
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceYttria-stabilized zirconia microspheres: novel building blocks for high-temperature photonics†
Elisabeth W.
Leib
a,
Robert M.
Pasquarelli
b,
Jefferson J.
do Rosário
b,
Pavel N.
Dyachenko
c,
Sebastian
Döring
a,
Anke
Puchert
a,
Alexander Yu.
Petrov
cd,
Manfred
Eich
c,
Gerold A.
Schneider
b,
Rolf
Janssen
b,
Horst
Weller
aef and
Tobias
Vossmeyer
*a
aInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, D-20146 Hamburg, Germany. E-mail: tobias.vossmeyer@chemie.uni-hamburg.de
bInstitute of Advanced Ceramics, Hamburg University of Technology (TUHH), Denickestraße 15, 21073 Hamburg, Germany
cInstitute of Optical and Electronic Materials, Hamburg University of Technology (TUHH), Eissendorfer Straße 38, 21073 Hamburg, Germany
dITMO University, 49 Kronverskii Ave., 197101, St. Petersburg, Russia
eThe Hamburg Centre for Ultrafast Imaging, Luruper Chaussee 149, 22761 Hamburg, Germany
fDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 2nd November 2015
Abstract
Zirconia-based ceramics cover a huge variety of applications, including refractories, electro- and bioceramics, fuel cells, catalysts, and many more. For various photonic applications considered for energy systems and heat management, zirconia microspheres are interesting building blocks due to their high refractive index, as well as their chemical and mechanical robustness. However, instabilities caused by thermally-induced phase transitions and grain growth at temperatures above ∼1000 °C preclude high-temperature applications of pure zirconia particles. Here, we present a synthetic route for yttria-stabilized zirconia microparticles with significantly improved thermal stability. With these particles we conducted the first study on their thermal stability as a function of the yttrium content and at temperatures up to 1500 °C. Using X-ray diffraction and scanning electron microscopy, the optimum Y content was determined to be 8–10%, which was marked by stabilization of the tetragonal or cubic phase and significantly attenuated grain growth. Furthermore, with diameters ranging from 2 to 5 μm, the particles covered a size range perfectly suited for photonic applications in the IR spectral range. To demonstrate this, photonic glass coatings were prepared with these particles and their IR reflectivity and microstructural stability was studied after subjecting them to various heating cycles. While heating beyond 1200 °C led to failure and delamination of undoped particle films, films doped with 6 and 10% Y displayed quite stable broadband IR reflection of up to 80% in the wavelength range from 1–5 μm, even after prolonged heating at 1400 °C. A detailed analysis of the X-ray diffraction patterns revealed that prolonged heating at 1400 °C resulted in phase decomposition due to Y segregation into Y-lean and Y-rich domains, confirming the presence of the solute-drag effect.
1. Introduction
Zirconia powders are currently being used as precursors for performance ceramics with a broad variety of applications including refractories,1 electro- and bioceramics,2,3 fuel cells4 and catalysts.5 In addition, well-defined zirconia microparticles with their high refractive index, high thermal stability and narrow size distributions are promising candidates for the building blocks of novel photonic materials suited for high temperature applications, such as advanced reflective thermal barrier coatings (TBC)6–8 and as absorbers/emitters in thermophotovoltaics (TPV).9 The width and spectral position of the reflection band of these materials can be adapted quite easily to application specific requirements by varying the particle arrangement and size, respectively.10–12 We have recently reported a first demonstration of photonic glasses assembled from undoped zirconia microspheres, which acted as efficient broadband reflectors in the infrared spectrum.7A sol–gel approach for the fabrication of zirconia powders with improved sintering behavior compared to conventional powders,13,14 and, shortly after, of submicron spherical zirconia and yttria-stabilized zirconia (YSZ) particles was developed in previous studies.15,16 The synthesis of zirconia microparticles was expanded on by Lerot et al.,17 Yan et al.18,19 and Widoniak et al.2,3 In our last study, we improved the synthesis of zirconia submicron and microparticles with respect to size uniformity and examined their high temperature stability. We observed that these non-stabilized particles disintegrate after heating to 850–1200 °C and that the particle stability is influenced by the interplay between excessive grain growth, a tetragonal-to-monoclinic phase transformation and microstrain appearing as a result of these processes.20
Bulk zirconia has three temperature dependent polymorphs. The thermodynamically stable room temperature phase is monoclinic (P21/c) and transforms firstly to the tetragonal phase (P42/nmc) at 1170 °C and to the cubic phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) at 2370 °C, before it melts at 2715 °C.21 Upon cooling tetragonal zirconia, it transforms back to the monoclinic phase via a martensitic mechanism, which is diffusionless and displacive in nature. The transformation is accompanied by a 4% volume expansion. The addition of aliovalent cations is known to favor the more symmetric tetragonal and cubic phases and thus suppress the destructive tetragonal-to-monoclinic phase transformation.22 Yttria has proven to be a very suitable dopant, as it displays a wide solid solubility range in zirconia and has a similar chemical and thermal stability. A content of 0.02 < x < 0.09 and 0.04 < x < 0.4 in (Y2O3)x(ZrO2)1−x retains the high temperature tetragonal and cubic phases at ambient conditions, respectively.23 This translates to a Y/(Y + Zr) molar ratio of 4–17% and 8–57%. In addition to the phase transformation, excessive grain growth and coalescence also led to the destruction of undoped zirconia microparticles at elevated temperatures.20 This challenge can likewise be addressed by yttria doping, which has been shown to decrease grain growth due to a solute drag effect.24
m) at 2370 °C, before it melts at 2715 °C.21 Upon cooling tetragonal zirconia, it transforms back to the monoclinic phase via a martensitic mechanism, which is diffusionless and displacive in nature. The transformation is accompanied by a 4% volume expansion. The addition of aliovalent cations is known to favor the more symmetric tetragonal and cubic phases and thus suppress the destructive tetragonal-to-monoclinic phase transformation.22 Yttria has proven to be a very suitable dopant, as it displays a wide solid solubility range in zirconia and has a similar chemical and thermal stability. A content of 0.02 < x < 0.09 and 0.04 < x < 0.4 in (Y2O3)x(ZrO2)1−x retains the high temperature tetragonal and cubic phases at ambient conditions, respectively.23 This translates to a Y/(Y + Zr) molar ratio of 4–17% and 8–57%. In addition to the phase transformation, excessive grain growth and coalescence also led to the destruction of undoped zirconia microparticles at elevated temperatures.20 This challenge can likewise be addressed by yttria doping, which has been shown to decrease grain growth due to a solute drag effect.24
As a result of the stable crystalline structure and small grain sizes, YSZ is used in high temperature applications such as thermal barrier coatings.6,8 YSZ nanoparticles with sizes ranging from 5 to 200 nm and Y/(Y + Zr) molar ratios between 2 to 17% have been reported.25–27 However, the characterization at high temperatures is typically limited to simple thermogravimetric analysis, monitoring the weight loss and crystallization temperatures of the particles. Only few studies on yttria-stabilized microparticles in a size range of 1–5 μm exist to date: Uchiyama et al.28 and Keshmiri et al.29 synthesized ≤1 μm particles with Y/(Y + Zr) ratios of ∼7% and ∼16%, respectively. Hua et al.30 employed a solvothermal method, leading to larger (1–3 μm) aggregates with a wide size distribution and a Y/(Y + Zr) ratio of 6%. However, these were isolated studies focusing on one attribute, a fixed composition or phase at temperatures up to 1000 °C. Furthermore, there has been no characterization of the morphological evolution of the particles with heating.
Recently, we introduced a simple and versatile approach for the synthesis of micrometer-sized YSZ particles.20 Here, we present the synthesis of Y-doped zirconia microparticles with a well-controlled composition over a broad Y/(Y + Zr) target range (4–18%). Their thermal stability was assessed by studying phase transitions and grain growth using ex situ XRD measurements after heating the samples at temperatures of up to 1500 °C. Furthermore, we assembled photonic glasses from these particles and demonstrate their improved durability with respect to potential applications as reflective thermal barrier coatings.
2. Experimental section
2.1 Materials
n-Butanol (99.5%) with a maximum water content of 0.1% was from Th.Geyer, n-propanol (99.7%) from Sigma-Aldrich, ethanol (99.5%) from Grüssing, eicosanoic acid (99%) from Sigma-Aldrich, zirconium n-propoxide (70 wt% in n-propanol) from Alfa Aesar, ABCR or Aldrich (stored in a glove box), yttrium iso-propoxide (90%) from ABCR (stored in a glovebox), zeolith molecular sieve (0.4 nm) from Merck, 0.20 μm and 0.10 μm Minisart-Plus syringe filters from Sartorius Stedim Biotech, demineralized water (ACS reagent grade) from Aldrich.2.2 Particle synthesis and deposition of photonic glasses
Yttria-stabilized zirconia particles with diameters of ∼2 μm (before heat treatment) were synthesized according to a sol–gel approach published previously by our group,20 which was based on the works of Yan et al.18,19n-Butanol with 0.1% water content at most was dried over 0.4 nm molecular sieves and freed from dust using a syringe filter (pore size 0.20 μm). The reaction was carried out in a 250 mL wide-mouth glass bottle. Yttrium was introduced to the reaction in the form of yttrium iso-propoxide, which was suspended in 10 mL dry n-propanol, mixed with 12.2 g (26.1 mmol) zirconium n-propoxide (70% in n-propanol) and homogenized by ultrasonication. The mixture containing the yttria and zirconia precursors was filtered through a syringe filter (pore size 0.10 μm) and added under vigorous stirring to a solution of 1.06 g (3.39 mmol) eicosanoic acid in 100 mL dried n-butanol at 50 °C. After 30 min, a freshly prepared solution of 1.7 mL (94.3 mmol) water in 87 mL n-butanol was added over the course of 60 s. As reported previously, the water content was increased (from 1.4 to 1.7 mL) in comparison to the synthetic approach for undoped zirconia particles.20 After an induction time of several minutes, the slightly yellow solution turned white. The glass bottle was transferred onto an analogue tube roller (SRT6 Stuart, roller size length × diameter: 340 mm × 30 mm) for an ageing time of 90 min at a rotation speed of 12 rpm. With tube diameters of 30 mm and a bottle diameter of 70 mm, this corresponded to approximately five bottle revolutions per minute. Finally, the particle suspension was poured into 200 mL of n-butanol at 0 °C, and the particles were separated by centrifugation (250–500 g) at 0 °C.In order to obtain particles with varying Y contents, the amount of added yttrium iso-propoxide was varied while keeping the amount of zirconium n-propoxide fixed, so that the Y/(Y + Zr) molar ratio was 4, 6, 8, 10, 12, 14, 16, and 18%. The respective samples are labelled Y4 to Y18. It was observed that with an increasing amount of yttrium iso-propoxide added, the particles became more and more agglomerated. To counteract this, the amount of water was decreased from sample Y10 onward. This is expanded on in the discussion. A detailed account of the amounts of water and yttrium iso-propoxide added can be found in the ESI,† in Table S1.
