suited model materials for such studies, particularly those that exhibit room temperature magnetic properties.
All magnetization modulation experiments were carried out in situ in a SQUID magnetometer at room temperature and in an applied field of 100 Oe (see CAD drawing of the home-built cell in Fig. S4, ESI†). Li half-cells were assembled by using LiTFSI in EMIM-TFSI as the electrolyte. An ionic liquid-based electrolyte was chosen because of the low vapor pressure, allowing measurements under reduced pressure. Fig. 5a shows a representative cyclic voltammogram between 3.2 and 1.4 V with respect to Li+/Li at a scan rate of 1 mV s−1 collected after some cycles when the performance reached a stable level (see also voltammetry sweeps in Fig. S5, ESI†).
 | ||
| Fig. 5 In situ tuning of magnetization via electrochemical Li insertion into mesoporous LFO thin films. (a) Cyclic voltammogram at a scan rate of 1 mV s−1. (b) Relative change in magnetization at room temperature measured in an applied field of 100 Oe and (c) corresponding potential (dashed line) and current (red line) versus the time. | ||
The mesoporous LFO films exhibit pronounced redox peaks at about 1.6 V (cathodic) and 1.9 V (anodic), thereby indicating the presence of distinct Li insertion sites. The overall shape of the cyclic voltammogram is consistent with that of other Fe-based oxides.41,57 A small cathodic current is observed between 3.2 and 1.9 V. According to experimental results and first-principles modelling,57,58 Li uptake in this potential range leads to the reduction of tetrahedral Fe3+ ions. Because Fe2+ ions have a larger radius, the tetrahedral sites become unfavorable, and thus they either exchange places with octahedral Li+ ions or hop to vacant neighboring (octahedral) sites.38,59 The latter scenario appears more likely since Li3Fe5O8 with space group P4332, which is apparently an intermediate in the electrochemical reaction of LFO with Li, has both Fe and Li ions residing solely on octahedral sites.
A schematic of the Li insertion/extraction into/from alpha-LFO is shown in Fig. 6. With increasing population of Fe ions on octahedral sites, the magnetic moment increases, albeit only slightly (from 1 to 2 in the ΔM/M vs. t curve in Fig. 5b). This is due to altered exchange interactions between the Fe ions in the spinel lattice. Their magnetic moments are coupled by superexchange interaction through intermediate oxygen ions. The inter-sublattice exchange, which is stronger than the intra-sublattice exchange, is known to be antiferromagnetic (antiparallel) in nature. However, because the sublattice magnetization is not equal due to the different population of Fe ions on tetrahedral and octahedral sites (Fe8tet[Li4Fe12]octO32 per unit cell), LFO shows ferrimagnetic behavior. Given that Fe2+ ions apparently only reside on octahedral sites, Li insertion into the parent material must lead to an increase in magnetic moment. Upon lowering the potential further, a relatively large cathodic peak is seen at about 1.6 V, and the Fe3+ ions on both the octahedral and tetrahedral sites are reduced. This reduction leads to a monotonic decrease in magnetic moment (see Fig. 5b). The lowest moment or, in other words, largest relative change in magnetization is found when the current becomes positive (anodic), as is evident from Fig. 5b (from 2 to 3 in the ΔM/M vs. t curve). Then, Li is extracted from the host lattice and the Fe2+ ions are re-oxidized on the reverse sweep, which is accompanied by an increase in magnetic moment until the “initial” state is recovered. As expected on the basis of the behavior during Li insertion, the steepest increase occurs between 3 and 4 in the ΔM/M vs. t curve, in line with the current response in Fig. 5c. The measured magnetization modulation is as high as 4% with 1 mV s−1 of scan rate. More importantly, the process is highly reversible, as can be seen from Fig. 5 and Fig. S5 (ESI†).
 | ||
| Fig. 6 Schematic of the topotactic Li insertion/extraction into/from LFO during potentiodynamic cycling in the 3.2 to 1.4 V range with respect to Li+/Li. (1) Tetrahedral Fe3+ ions are reduced and (2) likely hop to vacant octahedral sites. (3) With further Li insertion, Fe3+ ions on both the tetrahedral and octahedral sites are reduced, leading to a monotonic decrease in magnetic moment. (4) On reversing the potential sweep (change from cathodic to anodic current), Li is extracted from the spinel lattice and the “initial” state is recovered. | ||
Fig. 7 shows an STEM image of the lithiated LFO material after 20 cycles as well as selected-area electron diffraction (SAED) patterns collected before and after cycling. From these data, it is apparent that the mesoporous structure is little affected because the crystalline phase does not change upon lithiation. The latter can be clearly seen from the radial integration patterns, which also confirm that irreversible microstructural effects (notable grain growth, partial amorphization, etc.) during topotactic Li insertion/extraction can be ruled out.
 | ||
| Fig. 7 Electron microscopy of mesoporous LFO thin films in a lithiated state (1.4 V with respect to Li+/Li) after 20 cycles. (a) Low-magnification high-angle annular dark-field STEM image. (b) SAED before and after cycling and (c) corresponding radial integration patterns. The line pattern corresponds to ICSD reference card no. 35769 for alpha-LFO.7 | ||
Overall, the experimental results are consistent and agree qualitatively with the few data in existence, but further work is needed to confirm the hypotheses. However, we note that more detailed studies by Mössbauer spectroscopy or other state-of-the-art characterization techniques are difficult because of the low amount of material in polymer-templated mesostructured thin films.
Conclusions
In summary, we have shown, for the first time, that α-LiFe5O8 can be templated using large diblock copolymers to produce mesostructured thin films with cubic pore symmetry. As evidenced by different characterization techniques (TEM, GISAXS, XRD, XPS, etc.), the sol–gel derived material is well-defined and shows no signs of impurity phases after thermal crystallization, despite the large Fe excess. Likewise, no signs of major structural changes are observed. The magnetization results indicate ferrimagnetic ordering below 300 K. Cyclic voltammetry and ex situ electron microscopy analysis of the mesoporous α-LiFe5O8 thin film electrodes in two-electrode Li battery cells using an ionic liquid-based electrolyte indicate that the spinel ferrite can reversibly store Li in the potential range from 3.2 to 1.4 V – with the electrochemical reactions proceeding in a topotactic way – which allows control and tuning of the magnetization at room temperature. These results are exciting and show that the approach of reversibly modulating the bulk magnetic properties via facile Li insertion has potential for the development of novel device concepts and design principles.Acknowledgements
We thank Dr Marcus Rohnke and Dr Thomas Leichtweiss for assistance with the TOF-SIMS and XPS measurements, respectively. We also thank Dr Robert Kruk and Dr Subho Dasgupta for fruitful discussions. This work was partly carried out with the support of the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT). Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We thank Dr Matthias Schwartzkopf for assistance in using beamline P03. Financial support by the German Academic Exchange Service (T. B.) and the German Research Foundation (H. H., grant no. DA 1781/1-1 and HA 1344/34-1) is gratefully acknowledged.Notes and references
- M. Sugimoto, J. Am. Ceram. Soc., 1999, 82, 269–280 CrossRef CAS.
- V. G. Harris, IEEE Trans. Magn., 2012, 48, 1075–1104 CrossRef CAS.
- E. A. Schwabe and D. A. Campbell, J. Appl. Phys., 1963, 34, 1251–1253 CrossRef CAS.
- G. O. White and C. E. Patton, J. Magn. Magn. Mater., 1978, 9, 299–317 CrossRef CAS.
- H. J. Masterson, J. G. Lunney, D. Ravinder and J. M. D. Coey, J. Magn. Magn. Mater., 1995, 140, 2081–2082 CrossRef.
- F. O. Ernst, H. K. Kammler, A. Roessler, S. E. Pratsinis, W. J. Stark, J. Ufheil and P. Novak, Mater. Chem. Phys., 2007, 101, 372–378 CrossRef CAS.
- A. Tomas, P. Laruelle, J. L. Dormann and M. Nogues, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1615–1617 CrossRef.
- S. J. Marin, M. Okeeffe and D. E. Partin, J. Solid State Chem., 1994, 113, 413–419 CrossRef CAS.
- A. Ahniyaz, Solid State Ionics, 2002, 151, 419–423 CrossRef CAS.
- P. B. Braun, Nature, 1952, 170, 1123 CrossRef CAS.
- A. J. Pointon and R. C. Saull, J. Am. Ceram. Soc., 1969, 52, 157–160 CrossRef CAS.
- G. Bandyopadhyay and R. M. Fulrath, J. Am. Ceram. Soc., 1974, 57, 182–186 CrossRef CAS.
- P. Innocenzi and L. Malfatti, Chem. Soc. Rev., 2013, 42, 4198–4216 RSC.
- B. Smarsly and M. Antonietti, Eur. J. Inorg. Chem., 2006, 1111–1119 CrossRef CAS.
- C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS.
- Y. Deng, J. Wei, Z. Sun and D. Zhao, Chem. Soc. Rev., 2013, 42, 4054–4070 RSC.
- D. Gu and F. Schüth, Chem. Soc. Rev., 2013, 43, 313–344 RSC.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- D. Grosso, C. Boissière, B. Smarsly, T. Brezesinski, N. Pinna, P. A. Albouy, H. Amenitsch, M. Antonietti and C. Sanchez, Nat. Mater., 2004, 3, 787–792 CrossRef CAS PubMed.
- S. Y. Choi, M. Mamak, N. Coombs, N. Chopra and G. A. Ozin, Adv. Funct. Mater., 2004, 14, 335–344 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- G. A. Ozin, K. Hou, B. V. Lotsch, L. Cademartiri, D. P. Puzzo, F. Scotognella, A. Ghadimi and J. Thomson, Mater. Today, 2009, 12, 12–23 CrossRef CAS.
- T. von Graberg, P. Hartmann, A. Rein, S. Gross, B. Seelandt, C. Röger, R. Zieba, A. Traut, M. Wark, J. Janek and B. M. Smarsly, Sci. Technol. Adv. Mater., 2011, 12, 025005 CrossRef.
- C. Weidmann, K. Brezesinski, C. Suchomski, K. Tropp, N. Grosser, J. Haetge, B. M. Smarsly and T. Brezesinski, Chem. Mater., 2012, 24, 486–494 CrossRef CAS.
- P. Hartmann, D. Lee, B. M. Smarsly and J. Janek, ACS Nano, 2010, 4, 3147–3154 CrossRef CAS PubMed.
- C. Reitz, P. M. Leufke, R. Schneider, H. Hahn and T. Brezesinski, Chem. Mater., 2014, 26, 5745–5751 CrossRef CAS.
- C. Reitz, C. Suchomski, J. Haetge, T. Leichtweiss, Z. Jagličić, I. Djerdj and T. Brezesinski, Chem. Commun., 2012, 48, 4471–4473 RSC.
- C. Reitz, C. Suchomski, V. S. K. Chakravadhanula, I. Djerdj, Z. Jagličić and T. Brezesinski, Inorg. Chem., 2013, 52, 3744–3754 CrossRef CAS PubMed.
- M. Bagheri, M. A. Bahrevar and A. Beitollahi, Ceram. Int., 2015, 41, 11618–11624 CrossRef CAS.
- M. Gu, B. Yue, R. Bao and H. He, Mater. Res. Bull., 2009, 44, 1422–1427 CrossRef CAS.
- Y. Wang, H. Zhao, M. Li, J. Fan and G. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS.
- P. Leufke, R. Kruk, R. Brand and H. Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 094416 CrossRef.
- A. K. Mishra, A. J. Darbandi, P. M. Leufke, R. Kruk and H. Hahn, J. Appl. Phys., 2013, 113, 033913 CrossRef.
- J. Weissmüller, R. N. Viswanath, D. Kramer, P. Zimmer, R. Würschum and H. Gleiter, Science, 2003, 300, 312–315 CrossRef PubMed.
- C. Lemier, S. Ghosh, R. N. Viswanath, G.-T. Fei and J. Weissmüller, MRS Proc., 2011, 876, R2.6.
- C. Thiele, K. Dörr, L. Schultz, E. Beyreuther and W.-M. Lin, Appl. Phys. Lett., 2005, 87, 162512 CrossRef.
- S. Dasgupta, B. Das, M. Knapp, R. A. Brand, H. Ehrenberg, R. Kruk and H. Hahn, Adv. Mater., 2014, 26, 4639–4644 CrossRef CAS PubMed.
- T. Tsuchiya, K. Terabe, M. Ochi, T. Higuchi, M. Osada, Y. Yamashita, S. Ueda and M. Aono, ACS Nano, 2016, 10, 1655–1661 CrossRef CAS PubMed.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Müller-Buschbaum, M. Trebbin, S. Förster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- J. Haetge, C. Suchomski and T. Brezesinski, Inorg. Chem., 2010, 49, 11619–11626 CrossRef CAS PubMed.
- S. Y. An, I. B. Shim and C. S. Kim, J. Magn. Magn. Mater., 2005, 290, 1551–1554 CrossRef.
- W. B. White and B. A. DeAngelis, Spectrochim. Acta, Part A, 1967, 23, 985–995 CrossRef CAS.
- M. N. Iliev, V. G. Ivanov, N. D. Todorov, V. Marinova, M. V. Abrashev, R. Petrova, Y. Q. Wang and A. P. Litvinchuk, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 174111 CrossRef.
- A. K. Arora, M. Rajalakshmi, T. R. Ravindran and V. Sivasubramanian, J. Raman Spectrosc., 2007, 38, 604–617 CrossRef CAS.
- G. Gouadec and P. Colomban, J. Raman Spectrosc., 2007, 38, 598–603 CrossRef CAS.
- E. C. Stoner and E. P. Wohlfarth, Philos. Trans. R. Soc., A, 1948, 240, 599–642 CrossRef.
- W. C. Nunes, W. S. D. Folly, J. P. Sinnecker and M. A. Novak, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 014419 CrossRef.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120 CrossRef.
- L. Neel, Ann. Geophys., 1949, 5, 99–136 Search PubMed.
- L. A. de Picciotto and M. Thackeray, Mater. Res. Bull., 1986, 21, 583–592 CrossRef CAS.
- S. Permien, S. Indris, U. Schürmann, L. Kienle, S. Zander, S. Doyle and W. Bensch, Chem. Mater., 2016, 28, 434–444 CrossRef CAS.
- J.-M. Tarascon, P. Poizot, S. Laruelle, S. Grugeon and L. Dupont, Nature, 2000, 407, 496–499 CrossRef PubMed.
- X. Guo, X. Lu, X. Fang, Y. Mao, Z. Wang, L. Chen, X. Xu, H. Yang and Y. Liu, Electrochem. Commun., 2010, 12, 847–850 CrossRef CAS.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS.
- M. M. Thackeray, W. I. F. David and J. B. Goodenough, Mater. Res. Bull., 1982, 17, 785–793 CrossRef CAS.
- W. Zhou, Y. Wang, L. Zhang, G. Song and S. Cheng, Int. J. Electrochem. Sci., 2015, 10, 5061–5068 CAS.
- M. Catti and M. Montero-Campillo, J. Power Sources, 2011, 196, 3955–3961 CrossRef CAS.
- C. J. Chen, M. Greenblatt and J. V. Waszczak, J. Solid State Chem., 1986, 64, 240–248 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional SEM/TEM images, Raman analysis, XPS spectra, magnetic hysteresis curves, CAD drawing of the SQUID cell, and data from magnetization tuning. See DOI: 10.1039/c6tc02731h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |