
A microfluidic-based cell encapsulation platform to achieve high long-term cell viability in photopolymerized PEGNB hydrogel microspheres
Zhongliang
Jiang
,
Bingzhao
Xia
,
Ralph
McBride
 and
John
Oakey
*
and
John
Oakey
*
Department of Chemical Engineering, University of Wyoming, Laramie, WY 82071, USA. E-mail: joakey@uwyo.edu
First published on 25th November 2016
Abstract
Cell encapsulation within photopolymerized polyethylene glycol (PEG)-based hydrogel scaffolds has been demonstrated as a robust strategy for cell delivery, tissue engineering, regenerative medicine, and developing in vitro platforms to study cellular behavior and fate. Strategies to achieve spatial and temporal control over PEG hydrogel mechanical properties, chemical functionalization, and cytocompatibility have advanced considerably in recent years. Recent microfluidic technologies have enabled the miniaturization of PEG hydrogels, thus enabling the fabrication of miniaturized cell-laden vehicles. However, rapid oxygen diffusive transport times on the microscale dramatically inhibit chain growth photopolymerization of polyethylene glycol diacrylate (PEGDA), thus decreasing the viability of cells encapsulated within these microstructures. Another promising PEG-based scaffold material, PEG norbornene (PEGNB), is formed by a step-growth photopolymerization and is not inhibited by oxygen. PEGNB has also been shown to be more cytocompatible than PEGDA and allows for orthogonal addition reactions. The step-growth kinetics, however, are slow and therefore challenging to fully polymerize within droplets flowing through microfluidic devices. Here, we describe a microfluidic-based droplet fabrication platform that generates consistently monodisperse cell-laden water-in-oil emulsions. Microfluidically generated PEGNB droplets are collected and photopolymerized under UV exposure in bulk emulsions. In this work, we compare this microfluidic-based cell encapsulation platform with a vortex-based method on the basis of microgel size, uniformity, post-encapsulation cell viability and long-term cell viability. Several factors that influence post-encapsulation cell viability were identified. Finally, long-term cell viability achieved by this platform was compared to a similar cell encapsulation platform using PEGDA. We show that this PEGNB microencapsulation platform is capable of generating cell-laden hydrogel microspheres at high rates with well-controlled size distributions and high long-term cell viability.
Introduction
Bulk hydrogels are widely used to encapsulate live cells for a variety of applications. In tissue engineering, encapsulating and organizing cells within a three-dimensional hydrogel scaffold can direct the formation of desired tissue through the presentation of specific signaling cues.1,2 Recently, implantation of a cell-laden hydrogel scaffold has been developed as an alternative and appealing approach for controlled, sustainable in vivo growth factor delivery.3 Here, hydrogel scaffolds serve as a barrier to isolate implanted cells from the immune system, preventing immune-rejection4 while allowing bidirectional diffusion of oxygen, nutrients, and cell products and wastes.5 Advances in hydrogel scaffold development has facilitated cell encapsulation as a potential therapy for type 1 diabetes,6 cartilage7 and bone regeneration,8 osteoarthritis,9 Parkinsonian symptoms10 and cancer.11 A variety of cell encapsulants have been investigated including agarose,12 alginate,13 collagen,14 and hyaluronic acid.15 In recent years, PEG-based, free-radical photopolymerized hydrogels have been intensively studied and developed as a cell encapsulant due to their synthetic versatility and ability to spatially and temporally control hydrogel network properties.16–18 Also, PEG hydrogels have displayed excellent cytocompatibility for many cell types including mesenchymal stem cells (MSCs),19 β cells,20 fibroblasts,21 and neural cells.22Miniaturizing PEG hydrogels through microfabrication or microfluidics has emerged as a promising and versatile platform for biosensing,23 tissue engineering,24 drug delivery,25,26 and high throughput screening.27 The generation of structured hydrogels through bioprinting,28 photolithography,29 and liquid bridging30 have been demonstrated to effectively generate functional platforms for cell studies. As these approaches produce device-bound structures, they are useful for rather fundamental in vitro studies, but have limited translational potential. To increase microgel fabrication throughput and collection efficiency, cell encapsulation has also been demonstrated by stop flow lithography (SFL), a single-phase microfluidic technique for PEG microgel fabrication, at low throughput but with precise control over microgel shape and size.31 SFL combined with hydrolytically degradable hydrogel networks can produce a cell or drug delivery scaffold with tunable properties and degradation profiles.32 However, the throughput of SFL is ultimately limited by exposure area and polymerization kinetics.33,34 Microfluidic emulsification, a two-phase microfluidic technique capable of generating monodisperse aqueous droplets in continuous oil phase has improved microgel fabrication frequency to kHz,26 thus providing a potential tool for high throughput single cell encapsulation or screening.35–37 Sustained cell viability following photoencapsulation is a critical parameter for therapeutic cell delivery applications. However, even though high initial post-encapsulation cell viability can be achieved after encapsulating cells within PEGDA hydrogel microspheres,38,39 a dramatic decrease in cell viability is typically observed over longer time periods.45 Thus, an integrated solution that combines technology with materials chemistry is needed to successfully encapsulate live cells and maintain their high cell viability in microgels for weeks, in order to conduct long-term studies.
PEG-based free-radical photopolymerization strategies generally include either acrylate40 or thiol–ene41 chemistries allowing chain-growth or step-growth gelation mechanisms, respectively.42–44 Chain-growth polymerization (PEGDA), however, is almost completely inhibited by the presence of oxygen45 because the rate of radical quenching by oxygen is much faster than the rate of chain initiation and propagation.38,46 Only once oxygen is locally consumed can polymerization occur. Side effects of this radical scavenging include an accumulation of reactive oxygen species (ROS), which can induce elevated intracellular oxidative stress in cells thus cause damage to biomacromolecules such as DNA, proteins, and lipids.47,48 Conversely, step-growth polymerization of PEGNB, occurring between a thiol and vinyl group is not dramatically inhibited by oxygen. Upon photoinitiation, PEGNB can form a more homogenous hydrogel network with reduced network contraction,49 and thus reduced stress upon encapsulated cells than PEGDA. Perhaps most importantly, PEGNB has been shown to support increased post-encapsulation viability over PEGDA for certain cell types, suggesting its broad utility as a cell encapsulant.50 While the origins of PEGNB's increased cytocompatibility is unclear, studies have shown that PEGNB polymerization can be propagated by, thereby consuming, ROS.51,52 For cell types that are especially sensitive to ROS, reducing these deleterious side products of photopolymerization may be a contributing factor to observed increasing cell viability.
Here, we utilized a simply designed, flow-focusing microfluidic device to encapsulate cells within PEGNB droplets, which were then collected and exposed to UV light in bulk solution for photopolymerization. Utilizing this highly reproducible platform, we were able to exploit the benefits of microfluidics to produce uniform cell-laden hydrogel microspheres at high rates. By collecting cell-laden PEGNB droplets into a centrifuge tube and exposing it to a UV source for an extended time, we overcame the relatively slow polymerization kinetics of PEGNB,43 and achieved high post-encapsulation cell viability over the course of 30 days. Hydrogel microsphere uniformity and post-encapsulation cell viability has been compared between this platform and vortex-based cell encapsulation. Several operating parameters have been investigated in order to identify factors to which post-encapsulation cell viability is particularly sensitive. Relative to PEGDA, we have shown this microfluidic-based cell encapsulation platform is able to produce cell-laden PEGNB hydrogel microspheres with dramatically increased post-encapsulation cell viability.
Materials and methods
Initiator and macromer synthesis
Synthesis of the photoinitiator lithium acylphosphinate salt (LAP) was conducted according to an established protocol.53 Briefly, 2,4,6-trimethylbenzoyl chloride was added dropwise to a 100 mL round bottom flask containing an equimolar quantity of dimethyl phenylphosphonite (both from Sigma-Aldrich). The flask was continuously purged with nitrogen gas and stirred at room temperature overnight. A solution containing a four-fold (mol:mol) excess of LiBr dissolved in 80 mL 2-butanone (both from Sigma-Aldrich) was added to the Michaelis–Arbozov product from previous step, stirred, and heated to 50 °C until the formation of a precipitate was observed (10 minutes). The flask was then cooled to room temperature and allowed to stir for 4 hours. Product was dried under vacuum and rinsed with aliquots of 2-butanone to remove unreacted LiBr. The product LAP was recovered with a yield of >80%, and confirmed via H-NMR.Synthesis of the macromer 4-arm poly(ethylene glycol) tetranorbornene (PEGNB) was conducting according to established protocols.43,50,54 Briefly, 12 grams of 20 kDa 4-arm PEG (Creative PEGworks) was added to 20 mL of methylene chloride (MeCl, Sigma Aldrich) in a 40 mL scintillation flask, stirred until complete dissolution was achieved, and set aside. To a 50 mL rb-flask containing 10 mL MeCl was added N,N-dicyclohexyldiimide followed by the dropwise addition of 5-norbornene-2-carboxylic acid (5 and 10 molar excess to 4-arm PEG, respectively; both from Sigma Aldrich). The previously prepared 4-arm PEG solution was added to the 50 mL rb-flask, placed in an ice bath, and allowed to stir overnight under nitrogen. Ice cold diethyl ether (Sigma Aldrich) was added to the product-containing solution and the resultant precipitate was vacuum-filtered, recrystallized, and then subjected to Soxhlet extraction for 36 hours to remove undesired impurities. The white PEGNB product was dried overnight under vacuum. The product was recovered with 67% yield, and confirmed via H-NMR.
Microfluidic device fabrication
Microfluidic cross-flow devices were fabricated using standard PDMS soft lithography techniques.55 Briefly, polydimethylsiloxane (PDMS) was poured onto an SU-8 micropatterned silicon wafer, vacuumed to remove entrapped air, and cured in a 70 °C oven for at least 1 hour. PDMS replicas were cut and punched with a 20 G needle (Brico Medical Supplies, Inc., USA) to form inlets and outlets. PDMS replicas were treated with oxygen plasma and bonded to glass slides. To facilitate droplet formation, a hydrophobic microchannel surface was created by filling the device with Aquapel (PPG Industries) and flushing with air. For PEGNB droplet formation, channel dimensions (h × w) were 80 μm × 100 μm for the cell-containing aqueous phase and 80 μm × 30 μm for the oil phase. Double layer nitrogen-jacketed microfluidic devices for PEGDA particle production were fabricated as previously described.39Hydrogel-forming solution
PEGNB pre-polymer solutions were mixed to a final concentration of 10 wt% PEGNB (Mn ≈ 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da with 4 arms), 10 mM di-thiol linker (Mn ≈ 1500 Da, Sigma-Aldrich, USA), 0.1 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, photoinitiator). PEGDA pre-polymer solutions were mixed to a final concentration of 10 wt% PEGDA (Mn ≈ 3400 Da, JenKem Technology, USA), 0.1 wt% LAP, and 5 mM RGDS. The average molecular weight of each PEG chain was approximately conserved between PEGNB and PEGDA, and these materials have been shown to have similar mechanical properties52,56–58 as hydrogels with comparative compositions. To characterize hydrogel microsphere size, a fluorescent dye, 0.01 wt% thiolated Rhodamine B, was added to and copolymerized with the hydrogel-forming solution to directly label the network. This approach has been widely used to label hydrogel networks with Rhodamine B,59 where it is stable over long time periods. This dye is therefore appropriate to spatially label hydrogel networks60 and track their degradation over long periods of time.32
000 Da with 4 arms), 10 mM di-thiol linker (Mn ≈ 1500 Da, Sigma-Aldrich, USA), 0.1 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, photoinitiator). PEGDA pre-polymer solutions were mixed to a final concentration of 10 wt% PEGDA (Mn ≈ 3400 Da, JenKem Technology, USA), 0.1 wt% LAP, and 5 mM RGDS. The average molecular weight of each PEG chain was approximately conserved between PEGNB and PEGDA, and these materials have been shown to have similar mechanical properties52,56–58 as hydrogels with comparative compositions. To characterize hydrogel microsphere size, a fluorescent dye, 0.01 wt% thiolated Rhodamine B, was added to and copolymerized with the hydrogel-forming solution to directly label the network. This approach has been widely used to label hydrogel networks with Rhodamine B,59 where it is stable over long time periods. This dye is therefore appropriate to spatially label hydrogel networks60 and track their degradation over long periods of time.32
Cell culture
Human lung adenocarcinoma epithelial cells (A549s) were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technology, USA) with low glucose, and supplemented with 10% fetal bovine serum (FBS, Life Technology, USA), 1% PenStrep, and 0.2% Fungizone (Sigma-Aldrich, USA). A549s were cultured in a 37 °C, 5% CO2 incubator, with medium change every 3 days and subculture every 6 days. In preparation for cell encapsulation, A549s were detached from culture flask with 0.05% trypsin (Life Technology, USA), pelleted, and resuspended to a cell density of 5 × 107 cells per mL in culture medium. Medium density was adjusted to 1.06 g mL−1 by adding in 18.75% v/v OptiPrep (Sigma-Aldrich, USA).Microfluidic cell encapsulation
Prior to cell encapsulation in PEGNB (Fig. 1A), A549s (5 × 107 cells per mL) were suspended in hydrogel-forming solution, mixed, and injected into the microfluidic device via syringe pump. Flow rates for fluorocarbon oil (Novec 7500 with 2 wt% Pico-Surf (Dolomite) as surfactant to maintain surface tension, and prevent droplet aggregation) and aqueous phase were held constant at 10 μL min−1 and 2 μL min−1, respectively. Cell-laden droplets were collected into a 1.5 mL centrifuge tube, and polymerized into hydrogel microspheres in bulk by exposing to UV light for 20 seconds (100 mW cm−2, Lumen Dynamics Group Inc, S2000-XLA, Canada). Hydrogel microspheres were separated from oil by washing and centrifuging on a 10 μm strainer (PluriSelect, Germany) with 1 mL 0.1 wt% Pluronic F-68 (Sigma-Aldrich, USA) in PBS. The washing step was repeated once more to completely remove the oil. Cell-laden hydrogel microspheres were resuspended into culture medium containing 0.1 wt% F-68 in low-attachment 6-well plates (Corning Incorporated, USA).38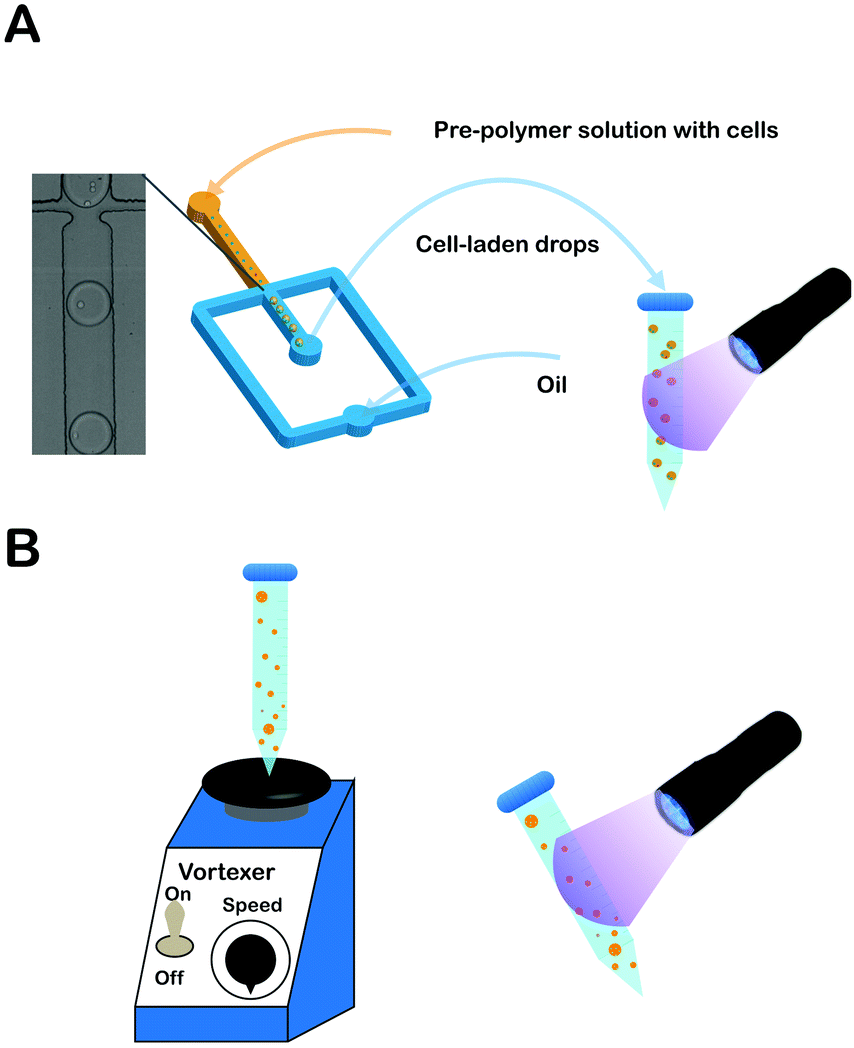 | ||
| Fig. 1 (A) Schematic of PEGNB hydrogel microsphere fabrication by emulsification in a cross-flow microfluidic channel or (B) by vortex, followed by exposure to ultraviolet radiation. | ||
For PEGDA encapsulation (Fig. 5C), A549s, PEGDA, and LAP with RGDS were loaded into three syringes and mixed within the microfluidic device at a constant flow rate of 0.33 μL min−1 for each individual solution. Fluorocarbon oil was injected into microfluidic device at 2 μL min−1 and 7 μL min−1 for the first layer and second layer, respectively. Nitrogen pressure in the jacketing was held at a constant pressure of 7 psi. Before exiting the device, droplets passed through a serpentine channel in the second layer of the device where they were polymerized by exposure to UV light (Mercury-100 W, Chiu Technical Corporation, USA) through a 20× microscope objective (Olympus). Cell-laden PEGDA hydrogel microspheres were separated from oil and cultured as described above.
Vortex-based cell encapsulation
A549s and PEGNB hydrogel-forming solution were mixed by vortex for 30 s on a vortexer (∼1000 osc min−1) (Scientific Industries Inc, USA). Fluorocarbon oil was then added to the aqueous solution and vortexed for 1 min to form emulsions, which were polymerized into hydrogel microspheres by exposure to 100 mW cm−2 UV light for 20 seconds (Fig. 1B). Hydrogel microspheres were separated and cultured using the same approach as described above.Bulk cell encapsulation
A549s were mixed within PEGNB or PEGDA hydrogel-forming solution and exposed to 100 mW cm−2 UV light for 20 seconds to form cell-laden hydrogel constructs. Cells encapsulated within bulk hydrogels were cultured in culture medium, which was changed every other day. The approximate volume of each construct was 60 μL and the density of cells within construct was held constant at ∼5 × 107 cells per mL.Cell viability measurement
Viability of encapsulated cells was measured using a membrane integrity assay (LIVE/DEAD Viability kit, Life Technologies, USA) that stains live cells green and dead cells red. Cell viability was imaged using an inverted fluorescence microscope (IX-71, Olympus, USA) and the images were merged in ImageJ. Cell viability was calculated from statistics generated from three images.Results and discussion
Hydrogel microsphere size distributions
About 100 hydrogel microspheres were imaged using fluorescence microscopy and their diameters were measured using ImageJ. Fluorescence images of hydrogel microspheres and size normal distributions are shown in Fig. 2. As expected, hydrogel microspheres produced by microfluidic devices range in diameter from 90 to 130 μm, exhibiting a much tighter size distribution than those produced by vortex with a diameter range from 30 to 140 μm. The polydispersity of microgel particle size distributions was calculated as the standard deviation of the diameter of particles divided by the mean diameter for both microfluidic and vortex fabrication methods. The microfluidic technique generated microgels with a 6.22% polydispersity while vortexing produced particle samples with an average polydispersity of 23.62%, Monodispersity of microgels generated by microfluidics has been shown to be increased by further reducing channel and particle size.26 Well-controlled size distributions holds enabling consequences for precision cell encapsulation61 and high throughput cell screening.37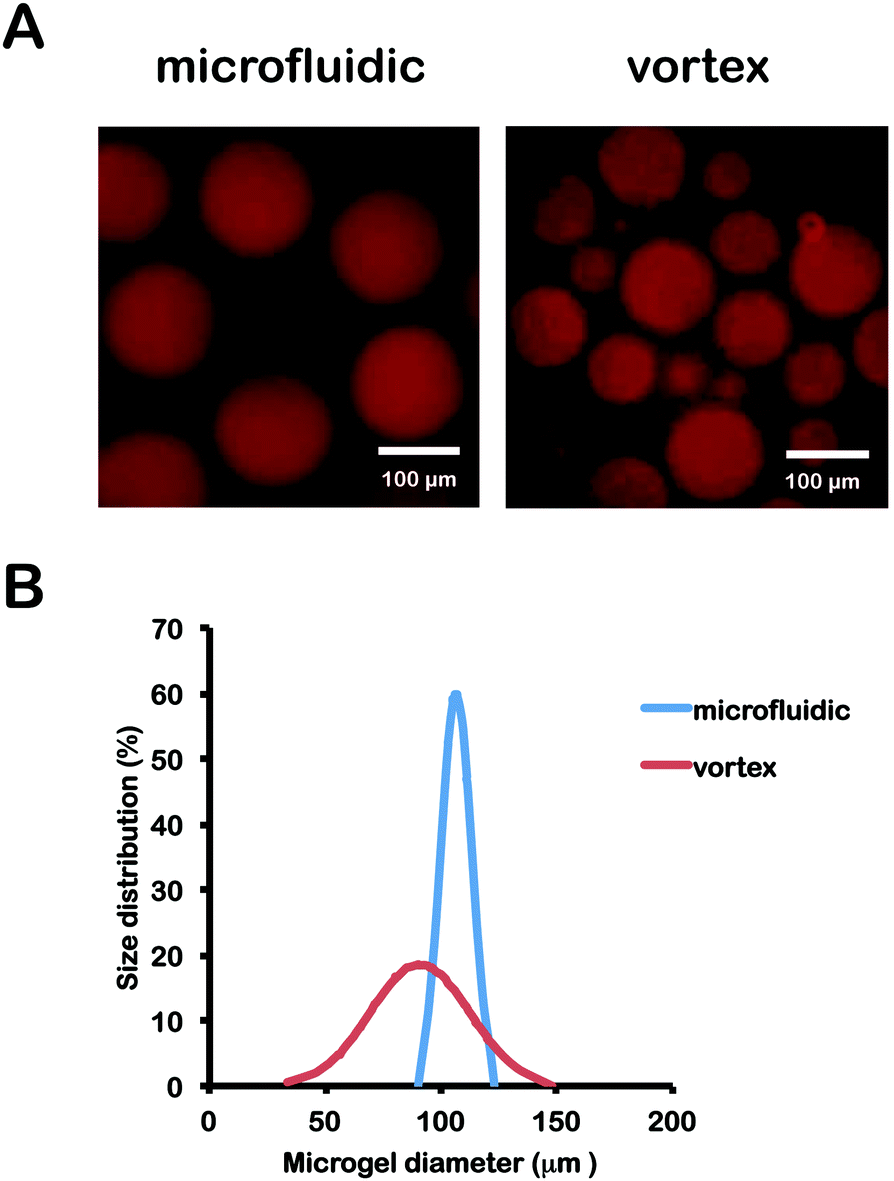 | ||
| Fig. 2 (A) Size distributions and images of PEGNB hydrogel microspheres made by emulsification in a cross-flow microfluidic devices or (B) by vortex. | ||
Cell microencapsulation and effect on cell viability
A549s were encapsulated and photopolymerized within PEGNB hydrogel microspheres through two approaches described above (Fig. 3B). These cell-laden particles were collected and cultured separately in a 37 °C, 5% CO2 incubator with medium change every other day. Cell viability assays were conducted over the course of 30 days by staining cell-laden particles with 2 μM calcein AM and 4 μM ethidium homodimer-1. Cells encapsulated by vortexing and within microfluidic devices shared similar trends in viability. High cell viability (>90%) was observed immediately following encapsulation into hydrogel microspheres, followed by gradually decreased cell viability over the observation period. Cell viability trends were similar for both encapsulation techniques, and neither fell below 40% (Fig. 3A). Notably, blank PEGNB hydrogel particles were fabricated without any modification to facilitate cell attachment or proliferation in this work. However, as previous work has indicated, protein or peptide modifications to the hydrogel network improve cell attachment62 or hydrogel degradation.63 Similar network modifications may be explored as a route to further increasing long-term post-encapsulation viability of cells encapsulated within PEGNB particles.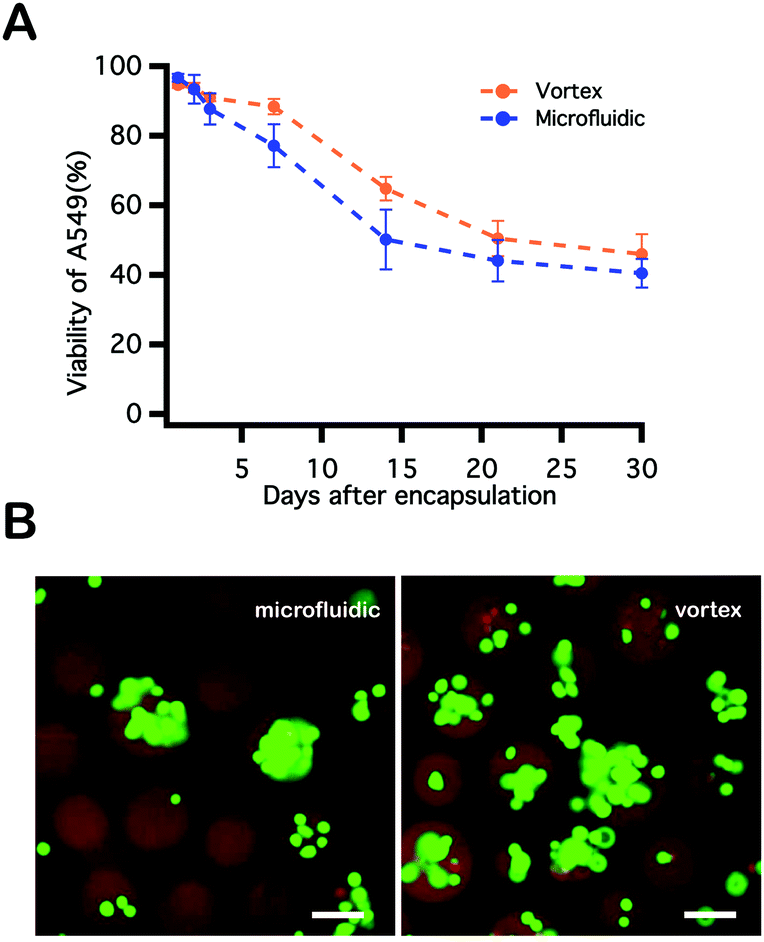 | ||
| Fig. 3 (A) Viability of A549 cells encapsulated within PEGNB microparticles by either vortex or microfluidic emulsification. Cell residence times in the hydrogel-forming solutions prior to photopolymerization were 1 minute and 10 minutes for the vortex and microfluidic methods, respectively. (B) Images of cell-laden hydrogel microspheres made by each method (scale bar = 100 μm). Viability of cells is indicated by live/dead staining assay (live and dead correspond to green and red, respectively). | ||
Influences on post-encapsulation cell viability
To identify parameters during microfluidic cell encapsulation that associate with cell damage and loss of viability, a series of control experiments were performed using cell viability as the primary metric. To replicate the logistics of encapsulation, A549s were transferred in culture medium to a syringe, where they were maintained at room temperature for 2 hours, with cell viability assayed every 0.5 hour. Little decrease in cell viability was observed over the course of 2 hours (Fig. 4A), indicating that long residence times in a syringe prior to delivery to the device is not deleterious to cells. To explore whether aspects of microfluidic encapsulation of cell into droplets affected cell viability, A549 in medium were injected into a microchannel and were either passed through or were pinched off into droplets by an oil phase. In the latter case, flow rates for the aqueous and oil phases were 2 μL min−1 and 10 μL min−1. Emulsions were broken by centrifugation using a 5 μm cell strainer. Cell viability was reduced by only about 4% as a result of traveling through the microfluidic device, and by an additional 5% by virtue of being pinched off by oil and encapsulated into droplets (Fig. 4B). A549s were next mixed into the PEGNB pre-polymer solution and maintained at room temperature without any UV exposure over a period of 2 hours. A notable decrease in cell viability was observed by staining these cells every 30 minutes (Fig. 4C), indicating that merely incubating cells in the PEG hydrogel-forming solution for extended periods of time represents a process that reduces cell viability. The effect of the elapsed time between cell encapsulation and hydrogel photopolymerization on post-encapsulation cell viability was also investigated. Cell-laden PEGNB droplets were collected for discrete durations, and then polymerized by exposure to UV light. Cells suffered a significant decrease in viability with increasing collection time (Fig. 4D). The time-dependent decrease in cell viability observed as a result of cell residence within hydrogel forming solutions may explain the slight increase in cell viability following encapsulation by vortexing over microfluidics (Fig. 3). Vortexing, like the polymerization of bulk hydrogel constructs, is an inherently batch process, which required only about two minutes between emulsion generation and the collection of polymerized cell-laden particles. Microfluidic encapsulation, on the other hand, is a continuous process that may require extended incubation within unpolymerized hydrogel forming solutions. As noted, this residence time, while unexplained, is deleterious to cells.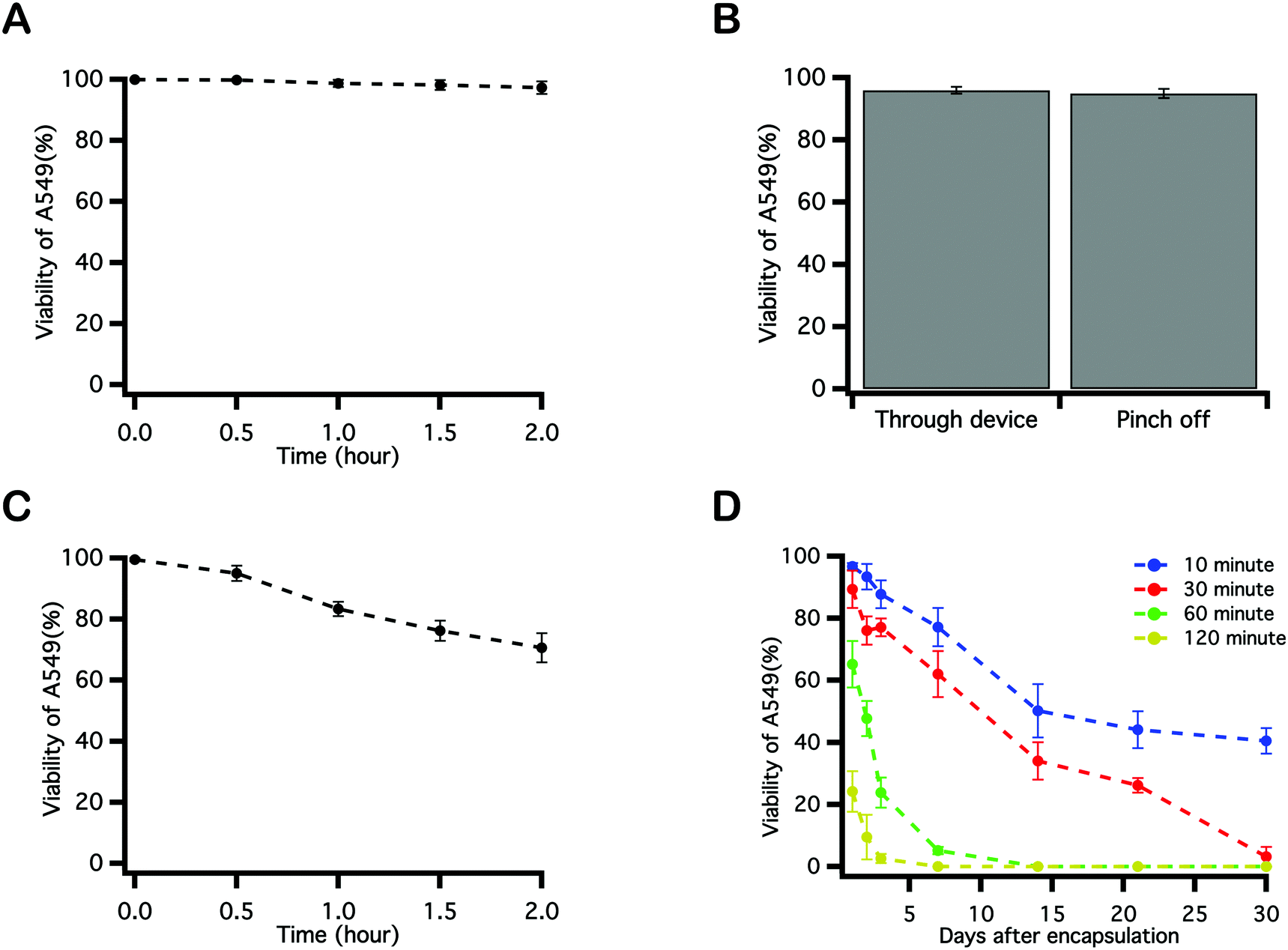 | ||