Particles with diameters of 4–5 μm (before heat treatment) for the fabrication of photonic glasses were synthesized by a modified approach: increasing the amount of eicosanoic acid to 1.31 g (4.19 mmol), decreasing the tube roller speed to 5–9 rpm, and increasing the ageing temperature for sample YPG10 to ∼70 °C by placing an infrared lamp (175 W) 25 cm over the reaction vessel. Furthermore, the stir time was extended beyond the induction time. This extended stirring in combination with the adjusted synthetic parameters led to larger particles. A complete list of all synthetic parameters is shown in Table S2 in the ESI.† Three particle samples with Y/(Y + Zr) ratios of 0%, 6% and 10% were prepared. They are referred to as samples YPG0, YPG6 and YPG10, respectively.
Photonic glasses were deposited via a drop casting process, as described previously.7 Briefly, the as-synthesized particles were pre-calcined at 450 °C for 3 h (heating and cooling rate of 5 °C min−1) in order to avoid crack formation due to particle shrinkage. It was previously shown that by 450 °C the main mass loss events had occurred and that the particle diameters remained stable after heating to higher temperatures.20 Suspensions of the pre-calcined microparticles in ethylene glycol (300 mg mL−1) were prepared via ultrasonication (Badelin, Sonorex Super RK 106) and drop-cast within a silicone ring fixed to single crystal sapphire substrates. The solvent was evaporated by heating the substrates on a hot plate at 150 °C. The resulting photonic glasses had areas of 4.5 cm2 with their thickness being controlled by the amount of applied suspensions.
2.3 Heating experiments and particle characterization
The particle samples were subjected to heat treatments in a muffle oven (L9/SKM, Nabertherm) and in a tube furnace (STF 16/100, Carbolite). The crystal phases were characterized by X-ray diffraction (XRD, Philips X'Pert PRO MPD) and the particle morphology and stability were assessed by scanning electron microscopy (SEM, EVO MA 10 and a Leo 1550 Gemini, both Zeiss). The particle composition was analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES, Spectro Model ARCOS spectrometer). Photonic glass films were heated in a tube furnace and their hemispherical diffuse reflectance was measured with a Fourier transform infrared (FTIR) spectrometer (Vertex 70, Bruker) equipped with a gold-coated integrating sphere accessory and an MCT detector. The heating rate profiles and details of these measurements are provided in the ESI† (Section S2).3. Results
3.1 Particle synthesis
Yttria-stabilized zirconia microparticles were synthesized according to a wet chemical approach developed in our group20 which was based on the works of Yan et al.18,19 In contrast to previously used approaches in which the zirconia precursor and the dopant precursor are separately added to the alcoholic solution,28,29 the precursors were premixed and homogenized before addition. Filtering this mixture to remove possible hydrolizates was found to decrease the degree of agglomeration of the particles. With these modifications, spherical particles were achieved with Y/(Y + Zr) molar ratios from 4 to 18%, which translates to an amount of x in (Y2O3)x(ZrO2)1−x between 0.02 and 0.10.An overview of the size distribution of the as-synthesized initial particles and SEM micrographs of the as-synthesized and heated particles are shown in Fig. 1 for selected yttria-stabilized particle samples. The undoped particle sample (Y0) is shown for comparison. The particles' thermal stability was evaluated from the microstructural changes and the degree of particle disintegration after heating.
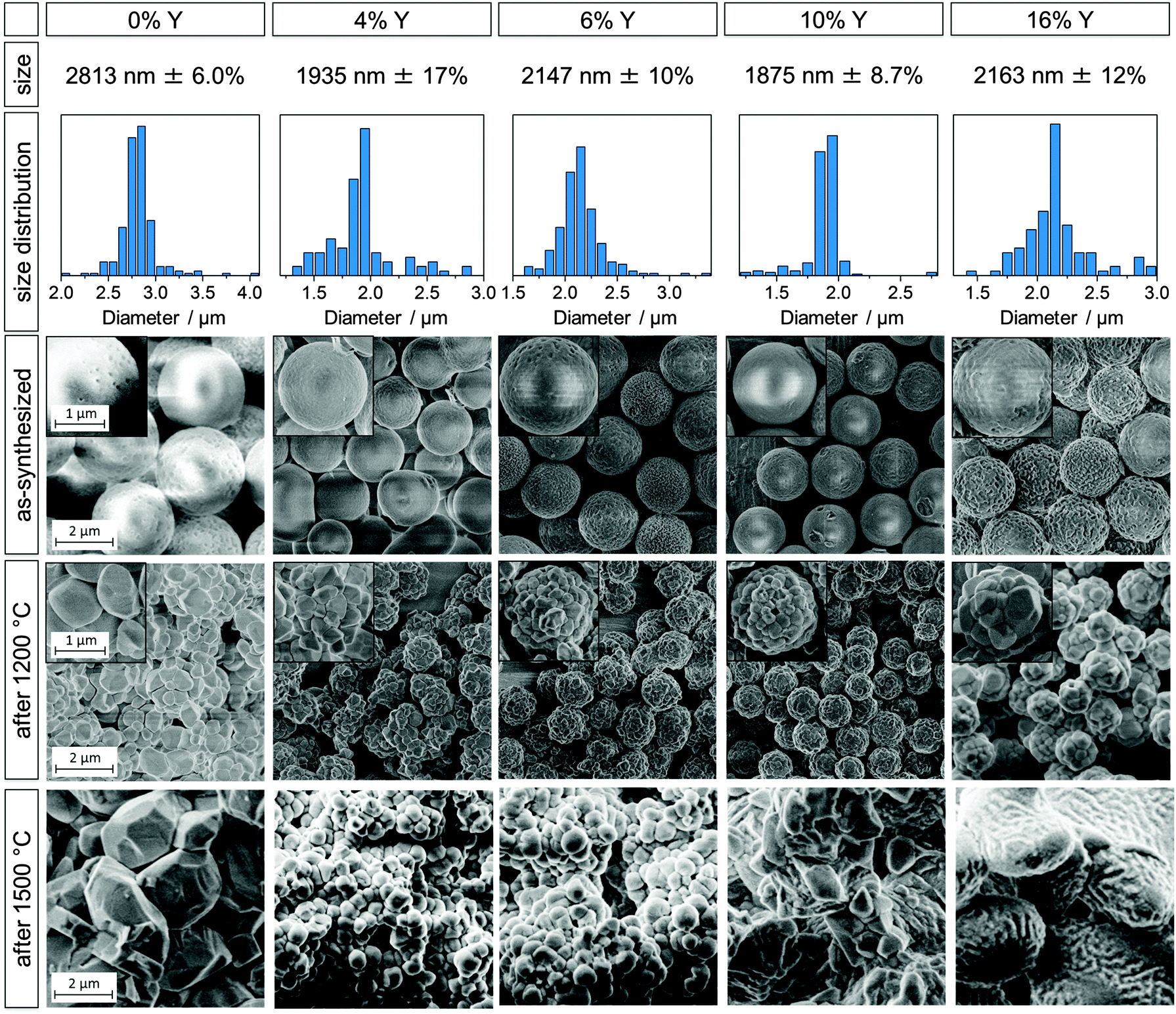 | ||
| Fig. 1 Undoped zirconia microparticle sample (0% Y) and yttria-stabilized zirconia particles with added yttrium amounts of 4, 6, 10, and 16% (from left to right). Diameter sizes with standard deviations (first row), size distributions (second row), SEM images of as-synthesized particles (third row), of particles heated at 1200 °C for 3 h (fourth row) and particles heated at 1500 °C for 3 h (fifth row). The undoped sample corresponds to B2.8 in prior work.20 Scale bars apply to all images and insets within a given row. | ||
Y0 corresponds to sample B2.8 in our previous study20 and was chosen as a comparison for the YSZ particles because of the similar particle sizes (2.8 μm for sample Y0 and ∼2 μm for samples Y4–Y18). The first, second and third row in Fig. 1 display the diameter, size distribution and SEM micrographs of the as-synthesized particle samples Y0, Y4, Y6, Y10 and Y16. SEM images of these samples after heating at 1200 and 1500 °C for 3 h are shown in the fourth and fifth row, respectively. SEM images and data of all YSZ samples Y4 to Y18 can be found in the ESI† (Fig. S3 and S4). A brief synopsis of particle compositions, diameters and size standard deviations for samples Y0 to Y18 is displayed in Table 1.
| Sample | Y0 | Y4 | Y6 | Y8 | Y10 | Y12 | Y14 | Y16 | Y18 |
|---|---|---|---|---|---|---|---|---|---|
| ICP-OES/% | 0 | — | 7.0 | 8.4 | 9.8 | 13.9 | 16.7 | — | — |
| EDX/% | 0 | 2.9 | 4.8 | 6.7 | 10.8 | 12.1 | 14.8 | 20.6 | 26.6 |
| d/nm | 2813 | 1935 | 2147 | 2336 | 1875 | 2142 | 2145 | 2163 | 2608 |
| σ/% | 6.0 | 17 | 10 | 6.0 | 8.7 | 10 | 9.8 | 12 | 12 |
The YSZ particle diameters were in the range of ∼2 μm, independent of the Y content, except for sample Y18 which had a diameter of 2.6 μm. This is comparatively smaller than the 2.8–4.3 μm, previously obtained for pure zirconia particles.20 In order to understand the reason behind this change in diameter, it is useful to consider the induction period of the different syntheses. The induction time marks the delay between the addition of water to the reaction mixture and a change of the solution from transparent to white. Hydrolysis and polymerization of the alkoxides results in the formation of 10–20 nm sized primary particles,3,17,31 which then quickly agglomerate to larger particles and cause a visible clouding of the reaction mixture. The induction period is correlated with the hydrolysis rate of the reaction, with a short induction time suggesting a quick hydrolysis and vice versa. A quick hydrolysis has previously been linked to smaller particle sizes.3,17,18 The induction times of the YSZ particles were comparatively short, ranging between 90 and 810 s, compared to the undoped sample Y0 with 1920 s. Most likely, the increased hydrolysis rate is a result of the higher reactivity of the yttria precursor relative to the zirconia precursor. The yttria precursor is favorably attacked by water molecules and growing polymeryzates due to steric effects, as yttrium iso-propoxide has only three organic ligands while zirconium n-propoxide has four. Additionally, the lower electronegativity of Y in comparison to Zr (1.22 and 1.33 in the Pauling scale) makes it more susceptible to nucleophilic attack. Also, the increased water content (as compared to the undoped particle synthesis) is expected to contribute to the accelerated hydrolysis and, thus, to the observed shortening of the induction time.
The as-synthesized YSZ particles were spherical with rather smooth surfaces and a low degree of agglomeration. Their size distribution was quite narrow, with standard deviations ranging between 6.0 and 17%, which is, however, higher than the 6–10% reported for the undoped particles.20 Given the aforementioned quicker hydrolysis of the yttria precursor, it was found that an increasing Y content lead to an increase in agglomeration and hence irregular particles. In order to slow down the hydrolysis rate, the water concentrations were decreased with increasing Y content, from 0.47 mol L−1 to 0.38 mol L−1 for Y18. Synthetic details, such as the amounts of yttrium iso-propoxide and water added to the syntheses can be found in the ESI† (Fig. S1 and Table S1).
The elemental composition was analyzed via EDX and, in the case of samples Y6–Y14, with optical emission spectroscopy (ICP-OES). The results are shown in Table 1 and Fig. 2. The uncertainty of the EDX was ±1 mol% and mainly due to the overlap of the Zr and Y L-emission peaks. For ICP-OES a double determination was carried out for each sample. The average uncertainty obtained from these determinations was found to be ±0.4 mol% and was most likely due to variations in the salt fusion dissolution of the particles. For samples Y4–Y14 the Y fractions obtained by ICP-OES or EDX were in reasonable agreement with the target values based on the amounts of precursors added. For the higher Y content particles, the EDX values were higher than expected compared to the added precursor amounts, with samples Y16 and Y18 being approximately 5 and 9 percentage points higher, respectively. This trend of increasing Y incorporation rates with higher Y contents was attributed to the higher reactivity of the yttrium iso-propoxide relative to the zirconium n-propoxide, which is also supported by the aforementioned increase in hydrolysis rates and shorter induction times.
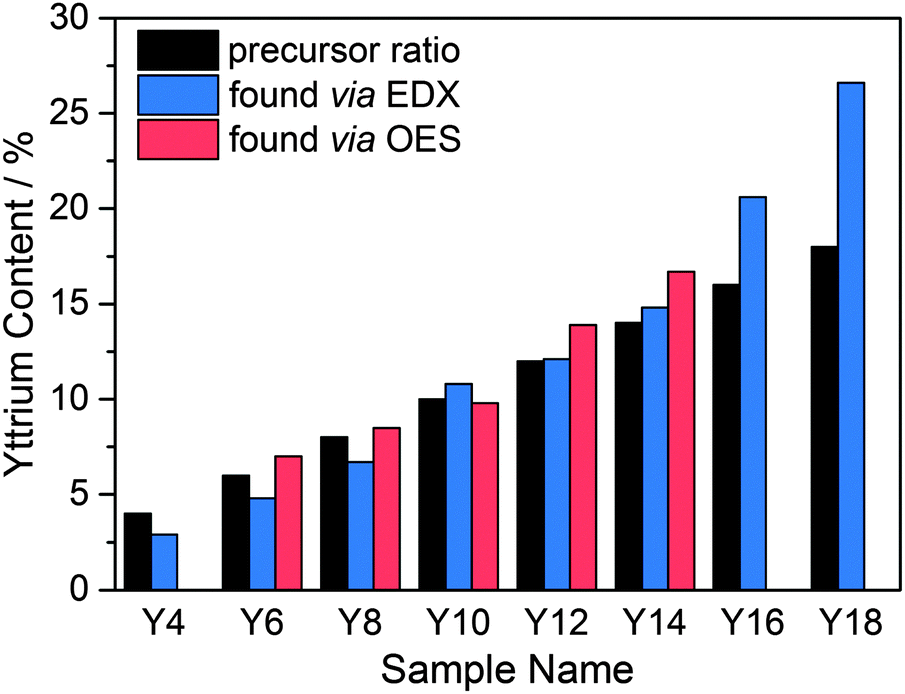 | ||
| Fig. 2 Target Y content (black) and Y contents measured via EDX (blue) and ICP-OES (red) for YSZ samples Y4 to Y18. | ||
The larger YSZ particles for the fabrication of photonic glasses were synthesized by a modified approach utilizing “extended stirring”. This refers to a period of a few minutes in which the particles are still vigorously stirred after the induction time has passed. The use of extended stirring in order to increase the particle size has previously been reported by Yan et al.18 They assumed that the external forces imposed by stirring caused the still small particles to coagulate irreversibly and thus resulted in overall larger particles. However, they acknowledged that any use of extended stirring greatly perturbs the system and can lead to agglomerated and irregularly shaped particles. We were able to counteract these detrimental effects by adapting several other synthetic parameters. Firstly, it was found that increasing the amount of eicosanoic acid by 25% lowered the extent of agglomeration in the particles. Secondly, the rotation speed of the analogue tube roller was decreased, which led to narrower size distributions. Finally, in the case of the higher Y content sample (YPG10), increasing the ageing temperature from room temperature to ∼70 °C in conjunction with the other two adjustments was necessary to achieve large diameter particles. These adaptations allowed for the synthesis of the samples YPG0, YPG6 and YPG10 with respective diameters of 4.3, 4.1 and 5.1 μm and size standard deviations ranging between 12–16%. SEM micrographs and particle size distributions of samples YPG0, YPG6 and YPG10 are shown in the ESI† (Fig. S2).
3.2 Crystalline phases
The addition of Y in molar ratios Y/(Y + Zr) of 4–17% and 8–57% has been shown to stabilize the tetragonal and cubic phase in bulk zirconia.23 Substituting a Zr4+ with an Y3+ cation results in the creation of oxygen vacancies. X-ray absorption experiments32 and ab initio33 and tight binding22 calculations indicate that it is not the dopant ions but rather the resulting crystal distortions around the oxygen vacancies that play a major role in the stabilization mechanism. Y3+ ions are next nearest neighbors to the oxygen vacancies, which associate with the Zr4+ ions and reduce their coordination numbers from 8 to 7.34 At low dopant concentrations, and thus a small amount of oxygen vacancies, they cause a distortion in the surrounding ions that translates into a tetragonal distortion of the oxygen sublattice. Subsequently, a tetragonally distorted unit-cell is formed. A further increase in the amount of oxygen vacancies causes local distortions that result in the formation of the cubic phase.22After confirming via EDX and ICP-OES that Y was incorporated in the microparticles (Section 3.1), it was to be examined whether Y doping led to a stabilization of the tetragonal or cubic phase and thus prevented the destructive phase transformation in the microparticles. For this purpose, their crystalline phases as a function of Y content and temperature were determined by XRD. In order to assess their high temperature stability under operating and sintering conditions, X-ray diffractograms were measured after heating at 1200 and 1500 °C for 3 h for samples Y4 to Y18 and are shown in Fig. 3. Additional measurements were performed for samples Y6 to Y14 after heating to 250, 450, 650 and 850 °C for 1 h at each temperature. X-ray diffractograms acquired for samples Y6 to Y14 after each heating experiment can be found in the ESI† (Fig. S8–S12).
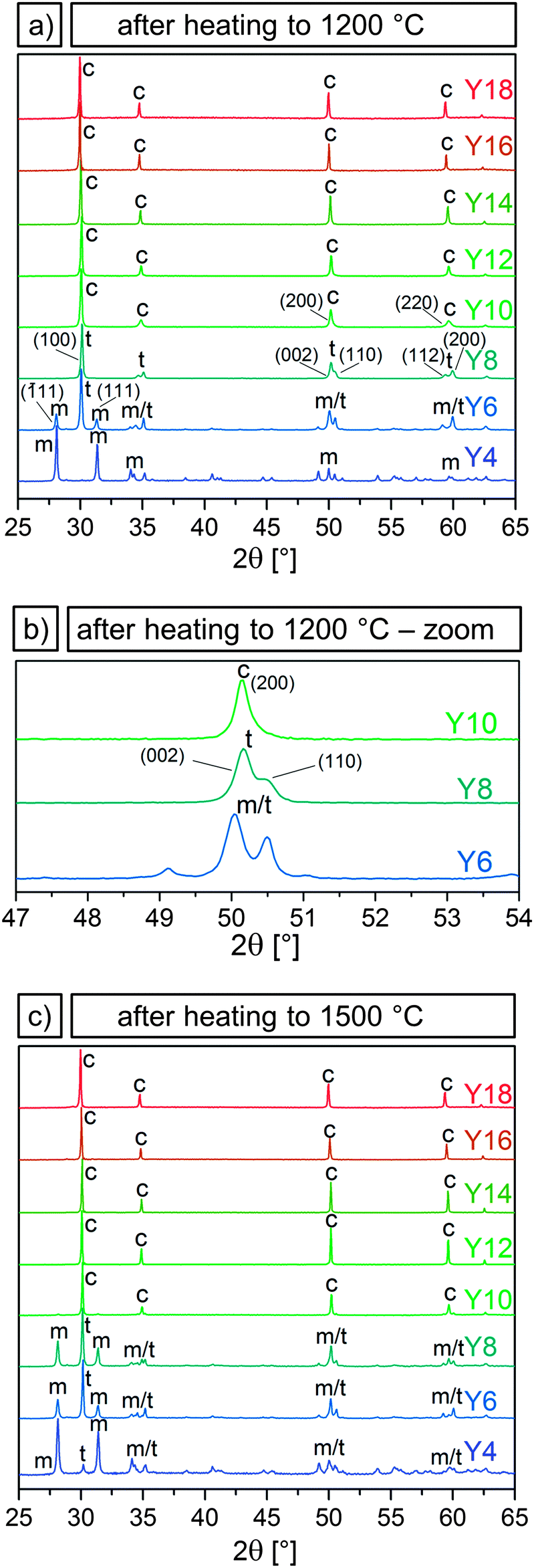 | ||
| Fig. 3 X-ray diffractograms of samples Y4–Y18 (bottom to top) after heating at 1200 °C for 3 h (a), an inset showing the transition from tetragonal to cubic for samples Y6–Y10 (b), and X-ray diffractograms for samples Y4–Y18 after heating at 1500 °C for 3 h (c). The tetragonal (t), monoclinic (m) and cubic (c) phases are indicated and labeled with their characteristic peaks. | ||
It was observed that the particle samples Y6–Y14 were still amorphous after heating to 250 °C and crystallized in the tetragonal or cubic phase after heating to 450 °C. Further heating to 650 and 850 °C led to a narrowing of the peaks with the phases remaining tetragonal or cubic. Peak broadening due to the small grain sizes made it impossible to distinguish between the tetragonal and cubic phase for these temperatures.
In Fig. 3 the presence of the characteristic (![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 11) and (111) peaks in the ∼30° region in sample Y4 after heating to 1200 and 1500 °C indicate a transition to the monoclinic phase, showing that an Y content of 3–4% is not sufficient to stabilize the tetragonal phase. Sample Y6 predominantly exhibited the tetragonal phase with a strong (100) peak and weak monoclinic (11) and (111) peaks, after heating to 1200 °C. The fractions shifted in favor of the monoclinic structure after heating to 1500 °C. Sample Y8 was fully tetragonal after 1200 °C but likewise showed the appearance of monoclinic peaks after heating to 1500 °C.
11) and (111) peaks in the ∼30° region in sample Y4 after heating to 1200 and 1500 °C indicate a transition to the monoclinic phase, showing that an Y content of 3–4% is not sufficient to stabilize the tetragonal phase. Sample Y6 predominantly exhibited the tetragonal phase with a strong (100) peak and weak monoclinic (11) and (111) peaks, after heating to 1200 °C. The fractions shifted in favor of the monoclinic structure after heating to 1500 °C. Sample Y8 was fully tetragonal after 1200 °C but likewise showed the appearance of monoclinic peaks after heating to 1500 °C.
Samples Y10–Y18 were determined to be cubic both after heating to 1200 and to 1500 °C. Virtually no monoclinic peaks were observed. The distinction between the tetragonal and the cubic phase is subtle, but can be made by examining the ∼50 and ∼60° region of the diffractograms. The reflexes (200) and (220) in the cubic system split into the two sets of (002) (110) and (112) (200) in the tetragonal system, as can be seen by comparing the diffractograms of samples Y8 and Y10 displayed in Fig. 3a. Fig. 3b shows the reflexes of these two samples, and of sample Y6, in the ∼50° region, where it can be seen that sample Y8, which displays a double peak, is tetragonal, while samples Y10, with a single peak, is cubic. With 8 and 10% Y corresponding to an x of 0.04 and 0.05, respectively, these results are in good agreement with the aforementioned 0.02 < x < 0.09 for the tetragonal and 0.04 < x < 0.4 for the cubic phase in (Y2O3)x(ZrO2)1−x. Therefore, it can be concluded that an Y/(Y + Zr) ratio of 6% is necessary to partially stabilize the tetragonal phase after heating to 1200 °C, while 10% Y or higher leads to a full stabilization of the cubic phase even after heating up to 1500 °C.
3.3 Grain size analysis
In our last study, we observed that an earlier crystallization and smaller initial grain sizes correlated with better high temperature stability of undoped zirconia particles.20 In bulk zirconia, Y doping slows down grain growth.24,35 In order to determine whether Y doping has similar effects on the microparticles, grain sizes were determined via the Scherrer equation. The FWHM (full width at half maximum) values were acquired from X-ray diffractograms measured after heating to 450, 650 and 850 °C. Grain sizes determined by the Scherrer method were validated by comparison to SEM grain size measurements for the 850 °C samples. (ESI,† Fig. S7). The grain sizes of samples Y6–Y14, together with the grain sizes of the undoped zirconia sample (Y0), are shown in Fig. 4a and Table 2.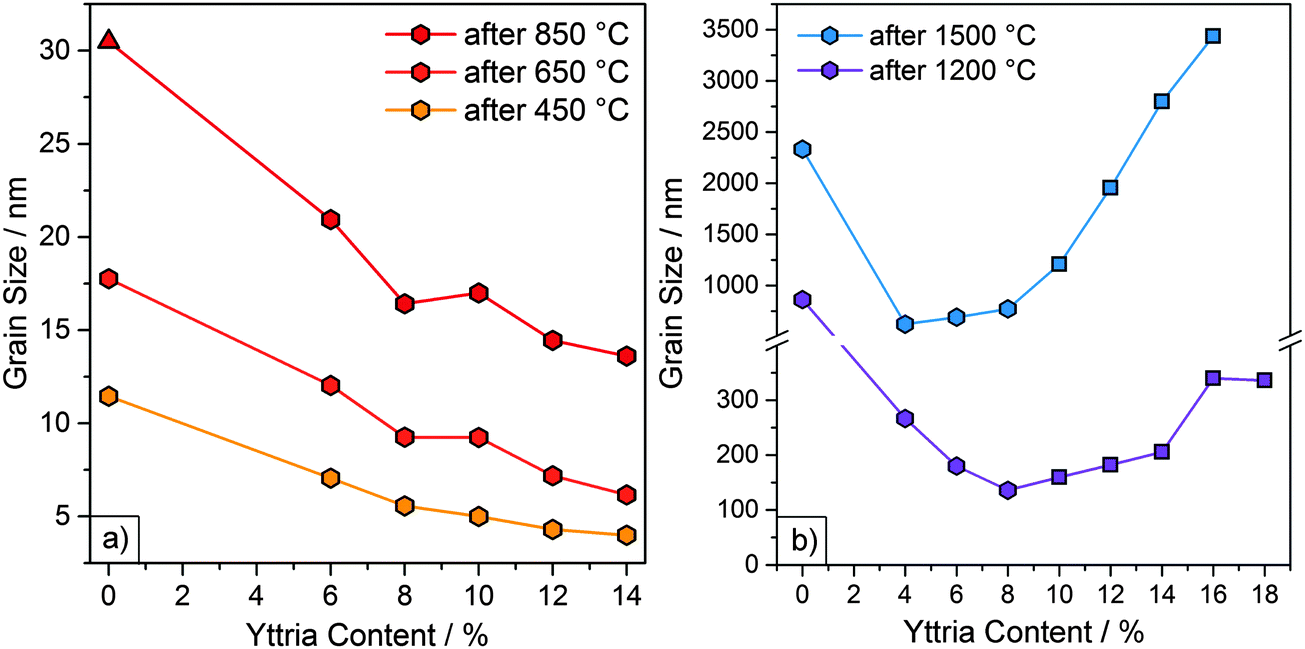 | ||
| Fig. 4 (a) Grain sizes (obtained via XRD) as a function of Y content for particle samples Y6 to Y14 and undoped particle sample (Y0)20 after heating at 450, 650 and 850 °C for 1 h. The monoclinic phase is represented by a triangle. All other samples were tetragonal or cubic. (b) Grain sizes (obtained from SEM micrographs) of samples Y4 to Y18 and undoped sample (Y0) after heating at 1200 and 1500 °C for 3 h. The cubic phase is represented by squares. All other samples were tetragonal or monoclinic. | ||
| Grain size (nm) after heating to | |||||
|---|---|---|---|---|---|
| 450 °C | 650 °C | 850 °C | 1200 °Ca | 1500 °Ca | |
| a Determined by SEM measurements of 50–100 grains. All other grains sizes were determined by the Scherrer method. | |||||
| Y0 | 11 | 18 | 31 | 863 ± 397 | 2332 ± 917 |
| Y4 | — | — | — | 267 ± 87 | 623 ± 118 |
| Y6 | 7 | 12 | 21 | 180 ± 74 | 691 ± 154 |
| Y8 | 6 | 9 | 16 | 136 ± 36 | 772 ± 244 |
| Y10 | 5 | 9 | 17 | 160 ± 54 | 1208 ± 682 |
| Y12 | 4 | 7 | 15 | 182 ± 64 | 1955 ± 889 |
| Y14 | 4 | 6 | 14 | 206 ± 73 | 2800 ± 1177 |
| Y16 | — | — | — | 340 ± 124 | 3436 ± 1215 |
| Y18 | — | — | — | 336 ± 124 | >4000 |
As the samples were still amorphous after heating to 250 °C, grain sizes could not be determined for this temperature. After heating to 450 °C both the YSZ and the undoped particles had crystallized. Initial grain sizes after heating to 450 °C were considerably smaller for YSZ particles (4–7 nm) than for undoped particles (11 nm). Furthermore, a decrease in the grain sizes with increasing Y content was observed. Y6, with the lowest Y content, consisted of the largest grains with 7 nm, followed by Y8 and Y10, with grain sizes of 6 and 5 nm. Sample Y12 and sample Y14 both had the smallest grain sizes of 4 nm.
The initial grain size can be attributed to various factors. Firstly, the incorporation of Y could provide nuclei for crystallization. Keramidas et al. applied Raman spectroscopy to show that amorphous zirconia is not necessarily truly amorphous but rather composed of 1.5–3.0 nm “pre-grains” in a tetragonal-like phase.36 Higher Y contents could result in an increased amount of nuclei and/or “pre-grains”, which then lead to smaller initial grain sizes. Secondly, the decrease in initial grain size can possibly be linked to a modified synthetic mechanism. The addition of yttrium iso-propoxide to the reaction and the resulting increase in hydrolysis rate may lead to a higher reactivity of the initial nanometer-sized primary particles. These primary particles then agglomerate quicker and at smaller diameters to form the microspheres. It would be these primary particles, which crystallize to form the observed initial grains. An increased Y content would subsequently lead to even smaller primary particles and thus smaller initial grain sizes.
A similar progression in grain size, with higher Y contents yielding smaller grain sizes, was observed upon further heating to 650 and 850 °C, resulting in grain sizes of 6–12 nm and 14–21 nm, respectively. For samples heated to 850 °C, the grain sizes were also measured via SEM. The particles consisted of grains with diameters of ≤25 nm, which is in good agreement with the range yielded through the Scherrer equation. Again, the grain sizes were considerably smaller than the 18 and 31 nm in the case of the undoped particles. The slower grain growth in the YSZ particles can be explained by the solute drag effect exhibited by the Y3+ ions. Previous studies using STEM, AES and XPS indicate that Y3+ ions in YSZ are not homogeneously distributed but segregate to the grain boundaries.35,37,38 Grain boundary migration is hindered as the boundary drags the dopants along with it, which results in slower grain growth.24 This solute drag effect is strongest for solutes with a large valence difference and large ionic radii relative to the parent cation.38 The ionic radii for Zr4+ and Y3+ are 86 and 104 pm, respectively.39
After heating to 1200 and 1500 °C, the grain sizes had grown larger than the ∼80 nm maximum grain size which can be calculated with the Scherrer equation due to the limiting factor of the instrument broadening. Therefore, the grain sizes were assessed from the SEM micrographs and are included in Fig. 4b and Table 2. Initially, the grain sizes decreased with increasing Y content due to the solute drag effect. However, this trend is discontinued at higher temperatures (≥1200 °C). After heating to 1200 °C, grain sizes decreased from sample Y0 to Y8 and then increased from sample Y10 onward. This increase coincided with the appearance of the cubic phase (marked by square symbols in Fig. 4b). After heating to 1500 °C, a decrease in grain size from sample Y0 to Y4 was observed, after which the grain size increased slowly at first (Y4 to Y8) and then very rapidly (from sample Y10 onward). The rapid increase in grain sizes again coincided with the appearance of the cubic phase.
This increase in growth rate can be attributed to a higher oxygen vacancy content and diffusivity, which enhances mass transport at elevated temperatures.35 This effect is particularly pronounced for the cubic phase, whose formation is based on a high amount of oxygen vacancies.22 For similar reasons, bulk, fully stabilized cubic zirconia (8YSZ, 15% Y) undergoes more grain growth than partially stabilized tetragonal zirconia (3YSZ, 6% Y).40 Higher grain boundary and volumetric diffusivities and lower activation energies have also been reported for fully stabilized zirconia compared to partially stabilized zirconia.40 The consequence of these differences on the resulting microstructure is evident by comparing the SEM images of sample Y6 (mostly tetragonal) to sample Y16 (cubic) heated at 1500 °C (Fig. 1), with the latter exhibiting significantly more sintering.
Another important factor for the assessment of the particle processability, e.g. for their deposition as reflective TBCs described below, is their stability to resuspension in solvents. The resuspendability of the particles is greatly influenced by the phase transformations and grain sizes. After heating to 850 °C samples Y6–Y14 were all fully suspendable in ethanol via sonication without destruction. However, after heating to 1200 °C, samples Y4 and Y6 were partially destroyed after resuspension, which coincided with the formation of the monoclinic phase. Meanwhile, samples Y16 and Y18 were destabilized by sintering processes which caused the particles to fuse and merge together. In contrast, samples Y8 to Y14 were fully stable to resuspension via sonication after heating to 1200 °C. Considering the factors of phase stability, resistance to grain growth and resuspendability, the optimum Y content was determined to be 8 to 10%.
In conclusion it could be shown that Y doping results in smaller initial grain sizes, which is likely explained by a modified synthetic mechanism, and slowed grain growth for Y contents up to 10%. At higher Y contents, the formation of the cubic phase and the resulting increase in mass transport led to an increase in grain sizes. Thus, while cubic zirconia may be stabilized from a phase perspective, it can be considered morphologically unstable at elevated temperatures for applications approaching 1500 °C.
3.4 Photonic glasses
The optical properties and thermal stability of photonic glasses assembled from pure zirconia and YSZ particles were studied as a function of operating temperature and time. As previously reported, in order to achieve broadband reflection in the infrared wavelength range between 1 and 5 μm, the calcined particles should have diameters of ∼3 μm.7 For this purpose, a modified synthetic procedure was developed for the synthesis of larger doped and undoped microspheres, as discussed in Section 3.1. This approach yielded particles with diameters between 4–5 μm, which translated to ∼3 μm after heat treatment. Microparticles with Y/(Y + Zr) molar ratios of 0, 6 and 10%, designated as YPG0, YPG6 and YPG10, were used for the assembly of photonic glass films. These films will henceforth be referred to as F0, F6 and F10, respectively. Films with Y contents of 6 and 10% were selected as these compositions straddled the tetragonal-stabilized region. Uniform, mostly crack-free films with a surface area of 4.5 cm2 and a thickness of ∼100 μm were yielded with the drop-cast procedure.With TBC surface temperatures expected to reach or even exceed 1400 °C in next generation turbines, further understanding is required to guide material design, especially to block radiative transport in TBCs.8 In order to examine their high temperature stability, the films were subjected to sequential heat treatments at 700, 1000, 1200, 1300 and 1400 °C for 3 h. Additionally, the films were heated at 1400 °C for a total of 24, 72 and 192 h in order to determine the effects of prolonged heating. After each heating cycle, the structural integrity, optical reflectance and crystalline phases of the films were examined by means of SEM, IR-spectroscopy and X-ray diffraction. The results of these measurements for selected heating cycles are shown in Fig. 5, 6 and 7, respectively. The complete set of images, spectra, and diffractograms can be found in the ESI† (Fig. S13–S19).
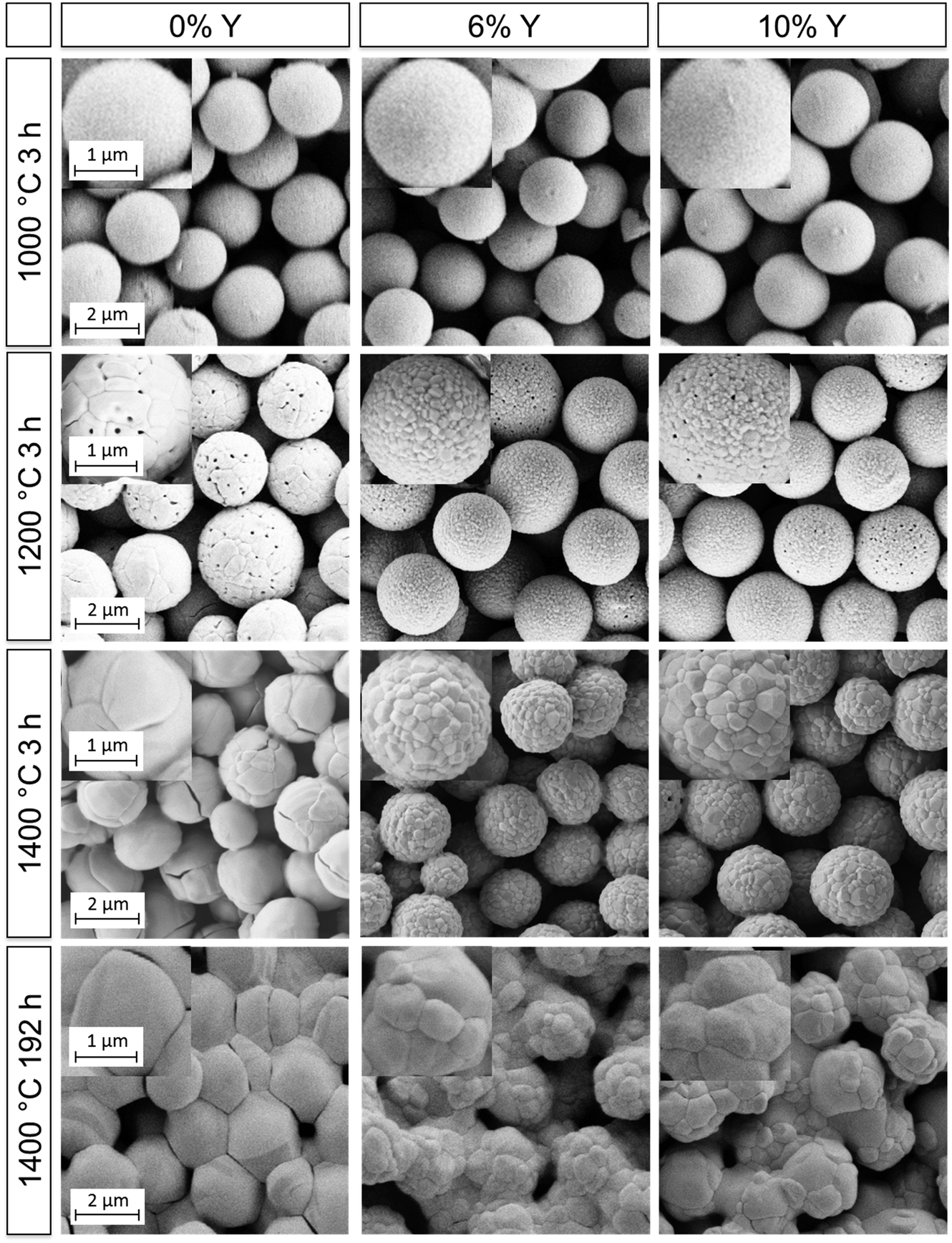 | ||
| Fig. 5 SEM micrographs of the films F0, F6 and F10 (left to right) after selected heating cycles. Scale bars apply to all images and insets within a given row. | ||
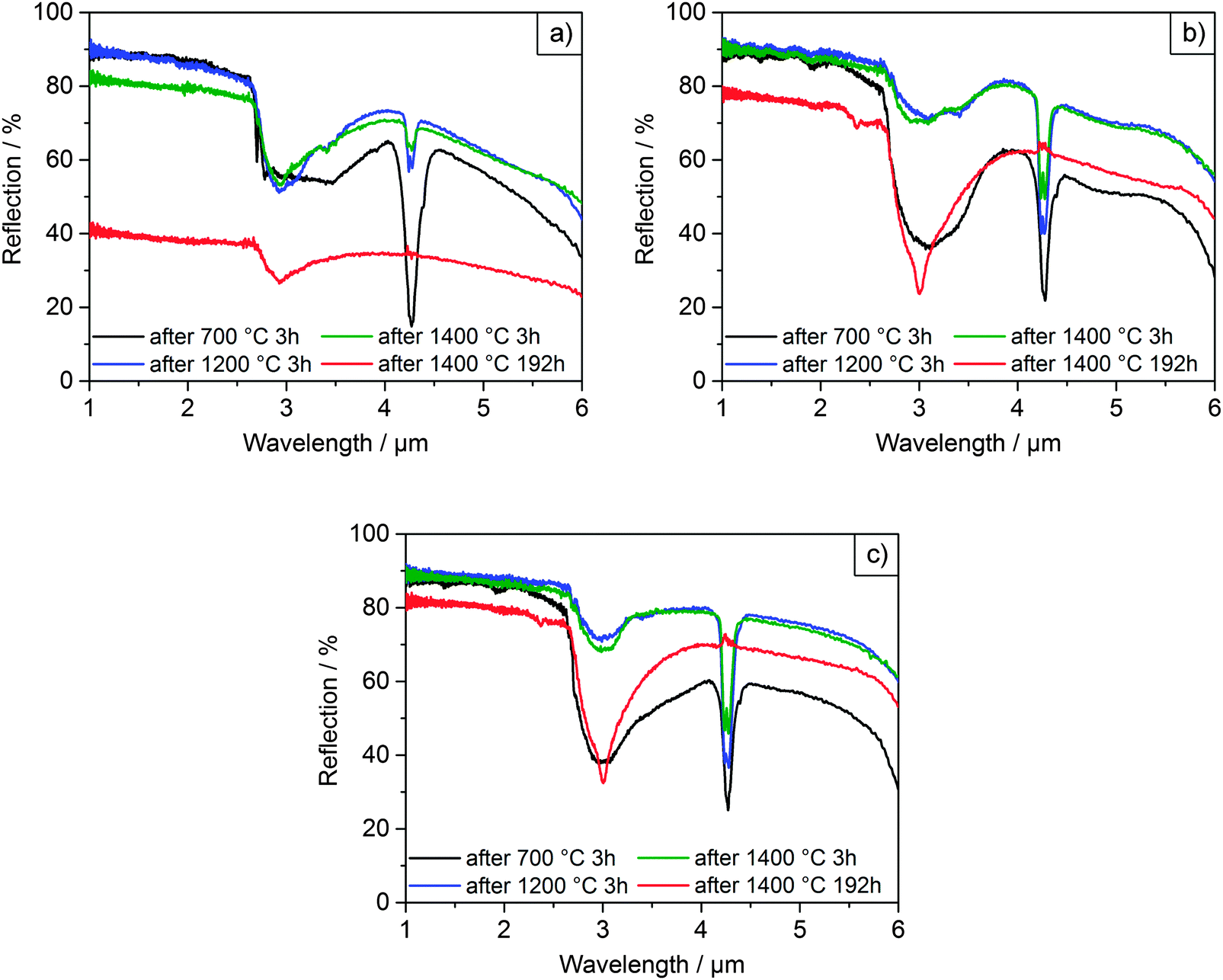 | ||
| Fig. 6 Diffuse reflectance spectra of films F0 (a), F6 (b) and F10 (c) after selected heating cycles. | ||
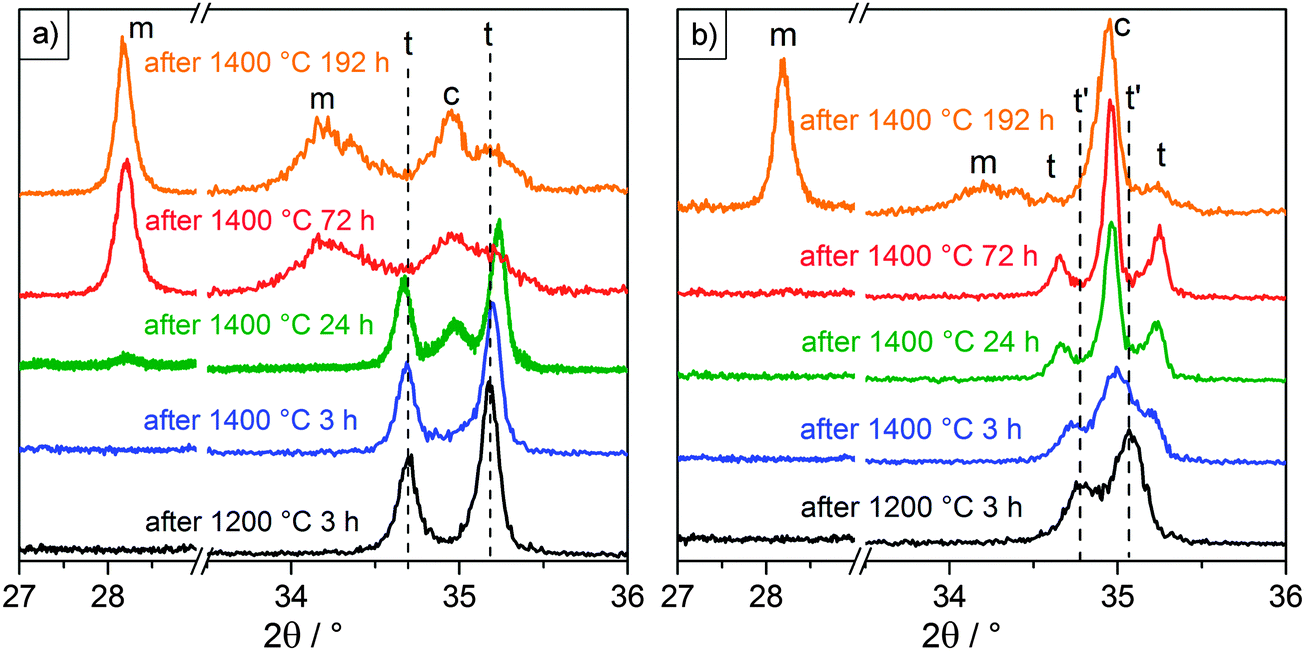 | ||
| Fig. 7 X-ray diffractograms of films F6 (a) and F10 (b) after selected heating cycles. 2θ is shown divided into two regions, one at low angles where the characteristic monoclinic (m) peak occurs and one at higher angles where the tetragonal (t) and cubic (c) peaks are resolved. The initial peak positions after heating at 1200 °C are indicated by dashed lines, starting as tetragonal for 6% Y and tetragonal prime (t′) for 10% Y. | ||
Initially, all films were heated at 700 °C for 3 h in order to remove organic residues. The films were intact with the microparticles displaying smooth surfaces after heating up to 1000 °C, as can be seen in Fig. 5. The initial spectra of all three films (Fig. 6) exhibited diffuse, broadband reflection in the infrared range from 1–6 μm with values as high as 85%. A broad drop in reflectance from 2.5–4.0 μm down to values of ∼40% for F6 and F10 and ∼60% for F0 at 700 °C was observed, which was attributed to absorption from OH functional groups adsorbed on the particles' surfaces. Another sharper drop at 4.2 μm with reflectivities as low as ∼20% was due to absorption from atmospheric CO2 within the optical path of the spectrometer. These findings are in good agreement with previous experiments conducted on films assembled from 2.6 μm undoped zirconia particles.7 After further heating up to 1200 °C, the overall reflectance of the films improved and the contribution of OH absorption was considerably reduced, especially for F6 and F10. Reflectance values ranged between 90–45% for F0, 90–55% for F6 and 90–60% for F10 in the 1–6 μm infrared region. In terms of their reflectance performance, there was no significant difference between the three films for operating temperatures up to 1200 °C. We expect that in applications under high temperature conditions neither the absorption from atmospheric CO2 nor absorption from adsorbed OH functional groups will play a role. Current CO2 absorption is accumulated in a one meter long optical path of the spectrometer and will not contribute in a 100 μm coating. OH groups will not be present at elevated temperatures. The observed transmission dips exist due to reabsorption of water at room temperature, as was shown in our previous publication.7
However, significant differences in stability between the undoped and YSZ films began to occur after heating at 1200 °C. The undoped F0 film started to show signs of macroscopic failure, characterized by crack formation and partial delamination of the film (exemplary photographs of the films are shown in the ESI† in Fig. S20). Given that the retained room temperature phase of the film was fully monoclinic after heating to 1000 °C, the failure was attributed to cycling through the monoclinic-tetragonal phase transition, which occurs at ∼1200 °C. The transformation back from the high temperature tetragonal phase to the monoclinic phase and the accompanying 4% volume expansion likely resulted in stresses and the observed failure. The problem became more pronounced at higher temperatures. The coating was nearly completely chipped off with cyclic heating at 1400 °C, to the point where it became impossible to accurately perform reflectance measurements. The delamination manifested as a significant decrease in the overall measured reflectance (Fig. 6a), with a maximum of 90% after heating to 1200 °C for 3 h, dropping to 80% after heating to 1400 °C for 3h and only 40% after heating to 1400 °C for 192 h. In contrast, the yttria-doped films F6 and F10 were quite stable and displayed only few cracks and very little delamination. They retained their reflectance performance even after heating at 1300 and 1400 °C for 3 h (Fig. 6b, c and Fig. S16, ESI†).
Additionally, the particle morphology changed after heating to 1200 °C and above. As shown in the SEM images in Fig. 5, the particles lost their smooth appearance and grains were clearly observable. Grain growth was most pronounced for the undoped particles of F0. Large grains on the order of 0.5–1.0 μm can be distinguished, as well as some crack formation between the grains. In contrast, the grain growth was less pronounced for the films assembled from YSZ particles (F6 and F10), exhibiting grain sizes below 300 nm. This attenuated grain growth in yttria-doped particles is in good agreement with the prior results (Section 3.3) and can be explained by the solute drag effect. Further heating to 1300 and 1400 °C for 3 h resulted in more pronounced grain growth, especially for sample F0 where the grains were now 1–2 μm in diameter, approaching the size of the microparticles themselves. Films F6 and F10 showed less pronounced grain growth with grain sizes up to 600 nm.
The lifetime and stability of the films against sintering was assessed with prolonged heating at 1400 °C for up to 192 h. The onset of sintering necks was observed in all films after 3 h at 1400 °C but was much more pronounced for the undoped particles. Growth of both the grains within and the necks between microparticles continued with longer heating. The resulting microstructures after 192 h at 1400 °C are shown in the lower row of Fig. 5, in which sintering of the particles and densification was clearly observed. While the grain sizes of the undoped film F0 were now in the order of or larger than the microparticle diameters, the grain sizes in films F6 and F10 had only increased up to 0.8 and 1 μm, respectively. Regardless of the retardation of grain growth, the YSZ films still exhibited necking and sintering of the particles similar to the undoped film. Thus, the inhibition of grain growth by yttria-doping failed to prevent sintering at long times at 1400 °C. The cause is attributed to a change in the phase of the material at this temperature from tetragonal to a mixture of cubic and monoclinic, which will be discussed in the following. Sintering notwithstanding, only a small drop in reflectance over the 1–6 μm range was measured for the YSZ films (Fig. 6b and c), ranging from 90–55% after 3 h at 1400 °C down to 78–45% after 192 h at 1400 °C in the case of F6 and from 90–60% down to 80–55% for F10.
After each heating cycle, the phases retained at room temperature were determined via X-ray diffraction measurements. The results are summarized in Table 3. Undoped film F0 remained in the monoclinic phase throughout the whole set of heat treatments. For films F6 and F10 a decomposition of the tetragonal phase was observed with heating at 1400 °C. In order to distinguish tetragonal from cubic, it is convenient to compare the presence of peak doublets indicative of the tetragonal phase to their corresponding peak singlets of the more symmetric cubic phase. This is the case for peaks in the regions of ∼35, ∼50, ∼60 and ∼75° 2θ. Fig. 7 shows the diffractograms of the YSZ films in the region of 35°. This region was selected because it displayed the highest intensity. The results shown were consistent with those measured at the higher angles, where peaks were more resolved but less intense. Fig. 7 also includes the region around 28°, where the characteristic monoclinic peak appears. The initial peak positions after heating at 1200 °C are included for reference in Fig. 7, indicated by dashed lines for clarity. The diffractograms after heating at 1300 °C were identical to those observed after heating at 1200 °C.
| Heat treatment | Phases at room temperaturea | |||
|---|---|---|---|---|
| Temperature (°C) | Time (h) | F0 | F6 | F10 |
| a Phases detected by XRD in trace amounts shown in parentheses. b For F6 and F10, phases t′, t, and c cannot be distinguished at 700 °C due to broadness of peak. | ||||
| 700 | 3 | t + m | —b | —b |
| 1000 | 3 | m + (t) | t | t′ |
| 1200 | 3 | m | t | t′ |
| 1300 | 3 | m | t | t′ |
| 1400 | 3 | m | t | t′ + t + c |
| 1400 | 24 | m | t + c + (m) | t + c |
| 1400 | 72 | m | m + c | t + c |
| 1400 | 192 | m | m + c | m + c + (t) |
After heating to 1000 °C sample F6 was tetragonal and remained that way until it was heated to 1400 °C for 24 h. This was determined from the distinct (002) and (110) peaks visible at 34.7 and 35.2° in Fig. 7a. After a total heating time of 24 h at 1400 °C the cubic (200) peak appeared at 35.0°, together with a very faint (11) monoclinic peak at 28.2°. Heating to 1400 °C for a total of 72 h led to the complete disappearance of the tetragonal peaks, with the cubic peak and very pronounced monoclinic peaks remaining. These observations are consistent with a previously documented decomposition mechanism of the tetragonal phase at high temperatures for plasma sprayed and electron-beam physical vapor deposited YSZ TBCs:41–43 upon heating, Y diffuses and segregates at the grain boundaries, which leads to a disproportionation of the material into Y-lean and Y-rich domains. When those respective domains reach critical Y concentrations, they transition – the Y-lean domains into the monoclinic phase upon cooling and the Y-rich domains into the cubic phase.
Heating film F10 at 1200 °C resulted in the appearance of two distinctive peaks at 34.8 and 35.1°. These peaks were more narrowly spaced from the tetragonal peaks observed at 34.7 and 35.3° for F6. Given its more saturated Y content, the deviation can be explained with the formation of a Y-rich tetragonal phase, referred to here as tetragonal prime (t′), which has slightly different cell parameters than the typical tetragonal (t) phase. Witz et al. reported the t′ and t phases to have Y/(Y + Zr) molar ratios of 7.6–8.6% and 4.3–5.4%, respectively.41 Further heating at 1400 °C for 3 h led to the appearance of the cubic peak at 35.0° and a slight shift of the t′ peaks in the direction of where the t peaks would be expected. This trend continued after heating at 1400 °C for a total of 24 and 72 h, with the cubic peak growing in intensity and the t′ peaks being replaced by the t peaks. A decomposition mechanism similar to the one observed in sample F6 can be formulated: Y segregation upon heating causes the initial t′ phase to partition into even more Y-rich domains and Y-lean domains. Again, the Y-rich domains transform into the cubic phase, while the Y-lean domains transition to the typical t phase with a comparably lower Y content. Depletion of Y caused a change in the c/a ratio of the crystalline unit cell and resulted in the observed broadening of the distance between the (002) and (110) peaks, as clearly seen in Fig. 7b. While the loss of the tetragonal phase fraction and formation of the monoclinic phase was delayed compared to F6, film F10 eventually decomposed into a mixture of cubic and monoclinic after 192 h at 1400 °C.
In general, both the 2 μm particles and 4–5 μm particles for the photonic glasses behaved similarly. Only a difference in phase was observed for particles near the tetragonal-cubic transition, with sample YPG10 initially crystallizing in the tetragonal prime phase while sample Y10 was characterized as cubic. This deviation may be attributed to the adaptations in the synthesis of the larger particles or an inability to resolve the peaks differentiating these phases. Further, after heating to 1200 °C the larger particles displayed somewhat more pronounced shape stability.
4. Conclusions
In summary, we have introduced a new synthetic approach for the fabrication of yttria-doped zirconia microparticles with Y contents ranging from 4 to 18%. Smooth, as-synthesized particles had diameters of ∼2 μm, which by adaptation of various synthetic parameters could be increased to 4–5 μm. The size standard deviations ranged between 6–17%. The particles' crystalline phases and thermal stability were assessed up to 1500 °C. Furthermore, reflective photonic glasses were assembled from doped and undoped 4–5 μm zirconia particles and their optical and physical properties at high temperatures were examined. In particular, we conclude the following:(1) The Y content of YSZ microparticles could be controlled over a wide range simply by adjusting the ratio of zirconia and yttria precursors in the reaction mixture.
(2) Y doping resulted in stabilization of the tetragonal phase, smaller initial grain sizes and slower grain growth for Y contents up to 10%. At higher Y contents, the formation of the cubic phase and accelerated mass transport led to increasing grain sizes. Considering the factors of phase stabilization, grain growth and particle resuspendability, the optimum Y content for stable particles was 8 to 10%.
(3) Films assembled from undoped or Y-doped 4–5 μm zirconia particles displayed broadband reflection up to 90% in the relevant infrared wavelength range between 1–5 μm at temperatures up to 1200 °C. The undoped films exhibited delamination, which was attributed to mechanical stress due to the martensitic tetragonal-to-monoclinic phase transformation, and resulted in significant loss of IR-reflectivity after repeated heating to 1400 °C. In contrast, the 6 and 10% Y-doped films were much more stable and retained their IR-reflectivity even after repeated heating to 1400 °C.
(4) Prolonged heating of the YSZ particles at 1400 °C for 192 h led to a phase separation into the monoclinic and cubic phases, most likely due to the formation of Y-lean and Y-rich domains. This observation confirms the segregation of the Y ions and hence the presence of the solute-drag effect.
In order to enable the assembly of YSZ microparticles into more ordered photonic materials our current research efforts are focusing on further narrowing the particles' size distribution. Moreover, by co-doping with various other metal cations we intend to further enhance the thermal stability of these particles and the photonic materials assembled thereof.
Author contributions
E.W.L. developed the synthetic strategy for the YSZ microparticles and contributed to major extent to their characterization by SEM and XRD measurements, evaluation of their thermal stability, and data interpretation. R.M.P contributed significantly to the interpretation of the SEM- and XRD data and was involved in the high temperature experiments and IR reflectivity measurements. J.J.R. prepared the photonic glass coatings and performed the high temperature experiments with these samples. He performed the XRD measurements and was involved in the IR reflectivity measurements of these coatings. Further, he contributed to the data interpretation regarding both sets of experiments. P.N.D. conducted the IR reflectivity measurements and contributed together with A.Y.P. to the experimental design and interpretation of respective optical data. S.D. and A.P. gave important input to improve the synthetic procedures regarding particle size control. M.E., G.A.S., R.J., H.W. and T.V. were involved as project leaders of the SFB 986 and were responsible for different parts of the study (M.E.: optical properties; G.A.S.: particle assembly; R.J.: high-temperature processes in ceramics; H.W., T.V.: particle synthesis and characterization). All project leaders contributed to the experimental design and data interpretation and by setting the scientific objectives. The manuscript was written by E.W.L. and R.M.P and finalized together with T.V. taking into account the input of all coauthors.Acknowledgements
The authors gratefully acknowledge financial support from the German Research Foundation (DFG) via SFB 986 “Tailor-Made Multi-Scale Materials Systems: M3”, projects C2, C4, C5, C6 and A1. We thank Frank Meyberg and the analytics department for conducting the ICP-OES measurements, Robert Schön for measuring HR-SEM images and Almut Barck for the ex situ XRD measurements. We also thank Nils Walter and Vi Quint Duong for their contributions regarding some experimental procedures. We thank Adam Steiger for assisting with proof reading the manuscript.Notes and references
- T. Ogihara, N. Mizutani and M. Kato, Ceram. Int., 1987, 13, 35–40 CrossRef CAS.
- J. Widoniak, S. Eiden-Assmann and G. Maret, Colloids Surf., A, 2005, 270–271, 329–334 CrossRef CAS.
- J. Widoniak, S. Eiden-Assmann and G. Maret, Eur. J. Inorg. Chem., 2005, 3149–3155 CrossRef CAS.
- J. H. Shim, C. Chao, H. Huang and F. B. Prinz, Chem. Mater., 2007, 19, 3850–3854 CrossRef CAS.
- T. Miller and V. Grassian, J. Am. Chem. Soc., 1995, 117, 10969–10975 CrossRef CAS.
- D. R. Clarke and C. G. Levi, Annu. Rev. Mater. Res., 2003, 33, 383–417 CrossRef CAS.
- P. N. Dyachenko, J. J. do Rosário, E. W. Leib, A. Y. Petrov, R. Kubrin, G. A. Schneider, H. Weller, T. Vossmeyer and M. Eich, ACS Photonics, 2014, 1, 1127–1133 CrossRef CAS.
- V. Shklover, L. Braginsky, G. Witz, M. Mishrikey and C. Hafner, J. Comput. Theor. Nanosci., 2008, 5, 862–893 CAS.
- P. N. Dyachenko, J. J. do Rosário, E. W. Leib, A. Y. Petrov, M. Störmer, H. Weller, T. Vossmeyer, G. A. Schneider and M. Eich, Opt. Express, 2015, 23, A1236–A1244 CrossRef CAS PubMed.
- G. von Freymann, V. Kitaev, B. V. Lotsch and G. A. Ozin, Chem. Soc. Rev., 2013, 42, 2528–2554 RSC.
- P. D. García, R. Sapienza, Á. Blanco and C. López, Adv. Mater., 2007, 19, 2597–2602 CrossRef.
- P. D. García, R. Sapienza and C. López, Adv. Mater., 2010, 22, 12–19 CrossRef PubMed.
- K. Mazdiyasni, Ceram. Int., 1982, 8, 42–56 CrossRef CAS.
- W. H. Rhodes, J. Am. Ceram. Soc., 1981, 64, 19–22 CrossRef CAS.
- B. Fegley and E. A. Barringer, Mat. Res. Soc. Symp. Proc., 1984, 32, 187–197 CrossRef CAS.
- B. Fegley, P. White and H. K. Bowen, Am. Ceram. Soc. Bull., 1985, 64, 1115–1120 CAS.
- L. Lerot, F. Legrand and P. De Bruycker, J. Mater. Sci., 1991, 26, 2353–2358 CrossRef CAS.
- B. Yan, C. V. McNeff, P. W. Carr and A. V. McCormick, J. Am. Ceram. Soc., 2005, 88, 707–713 CrossRef CAS.
- B. Yan, C. V. McNeff, F. Chen, P. W. Carr and A. V. McCormick, J. Am. Ceram. Soc., 2001, 27, 1721–1727 Search PubMed.
- E. W. Leib, U. Vainio, R. M. Pasquarelli, J. Kus, C. Czaschke, N. Walter, R. Janssen, M. Müller, A. Schreyer, H. Weller and T. Vossmeyer, J. Colloid Interface Sci., 2015, 448, 582–592 CrossRef CAS PubMed.
- R. H. J. Hannink, P. M. Kelly and B. C. Muddle, J. Am. Ceram. Soc., 2000, 83, 461–487 CrossRef CAS.
- S. Fabris, A. T. Paxton and M. W. Finnis, Acta Mater., 2002, 50, 5171–5178 CrossRef CAS.
- S. A. Ostanin and E. I. Salamatov, J. Exp. Theor. Phys. Lett., 2001, 74, 552–555 CrossRef.
- I.-W. Chen, Mater. Sci. Eng., A, 1993, 166, 51–58 CrossRef.
- H. Yang, J. Ouyang, X. Zhang, N. Wang and C. Du, J. Alloys Compd., 2008, 458, 474–478 CrossRef CAS.
- C. Guiot, S. Grandjean, S. Lemonnier, J.-P. Jolivet and P. Batail, Cryst. Growth Des., 2009, 9, 3548–3550 CAS.
- Y. Chang, S. Dong, H. Wang, K. Du, Q. Zhu and P. Luo, Mater. Res. Bull., 2012, 47, 527–531 CrossRef CAS.
- K. Uchiyama, T. Ogihara, T. Ikemoto, N. Mizutani and M. Kato, J. Mater. Sci., 1987, 22, 4343–4347 CrossRef CAS.
- M. Keshmiri and O. Kesler, Acta Mater., 2006, 54, 4149–4157 CrossRef CAS.
- Z. Hua, X. M. Wang, P. Xiao and J. Shi, J. Eur. Ceram. Soc., 2006, 26, 2257–2264 CrossRef CAS.
- T. Ogihara, N. Mizutani and M. Kato, J. Am. Ceram. Soc., 1989, 26, 421–426 CrossRef.
- P. Li, I.-W. Chen and J. E. Penner-Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10074–10081 CrossRef CAS.
- G. Stapper, M. Bernasconi, N. Nicoloso and M. Parrinello, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 797–810 CrossRef CAS.
- A. Navrotsky, J. Mater. Chem., 2005, 15, 1883–1890 RSC.
- J. A. Allemann, B. Michel, H.-B. Märki, L. J. Gauckler and E. M. Moser, J. Eur. Ceram. Soc., 1995, 15, 951–958 CrossRef CAS.
- V. G. Keramidas and W. B. White, J. Am. Ceram. Soc., 1974, 57, 22–24 CrossRef CAS.
- W. D. Kingery, J. Am. Ceram. Soc., 1974, 57, 1–8 CrossRef CAS.
- S.-L. Hwang and I.-W. Chen, J. Am. Ceram. Soc., 1990, 73, 3269–3277 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- J. Xu, S. R. Casolco and J. E. Garay, J. Am. Ceram. Soc., 2009, 92, 1506–1513 CrossRef CAS.
- G. Witz, V. Shklover, W. Steurer, S. Bachegowda and H. P. Bossmann, J. Am. Ceram. Soc., 2007, 90, 2935–2940 CrossRef CAS.
- J. A. Krogstad, S. Krämer, D. M. Lipkin, C. A. Johnson, D. R. G. Mitchell, J. M. Cairney and C. G. Levi, J. Am. Ceram. Soc., 2011, 94, 168–177 CrossRef.
- K. Matsui, H. Yoshida and Y. Ikuhara, Acta Mater., 2008, 56, 1315–1325 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, heating rate profiles, SEM and XRD data for the particles as well as SEM, XRD and IR-spectroscopic data for the photonic glass films. See DOI: 10.1039/c5tc03260a |
| This journal is © The Royal Society of Chemistry 2016 |