
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the electrochemical properties of silicon wafer by grafted-from micropatterned polymer brushes†
G.
Panzarasa‡
 *a,
G.
Soliveri
bc and
V.
Pifferi‡
*bc
*a,
G.
Soliveri
bc and
V.
Pifferi‡
*bc
aDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale “Amedeo Avogadro”, Viale T. Michel 11, 15100 Alessandria, Italy. E-mail: guido.panzarasa@uniupo.it
bDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: valentina.pifferi@unimi.it
cConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via Giusti 9, 50121 Firenze, Italy
First published on 26th November 2015
Abstract
Silicon wafer is the material of choice for microfabrication. However, the development of innovative on-chip electrochemical sensors using silicon is hampered by its poor electrochemical properties. In this article we demonstrate how grafted-from micropatterned polymer brushes dramatically enhance the electrochemical response of silicon electrodes. Our results are relevant not only for a deeper understanding of the structure and behavior of polymer brushes, but also for the combination of the versatility of surface-initiated polymerization and an innovative patterning technique (remote photocatalytic lithography) which paves the way for the fabrication of integrated devices.
Introduction
The development of reliable, fast and inexpensive signal-responsive material systems as sensing platforms to monitor target molecules is a flourishing field of investigation.1–3 The miniaturization and incorporation of such systems into microchip assemblies is highly desirable for the realization of compact, portable, implantable devices. Silicon wafer would be the substrate of choice for such applications, but even its nm-thick native oxide layer impairs its electrochemical response.4 To overcome this problem, a typical strategy involves the grafting of redox-active molecules onto the surface of hydride-terminated silicon, but this approach requires specific techniques such as handling of dangerous hydrofluoric acid for the removal of native oxide.5,6In this paper we report for the first time a potentially powerful approach for the fabrication of integrated lab-on-a-chip and microfluidic devices: we found that the electrochemical response of silicon wafer is dramatically improved when polymer brushes are grafted directly from its surface. Moreover, this behaviour is enhanced if the brushes are micropatterned.
Polymer brushes can be defined as dense arrays of chains tethered on a surface at one end.7 Thanks to their peculiar physico-chemical properties, they provide extraordinary opportunities for surface functionalization, generating thin films for integration into various devices.8,9 Being able to control the immobilization and preconcentration of analytes through specific interactions, thanks to the high concentration of functional groups exposed at the brush interface,10 polymer brushes can be used to significantly enhance the response of different kinds of sensors.11–13
Understanding the electrochemical properties of polymer thin films is of great importance for a wide range of technologies, from the development of biosensors to the miniaturization of batteries (battery-on-chip)14 for autonomous microelectronic devices and microsensors. Electrochemistry has already proven to be an useful tool to elucidate the internal structure of polymer brushes.15,16 The diffusion of probe molecules in and out of the macromolecular environments plays a key role in the response of polymer-based sensor materials or in the design of drug delivery systems. On a molecular level, the effect of the grafting density on the transport through thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) brushes grafted from gold substrates was investigated using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS).17 EIS has been used to determine the effectiveness of fluorinated poly(2-hydroxyethyl methacrylate) (PHEMA) films as barriers against the transport of redox species.18,19 Ion exchange inside polyelectrolyte brushes, in which the polymer chains have fixed electrostatic charges, is the subject of extensive study.20–23 However, a detailed understanding of their electrochemical behaviour has not been obtained yet. In addition, all of these studies have been performed for brushes grafted from highly conductive gold or indium tin oxide (ITO)-coated surfaces: not a single report is available in the literature for polymer brush-modified silicon electrodes.
Here, we would like to fill this gap by studying the electrochemical behaviour of homo- and co-polymer brushes made from 2-hydroxyethyl methacrylate (HEMA) and 2-aminoethyl methacrylate hydrochloride (AMA). Poly(HEMA) is a hydrophilic polymer with current uses in contact lenses, biocompatibilization and drug delivery.24 On the other hand, poly(AMA) is a cationic polymer which has already found promising applications in the biomedical field for the development of antimicrobial surfaces25 and for the bioconjugation of antigens and proteins, thanks to its primary amine groups allowing post-functionalization reactions.26
By means of cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) meaningful correlations were found between the charge transfer resistance, the length and the chemical composition of the brushes, revealing precious information about the structure and dynamics of the brushes themselves.
We then selected the most electrochemically-active brushes to perform micropatterning studies. Patterning is a fundamental tool for the engineering of surfaces, and micropatterned polymer brushes can display new useful features compared to bulk analogues.27,28 As expected, the electrochemical response of the patterned brushes was found to be dramatically different compared to that of non-patterned ones.
The crucial development reported in the present study demonstrates how, by choosing the thickness, composition and microstructuration of polymer brushes, we are able to tune the electrochemical behaviour of silicon wafer, an otherwise poor electrode material, with tremendous importance for the realization of integrated sensing devices.