| Fig. 4 Factors affecting post-encapsulation cell viability were investigated. (A) Viability of cells maintained in media within a syringe over time. (B) Viability of cells pumped through a device in culture medium with or without being formed into emulsion droplets. Flow rates for cells and oil were 2 μL min−1 and 10 μL min−1, respectively. (C) Viability of cells maintained within a hydrogel-forming media solution incubated within a syringe over time. (D) Viability of cells encapsulated into PEGNB microspheres via microfluidic emulsification, collected in a vial, and exposed to UV light following specific incubation periods in culture media. Viability was measured for 30 days. | ||
Comparison between PEGNB and PEGDA post-encapsulation cell viability
PEGDA has been widely proven as a viable material for cell encapsulation, and has recently been shown to support high cell viability immediately following encapsulation and photopolymerization in microdroplets.39 To evaluate the biocompatibility of PEGNB as a cell encapsulant relative to PEGDA under different encapsulation conditions, A549s were encapsulated into PEGNB or PEGDA bulk hydrogels and hydrogel microspheres. Following bulk encapsulation, cells in PEGNB hydrogels maintained a higher viability than those in PEGDA hydrogel (Fig. 5A and B), which is in agreement with previously reported results.50,64–66 For continuous cell microencapsulation, the high oxygen permeability of PDMS microfluidic devices,67 combined with the high oxygen solubility of the fluorocarbon oil used in these studies68 necessitates the use of a custom microfluidic design.38 This device, a nitrogen-jacketed double layer device (Fig. 5C) was introduced to circumvent oxygen inhibition of PEGDA39 by eliminating oxygen diffusion to the droplet from the PDMS device or oil. Immediately after encapsulation, high cell viability was achieved in both blank PEGNB and RGDS-modified PEGDA hydrogel microspheres. After incubating for 8 days, however, high cell viability was sustained in PEGNB particles while cell viability decreased dramatically to 0% on day 8 in PEGDA particles (Fig. 5D). These results reflect trends observed in bulk hydrogels and further suggest that PEGNB is an inherently more cytocompatible cell encapsulant than PEGDA and supports enhanced and sustained cell survival.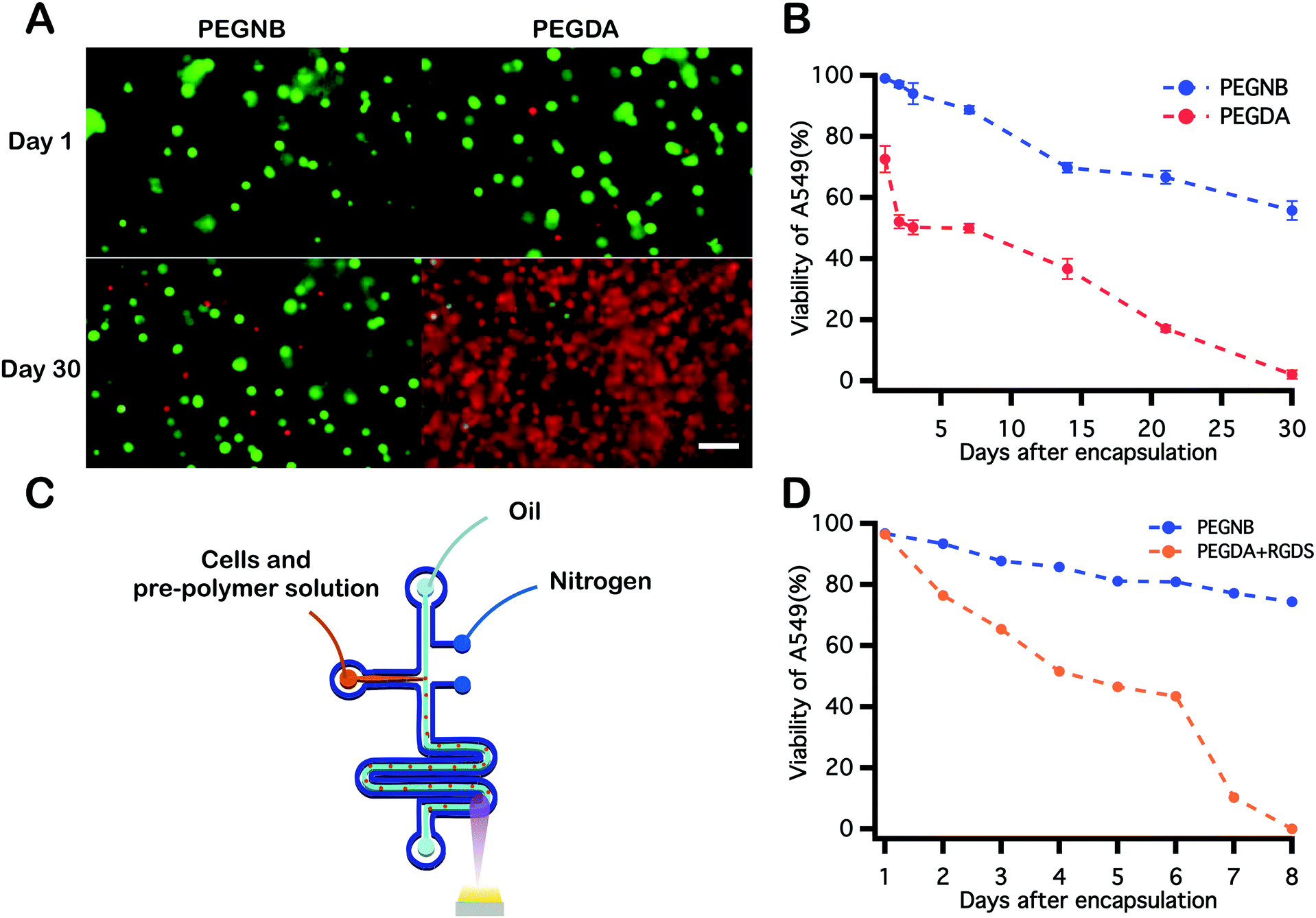 | ||
| Fig. 5 Comparison between observed viability for cells encapsulated within PEGNB and PEGDA microspheres via microfluidic emulsification. (A) Images of cells encapsulated into 10 wt% PEGNB and PEGDA bulk hydrogels. Cells were mixed with PEG hydrogel-forming solution followed by exposure to UV light to polymerize the cell-laden bulk structure. (B) Cell viability was quantified for 30 days following encapsulation in bulk hydrogels. (C) Schematic illustration of the nitrogen jacketed microfluidic device used for cell encapsulation into PEGDA hydrogel microspheres. (D) Quantification of post-encapsulation viability of cells within PEGNB and RGDS-modified PEGDA hydrogel microspheres. Scale bar = 100 μm. | ||
Conclusions
In this work, we have introduced a microfluidic-based cell encapsulation platform that allows PEGNB to be employed as the cell encapsulant. This approach was compared with and found to be superior to vortex-based cell encapsulation in terms of controlling hydrogel microsphere size and uniformity. We have shown that highly monodisperse, uniform cell-laden microgels with average diameters of approximately 110 μm can be generated consistently at high rates. Post-encapsulation cell viability, which is regarded as a primary parameter in this study, can be maintained at a relatively high level in the course of 30 days. Several experimental parameters have been investigated in order to tune cell viability after microencapsulation. Notably, a correlation was identified between post-encapsulation cell viability and droplet collection time, indicating cell viability decreases in a time-dependent manner until photopolymerization is conducted. Finally, the cytocompatibility of PEGNB was compared with PEGDA under bulk and microparticle cell encapsulation conditions. Cells encapsulated within PEGNB were found to exhibit excellent long-term survival, far outperforming PEGDA-encapsulated cells despite equivalent viabilities immediately following encapsulation. The microfluidic approach demonstrated here will enable PEGNB to be formed into microparticles that display the same relative advantages over PEGDA that have been observed in bulk hydrogels.Acknowledgements
This work was supported by the NSF Faculty Early Career Development (CAREER) Program (BBBE 1254608) and by the NIH-funded Wyoming IDeA Networks of Biomedical Research Excellence program (P20RR016474 and P20GM103432).References
- S. Ansari, C. Chen, X. Xu, N. Annabi, H. H. Zadeh, B. M. Wu, A. Khademhosseini, S. Shi and A. Moshaverinia, Ann. Biomed. Eng., 2016, 1–13 Search PubMed.
- S. J. Bryant, R. J. Bender, K. L. Durand and K. S. Anseth, Biotechnol. Bioeng., 2004, 86, 747–755 CrossRef CAS PubMed.
- M. Kar, Y.-R. V. Shih, D. O. Velez, P. Cabrales and S. Varghese, Biomaterials, 2016, 77, 186–197 CrossRef CAS PubMed.
- P. E. Lacy, O. D. Hegre, A. Gerasimidi-Vazeou, F. T. Gentile and K. E. Dionne, Science, 1991, 254, 1782–1784 CAS.
- A. Murua, A. Portero, G. Orive, R. M. Hernández, M. de Castro and J. L. Pedraz, J. Controlled Release, 2008, 132, 76–83 CrossRef CAS PubMed.
- A. J. Vegas, O. Veiseh, M. Gürtler, J. R. Millman, F. W. Pagliuca, A. R. Bader, J. C. Doloff, J. Li, M. Chen, K. Olejnik, H. H. Tam, S. Jhunjhunwala, E. Langan, S. Aresta-Dasilva, S. Gandham, J. J. McGarrigle, M. A. Bochenek, J. Hollister-Lock, J. Oberholzer, D. L. Greiner, G. C. Weir, D. A. Melton, R. Langer and D. G. Anderson, Nat. Med., 2016, 22, 306–311 CrossRef CAS PubMed.
- J. Lam, E. C. Clark, E. L. S. Fong, E. J. Lee, S. Lu, Y. Tabata and A. G. Mikos, Biomaterials, 2016, 83, 332–346 CrossRef CAS PubMed.
- S. Subramaniam, Y.-H. Fang, S. Sivasubramanian, F.-H. Lin and C.-P. Lin, Biomaterials, 2016, 74, 99–108 CrossRef CAS PubMed.
- X. Guo, H. Park, S. Young, J. D. Kretlow, J. J. van den Beucken, L. S. Baggett, Y. Tabata, F. K. Kasper, A. G. Mikos and J. A. Jansen, Acta Biomater., 2010, 6, 39–47 CrossRef CAS PubMed.
- M. D. Lindner, S. R. Winn, E. E. Baetge, J. P. Hammang, F. T. Gentile, E. Doherty, P. E. Mcdermott, B. Frydel, M. D. Ullman, T. Schallert and D. F. Emerich, Exp. Neurol., 1995, 132, 62–76 CrossRef CAS PubMed.
- A. Monette, C. Ceccaldi, E. Assaad, S. Lerouge and R. Lapointe, Biomaterials, 2016, 75, 237–249 CrossRef CAS PubMed.
- P. D. Benya and J. D. Shaffer, Cell, 1982, 30, 215–224 CrossRef CAS PubMed.
- A. I. Chou and S. B. Nicoll, J. Biomed. Mater. Res., Part A, 2009, 91, 187–194 CrossRef PubMed.
- C. M. Rubert Pérez, A. Panitch and J. Chmielewski, Macromol. Biosci., 2011, 11, 1426–1431 CrossRef PubMed.
- S.-Y. Choh, D. Cross and C. Wang, Biomacromolecules, 2011, 12, 1126–1136 CrossRef CAS PubMed.
- A. Priola, G. Gozzelino, F. Ferrero and G. Malucelli, Polymer, 1993, 34, 3653–3657 CrossRef CAS.
- D. Seliktar, Science, 2012, 1–6 Search PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 59–63 CrossRef CAS PubMed.
- D. S. W. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS PubMed.
- L. M. Weber, K. N. Hayda, K. Haskins and K. S. Anseth, Biomaterials, 2007, 28, 3004–3011 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- M. J. Mahoney and K. S. Anseth, Biomaterials, 2006, 27, 2265–2274 CrossRef CAS PubMed.
- V. K. Yadavalli, W.-G. Koh, G. J. Lazur and M. V. Pishko, Sens. Actuators, B, 2004, 97, 290–297 CrossRef CAS.
- Y. Lu, G. Mapili, G. Suhali, S. Chen and K. Roy, J. Biomed. Mater. Res., Part A, 2006, 77, 396–405 CrossRef PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CAS.
- C. Siltanen, M. Yaghoobi, A. Haque, J. You, J. Lowen, M. Soleimani and A. Revzin, Acta Biomater., 2016, 1–8 Search PubMed.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS PubMed.
- Y. Ma, Y. Ji, G. Huang, K. Ling, X. Zhang and F. Xu, Biofabrication, 2015, 7, 1–9 CrossRef PubMed.
- Y. Li, G. Huang, B. Gao, M. Li, G. M. Genin, T. J. Lu and F. Xu, NPG Asia Mater., 2016, 8, e238–e239 CrossRef CAS.
- L. Wang, M. Qiu, Q. Yang, Y. Li, G. Huang, M. Lin, T. J. Lu and F. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 11134–11140 Search PubMed.
- P. Panda, S. Ali, E. Lo, B. G. Chung, T. A. Hatton, A. Khademhosseini and P. S. Doyle, Lab Chip, 2008, 8, 1056–1061 RSC.
- D. K. Hwang, J. Oakey, M. Toner, J. A. Arthur, K. S. Anseth, S. Lee, A. Zeiger, K. J. Van Vliet and P. S. Doyle, J. Am. Chem. Soc., 2009, 131, 4499–4504 CrossRef CAS PubMed.
- D. Dendukuri, S. S. Gu, D. C. Pregibon, T. A. Hatton and P. S. Doyle, Lab Chip, 2007, 7, 818–828 RSC.
- K. Nishi, K. Fujii, Y. Katsumoto, T. Sakai and M. Shibayama, Macromolecules, 2014, 47, 3274–3281 CrossRef CAS.
- A. Reece, B. Xia, Z. Jiang, B. Noren, R. McBride and J. Oakey, Curr. Opin. Biotechnol., 2016, 40, 90–96 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed.
- K. Krutkramelis, B. Xia and J. Oakey, Lab Chip, 2016, 1–9 Search PubMed.
- B. Xia, K. Krutkramelis and J. Oakey, Biomacromolecules, 2016, 17, 2459–2465 CrossRef CAS PubMed.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Macromolecules, 2003, 36, 3861–3873 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- A. M. Kloxin, C. J. Kloxin, C. N. Bowman and K. S. Anseth, Adv. Mater., 2010, 22, 3484–3494 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, Adv. Funct. Mater., 2009, 19, 2325–2331 CrossRef CAS PubMed.
- A. K. O'Brien and C. N. Bowman, Macromol. Theory Simul., 2006, 15, 176–182 CrossRef.
- D. Dendukuri, P. Panda, R. Haghgooie, J. M. Kim, T. A. Hatton and P. S. Doyle, Macromolecules, 2008, 41, 8547–8556 CrossRef CAS.
- H. Wiseman and B. Halliwell, Biochem. J., 1996, 313, 17–29 CrossRef CAS PubMed.
- E. Cabiscol, J. Tamarit and J. Ros, Int. Microbiol., 2010, 3, 3–8 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C.-C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- A. K. O'Brien, N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2007–2014 CrossRef.
- J. J. Roberts and S. J. Bryant, Biomaterials, 9999, 34, 9969–9979 CrossRef PubMed.
- T. Majima, W. Schnabel and W. Weber, Makromol. Chem., 1991, 192, 2307–2315 CrossRef CAS.
- H. Shih and C.-C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- K. Chatterjee, S. Lin-Gibson, W. E. Wallace, S. H. Parekh, Y. J. Lee, M. T. Cicerone, M. F. Young and C. G. Simon Jr, Biomaterials, 2010, 31, 5051–5062 CrossRef CAS PubMed.
- S. Lin-Gibson, S. Bencherif, J. A. Cooper, S. J. Wetzel, J. M. Antonucci, B. M. Vogel, F. Horkay and N. R. Washburn, Biomacromolecules, 2004, 5, 1280–1287 CrossRef CAS PubMed.
- W. Xi, H. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159–6165 CrossRef CAS PubMed.
- H. Lee, S. J. Shapiro, S. C. Chapin and P. S. Doyle, Anal. Chem., 2016, 88, 3075–3081 CrossRef CAS PubMed.
- D. Velasco, E. Tumarkin and E. Kumacheva, Small, 2012, 8, 1633–1642 CrossRef CAS PubMed.
- F. Yang, C. G. Williams, D.-A. Wang, H. Lee, P. N. Manson and J. Elisseeff, Biomaterials, 2005, 26, 5991–5998 CrossRef CAS PubMed.
- S. B. Anderson, C.-C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS PubMed.
- Z. Mũnoz, H. Shih and C.-C. Lin, Biomater. Sci., 2014, 2, 1063–1072 RSC.
- C. Nuttelman, M. Tripodi and K. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS PubMed.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2004, 68, 773–782 CrossRef PubMed.
- H. Shiku, T. Saito, C.-C. Wu, T. Yasukawa, M. Yokoo, H. Abe, T. Matsue and H. Yamada, Chem. Lett., 2006, 35, 234–235 CrossRef CAS.
- J. G. Riess and M. Le Blanc, Pure Appl. Chem., 1982, 54, 2383–2406 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |