
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards solution processable air stable p-type organic semiconductors: synthesis and evaluation of mono and di-fluorinated pentacene derivatives†
Jaroslaw M.
Wasikiewicz
a,
Laila
Abu-Sen
a,
Andrew B.
Horn
a,
Jacobus M.
Koelewijn
b,
Adam V. S.
Parry
a,
John J.
Morrison
*a and
Stephen G.
Yeates
a
aOrganic Materials Innovation Centre, School of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: john.morrison@manchester.ac.uk
bvan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands
First published on 11th July 2016
Abstract
The synthesis of a series of mono and di-fluorinated TIPS pentacene compounds for application as organic semiconductors is reported. Namely the novel 1-fluoro-6,13-bis[triisopropylsilyl-ethynyl]pentacene (1-fluoro-TIPS-pentacene), 2-fluoro-TIPS-pentacene and the recently reported 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 syn:anti isomeric mixtures of 1,(8:11)-difluoro-TIPS-pentacenes and 2,(9:10)-difluoro-TIPS-pentacenes. For the first time these materials have been subjected to a study of the relationship between molecular structure and thermal/photochemical stability. The changes in the pentacene HOMO via alteration of fluorine substitution is studied and related to environmental stability. As predicted, it was found that increasing the number of electron-withdrawing fluorine substituents increases stability to photodegradation. It was also observed that stability increases (and HOMO decreases) when fluorine is present on the 1- and 1,(8:11)-positions closer to the central rings rather than in the 2- and 2,(9:10)-positions. Comparative transistor measurements for the fluoro-derivatives demonstrated competitive device performance in comparison to underivatised TIPS-pentacene. Full characterisation and NMR assignment of these materials has been accomplished by comparison over the series for the first time.
:1 syn:anti isomeric mixtures of 1,(8:11)-difluoro-TIPS-pentacenes and 2,(9:10)-difluoro-TIPS-pentacenes. For the first time these materials have been subjected to a study of the relationship between molecular structure and thermal/photochemical stability. The changes in the pentacene HOMO via alteration of fluorine substitution is studied and related to environmental stability. As predicted, it was found that increasing the number of electron-withdrawing fluorine substituents increases stability to photodegradation. It was also observed that stability increases (and HOMO decreases) when fluorine is present on the 1- and 1,(8:11)-positions closer to the central rings rather than in the 2- and 2,(9:10)-positions. Comparative transistor measurements for the fluoro-derivatives demonstrated competitive device performance in comparison to underivatised TIPS-pentacene. Full characterisation and NMR assignment of these materials has been accomplished by comparison over the series for the first time.
Introduction
Pentacene has long been considered to be a benchmark p-type organic semiconductor due to its high hole field effect mobility of μFET = 1.1 cm2 V−1 s−1 on hard alumina, 3.3 cm2 V−1 s−1 on a soft polymer substrate and 35 cm2 V−1 s−1 for a single crystal at room temperature.1 The main drawbacks however, are its insolubility in common solvents and low photo-oxidative stability. 6,13-Bis(triisopropylsilylethynyl)pentacene (TIPS-pentacene) 1 is a benchmark material due to both its improved solvent solubility and photo-oxidative stability and promising organic field effect mobility.2 Recent reports on the subtle interplay of solvent selection and processing conditions have shown that μFET = 2.7 cm2 V−1 s−1 can be achieved.3 To date many structural pentacene adaptations have been investigated to increase transistor performance,4 however, these derivatives are still prone to photochemical degradation during processing.Appropriate fluorine substitution of the pentacene ring is an attractive strategy to maintaining high mobility but with enhanced photochemical stability due to both its small steric size, comparable to hydrogen, and high electronegativity.5 Tetra (1,2,3,4) and octa (1,2,3,4,8,9,10,11) fluorination of TIPS-pentacene has been shown to lower the HOMO to a level where ambipolar and electron transport behaviour are observed. However, such studies generally indicate lower device performance and lifetime.6 More recent studies have considered difluorinated TIPS-pentacene, 7, both for texture control and self-patterning of halogenated surfaces7 but also for enhanced charge transport properties and improved photochemical stability, 9.8 In this contribution we take the opportunity to report more fully on the synthesis of 7 and 9 providing a full NMR characterisation of the syn- and anti-isomeric mixtures. Further we extend this study to consider mono-fluorinated TIPS-pentacene, 6 and 8, where the issue of isomeric mixtures is absent and take the opportunity to contribute further on the extent and position of fluorination and aryl-fluoroaryl interactions on photochemical stability and device performance.9
Experimental
Materials, instrumentation and methods
Thermal gravimetric analysis was performed on TA Instruments TGA 5000 analyser at a heat rate 10 °C min−1 with average sample weight of 2 mg.Differential Scanning Calorimetry measurements were carried out on a PerkinElmer Diamond calorimeter at the scanning rate of 10 °C min−1 with average sample weight of 1 mg for 30–300 °C cycles and 5 mg for 25–150 °C scans.
Cyclic voltammetry scans were performed using BASI C3 Cell Stand, Bioanalytical Systems Inc, with Pt wire and Pt spiral as counter and working electrode respectively and Ag/AgNO3 in DMF as a reference electrode. Samples were measured in DCM solution in 0.1 M tetrabutylammoniumhexafluorophosphate as a supporting electrolyte at samples concentration of 1 × 10−3 M.
UV-VIS and fluorescence spectra were collected by Varian UV-VIS-NIR 5000 Spectrometer and Cary Eclipse Fluorescence Spectrometer respectively. Scan rate was 150 nm min−1, with solvent baseline subtracted and sample concentration of 5 × 10−5 mol dm−3.
Optical microscopy images were taken using Olympus C7070WZ digital camera placed on the top of Olympus microscope. Samples were placed on Linkam hot stage and heated up from RT to 150 °C held for 5 min and cooled down to RT at rates 10 °C min−1.
X-Ray Diffraction (XRD) data of powder samples was collected on Philips X'PERT diffractometer with multichannel detector (X'Celerator) at the scanning rate, 2° min−1; 2Θ angle scanning range, 4–50°. Heating and cooling rates between the temperature scans were 10 °C min−1.
ACPI mass spectra were taken on Micromass Platform II spectrometer (ESI†).
NMR spectra were taken at 400 and 500 MHz using Bruker Advance spectrometers. 13C data was obtained at 100 MHz and all signals found are listed prior to assignment. C–H signals are differentiated by DEPT 90. Full assignments including H–F and C–F couplings are reported in the ESI.†
Thin films and devices were prepared on heavily doped silicon (n++) substrates with 290 nm of thermally grown silicon dioxide. The substrates were cleaned by washing and sonication in acetone, propan-2-ol and methanol (Sigma Aldrich) followed by UV-ozone treatment. The surfaces of the substrates were then treated with octadecyltrichlorosilane (OTS) monolayers to reduce the surface energy, passivate traps and improve thin film growth. OTS treatment was done by spin coating of an OTS solution from chloroform (Sigma Aldrich) (3 mM), 3 times in ambient conditions. Unreacted OTS was removed by sonication in chloroform and dried at 105 °C. OTS treated substrates were all washed with organic solvents and dried with nitrogen before being transferred to the vacuum chamber for thin film deposition.
Evaporation of the organic semiconductors was performed in a modified Edwards Auto306 vacuum evaporator with a base pressure of 5 × 10−7 mbar. Nominally, 50 nm of compounds 1,6,7,8 and 9 was deposited onto substrates held at 40 °C at a rate of 0.05 nm s−1.
To create thin film transistors, the as prepared substrates were transferred to a separate Edwards Auto500 vacuum evaporator (base pressure 3 × 10−7 mbar) for gold evaporation. Nominally, 50 nm of gold was deposited on top of the organic layers at a rate of 0.1 nm s−1 through a shadow mask.
Thin films were characterised using a Park XE100 atomic force microscope in tapping mode and a Bruker D8 discover X-ray diffractometer for out-of-plane X-ray diffraction. Transfer and output curves were obtained and device performance was analysed in the saturation regime using the standard equation.
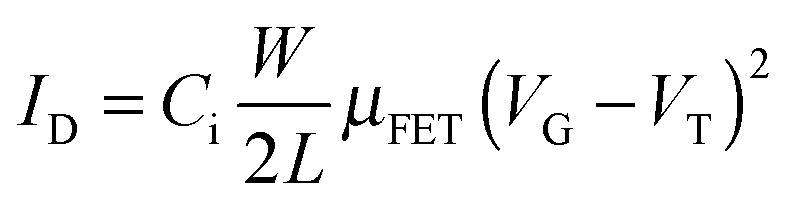
Photo-oxidative kinetic degradation measurements were performed on purified TIPS-pentacenes dissolved in solvent (Sigma Aldrich), which had previously been exposed to air for 24 hours by loosening the cap to allow for free exchange of air, over a range of concentrations and formulations. UV visible spectra were obtained on a Nicolet Evolution 300 spectrometer using 1 cm quartz cells. The cells were protected from light until each experiment began, at which point an initial spectrum was recorded to define A0. The cells were placed on a laboratory bench under conventional 60 W, SP35 fluorescent lighting (General Electric, 2850 lm). The solutions were repeatedly scanned at prescribed intervals until less than 5% of the starting acene remained. For concentrations below 1 × 10−5 M, UV-visible spectra were recorded without further dilution. For solutions at higher concentrations, samples were degraded under the same conditions and further diluted to 1 × 10−5 M immediately prior to the recording of spectra.
Synthesis
Common chemical reagents and solvents were purchased from Sigma Aldrich and used without any further purification. Dry THF was obtained by passing HPLC grade THF through Innovative Technology Solvent Purification System Inc. Brominated fluoroxylenes 10 and 11 were synthesised as detailed in the ESI.† 1,4-Anthracenedione was purchased from ACBR, GmbH KG, 3-fluoro-o-xylene and triisopropylsilyl-acetylene from Fluorochem Ltd.Synthesis of quinones
Synthesis of 6,13-bis[isopropylsilyl-ethynyl] (TIPS) derivatives
Other TIPS derivatives shown in Scheme 1 were obtained following the above procedure, giving:
 | ||
| Scheme 1 Synthetic route to the fluoropentacenes. (i) KI, DMA, 90 °C, 48–66 h. (iia) RLi, THF, 0 °C to RT, 24 h, (iib) Sn(II)Cl2·2H2O, AcOH, acetone, RT, 24 h. | ||
Results and discussion
Synthesis
The most common synthetic route to substituted pentacenes is by coupling cyclohexanedione with a phtalaldehyde derivative to obtain the relevant quinone,10 followed by further substitution to the ketones and eventual aromatisation. Alternatively, quinones can be obtained through reduction of phtalic acid derivatives to diols followed by bromination11 and cyclisation with p-benzoquinone.12 Here, we utilise synthesis of fluoro-quinones directly from brominated fluoro-o-xylenes and commercially available quinones 12 and 13. Although this Cava13 reaction usually generates products with a low yield (20–30%) from benzoquinone, unlike other common methods it is a straight forward one or two step process, depending on commercial availability of the appropriate bromide.Direct bromination of o-xylenes under UV irradiation with NBS11 in CCl4 provides a fast route to low cost starting materials. Unfortunately on writing the use of CCl4 is becoming more restricted due to its environmental impact. To counter this we also employed a ‘Green Chemistry’ route utilising methyl acetate as solvent14 along with a simple 60 W incandescent light source and found equally good bromination results. In the case of 4-fluoroxylene tetrabromination was readily accomplished however for 3-fluoroxylene we found the majority of product to be tri-brominated despite large excesses of NBS, extended reaction times and prolonged reflux being employed. Fortunately the following Cava cyclisation reaction with quinones will operate provided one or more halides are present on each benzyl unit.
In the case of the novel mono-fluoro-quinones 2 and 4 we obtained product yields of 60–80% from coupling reaction of brominated fluoroxylenes 10 and 11 with 1,4-anthracenedione 12 and lower yields (20–30%) for the difluoroquinones 3 and 5.
On the synthesis of difluoro-TIPS pentacenes 7 and 9 the final products are mixtures of syn and anti isomers in a 50/50 ratio and although several attempts were made we were not able to separate them to extract a single isomer by chromatography.
Anthony et al.7 reports some electronic characteristics of 7 and 9 however no details of the synthesis or chemical characterisation of these materials has been published until upon writing this manuscript where Anthony et al.8 reports briefly upon a synthetic pathway to 9. This report again contains no chemical characterisation which is likely due to the difficulties in reporting the complex nature of the NMR spectra obtained from such fluorinated aromatic systems. Brown et al.15 reports the synthesis of difluoro-TIPS 7 by a four step procedure via reduction of 3-fluorophthalic acid to the diol followed by Swern oxidation to the dialdehyde then condensation with cyclohexadione to give the quinone. Further reaction with lithium acetylide as reported here concurrently gives rise to an inseparable mixture of syn and anti isomers.
In Browns report nineteen 13C signals are reported for the mixture 7 with the furthest two downfield at 160.27 and 157.27 ppm. We have identified these signals as the C1 and C8:10 (syn and anti) carbons which are each split by 255.3 Hz due to the 1-(8:11) fluoro substitution. On careful analysis of our 13C spectra we find 49 signals for 7 which are attributed to a combination of long range (up to 4 bond or in some cases 5) C–F coupling and the presence of the mixed syn and anti isomers. These factors, combined with the asymmetry of the monofluoro TIPS-pentacene compounds 6 and 8 make for complex NMR spectra which can be used to advantage. As we have a series of related isomers comparison between the four can reveal subtle differences between each, particularly where similar C–F coupling constants are involved. Using this combined with some good reference data on monofluorinated polycylic aromatics16 has allowed us to complete almost comprehensive 1H and 13C assignments for all the molecules synthesised. These complete assignments are included in tabular form in the ESI.†
Thermal properties of products
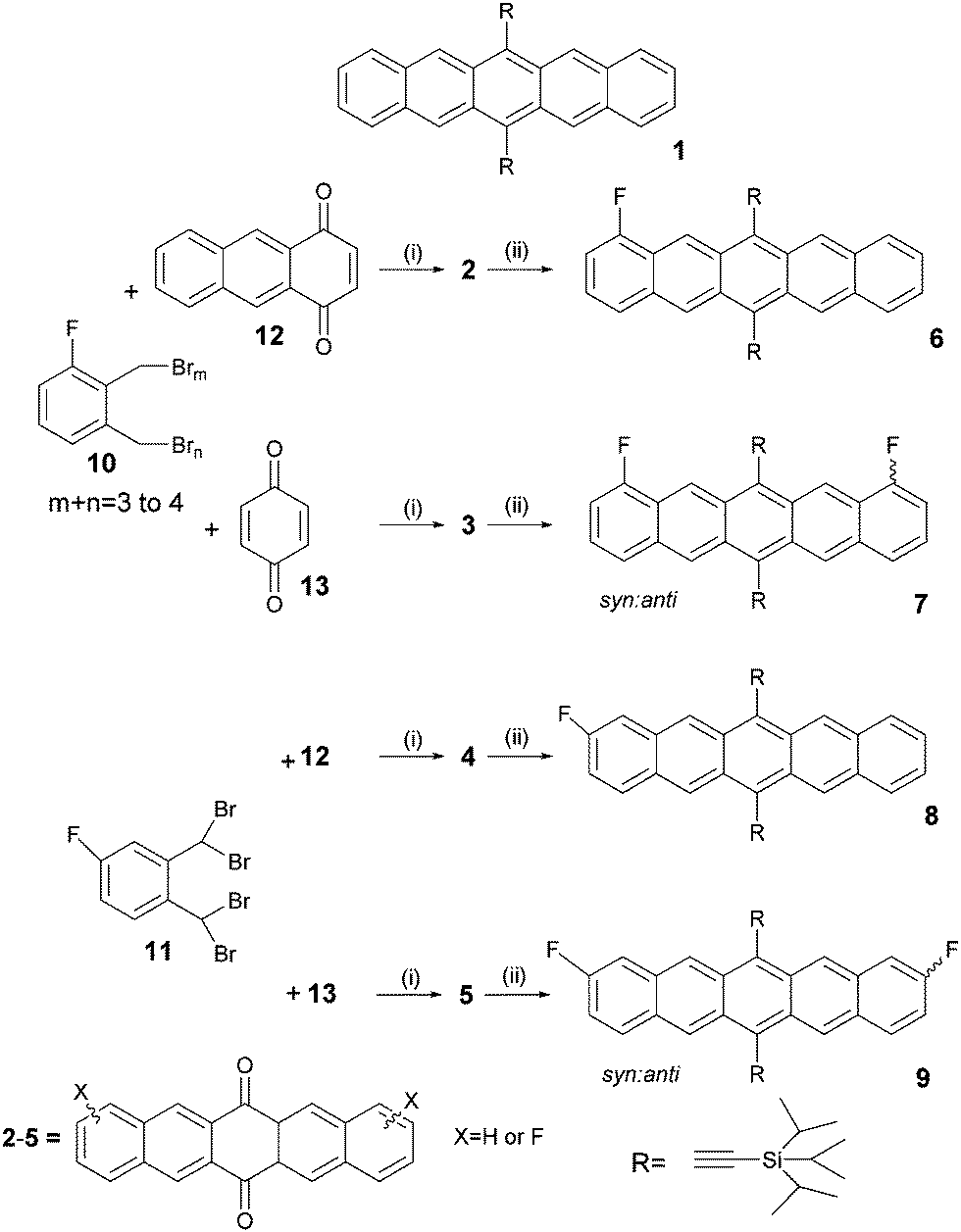
Thermal analysis of all molecules revealed a increase in melting point of the four TIPS-fluoro derivatives up to 279 °C compared with 250 °C for TIPS-pentacene. In all cases melting was imidiately followed by degradation as indicated by an exothermic peak (Fig. 1).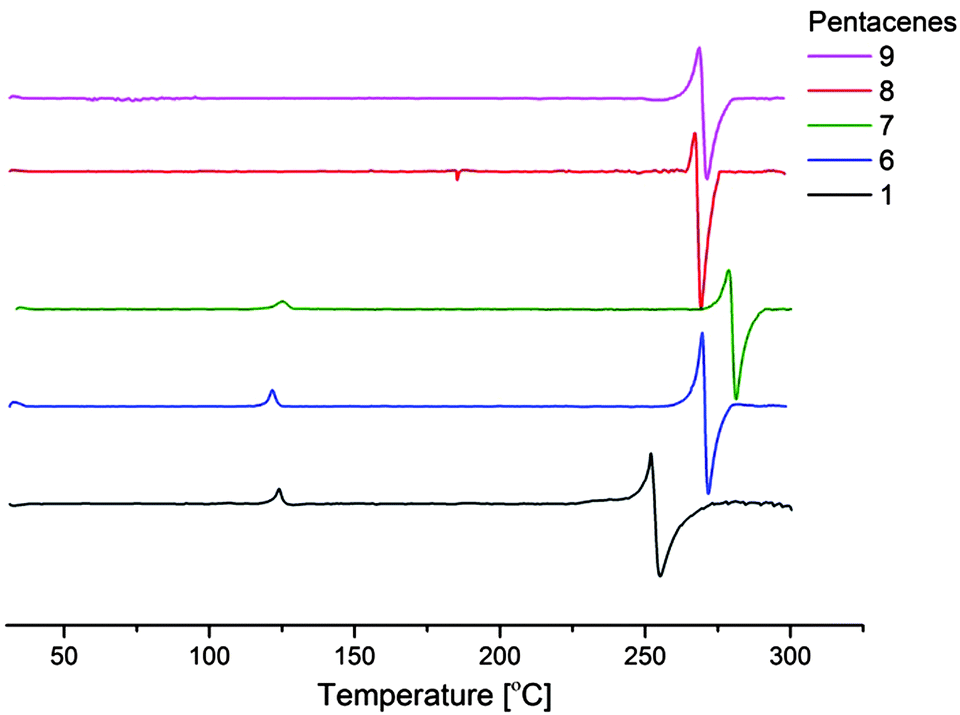 | ||
| Fig. 1 DSC scans of pentacenes 1, 6, 7, 8 and 9. | ||
Anthony et al.17 reports that 1 undergoes a reversible phase transition at 124 °C which has believed to be due to a thermal reorganisation of the triisopropylsilyl groups. As this is observed to cause cracks in TIPS-pentacene crystals, operation of devices over this transition is likely to compromise device performance.
For fluoro pentacenes 6 and 7 as well as for TIPS-pentacene a fully reversible thermal transition at around 125 °C was detected by DSC and XRD powder diffraction (see ESI†), but for 8 and 9 only by XRD powder diffraction. This suggests that even for these molecules there is still a structural reorganisation in the molecular packing at elevated temperature, however, the change is too small to be detected by calorimetry measurements. This appears to indicate that the presence of fluorine substitution with its small steric size has little impact upon crystal packing throughout this series although it might be interesting to conduct further studies on 8 and 9 which appear to have lower energy transitions.
Electrochemistry
Extracting data received from UV-VIS spectroscopy (ESI†) and cyclic voltammetry (Fig. 2) the HOMO levels and band gaps of the products were established. The HOMO–LUMO energy gap for TIPS-pentacene (1) and the inner (1,(8:11)-positions) fluoro-substituted, 6 and 7 derivatives is the same 1.8 eV, but slightly lower (less than 0.1 eV) for outer fluoro-substituted, 8 and 9. The reported measured value for TIPS-pentacene is 1.60–1.7 eV with a computational simulation at 1.5 eV.18 A small difference in measured values can be explained by the fact that the optical energy gap is an approximation not an absolute value.18
 | ||
| Fig. 2 Cyclic voltammetry of compounds 1, 6, 7, 8, and 9 measured in DCM solution at 1 × 10−3 mol dm−3. | ||
On estimation of HOMO levels using the equation19EHOMO = −(E[onset,ox vs. Fc/Fc+] + 5.1) (eV) where Fc/Fc+ corresponds to the HOMO of ferrocene (latterly measured in situ as 0.40 eV) all the fluoro-derivatives show lower energy levels compared to TIPS-pentacene (Table 1).
| Compound | 1 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|
| E onset ox (eV) | 0.45 | 0.50 | 0.52 | 0.48 | 0.50 |
| HOMO (eV) | −5.15 | −5.20 | −5.22 | −5.18 | −5.20 |
The most notable result here shows that fluorination on the 1-position, 6, and 1,(8:11)-positions, 7, closer to the centre of the molecule, has the greatest influence on the derivatives HOMO levels. It would be interesting to investigate this further by fluorination of the 5,7,12 or 14 positions (or combinations) if a rational synthetic strategy could be devised.
Photochemical degradation studies
The photostability of TIPS-pentacene in solution has been shown to depend upon a number of factors, including TIPS-pentacene concentration, competition between solute–solute, solvent–solute and solvent–solvent interactions, and the concentration of dissolved oxygen.20 At low concentration (<1 × 10−3 M), the major decomposition channel is activation of dissolved oxygen by excited state (singlet) TIPS-pentacene, with a subsequent reaction to create predominantly a 6–13 endoperoxide. The half-life, t1/2, for photochemical degradation can be obtained from the decay of the main S0–S1 visible absorption at λmax (typically ∼643 nm) versus time under constant illumination. Since the reaction is pseudo-first order at low concentration. Depending on the excited O2 + TIPS-pentacene reaction, with the excited O2 derived from a single TIPS-pentacene excitation in excess O2, the decay constant k1 can be obtained from the slope of a plot of ln(At/A0) versus time t, where A0 is the initial absorbance at λmax and At is its evolution with time. Note that low concentration is also necessary to avoid potential non-linear behaviour during exposure at high concentrations due to the high optical density of the solutions. Representative curves are shown in Fig. 3 for the series of fluorinated TIPS-pentacenes in chloroform. A similar plot for the series in toluene is shown in the ESI.† Rate (decay) constants are obtained from slopes of these curves via linear regression and are tabulated in Table 2 alongside those for toluene.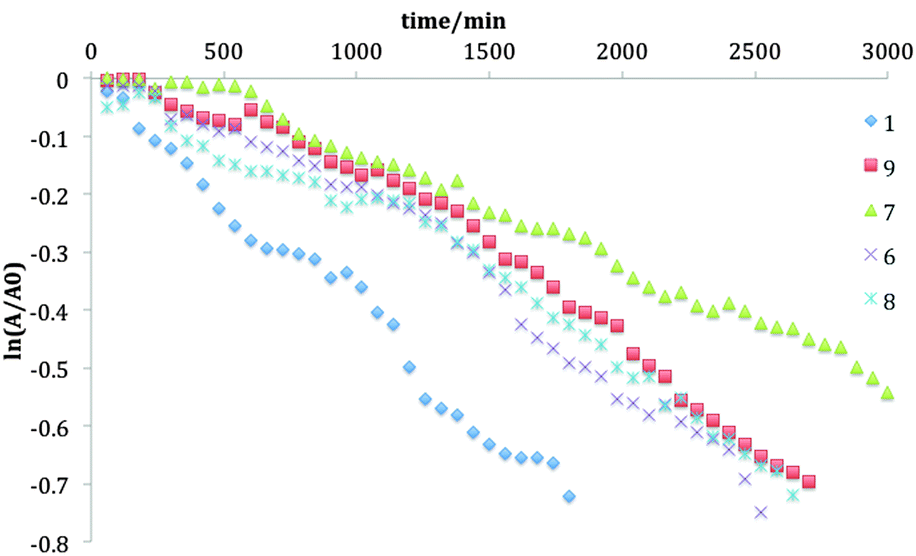 | ||
| Fig. 3 Plots of ln(At/A0) versus time t at 1 × 10−5 M in chloroform at room temperature of compounds 1, 6, 7, 8, and 9. | ||
| Chloroform | Toluene | |||||
|---|---|---|---|---|---|---|
| k 1/10−4 min−1 | R 2 | t 1/2/min | k 1/10−4 min−1 | R 2 | t 1/2/min | |
| 1 | 4.0 | 0.98 | 1700 | 6.1 | 0.97 | 1150 |
| 6 | 2.7 | 0.97 | 2350 | 3.7 | 0.98 | 1850 |
| 7 | 2.0 | 0.99 | 3450 | 2.3 | 0.92 | 2900 |
| 8 | 2.6 | 0.97 | 2700 | 4.7 | 0.97 | 1450 |
| 9 | 2.7 | 0.97 | 2550 | 4.5 | 0.98 | 1550 |
Photodegradation t1/2 for all the fluorinated derivatives are shorter in toluene compared to those in chloroform, for reasons described elsewhere.20 The inner difluorinated compound 7 was found to be most stable in both solvents, and all derivatives are more photostable than TIPS-pentacene. A positive correlation exists between the HOMO level and the lifetime in both solvents, as shown in Fig. 4, supporting the hypothesis that tailoring the HOMO level is a key factor in controlling the photochemical stability of TIPS-pentacene derivatives.
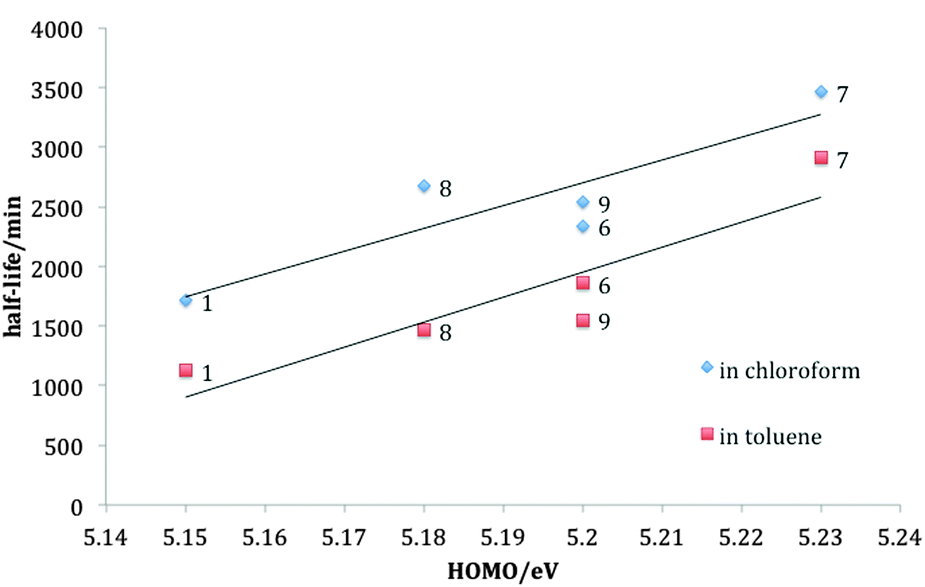 | ||
| Fig. 4 Plots half life versus HOMO level at 1 × 10−5 M for TIPS-pentacene 1 and fluorinated derivatives 6–9 in chloroform and toluene at room temperature. | ||
Transistor measurements
Despite the rationale of synthesising fluorinated TIPS pentacene to provide solution processable OSC materials, some reports on transistor measurements using tetra and octa-fluoro TIPS pentacenes have relied on using devices where the OSC has been sublimed.6 Anthony et al.7 reports success on solution based transistor fabrication using pentafluorobenzenethiol as a surface treatment prior to applying the single anti-isomers of 7 and 9. However, as we have not found any information as to how these have been achieved for comparative purposes transistors were sublimed. As such, all compounds were subject to the exact same processing and testing conditions (see ESI† for experimental details and full characterization). Table 3 and Fig. 5 show the average mobility extracted from devices made of compounds 1, 6, 7, 8 and 9. All of the devices have an on/off ratio greater than 1 × 104 and threshold voltages in the range of 0 to −4 V as displayed in the ESI† (Fig. S9).| Compound | #Devices | μ Mean/cm2 V−1 s−1 (×10−2) | μ Min/cm2 V−1 s−1 (×10−2) | μ Max/cm2 V−1 s−1 (×10−2) |
|---|---|---|---|---|
| 1 | 18 | 3.5 ± 0.8 | 2.0 | 4.9 |
| 6 | 16 | 2.2 ± 0.5 | 1.5 | 3.1 |
| 7 | 8 | 2.9 ± 0.3 | 2.2 | 3.3 |
| 8 | 7 | 3.8 ± 0.4 | 3.3 | 4.2 |
| 9 | 7 | 6.5 ± 0.5 | 4.8 | 8.1 |
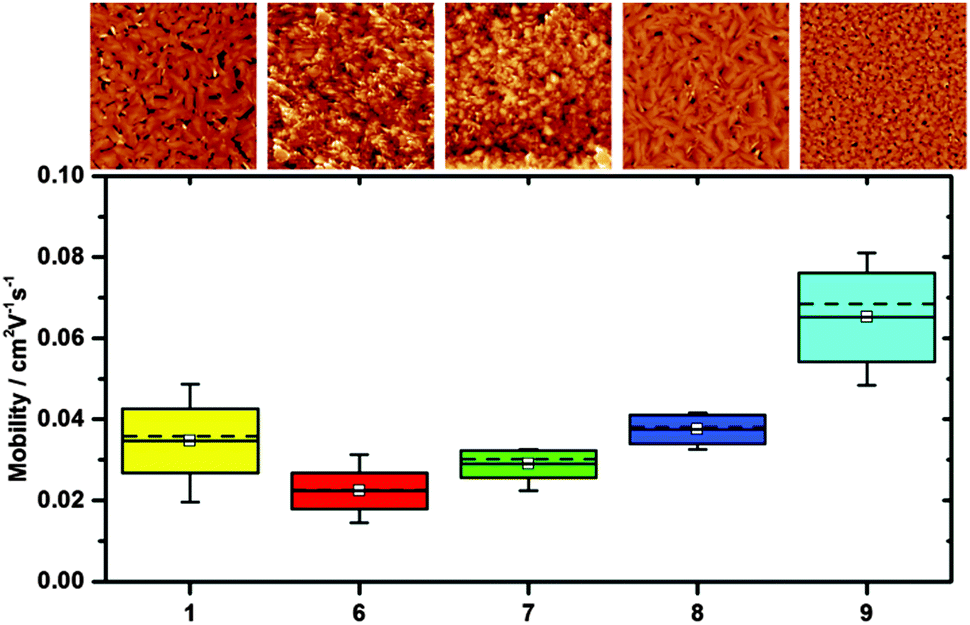 | ||
| Fig. 5 Mobility, mean-solid line, box represents the standard deviation and the whiskers the minimum and maximum values. The dotted line is the median value. | ||
All the novel compounds show performance comparable to the standard TIPS-pentacene derivative (1) with compound 9 demonstrating a slightly higher hole mobility. All materials have very similar thin film crystal structure (Fig. S7, ESI†) and the increased performance could be attributed to the more favorable thin film morphology (Fig. 5) in comparison to the other materials. Compound 9 forms small, well-interconnected crystallites unlike TIPS-pentacene where the larger crystals are separated by large voids, which could be detrimental to the charge transport. In conjunction with the stability of these fluorinated compounds, the favorable device performance and solubility for solution processed devices make them a promising class of material for organic thin film devices.
Conclusions
We observe a close link between the electrochemical measurement of a series of soluble fluorinated pentacenes HOMO's and their photochemical stability in solution. Where substitution of fluorine is present in the 1,(8:11)-positions 7 the photostability is greatly increased. This is believed to be due to the greater proximity of the electronegative fluorine to the central rings of the molecule where oxidation is most likely to take place and is supported by the correlation between this observation and the larger downward alteration of the pentacene HOMO.
Conversely, fluorine substitution further from the parent molecules core on the 2-position 8 and 2,(9:10)-positions 9 gives rise to smaller differences in the pentacene HOMO. Thermal studies show that the fluorinated derivatives tend to be more robust than the parent TIPS-pentacene 1.
Acknowledgements
JJM was supported by funding from the EPSRC (UK) via a Flagship Grant (FS/01/01/10) from the Innovative Electronic Manufacturing Research Centre, Loughborough. ASVP was supported by EPSRC EP/L019728/1. LA was supported by the Libyan Ministry of Education and partial sponsorship by Merck Chemicals Limited. The UoM thanks the EPSRC (EP/K039547/1) for the provision of Bruker NMR spectrometers and an Agilent SuperNova X-ray diffractometer. High resolution APCI, ASAP mass spectrometry was conducted by the EPSRC UK National Mass Spectrometry facility at Swansea university.Notes and references
- (a) O. D. Jurchescu, J. Baas and T. T. M. Palstra, Appl. Phys. Lett., 2004, 84, 3061–3063 CrossRef CAS; (b) T. W. Kelley, L. D. Boardman, T. D. Dunbar, D. V. Muyres, M. J. Pellerite and T. P. Smith, J. Phys. Chem. B, 2003, 107, 5877–5881 CrossRef CAS.
- (a) J. E. Anthony, D. L. Eaton and S. R. Parkin, Org. Lett., 2002, 4, 15–18 CrossRef CAS PubMed; (b) J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed; (c) J. Chen, J. Anthony and D. C. Martin, J. Phys. Chem. B, 2006, 110, 16397–16403 CrossRef CAS PubMed.
- (a) G. Giri, S. Park, M. Vosgueritchian, M. M. Shulaker and Z. Bao, Adv. Mater., 2014, 26, 487–493 CrossRef CAS PubMed; (b) G. Murtaza, I. Ahmad, H. Chen and J. Wu, Synth. Met., 2014, 194, 146–157 CrossRef CAS.
- G. R. Llorente, M. B. Dufourg-Madec, D. J. Crouch, R. G. Pritchard, S. Ogier and S. G. Yeates, Chem. Commun., 2009, 3059 RSC.
- (a) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato and S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138–8140 CrossRef CAS PubMed; (b) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Inoue and S. Tokito, Mol. Cryst. Liq. Cryst., 2006, 444, 225–232 CrossRef CAS.
- (a) C. R. Swartz, S. R. Parkin, J. E. Bullock, J. E. Anthony, A. C. Mayer and G. G. Malliaras, Org. Lett., 2005, 7, 3163 CrossRef CAS PubMed; (b) A. V. Subhas, J. Whealdon and J. Schrier, Comput. Theor. Chem., 2011, 966, 70–74 CrossRef CAS; (c) C.-H. Li, C.-H. Huang and M.-Y. Kuo, Phys. Chem. Chem. Phys., 2011, 13, 11148–11155 RSC.
- (a) O. D. Jurchescu, M. Feric, B. Hamadani, D. Mourey, S. Gundlach, S. Subramanian, B. Purushothaman, J. E. Anthony, T. Jackson and D. Gunlache, ECS Trans., 2008, 16, 283–289 CAS; (b) H.-Y. Tseng, B. Purushothaman, J. E. Anthony and V. Subramanian, Org. Electron., 2011, 12, 1120–1125 CrossRef CAS; (c) H. Zhang, Y. Yao, M. M. Payne, J. E. Anthony and J. W. Brill, Appl. Phys. Lett., 2014, 105, 073302 CrossRef; (d) J. W. Ward, R. Li, A. Obaid, M. M. Payne, D.-M. Smilgies, J. E. Anthony, A. Amassian and O. D. Jurchescu, Adv. Funct. Mater., 2014, 24, 5052–5058 CrossRef CAS.
- C. H. Kim, H. Hlaing, M. B. Payne, S. R. Parkin, J. E. Anthony and I. Kymissis, ChemPhysChem, 2015, 16, 1251–1257 CrossRef CAS PubMed.
- (a) M. Weck, A. R. Dunn, K. Matsumoto, G. W. Coates, E. B. Lobkovsky and R. H. Grubbs, Angew. Chem., Int. Ed., 1999, 38, 2741–2745 CrossRef CAS; (b) F. Ponzini, R. Zagha, K. Hardcastle and J. S. Siegel, Angew. Chem., Int. Ed., 2000, 39, 2323–2325 CrossRef CAS; (c) R. E. Gillard, J. F. Stoddart, A. J. P. White, B. J. Williams and D. J. Williams, J. Org. Chem., 1996, 61, 4504–4505 CrossRef CAS PubMed.
- (a) H. Meng, M. Bendikov, G. Mitchell, R. Helgeson, F. Wudl, Z. Bao, C. Siegrist and C.-H. Kloc, Adv. Mater., 2003, 15, 1090–1093 CrossRef CAS; (b) D. F. Perepichka, M. Bendikov, H. Meng and F. Wudl, J. Am. Chem. Soc., 2003, 125, 10190–10191 CrossRef CAS PubMed.
- (a) J. E Antony, G. A. Caldwell, R. S. Clough, J. C. Novack, M. M. Payne, D. H. Redinger and D. E. Vogel, WO 2009155106, 2009; (b) Z. Liang, Q. Tang, J. Liu, J. Li, F. Yan and Q. Miao, Chem. Mater., 2010, 22, 6438 CrossRef CAS.
- S. Mataka, G. B. Liu, T. Sawada, M. Kurisu and M. Tashiro, Bull. Chem. Soc. Jpn., 1994, 67, 1113–1119 CrossRef CAS.
- M. P. Cava, A. A. Deana and K. Muth, J. Am. Chem. Soc., 1959, 81, 6458–6460 CrossRef CAS.
- C. H. M. Amijs, G. P. M. van Klink and G. van Koten, Green Chem., 2003, 5, 470–474 RSC.
- B. A. Brown, J. Veres, R. M. Anemian, R. Williams, R. Thomas, S. D. Ogier and S. W. Leeming, PCT Int. Appl., WO2005055248 A220050616, 2005 Search PubMed.
- B. F. Lutnaes, G. Luthe, U. A. T. Brinkman, J. E. Johansen and J. Krane, Magn. Reson. Chem., 2005, 43, 588–594 CrossRef CAS PubMed.
- J. Chen, J. Anthony and D. C. Martin, J. Phys. Chem. B, 2006, 110, 16397–16403 CrossRef CAS PubMed.
- (a) O. Lobanova Griffith, J. E. Anthony, A. G. Jones and D. L. Lichtenberger, J. Am. Chem. Soc., 2010, 132, 580–586 CrossRef PubMed; (b) O. Ostroverkova, S. Shcherbyna, D. G. Cooke, R. F. Egerton, F. A. Hegmann, R. R. Tykwinski, S. R. Parkin and J. E. Anthony, J. Appl. Phys., 2005, 98, 033701/1 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- L. Abu-Sen, J. J. Morrison, A. B. Horn and S. G. Yeates, Adv. Opt. Mater., 2014, 2, 636–640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data and NMR interpretation. See DOI: 10.1039/c6tc01221c |
| This journal is © The Royal Society of Chemistry 2016 |