Experimental
Materials and methods
All the chemicals were of reagent grade, purchased from Aldrich and used as received unless otherwise stated. Hydroxyethyl methacrylate (HEMA) was filtered through an inhibitor-remover column and stored at +4 °C until use. 2-Aminoethyl methacrylate hydrochloride (AMA, 90%, contains ∼500 ppm phenothiazine as a stabilizer) was used as received or purified by washing with inhibitor-free THF (for details, refer to the ESI†). The ATRP initiator (3-(2-bromoisobutyramido)propyl)triethoxysilane (BIB-APTES) was synthesized and used to functionalize silicon substrates as previously described.26 Water obtained from a Millipore MilliQ purification system (resistivity ≥ 18.2 MΩ cm−1) was thoroughly used. Silicon (100) wafers, single-polished, n-type, phosphorus doped, 3–6 Ω cm, with a native oxide layer of 1.5 ± 0.2 nm-thickness (ellipsometric), were purchased from Ultrasil Corporation.The thickness of polymer brushes was measured using a Filmetrics F20 reflectometer. Each data was the result of three measurements on different spots of a same sample. Scanning electron microscopy (SEM) was performed, on samples fixed on a conducting carbon tape, using a Jeol JSM 7600f Schottky field emission scanning electron microscope. A potentiostat/galvanostat Autolab PGSTAT204 (Metrohm, The Netherlands) equipped with a FRA module was employed to perform electrochemical measurements using a standard three-electrode cell with a saturated calomel, a Pt wire and a modified silicon wafer as reference, counter and working electrodes, respectively. Aqueous 0.1 M KCl was used as the supporting electrolyte. Cyclic voltammetry (CV) was performed by scanning the potential between −0.1 V and+0.4 V (SCE) (scan rate 0.1 V s−1). Electrochemical impedance spectroscopy (EIS) was carried out at −0.1 V, +0.1 V and +0.25 V (SCE) in the presence or in the absence of 3 mM K4Fe(CN)6 as a redox probe. Impedance data were fitted by using ZView 2.0 software.
Grafting-from of homo- and copolymer brushes by SI-ATRP
Surface-initiated ATRP was performed on both non-patterned and patterned substrates. To ensure reproducibility each series was polymerized in the same batch.PHEMA brushes were obtained using the following recipe: 0.0214 g (0.074 mmol) of tris(2-pyridylmethyl)amine (TPMA), 0.002 g (0.009 mmol) of CuBr2 and 0.023 g (0.13 mmol) of ascorbic acid were combined in a Schlenck flask under nitrogen and dissolved under stirring in 15.4 mL of a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v methanol–water mixture previously degassed by bubbling nitrogen, then 4.6 mL (0.038 mol) of degassed HEMA were added. Each substrate, contained in a Schlenck flask, was immersed in 5 mL of the resulting mixture under nitrogen and polymerization was allowed to proceed at 30 °C for the desired time. After polymerization the substrates were washed with ethanol, gently sonicated in the same solvent and dried under a nitrogen stream.
:1 v/v methanol–water mixture previously degassed by bubbling nitrogen, then 4.6 mL (0.038 mol) of degassed HEMA were added. Each substrate, contained in a Schlenck flask, was immersed in 5 mL of the resulting mixture under nitrogen and polymerization was allowed to proceed at 30 °C for the desired time. After polymerization the substrates were washed with ethanol, gently sonicated in the same solvent and dried under a nitrogen stream.
PAMA brushes were obtained using the same protocol described for PHEMA brushes, using 6.3 g (0.038 mol) of AMA and 20 mL of 4:1 v/v methanol–water mixture solution. After polymerization the substrates were washed with water, gently sonicated in the same solvent and dried under a nitrogen stream.
PHEMA-co-PAMA copolymer brushes were obtained using the same basic procedure for PHEMA brushes. For achieving the 80:20 HEMA–AMA monomer ratio, 3.6 mL (0.03 mol) of HEMA, 1.36 g (0.0082 mol) of AMA and 17 mL of methanol–water mixture were combined. For achieving the 50:50 HEMA–AMA monomer ratio, 2.3 mL (0.019 mol) of HEMA and 3.41 g (0.0206 mol) of AMA were combined with 18 mL of methanol–water. Polymerization proceeded at 30 °C for a chosen amount of time, the workup was the same as previously described.
Micropatterning by remote photocatalytic lithography
Patterning by remote photocatalytic lithography was achieved following our previously reported procedure.29 Transparent and mechanically robust anatase films with high photoactivity were deposited on glass slides from an acidic TiO2 sol using the electrochemically-assisted technique previously reported by us.30 Before use, the titania-coated glass slides were cleaned by rapid sonication in water followed by UV irradiation for 1 h. Remote photocatalytic lithography was performed by irradiating with 365 nm near-UV light from a Jelosil HG500 halogen lamp (230 V, 500 W, effective power density from 40 cm: 57.5 mW cm−2 between 280 and 400 nm) the initiator-functionalized silicon substrates through the titania-coated glass, with the photocatalytic oxide facing the substrate, separated from the surface by a 100 μm-thick Teflon spacer. Micropatterning was achieved using TEM grids (Gilder Grids, nickel, d 3.05 mm, square mesh) or hollow, circular grid supports (dext 3 mm, dhole 1 mm, from this point onwards referred to as “donuts”) as contact photomasks. To obtain the best pattern resolution, the initiator-functionalized substrates were subjected to remote photocatalytic lithography for 5 h.Results and discussion
Grafting-from polymer brushes by SI-ATRP
The grafting-from technique allows the direct growth of polymer chains by starting polymerization from an initiator-functionalized surface. If a controlled radical polymerization method such as Atom Transfer Radical Polymerization (ATRP) is used, brushes with increasing thickness can be obtained simply by varying the time of polymerization. The mechanism of ATRP is based on the reversible formation of radicals by halogen atom abstraction from an alkyl halide (typically a secondary or tertiary bromide) by a copper(I)–ligand complex. The latter breaks the C–Br bond of the initiator and the resulting carbon–carbon-centered radical attacks a monomer molecule thereby starting the chain growth. At the same time, copper(I) oxidizes to copper(II). Since this process is reversible Cu(II) acts as a deactivator, reducing the overall concentration of active radicals and thus the occurrence of termination events.31 In the Activators Regenerated by Electron Transfer (ARGET) ATRP, Cu(II) is continuously reduced to active Cu(I) by a reducing agent (Scheme 1) and thus only ppm amounts of copper salt are required, which is beneficial when working with metal-complexing monomers such as AMA. In the present work, polymer brushes were grown at 30 °C in a 4:1 v/v methanol–water mixture using a CuBr2/tris(2-pyridylmethyl)amine (TPMA) catalyst in the presence of an excess of ascorbic acid as a reducing agent.29 The following molar ratios were employed: CuBr2/TPMA/ascorbic acid/monomer = 0.036:0.3:0.5:150. The solvent mixture was chosen to ensure the complete solubilisation of monomers, also water is known to speed up the polymerization (“water-accelerated ATRP”).32,33 More importantly, under these conditions we were able to polymerize commercial AMA without the need to remove the added phenothiazine, which is considered a powerful inhibitor for conventional free-radical polymerization (more details are available in the ESI,† pp. S3–S5).
 | ||
| Scheme 1 (a) Schematic mechanism of ARGET ATRP. (b) Depiction of the grafting-from of polymer brushes from an ATRP initiator-functionalized surface. | ||
The kinetics of polymerization (Fig. S1, ESI†) showed a pseudo-linear increase of the brush thickness as a function of the polymerization time suggesting that good control was attained. The evolution of brush thickness was observed to slow down as a result of the increased fraction of AMA in the polymerization feed: this behaviour could be attributed to the so-called “halogen exchange effect”.34 If chloride ions are added in a concentration significantly higher than bromide, as for AMA homopolymerization in the present case (Cl/Br molar ratio >4000), the kinetics will be slowed down due to a reduced rate of radical formation, C–Cl being more stable compared to the C–Br bond.
Micropatterning by remote photocatalytic lithography
Patterning of polymer brushes at the micrometer or sub-micrometer scale can be achieved by a number of techniques. They are divided into several categories according to their main working principle: photolithography, colloidal lithography, soft lithography, direct writing techniques, and electron and particle-beam lithography, to name a few.35 Photolithography is now a mature and versatile approach, thanks to the major developments achieved during the last few decades. Among these, photocatalytic lithography has already demonstrated its great potential for surface patterning.36–38 We previously demonstrated the suitability of remote photocatalytic lithography as a versatile means for the patterning of polymer brushes.29,39 In the present work, this approach has been preferred compared to classic photolithography both to avoid the possible contamination from a residual photoresist and to make the procedure easier and faster. According to a generally accepted mechanism, when titanium dioxide is irradiated with UV light, electron–hole pairs are generated and split; the adsorbed water and oxygen were transformed into reactive oxygen species (ROS).40 Such ROS can migrate into the surrounding atmosphere to another surface from a considerable distance, of hundreds of microns, where they degrade adsorbed organic molecules.41Here, the target was the initiator-functionalized silicon wafer (Fig. 1a). As demonstrated in our previous work, the initiator molecules are completely removed from the exposed surfaces while those protected by the photomask retain their ability to start polymerization.
 | ||
| Fig. 1 (a) Scheme of remote photocatalytic lithography. (b) Photographs of three representative samples, non-patterned and patterned. (c,d) Optical microscopy images of donut-patterned (DONUT) and grid-patterned (GRID) brushes, respectively. | ||
The resulting pattern was then amplified into polymer brushes by performing SI-ATRP (Fig. 1b, c and Fig. S4, ESI†). Two different kinds of photomasks (the DONUT, to obtain a macropatterning and the TEM grid, to obtain a micropatterning) were employed to evaluate the impact of macro- and micro-structuration on the electrochemical performance of brush-decorated electrodes.
Electrochemical study of non-patterned polymer brushes
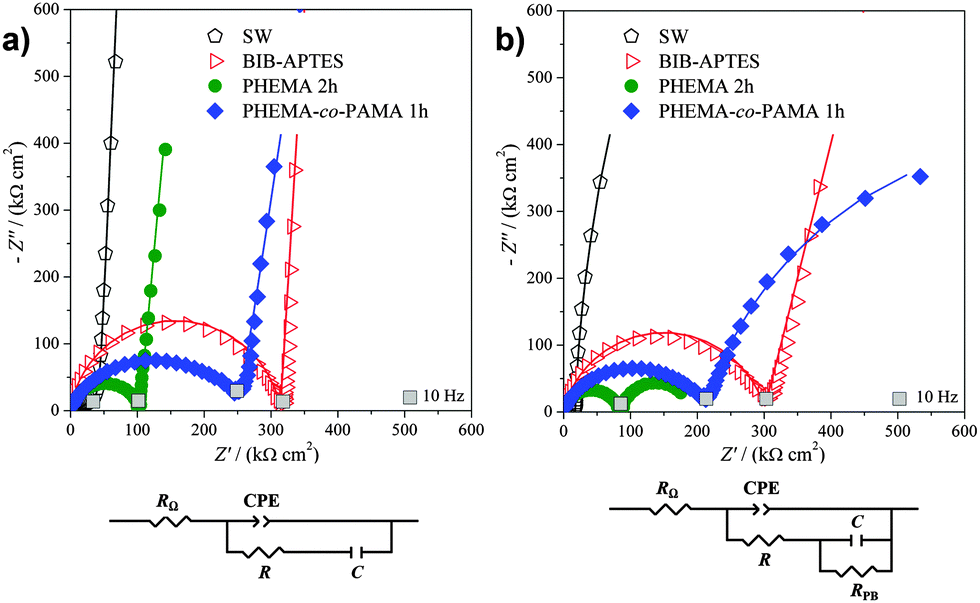 | ||
| Fig. 2 Complex plane plots measured for bare (SW) and initiator-functionalized (BIB-APTES) silicon wafer and for two representative brushes, along with the equivalent electrical circuits used for the interpretation of EIS plots, obtained (a) in the absence and (b) in the presence of a redox probe. | ||
The data were fitted using an equivalent circuit (Scheme 1a) as already reported in the literature for similar samples.23,43,44 The circuit is made of the electrolyte resistance RΩ in series with a constant phase element (CPE) (accounting for the film and silicon native oxide capacitance) and a resistance R (comprising the resistance of the silicon substrate and the polymeric film) in parallel (together represented by the semicircle). In series with the last element, a capacitance (C) can be found, representing the double layer and the substrate capacitance (described by the vertical straight line). The constant phase element CPE served to model the non-homogeneity of the electrode surface (giving a depressed semicircle). It is defined as (eqn (1)):
| CPE = [(CCPEiω)α]−1 | (1) |
| Sample | R (kΩ cm2) | C CPE (μF cm−2) | α |
|---|---|---|---|
| SW | 19 | 0.04 | 0.63 |
| BIB-APTES | 315 | 0.0006 | 0.90 |
| PHEMA | 103 | 0.007 | 0.85 |
| PHEMA-co-PAMA 80:20 |
255 | 0.01 | 0.71 |
The reported R values are in accordance with the trend of the semicircle diameters shown in Fig. 2a. Bare silicon wafer (“SW”) and initiator-functionalized silicon wafer (“BIB-APTES”) showed the lowest and highest values of resistance R, respectively, suggesting that the initiator layer behaves as a strong electrical insulator. The resistance R for the brushes presents intermediate values, thanks to the hydrophilic and polarizable groups, which increased their conductivity. Moreover, PHEMA brushes are more conductive with respect to PHEMA-co-PAMA 80:20 ones.
Bare silicon and initiator-functionalized silicon showed the highest and lowest values of CCPE, respectively. When the electrode surface is covered with an insulator layer (e.g. the BIB-APTES layer), the overall capacitance decreases because the capacitance of the insulating layer sums to that of the native oxide. PHEMA brushes, as well as the (PHEMA-co-PAMA) brushes, had lower insulating properties, thanks to hydrophilic –OH groups and electrically charged primary amine groups.
As previously mentioned, the α values can give information about surface homogeneity. The low α values found for bare silicon and the copolymer brushes indicate a lower surface homogeneity, due to the presence of native oxide or a non-homogeneous polymer. On the other hand, the initiator layer and PHEMA brushes are characterized by really homogeneous surfaces, giving α values very close to 1.
The EIS data trends for bare and initiator-functionalized silicon wafer were unaffected by the addition of the redox probe potassium ferrocyanide (Fig. 3b), confirming the electrochemical inertness of those samples. Contrarily, the polymer brush impedance at low frequencies showed important changes: the vertical straight line became another semicircle, requiring the introduction of RPB, the resistance to probe electron transfer, in parallel to C (Scheme 1b). This indicates that the reaction of the redox probe took place efficiently at brush-modified electrodes.20–23,44,45 The phenomenon could be due to interactions between the negatively charged probe with polar –OH (PHEMA) and positively charged –NH3 groups (PHEMA-co-PAMA).
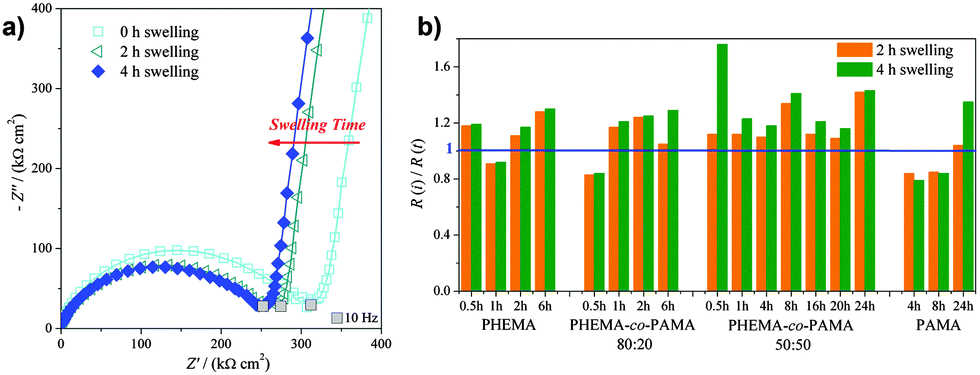 | ||
| Fig. 3 (a) Complex plane plots showing the swelling of 1 h-polymerized PHEMA-co-PAMA 80:20 brushes. (b) Comparison of the swelling behaviour of all the investigated brushes. | ||
From an electrochemical point of view, the swelling-collapse behaviour will affect the charge-transfer processes, the interface resistance and the capacitance of the brushes. EIS allows following this process as a function of time, obtaining information about the mobility and the internal structure of polymer chains.44
To monitor the swelling process (a representative complex plane plot is shown in Fig. 3a), EIS spectra were immediately recorded after immersion in the supporting electrolyte and then after 2 and 4 hours of immersion. The only detectable change was the decrease in the diameter of the semicircle, corresponding to a decrease of R, indicating an increase in polymer permeability with swelling. Fig. 3b presents the swelling behaviour for all the analysed brushes, reported as the ratio between the initial R(i) and the time-dependent R(t). The straight line at R(i)/R(t) = 1 corresponds to the value registered immediately after immersion. The data shown suggested that swelling reached completion after the first 2 h. For times longer than 4 h the impedance spectra were superimposable. This time was considered as the optimum time to reach complete swelling and allow equilibration of the brushes with the electrolyte.
 | ||
| Fig. 4 Behaviour of R as a function of thickness: (a) the complex plane plot for a representative 1 h-polymerized PHEMA-co-PAMA 80:20 brush and (b) comparison among the series. These data were obtained without the redox probe. | ||
 | ||
| Fig. 5 Behaviour of the RPB as a function of thickness: (a) the complex plane plot for representative PHEMA-co-PAMA brushes and (b) comparison among the series. These data were obtained in the presence of the redox probe (3 mM). | ||
Fig. 4a shows the complex plane spectra of the complete series of PHEMA-co-PAMA 80:20 brushes as an example: only the diameter of the semicircle increased with increasing thickness, without relevant changes in the other parameters. A higher thickness can be related to higher values of R, since the electrostatic charge carriers (ions, according to a generally accepted theory46) face a longer way to reach the electrode surface. In Fig. 4b the R values are reported for all the series to allow a comparison. PAMA and PHEMA-co-PAMA 50:50 brushes had the lowest values: apparently, the high density of charged groups helping the movement of ions inside the brushes was a more determinant factor than thickness. The R values of the PHEMA-co-PAMA 50:50 brushes seemed to be independent of thickness, probably due to a random distribution of the two monomers. PHEMA brushes showed an intermediate behaviour, while the highest values were found for the PHEMA-co-PAMA 80:20 ones. In general, R increased proportionally to the thickness, probably because longer chains impaired diffusion.
EIS spectra were then collected in the presence of the redox probe. According to previous swelling studies, the brushes were allowed to completely swell for 4 h, then ferrocyanide (3 mM) was added to the supporting electrolyte and the analysis was carried out after additional 2 h to allow the probe diffusion in the brushes and any other possible interaction to take place. While for PHEMA the interaction with the redox probe is supposed to occur simply by diffusion, PAMA and PHEMA–PAMA copolymer brushes can be considered as polyelectrolytes due to the presence of positively charged primary amine groups, which can facilitate their interaction with the anionic redox probe and consequently the electron transfer. According to Huck et al.,46 one molecule of ferrocyanide would be supposed to coordinate with four amine groups, at last for PAMA brushes. For the 80:20 and 50:50 copolymer brushes, different degrees of coordination would be possible due to the more or less intra- and inter-chain proximity of AMA units (we assume that the copolymers are random).
As previously discussed, in the complex plane plot at low frequencies the vertical straight line becomes a semicircle for all the polymers, indicating that the brushes interact with the probe: a lower resistance RPB means an easier reaction of the probe at the electrode. All the series were found to interact with the probe, but different RPB values were obtained depending on the brush composition and thickness. Fig. 5a shows the PHEMA-co-PAMA 80:20 series as an example, where the lowest resistances were obtained for intermediate thicknesses, probably as a result of a compromise between the quantity of charged groups that can interact with the probe and the thickness. Fig. 5b reports all the obtained RPB values. The lowest resistance values were displayed by the thickest PHEMA brushes. Probably, probe diffusion was facilitated by the homogeneous distribution of polar but neutral –OH groups while the thicker polymer layer allowed storing a higher concentration of probe, which in turn was forced to react at the electrode giving a low resistance. PAMA brushes exposed at the surface only charged amine groups, promoting strong interactions, which at the same time increased the overall resistance due to local collapse after the coordination of the probe with amine groups, blocking the diffusion of other ferrocyanide molecules. For the PHEMA-co-PAMA 80:20 series higher resistances were obtained in comparison to PHEMA, but the best situation was reached for intermediate polymerization times, probably thanks to a sufficient density of charged groups, while the electron transfer was not complicated by excessive chain length. Similar or even lower values were obtained in comparison with PAMA, even more considering the thicknesses, indicating how the presence of only amine groups was not so favourable to obtain low resistance values. In particular in the case of high thicknesses, brushes with a low density of amine groups were more desirable. The PHEMA-co-PAMA 50:50 series displayed the highest RPB values: it is likely that those brushes suffered from the so-called hydrophobic or ion-pairing collapse, which has been described for similar polyelectrolytes, e.g. poly[2-(methacryloyloxy)ethyl]trimethylammonium chloride (PMETAC). Such transition between the extended and collapsed conformational states could be due to a strong interaction between the cationic groups of the brushes with scarcely hydrated, large and highly polarizable ions such as ClO4− or, in this case, [Fe(CN)6]4−. The hydrophobic collapse typically results in a large water loss for the brush, thus increasing the overall electrical resistance.23 However, for higher thicknesses the resistance values decrease, probably due to the increase in the number and availability of interaction sites for the probe.
The influence of patterning on the electrochemical properties of polymer brushes
The previously described electrochemical analyses allowed us to select two types of brushes to study the effect of patterning: 1 h-polymerized PHEMA-co-PAMA 80:20 brushes and 2 h-polymerized PHEMA brushes were chosen due to their desirable properties, especially low resistance to probe reaction. For each brush type, three samples were prepared: a non-patterned one, a macropatterned one with a donut shape (“DONUT”, Fig. 1c) and a micropatterned one with a TEM grid (“GRID”, Fig. 1d and Fig. S4, ESI†).
The electrochemical response of electrodes modified with patterned polymer brushes was dramatically different compared to that of the non-patterned ones. Significant changes were observed in their EIS plots for both the low and high frequency zones suggesting different (and more efficient) interaction mechanisms with the redox probe.
According to Fig. 6a, for PHEMA series there was a clear increase in the first semicircle diameter as a function of patterning, reflecting changes in the film resistance (R) of the surface. The trend observed for R, PHEMA(non-patterned) < DONUT < GRID, was plausible considering the reduced amount of active polymer for the patterned samples (of the order of 8 μg cm−2 for non-patterned brushes and 0.5 μg cm−2 for patterned brushes), and the correspondingly decreased permeability. On the other hand, the values obtained for the resistance toward the probe reaction RPB, reported in Table 2 for both the oxidation (+0.1 V) and reduction (+0.25 V) potentials of the redox probe, were very similar for the non-patterned and DONUT samples, but were slightly increasing for the GRID sample. Since the resistance values remained in the same order of magnitude, this difference was probably due to an overall increase in disorder as a result of micropatterning.
 | ||
| Fig. 6 Complex plane plots showing the effect of patterning in the presence of the redox probe (3 mM). | ||
| Sample | PHEMA | PHEMA-co-PAMA 80:20 |
||
|---|---|---|---|---|
| +0.1 V | +0.25 V | +0.1 V | +0.25 V | |
| Non-patterned | 1413 | 91 | 3190 | 160 |
| DONUT | 1289 | 88 | 1048 | 104 |
| GRID | 1759 | 172 | 623 | 52 |
The situation observed for the PHEMA-co-PAMA 80:20 brushes is far more different. The trend of the first semicircle diameters is GRID < PHEMA-co-PAMA(non-patterned) < DONUT, which was different from that observed for PHEMA samples. The highest R value was measured for the DONUT sample, probably due to the reduced overall density of charged groups, which improved ion permeability, compared to the non-patterned sample. Interestingly, the lowest R value was obtained for the GRID sample, since in this case a high concentration of charged groups was directly exposed to the electrolyte, enhancing the lateral diffusion. The RPB reported in Table 2 assigned the lowest value to the GRID sample, followed by the DONUT. It is noteworthy that these values were consistently lower compared to those obtained for the corresponding non-patterned PHEMA-co-PAMA sample, confirming the relevance of patterning to increase the redox probe uptake ability of the brushes. Moreover, GRID also showed a lower resistance than non-patterned PHEMA, which had the lowest resistance among all the non-patterned samples.
All these results clearly demonstrate the power of patterning and especially micropatterning to control the values of probe reaction resistance. We hypothesize that its profound effect on the overall electrochemical response of polymer brushes could be the result of a combination of factors. First: micropatterned polymer brushes, compared to the non-patterned ones, display a more open surface towards the surrounding electrolyte (due to a wider perimeter border between the brush-covered and uncovered areas), which would result in more efficient diffusion processes of the redox probe. Second: for the same reason, the presence of microstructures would increase the density of functional groups directly exposed to the electrolyte, promoting their interaction with the probe.
Conclusions
The need for on-chip electrochemical sensors calls for innovative microfabrication strategies. In this article we demonstrated for the first time that the silicon wafer modified with grafted-from polymer brushes holds all the premises for the development of integrated electrochemical microsensors. We employed PHEMA, PAMA and their copolymers as a proof-of-concept. However, it is possible to tune the characteristics of the brushes e.g. to make them negatively charged, less or more hydrophilic, to confer antifouling properties (especially for biological applications) simply by choosing the appropriate monomer. More importantly, this means that the system can be designed for specific target molecules. Hydrophilic polymer brushes allowed a typical redox probe, ferrocyanide, to react at an otherwise electrochemically inert surface, suggesting that the grafted chains behaved as “tentacles” to capture the redox probe and holding it in the proximity of the electrode surface. Moreover, such behaviour was found to be dramatically enhanced for micropatterned polymer brushes. This is another unprecedented result which paves the way for miniaturization and on-chip applications, especially considering the remote photolithography approach employed for the micropatterning: mild, dry and highly compatible with conventional microfabrication processes.Acknowledgements
The authors want to thank Dr Gianluigi Marra (Eni Donegani Research Centre for Renewable Energies and Environment, Novara) for kindly providing SEM images. Prof. Silvia Ardizzone and Prof. Luigi Falciola (Università degli Studi di Milano, Department of Chemistry) are especially acknowledged for their support and helpful comments for manuscript preparation.References
- U. Guth, W. Vonau and J. Zosel, Meas. Sci. Technol., 2009, 20, 042002 CrossRef.
- G. Soliveri, V. Pifferi, G. Panzarasa, S. Ardizzone, G. Cappelletti, D. Meroni, K. Sparnacci and L. Falciola, Analyst, 2015, 140, 1486–1494 RSC.
- V. Pifferi, G. Soliveri, G. Panzarasa, S. Ardizzone, G. Cappelletti, D. Meroni and L. Falciola, RSC Adv., 2015, 5, 71210–71214 RSC.
- S. Xun, X. Song, L. Wang, M. E. Grass, Z. Liu, V. S. Battaglia and G. Liu, J. Electrochem. Soc., 2011, 158, A1260 CrossRef CAS.
- S. Ciampi, P. K. Eggers, G. Le Saux, M. James, J. B. Harper and J. J. Gooding, Langmuir, 2009, 25, 2530–2539 CrossRef CAS PubMed.
- R. D. Rohde, H. D. Agnew, W. S. Yeo, R. C. Bailey and J. R. Heath, J. Am. Chem. Soc., 2006, 128, 9518–9525 CrossRef CAS PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- S. Minko, Polym. Rev., 2006, 46, 397–420 CAS.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- J.-K. Chen, Z.-Y. Chen, H.-C. Lin, P.-D. Hong and F.-C. Chang, ACS Appl. Mater. Interfaces, 2009, 1, 1525–1532 CAS.
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC.
- M. Welch, A. Rastogi and C. Ober, Soft Matter, 2011, 7, 297–302 RSC.
- J.-K. Chen, J.-H. Wang, C.-C. Cheng, J.-Y. Chang and F.-C. Chang, Appl. Phys. Lett., 2013, 102, 151906 CrossRef.
- Y. Wang, M. Hung, C.-H. Lin, H. Lin and J. Lee, Chem. Commun., 2011, 47, 1249–1251 RSC.
- Electropolymerization: Concepts, Materials and Applications, ed. S. Cosnier and A. Karyakin, Wiley, 2010 Search PubMed.
- Functional Polymer Films, ed. W. Knoll and R. C. Advincula, Wiley, 2011, vol. 1–2 Search PubMed.
- T. A. García, C. A. Gervasi, M. J. Rodríguez Presa, J. I. Otamendi, S. E. Moya and O. Azzaroni, J. Phys. Chem. C, 2012, 116, 13944–13953 Search PubMed.
- M. R. Bantz, E. L. Brantley, R. D. Weinstein, J. Moriarty and G. K. Jennings, J. Phys. Chem. B, 2004, 108, 9787–9794 CrossRef CAS.
- E. L. Brantley, T. C. Holmes and G. K. Jennings, J. Phys. Chem. B, 2004, 108, 16077–16084 CrossRef CAS.
- E.-Y. Choi, O. Azzaroni, N. Cheng, F. Zhou, T. Kelby and W. T. S. Huck, Langmuir, 2007, 23, 10389–10394 CrossRef CAS PubMed.
- C. Combellas, F. Kanoufi, S. Sanjuan, C. Slim and Y. Tran, Langmuir, 2009, 25, 5360–5370 CrossRef CAS PubMed.
- T. Alonso-García, M. J. Rodriguez-Presa, C. Gervasi, S. Moya and O. Azzaroni, Anal. Chem., 2013, 85, 6561–6565 CrossRef PubMed.
- T. Alonso-Garcia, C. A. Gervasi, M. J. Rodriguez-Presa, E. Gutierrez-Pineda, S. E. Moya and O. Azzaroni, J. Phys. Chem. C, 2013, 117, 26680–26688 CAS.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- A. E. Madkour, J. M. Dabkowski, K. Nüsslein and G. N. Tew, Langmuir, 2009, 25, 1060–1067 CrossRef CAS PubMed.
- Y. Zhang, N. Islam, R. G. Carbonell and O. J. Rojas, ACS Appl. Mater. Interfaces, 2013, 5, 8030–8037 CAS.
- M. E. Welch and C. K. Ober, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1457–1472 CrossRef.
- T. Chen, I. Amin and R. Jordan, Chem. Soc. Rev., 2012, 41, 3280–3296 RSC.
- G. Panzarasa, G. Soliveri, K. Sparnacci and S. Ardizzone, Chem. Commun., 2015, 51, 7313–7316 RSC.
- G. Maino, D. Meroni, V. Pifferi, L. Falciola, G. Soliveri, G. Cappelletti and S. Ardizzone, J. Nanopart. Res., 2013, 15, 2087–2097 CrossRef.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- W. Huang, J. B. Kim, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 1175–1179 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- C. H. Peng, J. Kong, F. Seeliger and K. Matyjaszewski, Macromolecules, 2011, 44, 7546–7557 CrossRef CAS.
- J. K. Chen and C. J. Chang, Materials, 2014, 7, 805–875 CrossRef CAS.
- N. Blondiaux, S. Zürcher, M. Liley and N. Spencer, Langmuir, 2007, 23, 5329–5334 CrossRef PubMed.
- H. Notsu, W. Kubo, I. Shitanda and T. Tatsuma, J. Mater. Chem., 2005, 15, 1523–1527 RSC.
- K. Komori, J. Nada, M. Nishikawa, H. Notsu, T. Tatsuma and Y. Sakai, Anal. Chim. Acta, 2009, 653, 222–227 CrossRef CAS PubMed.
- G. Panzarasa, G. Soliveri, S. Ardizzone and K. Sparnacci, Mater. Today, 2015, 2, 4183–4189 CrossRef.
- O. Carp, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- W. Kubo and T. Tatsuma, J. Am. Chem. Soc., 2006, 128, 16034–16035 CrossRef CAS PubMed.
- G. Soliveri, V. Pifferi, R. Annunziata, L. Rimoldi, V. Aina, G. Cerrato, L. Falciola, G. Cappelletti and D. Meroni, J. Phys. Chem. C, 2015, 119, 15390–15400 CAS.
- R. N. Vyas and B. Wang, Int. J. Mol. Sci., 2010, 11, 1956–1972 CrossRef CAS PubMed.
- F. Zhou, H. Hu, B. Yu, V. L. Osborne, W. T. S. Huck and W. Liu, Anal. Chem., 2007, 79, 176–182 CrossRef CAS PubMed.
- M. R. Presa and L. Gassa, Anal. Chem., 2009, 81, 7936–7943 CrossRef PubMed.
- E. Spruijt, E. Choi and W. Huck, Langmuir, 2008, 24, 11253–11260 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and data. See DOI: 10.1039/c5tc03367e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |