
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser-synthesized ligand-free Au nanoparticles for contrast agent applications in computed tomography and magnetic resonance imaging†
Teresa
Simão
a,
Pascale
Chevallier
bc,
Jean
Lagueux
b,
Marie-France
Côté
b,
Christoph
Rehbock
d,
Stephan
Barcikowski
d,
Marc-André
Fortin
*bce and
Daniel
Guay
*a
aInstitut National de la Recherche Scientifique (INRS), Centre Énergie Matériaux Télécommunications, 1650 Lionel-Boulet Boulevard, Varennes (QC), J3X 1S2, Canada
bCentre de Recherche du Centre Hospitalier Universitaire de Québec (CR-CHUQ), Axe Médecine Régénératrice, Quebec City (QC), G1L 3L5, Canada
cCentre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, Quebec City (QC), G1V 0A6, Canada. E-mail: Marc-Andre.Fortin@gmn.ulaval.ca
dTechnical Chemistry I, University of Duisburg-Essen and Center for NanoIntegration Duisburg-Essen CENIDE, Universitaetsstrasse 7, 45141 Essen, Germany
eDépartement de Génie des Mines, de la Métallurgie et des Matériaux, Université Laval, Quebec City (QC), G1V 0A6, Canada
First published on 9th September 2016
Abstract
In recent years, pulsed laser ablation in liquids (PLAL) has emerged as a new green chemistry method to produce different types of nanoparticles (NPs). It does not require the use of reducing or stabilizing agents, therefore enabling the synthesis of NPs with highly-pure surfaces. In this study, pure Au NPs were produced by PLAL in aqueous solutions, sterically stabilized using minimal PEG excess, and functionalized with manganese chelates to produce a dual CT/MRI contrast agent. The small hydrodynamic size (36.5 nm), low polydispersity (0.2) and colloidal stability of Au NPs@PEG-Mn2+ were demonstrated by DLS. The particles were further characterized by TEM, XPS, FTIR and 1H NMR to confirm the purity of the Au surfaces (i.e. free from the common residual chemicals found after NP synthesis) and the presence of the different surface molecules. The potential of these particles as contrast agents for CT/MRI was assessed in vivo (e.g. chicken embryo). Au NPs@PEG-Mn2+ also demonstrated strong blood retention for at least 90 minutes following intravenous injection in mouse models. The promising performance of PEGylated PLAL-synthesized Au NPs containing manganese chelates could open new possibilities for the production of purer dual imaging contrast agents based on Au colloids.
1. Introduction
Research on biomedical applications of gold nanoparticles (Au NPs) has increased significantly in the last decade. Several formulations are now in clinical trials for therapy of resistant tumors in the head and neck, while others are used in biomedical technologies and pre-clinical studies.1 In the latter context, Au NPs have emerged as a viable alternative to iodine-based contrast agents (CAs) for computed tomography (CT) in animal research, and clinical applications of these products are currently under consideration. Au NPs are advantageous for this application since (i) Au atoms have a high atomic number (Z = 79), and their very high electron density strongly interacts with impinging X-ray photons; (ii) Au NPs are biocompatible and chemically stable in biological media; (iii) the size, shape and surface properties of the Au NPs lend themselves to versatile modifications.2–4 Therefore, when adequately covered with biocompatible antifouling materials, Au NPs show long blood retention times, minimizing the need for repeated contrast injections.Numerous examples of the performance of Au NPs as CT CAs are available in the literature. For example, Nebuloni et al. compared different pre-clinical (e.g. blood pool) CAs as well as a clinically-approved, iodine-based CA and concluded that Au NPs (i.e. Aurovist™ – 15 nm) provided the highest contrast resolution for the visualization of vasculature, using the lowest injected volume and one of the lowest molar concentrations of attenuating metal elements.5 Au NPs are also a versatile tool to study tumor vascular permeability (e.g. in anti-angiogenic therapies),6 and have been advantageously used to measure the lumen diameter of the main blood vessels (e.g. carotid arteries) in micro-CT images.7
Despite the fact that CT has high spatial and temporal resolutions as well as a relatively low cost, it has a low soft tissue contrast resolution, a limited capacity to perform functional studies, and uses ionizing radiation for image acquisition.8,9 On the other hand, MRI is free of ionizing radiation, which represents a significant advantage in the frequent follow-up of recurrent diseases. Also, MRI CAs based on paramagnetic elements such as Gd3+ and Mn2+ are detected at the sub-millimolar level (i.e. more than one level of magnitude lower than Au-based CT CAs).10 Finally, MRI is much more efficient than CT for the imaging of pathological soft tissues with subtle density differences (e.g. prostate tumors and breast tumors).11,12 Dual CT/MRI nanoparticulate CAs could find promising applications as brachytherapy sources for prostate and breast cancer, for radiosensitizing applications, as well as in radiotherapeutic planning by imaging (CT/MRI).13–15
However, to reach the detection thresholds of CT, large amounts of Au NPs must be injected into the blood and the particles must be very small to facilitate renal excretion. As a result of their small diameter, the total surface area developed by these Au NPs remains large, and scant traces of contamination or residual chemical from the synthesis step may lead to toxicity effects not necessarily associated with the Au NPs core. Accordingly, it would be highly desirable to synthesize Au NPs whose surface are ligand-free.
The preparation of Au NPs with a bare surface is also important for their stabilization in biological media and for the development of a multimodal CA. Indeed, one of the most critical steps in preparing Au NPs with long blood retention times for CT applications is the grafting of biocompatible molecules at the particle surface. Polyethylene glycol (PEG) features prominently among the biocompatible molecules used to create steric repulsion between particles, while minimizing agglomeration in biological media.16–18 This class of polymers is also well-known for its capacity to prevent protein adsorption at the surface of NPs, and it is therefore widely used to stabilize Au NPs in biological media.2,19 PEG is most often grafted at the Au NPs surface by using thiol-containing (–SH) heterobifunctional molecules.2,16,17 Thiols are well-known to bind strongly to Au.20 Strong, dense and lasting PEG grafting at the Au NP surface is a major requirement for vascular CT studies. However, the interaction of the thiol group of PEG to the Au surface can be affected by the presence of chemicals at the surface of NPs prepared by most common methods.
For instance, the Turkevich method, the most widely-used Au NP synthesis route, is based on the reduction of Au3+ ions by citrate molecules in hot aqueous solutions.2 The resulting Au NPs appear to be coated with citrate and acetoacetate molecules as well as chloride anions. These molecules are strongly adsorbed on the Au NPs surface due to stabilization through intermolecular interactions, such as hydrogen bonding between carboxylic groups of nearby citrate molecules.21 Citrate-coated Au NPs are not particularly stable in biological medium, as decreasing citrate concentrations in the fluid environment may lead to the detachment of these stabilizing ligands; moreover, citrate-coated NPs are strongly negatively charged, which triggers the adhesion of proteins and results in rapid opsonization of the injected NPs. Thereby, citrate molecules at the surface of Au NPs must be ligand-exchanged by PEG to provide steric stabilization.2,22 Synthesis of Au NPs in polyol solvents have also been developed, but the use of organic solvents and extensive washing steps to remove excess reactants and by-products limit the efficient and rapid grafting of PEG.2,23 In addition, several reviews have been written on the toxicity effects of Au NPs,24–26 and each one of these mention the impact of residual chemicals on the cytotoxic effects caused by Au NPs. Among the different reducing agents and additives that are used to generate Au NPs of either fine or narrow size distributions, figures cetyltrimethylammonium bromide (CTAB). CTAB is increasingly being used in Au NP's synthesis, as it facilitates the development of single or multi-step syntheses of narrow particle size distributions of various size and shapes. Unfortunately, a contaminant such as CTAB is considerably toxic to cells, even at low doses. For instance, Alkilany et al. have shown that CTAB, induces strong cytotoxicity when it detaches from the NP surface.27 Connor et al. also studied the toxicity of 18 nm Au NPs to K562 cells. CTAB-coated Au NPs were already toxic at 0.05 mM. Clearly, in this case the toxicity of the conjugate results entirely from the presence of CTAB on the particle surface, which must be removed before presentation to cells.28 Niidome et al. used Au nanorods obtained with a protocol of synthesis induced by CTAB-coated seed in a CTAB-containing growth solution. They demonstrated that CTAB-coated rods were highly cytotoxic on HeLa cells, at a concentration as low as 0.05 mM (80% of dead cells). However, when CTAB was replaced with PEG-SH, the authors observed 95% of viable cells, with the nanorod concentration being 0.5 mM.29 In summary, the available data permit us to draw the justified conclusion that Au NPs as clean as possible must be used prior to PEGylation, in order to limit as much as possible chemical interferences with cells. Overall, the efficient grafting of PEG on the Au NPs prepared by either methods requires the elimination of most chemical contaminants at their surfaces, but this is very difficult to achieve with centrifugation, dialyses, and other NP purification procedures.21,30 Also, each centrifugation or dialysis step presents a significant risk of losing large fractions of the stabilized Au NPs through agglomeration or adherence to the vials. Developing ligand-free and well-controlled synthesis techniques to produce ultra-small Au NPs would provide more efficient PEG grafting procedures as well as remove the residual presence of toxic reducing agents from NPs to be used as CT contrast media, which can be injected at concentrations that range from 0.01 to 2.7 g kg−1 depending on the application.31,32 Given the high dosage of NPs needed to produce significant contrast-enhancement levels in CT, it is necessary to develop CT CAs whose chemical compositions are as simple as possible and contain a minimal presence of molecules that might affect the protein corona and introduce pharmacokinetic variability.21,33,34
Bare surface Au NPs may be produced by pulsed laser ablation of metal targets in liquid (PLAL). This technique allows the production of Au NPs in water without the use of reducing or stabilizing chemicals.35 Consequently, the surface of the NPs is free of ligands and may easily be grafted with other molecules. This also allows a higher surface coverage of biocompatible, steric repulsion ligands (e.g. PEG-SH) in comparison with NPs synthesized through conventional chemical synthesis.36 Overall, Au NPs synthesized by PLAL could improve the biocompatibility of nanomaterials for biomedical applications, as well as eliminate tedious ligand exchange and solvent purification steps.
In this study, we explored the potential of Au NPs synthesized by PLAL to be used as a CA for dual imaging by CT and MRI. The bare Au NP surfaces allow for very efficient PEG grafting per unit surface of Au NPs. The particles are covered with PEG to achieve colloidal stability in biological fluids and to prevent protein adsorption. In order to enhance MRI contrast, the PEG-stabilized Au NPs were further functionalized with manganese-chelates, then characterized for CT and MRI contrast properties both in vitro and in vivo. Therefore, the method developed was suitable to produce a NP-based CA appropriate for dual imaging by CT/MRI, using a simple and aqueous based procedure, which presents a significant advantage compared with previously reported methods for the synthesis of “positive” MRI/CT CAs.
2. Results
In this study, we synthesized small Au NPs by PLAL in NaOH-supplemented water (Scheme 1A) without the use of stabilizing ligands typical of Au NP colloidal synthesis. NP functionalization was performed in three steps. First, pure Au NPs were modified with PEG-SH (Scheme 1B) to obtain a colloid that is sterically stabilized and consequently more resistant to changes in the solution's ionic strength.37 Second, Au NPs@PEG reacted with DMSA-DTPA (Scheme 1C). Finally, the excess of DTPA-DMSA was removed and Mn2+ was added for chelation by the DTPA grafted on the NPs (Scheme 1D).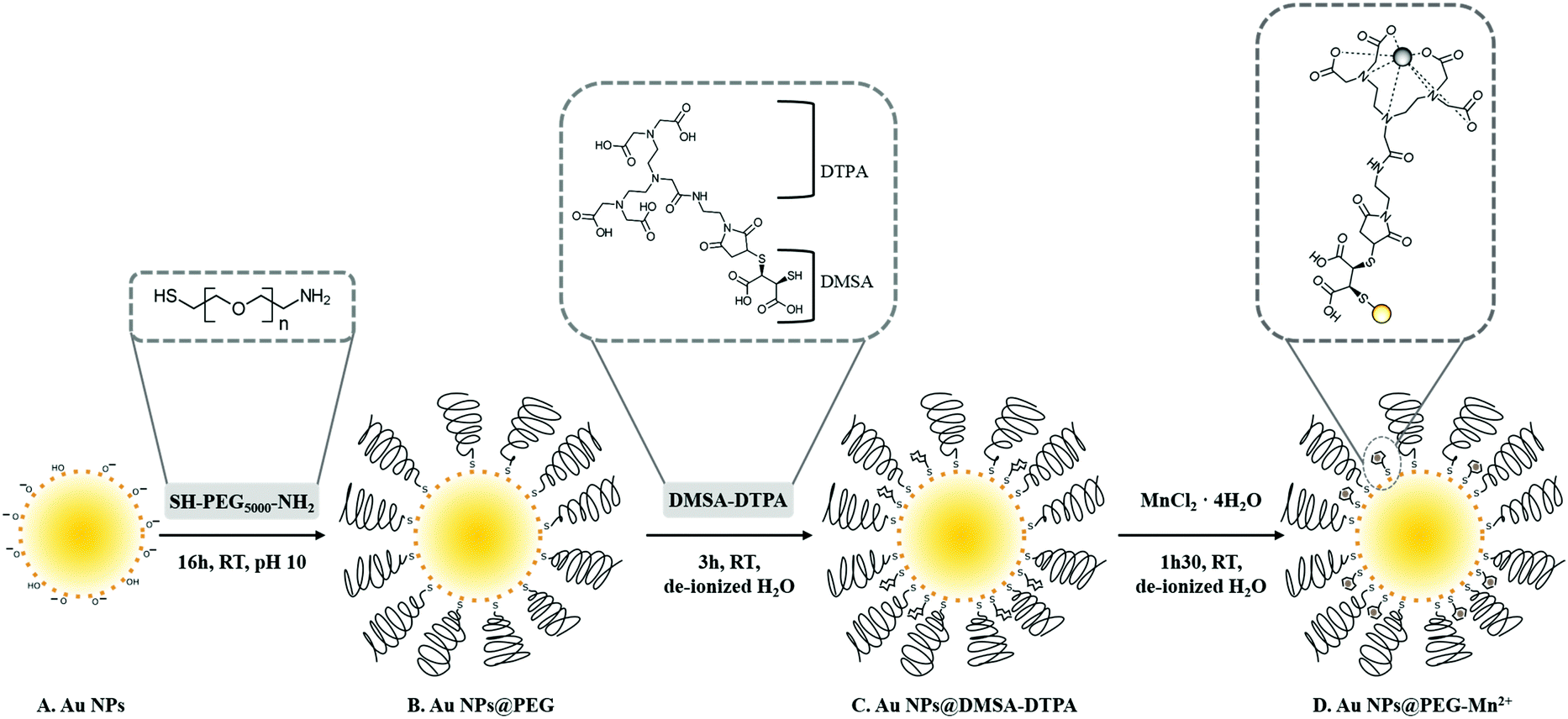 | ||
| Scheme 1 Procedure for the synthesis of Au NPs@PEG-Mn2+. PLAL-synthesized Au NPs with bare surfaces (A) were coated with PEG-SH (B) and functionalized with DMSA-DTPA (C). Mn2+ was chelated by DTPA to form Au NPs@PEG-Mn2+ (D). | ||
2.1 Characterization of Au NPs@PEG-Mn2+
The molecular changes resulting from each of the functionalization steps (Scheme 1) were investigated by FTIR and XPS analysis. FTIR spectra (Fig. 1) showed that, after drying, pure Au NPs are in contact with molecules bearing carbonyl (at 1668 and 1741 cm−1), carboxylate (in the region between 1338 and 1396 cm−1, and at 1568 cm−1) and C–Hx (at 2852 and 2924 cm−1) groups. Part of these later peaks most probably originate from traces of methanol used to clean the crystal. The carbonate groups were mostly absent after the addition of PEG-SH, as the spectrum of Au NPs@PEG mainly demonstrated vibration modes related to the PEG backbone chain, such as the C–O–C band (1113 cm−1).38 Subsequently, DMSA and DTPA grafting was confirmed by the presence of vibration modes related to carboxyl (1705 cm−1) and carboxylate (1599 and 1406 cm−1) functional groups.39 The other observed vibration bands were attributed to the presence of PEG-SH. Regarding the Au NPs@PEG-Mn2+, the spectrum was similar to that of Au NPs@DMSA-DTPA, which confirmed the presence of DMSA-DTPA and PEG-SH on the Au NPs’ surface. The only difference between the two spectra was a shift of the carboxylate asymmetric vibration mode from 1631 to 1599 cm−1, which may be attributed to the coordination of metal ions.40 In this case, it indicated that Mn2+ was chelated by DTPA carboxylate groups.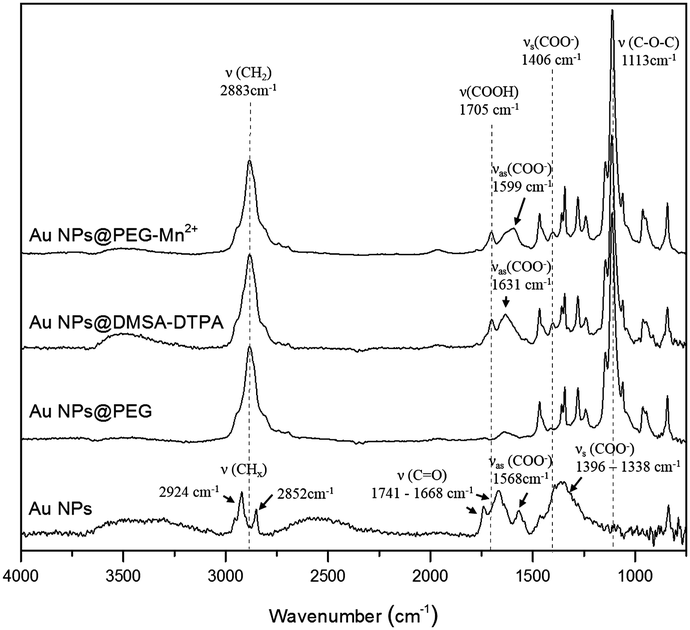 | ||
| Fig. 1 FTIR spectra of Au NPs after each step of the functionalization procedure. | ||
High resolution XPS spectra of O 1s for Au NPs showed peaks related to the presence of hydroxyl and of carbonyl groups (Fig. 2; Au NPs – O 1s peak). These results are consistent with the data from the HR spectra of C 1s (Fig. 2; Au NPs – C 1s peak) which again revealed peaks ascribed to carbonyl, carboxylate and carbonate (most probable sodium carbonate) functional molecules.41,42 After functionalizing the Au NPs with PEG-SH, the peaks related with the presence of hydroxyl and carbonyl groups disappeared in the HR spectra of O 1s and only one peak associated to oxygen in C–O–C bonds was observed (Fig. 2; Au NPs@PEG peaks). Similar results were demonstrated in the C 1s HR spectra, where the peak related with carbonate groups was absent, and the main carbon peak was attributed to C–O–C bonds, thus indicating the presence of PEG molecules.42 The C–O–C/O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ratio, 17.4 (Table 1, relative elemental concentration of C 1s chemical bonds), indicated a large excess of PEG molecules compared to molecules containing carboxyl groups.
O ratio, 17.4 (Table 1, relative elemental concentration of C 1s chemical bonds), indicated a large excess of PEG molecules compared to molecules containing carboxyl groups.
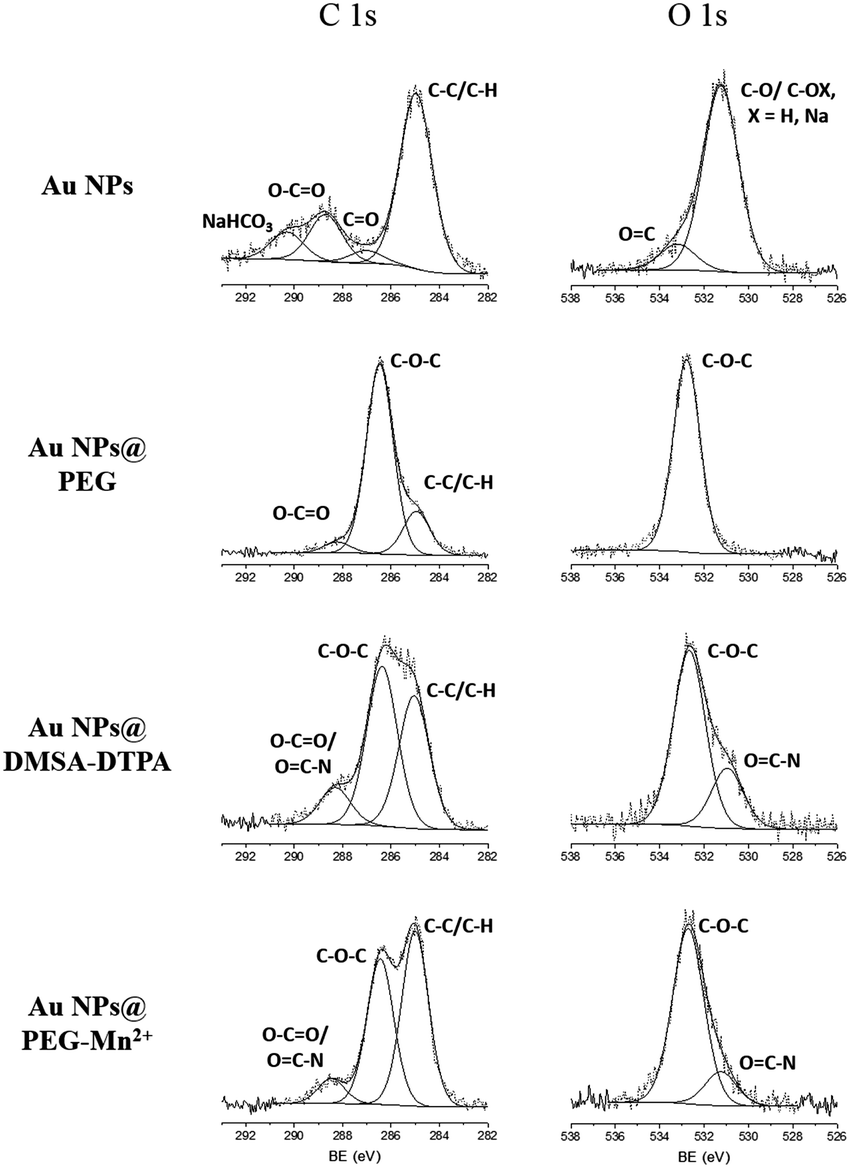 | ||
| Fig. 2 High-resolution XPS spectra of C 1s and O 1s obtained for the Au NPs at various steps of the functionalization procedure. | ||
| Sample | Total elemental concentration of C 1s (%) | Binding energy (eV)/C 1s chemical bonds | Relative elemental concentration of C 1s chemical bonds (%) | Total elemental concentration percentage (%) of O 1s | Binding energy (eV)/O 1s chemical bonds | Relative elemental concentration of O is chemical bonds (%) |
|---|---|---|---|---|---|---|
| a X = H, Na. | ||||||
| An NPs | 72.7 | 285.0 (C–C/C–H) | 48.5 | 27.4 | ||
| 287.0 (CO) | 3.5 | 531.2 (C–O, C–OXa) | 24.0 | |||
| 288.7 (O–CO) |
13.1 | 533.2 (OC) |
3.4 | |||
| 290.3 (NaHCO3) | 7.6 | |||||
| Au NPs@PEG | 76.0 | 285.0 (C–C/C–H) | 13.6 | 24.0 | 532.8 (C–O–C) | 24.0 |
| 286.5 (C–O–C) | 59 | |||||
| 288.2 (O–CO) |
3.4 | |||||
| An NPs@DMSA-DTPA | 79.2 | 285.0 (C–C/C–H) | 31.9 | 20.8 | 530.9 (OC–N) |
|
| 286.3 (C–O–C) | 38.4 | 532.6 (C–O–C) | 5.3 | |||
| 288.2 (O–CO/OC–N) |
8.9 | 15.5 | ||||
| Au NPs@PEG-Mn2+ | 80.7 | 285.0 (C–C/C–H) | 40.9 | 19.2 | 531.3 (OC–N) |
|
| 286.5 (C–O–C) | 34.0 | 532.7 (C–O–C) | 3.1 | |||
| 288.5 (O–CO/OC–N) |
5.8 | 16.1 | ||||
At this point in the functionalization process, it was not possible to observe the presence of N or S in the survey (data not shown), due to the low percentage of these atoms on the Au NPs (e.g. there is only one N or S atom for each of the 224 C atoms). After the addition of DMSA-DTPA, the C–O–C/(O–CO and OC–N) ratio decreased to 4.3 (Table 1 and Fig. 2, Au NPs@PEG-DMSA-DTPA), reflecting a stronger presence of molecules containing carboxyl groups and N atoms (i.e. DMSA and DTPA). In addition, a peak related to the presence of OC–N bonds appeared in the spectrum of O 1s. In the survey spectrum (not shown), N and S peaks were visible, in agreement with the addition of DTPA (e.g. contains 5 N atoms – Scheme 1) and DMSA (e.g. contains 2 S atoms – Scheme 1). Following the dialysis step and complexation of DTPA with Mn2+ (Fig. 2; Au NPs@PEG-Mn2+), similar HR spectra were recorded for C 1s and O 1s. A slight increase in the C–O–C/(O–CO and OC–N) ratio was noted (to 5.9), reflecting the elimination of excess DMSA-DTPA while confirming the strong presence of both PEG and DMSA-DTPA at the Au NPs’ surface.
As-synthesized Au NPs were assessed for size and polydispersity using UV-vis absorption spectroscopy (Fig. 3A), DLS (Fig. 3B) and TEM (Fig. 4A). UV-vis absorption spectroscopy demonstrated a surface plasmon resonance (SPR) peak at 514 nm, which suggested the Au NPs were smaller than 10 nm.43,44 Moreover, absorption in the near infrared region of the spectrum (800 nm) was minimal, indicating that few aggregates or large-sized particles were present.45 The inset of Fig. 3A evidenced a red shift limited to 2–3 nm after each modification (i.e. addition of PEG, followed by DMSA-DTPA and Mn2+). The shift in the SPR band is related to changes in the dielectric constant close to the Au NPs’ surface.46 This observation indicated that the molecular composition at the Au NPs’ surface changed with each step.
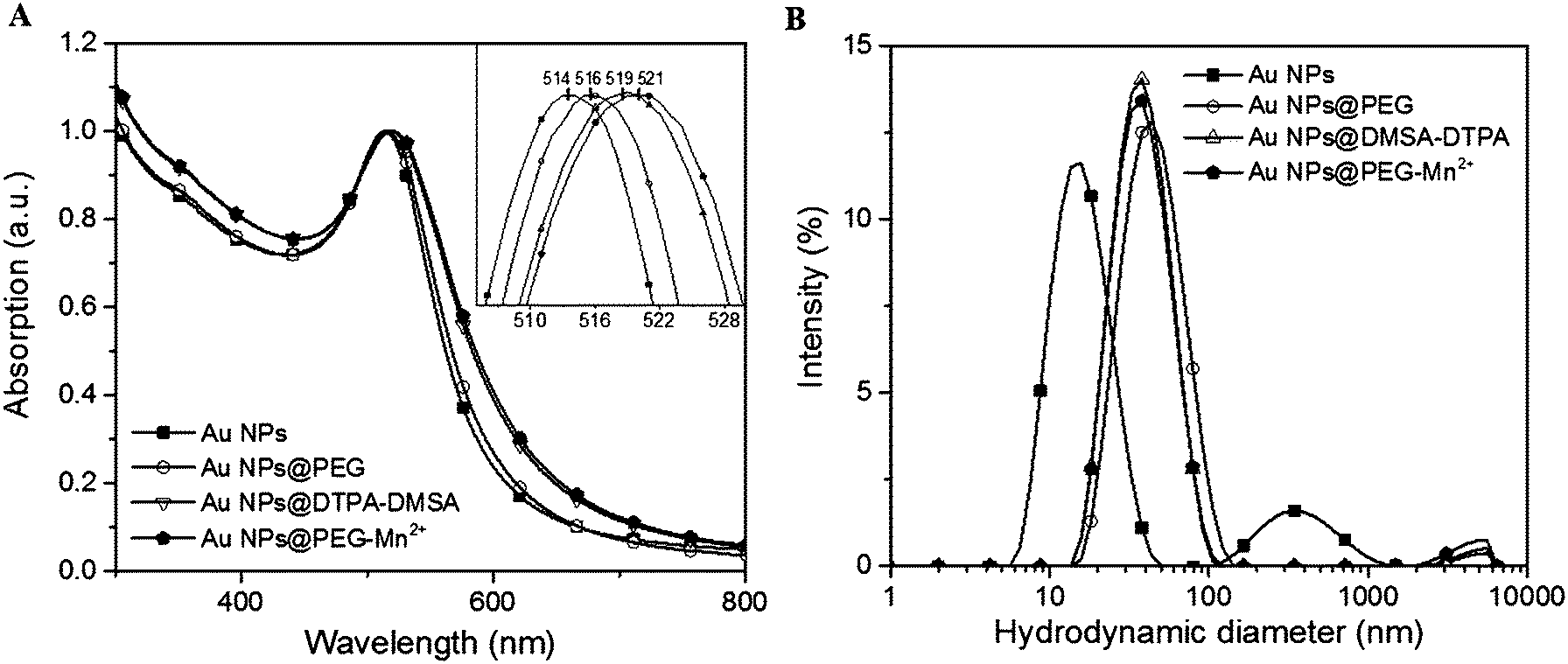 | ||
| Fig. 3 Effect of Au NPs surface modification on (A) UV-vis absorption spectra (inset graphic shows the shift in the SPR peak position), (B) DLS intensity-weighted size distribution profiles. | ||
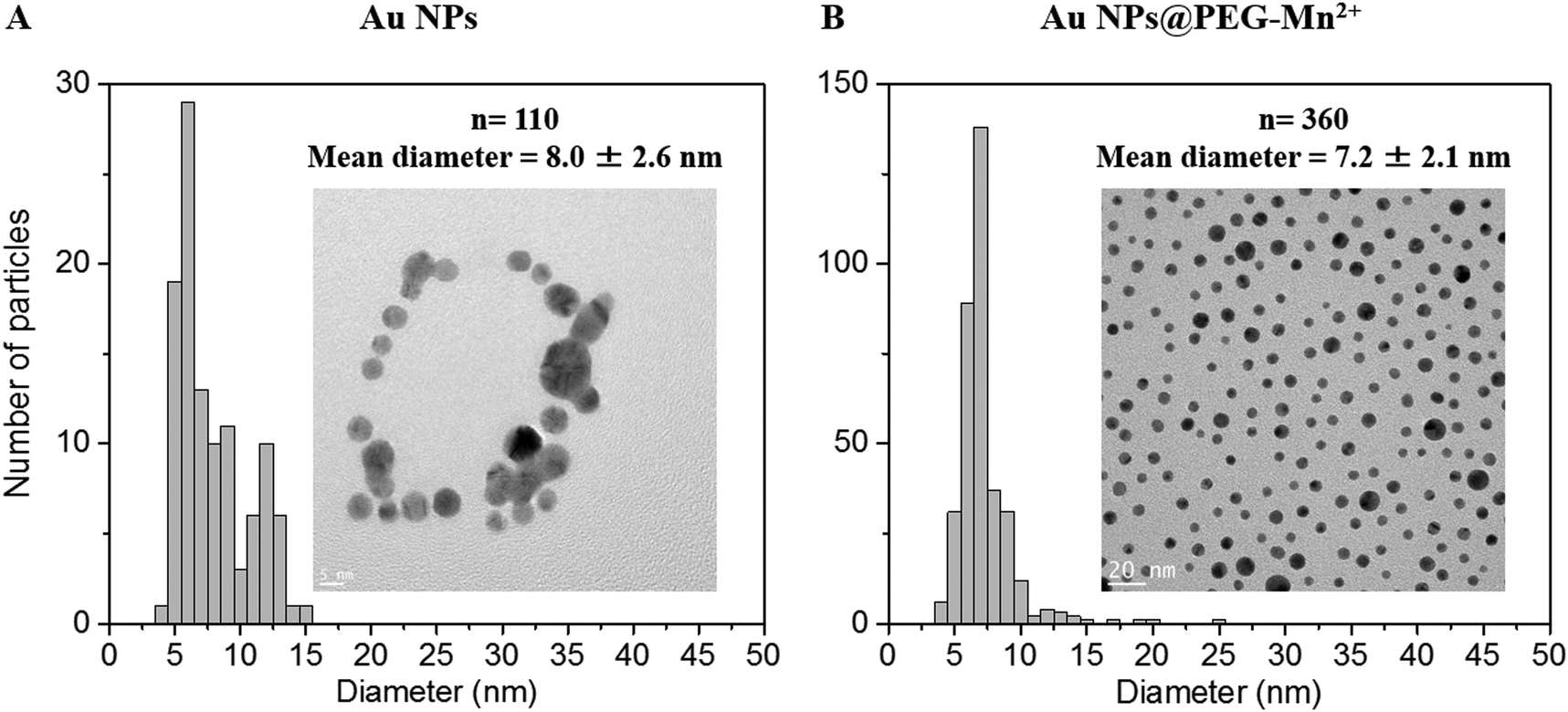 | ||
| Fig. 4 TEM images and size distributions of Au NPs prior to and after functionalization are shown in (A) and (B), respectively. | ||
Dynamic light scattering measurements of Au NPs showed a bimodal size distribution centered at ca. 17.5 and 350 nm. This indicated that there were some aggregates or agglomerates. However, as DLS is highly sensitive to large particles (the scattering power being proportional to d6, d being the diameter of the particle), only minute amounts of large aggregates or agglomerates can be responsible for the shape of the DLS curve. Indeed, the hydrodynamic diameter (HD) of Au NPs measured by DLS was 17.5 nm and 9.1 nm using intensity and number-weighting, respectively (PdI = 0.3; polydispersity index values are provided in the ESI,† Table S1). This last value agreed with the 8 nm average diameter obtained by TEM (Fig. 4A).
The presence of PEG (Fig. 3B and Table S1, ESI†) increased the HD of the Au NPs to 40.9 nm. Addition of DMSA-DTPA and Mn2+ led to a slight decrease in HD (34.1 and 36.5 nm, respectively). The major increase in HD was related to the size of the PEG chains grafted onto the surface of the Au NPs (e.g. 17.5 nm for Au NPs to 40.9 nm for Au NPs@PEG).47 This size difference (i.e. 23.4 nm) suggested a PEG layer with a thickness of approximately 11.7 nm. According to G. Zhang et al., who coated 20 nm Au NPs with 5 kDa PEG-SH, the polymeric layer showed a thickness of 13.8 nm, which is similar to the results obtained in this study.47 The Au NPs@PEG-Mn2+ were also visualized by TEM (Fig. 4B) and showed Au cores of 7.2 ± 2.1 nm without significant evidence of aggregation.
Furthermore, it was observed that before the addition of PEG, the pure Au NPs aggregated after drying on the TEM grid (Fig. 4A). However, the same phenomenon was not observed for Au NPs@PEG-Mn2+, as the particles remained evenly spaced after drying (Fig. 4B), in accordance with the presence of PEG molecules surrounding the NPs. The random coil model for a 5 kDa PEG-SH molecule estimates an end-to-end length of approximately 8 nm.48 The average distance between the Au NPs observed by TEM was 8.6 nm (n = 108), which was in agreement with the expected length for the PEG5 kDa molecule. In addition, this observation suggested that the PEG chains located on different Au NPs intercalated into each other during the drying procedure.
The net charge of NPs is a significant characteristic for CT CAs, as it not only influences the colloidal stability, but also the biodistribution and uptake by the cells.49 The zetapotential of Au NPs, Au NPs@PEG, Au NPs@DMSA-DTPA and Au NPs@PEG-Mn2+ were measured (Table 2). As expected, Au NPs at pH 10 have an average zetapotential of −28.5 mV, which explained the long-term shelf-storage colloidal stability of these NPs. PEG grafting onto the Au NPs’ surface (i.e. Au NPs@PEG) led to a positively-charged NP surface in deionized water (22.3 mV), in agreement with the presence of amine moieties. Following addition of DMSA-DTPA, a change in surface charge was observed as the zetapotential became slightly negative (−3.9 ± 1.8 mV). This was caused by the negative charge of deprotonated carboxyl groups present in the DMSA-DTPA molecules. On the other hand, the complexation with Mn2+ raised again the zetapotential to positive values (5.8 ± 0.1 mV).
| Sample | Electrophoretic light scattering | ||
|---|---|---|---|
| Zeta potential (mV) | Solvent | pH | |
| An NPs | −28.5 ± 3.3 | 3.3 mM NaOH | 10 |
| Au NPs@PEG | 22.3 ± 2.7 | De-ionized H2O | 7 |
| An NPs@DMSA-DTPA | −3.9 ± 1.8 | De-ionized H2O | 7 |
| Au NPs@PEG-Mn2+ | 5.8 ± 0.1 | De-ionized H2O | 7 |
2.2 Colloidal stability studies
The colloidal stability of Au NPs@PEG-Mn2+ was measured up to seven days in de-ionized water and 154 mM of NaCl, as well as up to three days in complete cell medium. The Au NPs@PEG-Mn2+ dispersed in de-ionized water did not show any change in HD over seven days (Fig. 5A).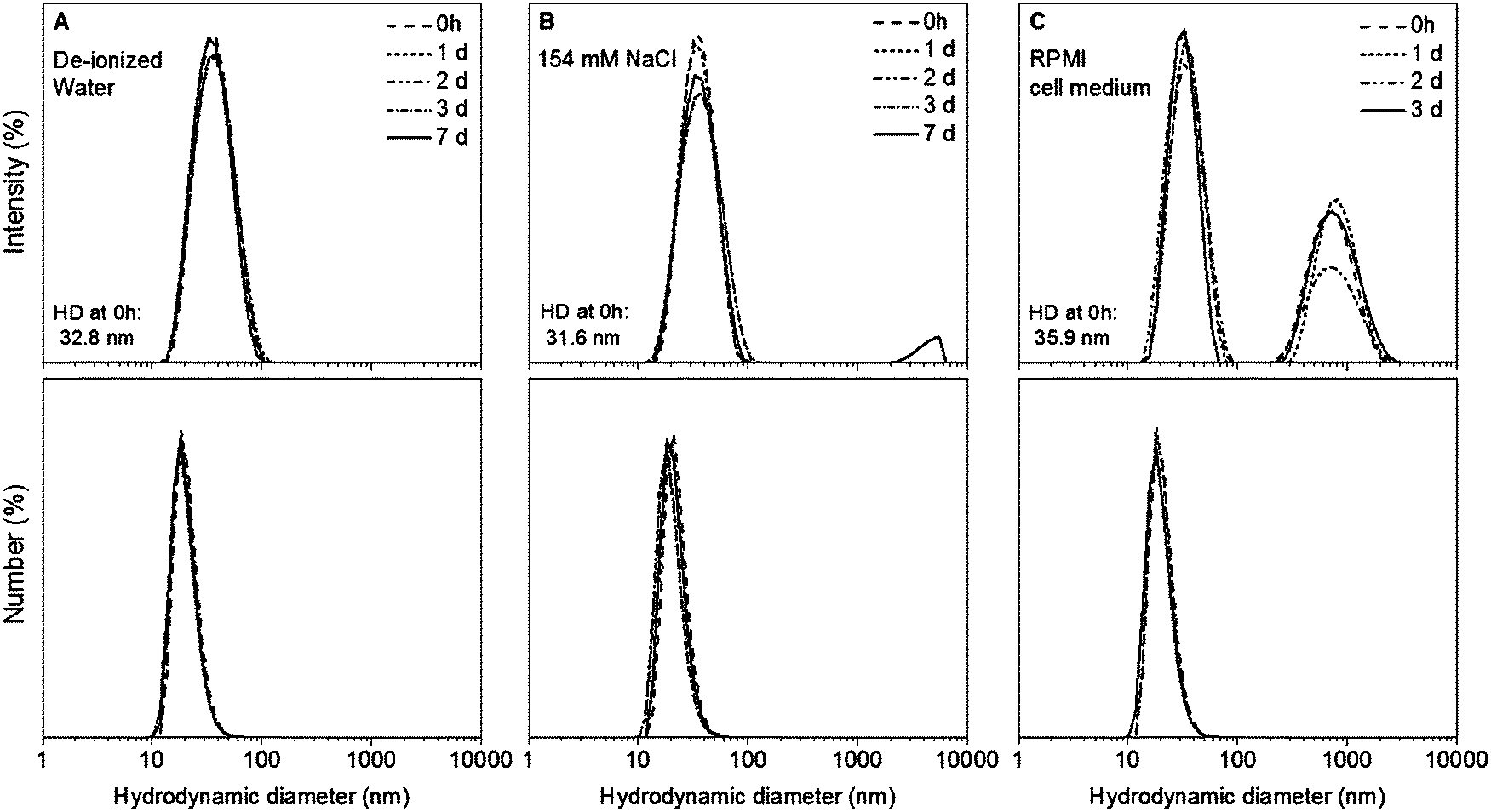 | ||
| Fig. 5 Intensity- and number-weighted size distribution plots obtained by DLS at different time points during the colloidal stability study of Au NPs@PEG-Mn2+ dispersed in de-ionized water (A), 154 mM of NaCl (B) and RPMI 1640 cell medium (C). | ||
The HD of the sample dispersed in saline remain unchanged during three days. However at seven days a minute presence (∼1%) of agglomerates was observed in intensity-weighted DLS measurements (Fig. 5B). When the Au NPs@PEG-Mn2+ were dispersed in cell medium, the HD of the majority of the NPs was slightly higher (35.9 nm) than the one observed in the sample dispersed in de-ionized water (32.8 nm) and saline solution (31.6 nm). Suspension of Au NPs@PEG-Mn2+ in cell medium also induced the formation of a small percentage (∼5%) of agglomerates (peak at ∼700 nm in intensity-weighted DLS measurements). Nevertheless, the size and polydispersity of this colloid were constant for three days (Fig. 5C).
The formation of minute amount of agglomerates could be due to a small fraction of Au NP surfaces that are not completely covered by PEG that could lead to aggregation in the presence of a strongly ionic solution and/or the presence of large quantities of proteins in complete cell culture medium that can induce the agglomeration of Au NPs due to adsorption and/or cross-linking between particles.
2.3 Cell toxicity of Au NPs@PEG and Au NPs@PEG-Mn2+
The effect of Au NPs@PEG and Au NPs@PEG-Mn2+ on the viability of PC3 cells was measured by the resazurin assay (mitochondrial function).50 To do so, long incubation times (48 hours and 72 hours) were used to place the cells in contact with highly-concentrated NPs suspensions. After 48 h of incubation with the Au NPs@PEG-Mn2+, the viability of PC3 cells remained unaffected up to 0.77 mM Au. However, a strong inflexion of the cell viability was observed (Fig. 6A) at concentrations above 1.75 mM and for both 48 hours and 72 hours of incubation time. A comparative study was performed with Au NP@PEG, which demonstrated the cells’ very high tolerance for PEG-stabilized Au NPs (up to 7 mM Au, Fig. 6B), even at strongly positive surface charges (Table 2). These results indicate that DMSA-DTPA-Mn2+ complexes interfere with the cells and may affect their viability at moderate and high concentrations. As the Au NPs synthesized in this study are very small, these results indicate the surface concentration of DMSA-DTPA-Mn2+ should be kept to low concentrations.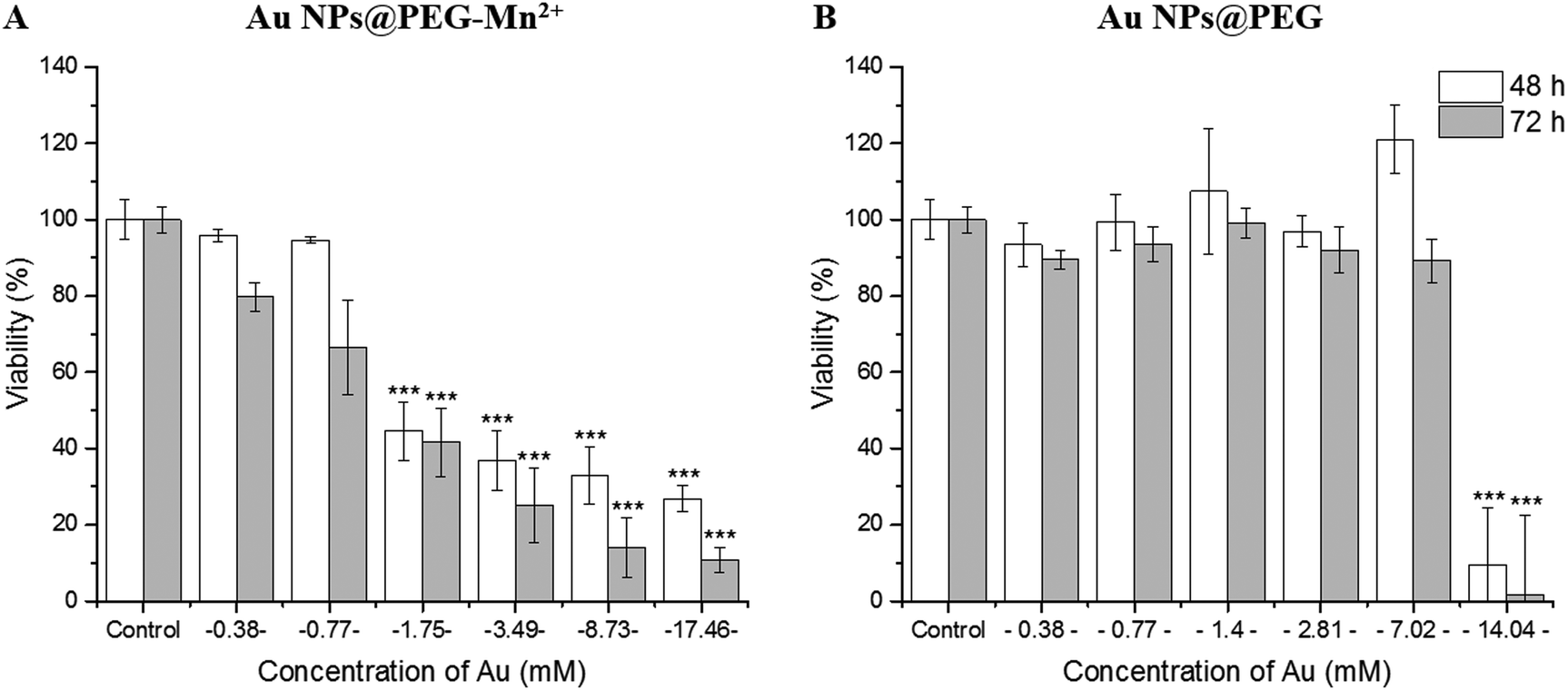 | ||
| Fig. 6 Resazurin cell viability assay on PC3 cancer cells incubated for 48 hours and 72 hours with increasing concentrations of Au NPs@PEG-Mn2+ (A) or Au NPs@PEG (B). The significance between the samples is indicated in the plots as follows: ***P < 0.001. | ||
2.4 In vitro CT and MRI contrast potential assays
The Au NPs@PEG-Mn2+ solutions imaged with CT showed a linear increase in contrast compared with water, from 10.0 to 66.4 mM Au (Fig. 7A). Solutions imaged by 1.0 T MRI produced a clear contrast enhancement in the 0.2–1.5 mM Mn2+ concentration range (Fig. 7A). The same solutions were measured in proton NMR to assess the impact of Mn2+ on the T1 and T2 relaxation times of water. The slope of the linear regression line (Fig. S1, ESI†) provides the relaxivity values (r1 and r2), which are used for in vitro comparison of the performance of different MRI CAs. In short, to induce strong “positive” contrast enhancement, the required relaxivities would consist of high r1 with r2 as low as possible, although always higher than r1. Solutions of Au NPs@PEG-Mn2+ had a longitudinal relaxivity (r1) of 4.8 mM−1 s−1 and a relaxometric ratio (r2/r1) of 3.6 which point to a strong “positive” CA for a Mn2+-based product.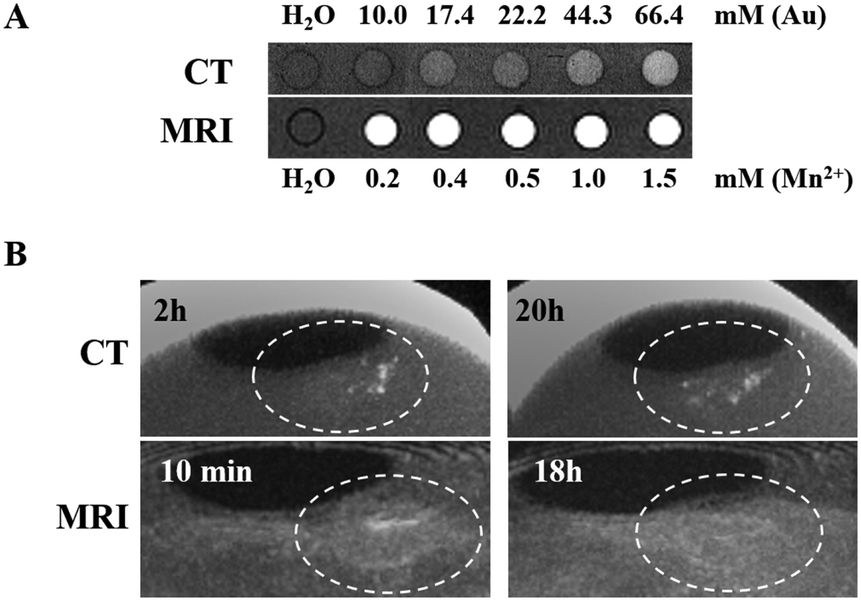 | ||
| Fig. 7 (A) CT (40 kVp) and MR (1.0 T) in vitro imaging of Au NPs@PEG-Mn2+. (B) In vivo contrast enhancement study of Au NPs@PEG-Mn2+ in CT and MRI using PC3 CAM tumors. The dashed white circle outlines the tumor area. The CT and MRI images were acquired after the intratumoral injection of Au NPs@PEG-Mn2+ at different time points. | ||
2.5 In vivo CT and MRI contrast-enhancement assay in the CAM tumor model
The dual MRI/CT contrast properties of Au NPs@PEG-Mn2+ were assessed in the CAM tumor model (intratumoral injections) (Fig. 7B). A clear CT contrast was observed in the injection sites at t = 2 hours and t = 20 hours. The latter result suggested a slight diffusion of Au NPs away from the main injection site. Injections were also accessed using MRI (at t = 10 min and 18 hours). The strong signal-enhancement at t = 10 min faded out at 18 hours; the signal decrease at t = 18 hours (MRI) appears to co-register with the signs of diffusion/concentration dilution of Au NPs@PEG-Mn2+ evidenced by CT. Overall, these studies confirmed the capacity of Au NPs@PEG-Mn2+ to provide a clear contrast enhancement in both CT and MRI, without evidence of strong aggregation upon injection in vivo (i.e. the NPs diffuse after a few hours in the tumor).2.6 In vivo vascular CT imaging with Au NPs@PEG-Mn2+
The performance of Au NPs@PEG-Mn2+ as vascular CAs was assessed in the mouse model. The Au NPs@PEG-Mn2+ provided a clear contrast enhancement in the heart and major blood vessels up to 90 minutes after the injection (Fig. 8A). Other tissues, such as the kidney, liver and spleen parenchyma showed a lower but still significant contrast enhancement. This contrast enhancement reached stability after 15 min and did not increase further, notably in the liver and spleen. This is an indication of NP blood retention. The image contrast obtained in the various monitored tissues and vessels was relatively constant throughout the study (Fig. 8B). The mice did not show any adverse reaction to the injection of Au NPs@PEG-Mn2+ during the experiment. A comparative assessment was performed with commercial CA AurovistTM, which showed a similar blood retention profile (Fig. S2, ESI†).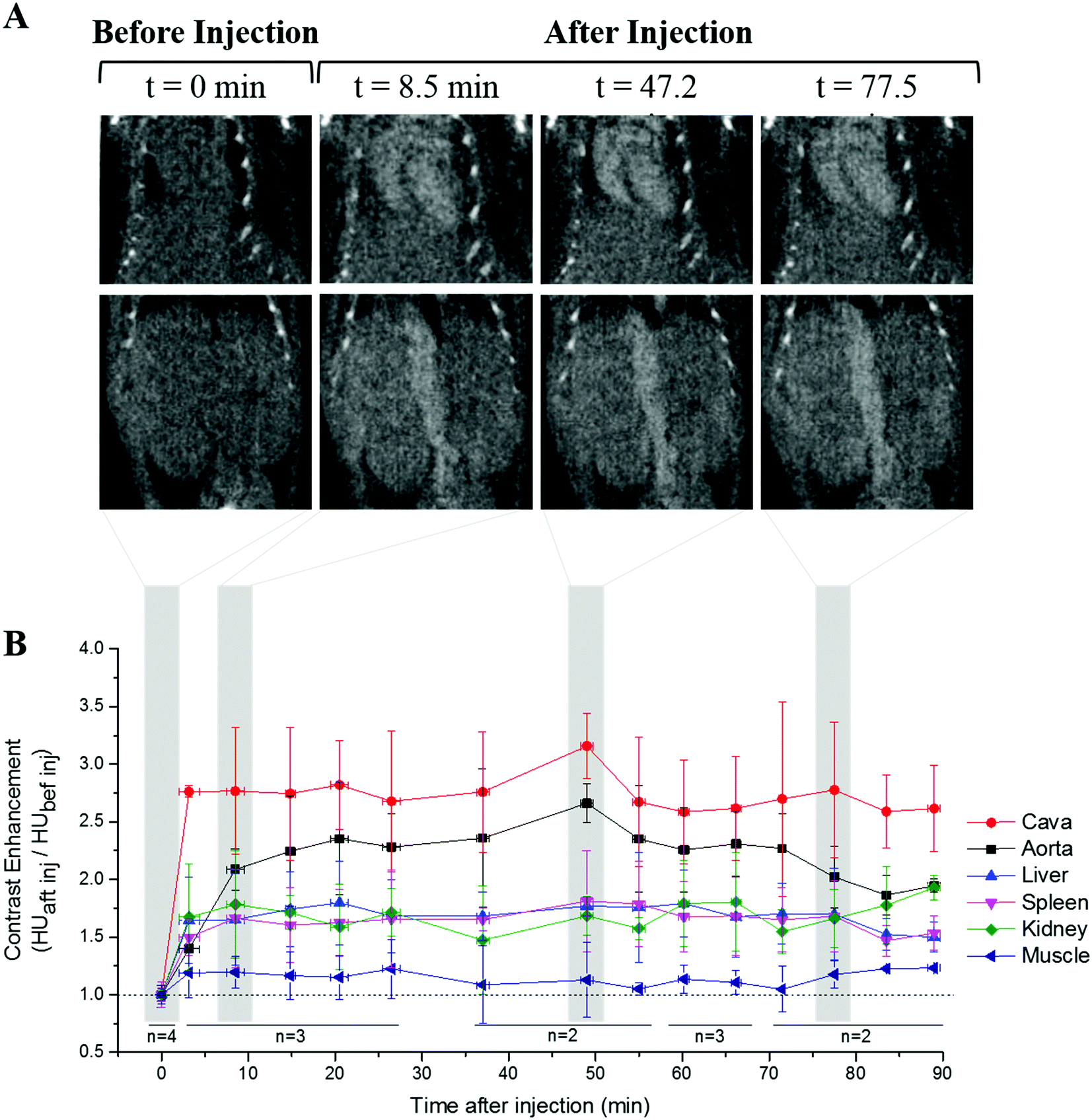 | ||
| Fig. 8 Dynamic contrast enhancement CT studies of Au NPs@PEG-Mn2+ using healthy CD-1 mice. Images show coronal sections of thoracic and abdominal regions in a representative mouse injected with Au NPs@PEG-Mn2+. The graph displays the contrast enhancement before (e.g. 0 min), and at different time points following the injection. | ||
3. Discussion
PLAL-synthesized Au NPs formed a stable colloid with a narrow size distribution and were efficiently grafted with PEG-SH. The particles were also labeled with Mn2+, a paramagnetic element which provides a complementary visibility during MRI, a key imaging modality in the diagnosis and staging of prostate and breast cancers. In the present study, we used Mn2+ instead of Gd3+, because Gd3+ has, in the recent years, been closely associated with the occurrence of nephrogenic systemic fibrosis (NSF).51 In the past, much less work has been carried out on the complexation of Au NPs with the element Mn2+. However, due to increasing concern related to the possible toxic effects (e.g. NSF) generated by the de-chelation of Gd3+, there has been a resurgence of interest for the use of Mn2+ in vascular contrast agents. In sub-10 nm Au NP-based nanoparticulate contrast agents for CT vascular imaging, Mn2+ could be beneficial, as it is less toxic. Indeed, the most conventional method used to prepare Au NPs for biomedical applications is direct reduction of Au3+ ions with sodium citrate.16,31,52,53 Studies have demonstrated that the initial stabilizers (e.g. citrates) are hardly completely removed from the Au NPs’ surface; this is true even when the compounds selected to be exchanged with citrates bear thiol functions, known for their high affinity for noble metal atoms.21,54 Given the small size of Au NPs for CT contrast media applications (<15 nm), concerns were raised relating to the potential impacts of traces of initial stabilizers on cell activity and cytotoxicity.55 In addition, such traces are likely to have also an impact in biodistribution during in vivo studies, as NPs’ surface properties are known to dictate their fate due to the protein corona formed.34,49For the above reasons, PLAL is a clearly advantageous method for the production of Au NPs for biomedical applications. In this study, stable Au NPs with a relatively small size (<10 nm) and narrow size distribution were obtained by adding a limited amount of NaOH in the ablation solution. PLAL-synthesized Au NPs are electrostatically stabilized by Au–OH and Au–O− species formed during the ablation.45,56,57 The alkaline medium favors the deprotonation of the Au–OH species, which maximizes the amount of Au–O− groups on the Au NPs’ surface. Consequently, the electrostatic repulsion between the particles increases, which minimizes nuclei coalescence during the growth of the NPs and results in a lower size distribution as well as improved long-term stability.56,58 Following synthesis, the Au colloids were centrifuged to eliminate most of the target fragments and large NPs that were inevitably formed due to the mechanisms involved in the formation of NPs synthesized by PLAL.57
In this study, the PEG molecular weight used was 5 kDa as it offered the best compromise between a final HD that promotes an extended blood half-life, and a viscosity suitable for in vivo administration of Au concentrations on the order of tens of mg mL−1.37,59 The PEG molecules also have a terminal amine function, so that modification with targeting ligands could be performed in later studies. The functionalization of the Au NPs surface with PEG was performed in alkaline medium to maintain electrostatic repulsion among Au NPs, and the incubation was left to continue overnight to maximize the packaging and ordering of the PEG molecules at the surface of Au NPs.60
Considering that the average footprint of thiolated PEG5kDa in an 8 nm AuNP is approximately 0.4 nm2,30,46,48 it was calculated that each Au NP has approximately 500 binding sites. Thus, the PEG molecules were added with double the excess. This amount was at least 1.8 times lower than the ligands excess reported in the literature. In fact, most studies perform ligand exchange with citrate-stabilized Au NPs using 8 to 40 times excess of ligands.30,47,61,62 In this study, it was observed that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 ratio (AuNP:PEG molecules) was sufficient to stabilize the Au NPs, and a HD of 37.9 nm was obtained (data not shown). In addition, by using a 1:1000 ratio, it was possible to reach HD values in the order of 40.9 nm.
:500 ratio (AuNP:PEG molecules) was sufficient to stabilize the Au NPs, and a HD of 37.9 nm was obtained (data not shown). In addition, by using a 1:1000 ratio, it was possible to reach HD values in the order of 40.9 nm.
Thus, while the 1:500 ratio induced approximately two times increase of the HD compared to as-synthesized Au NPs, the 1:1000 ratio created only a minor further increase (∼8%) of the HD. This means that the surface of the Au NPs is almost saturated with PEG molecules when using the 1:500 ratio. Therefore, with Au NPs produced by PLAL, it is possible to form a PEG monolayer by incubating the NPs with equimolar or a slightly higher excess of ligands. This observation agrees with previous studies, which reported that laser-synthesized Au NPs demonstrated higher surface coverage of thiolated aptamers compared with Au NPs functionalized with other nucleic acids through ligand exchange.36
The purity of Au NPs surfaces was assessed with FTIR and XPS. Carbonyl and carboxylate groups were identified at the surface of pure Au NPs. A previous study has also revealed the presence of these groups on PLAL-synthesized Au NPs. The authors suggested that these molecules originate in the atmospheric CO2 dissolved in water or may be formed when exposing Au NPs to air, after drying the sample for analysis.56 Our studies confirmed that the presence of these functional groups does not impede the efficient and strong functionalization of Au NP surfaces with PEG-SH molecules. In fact, Kim et al. synthesized Au NPs via citrate reduction and coated them with 5 kDa PEG-SH. XPS analysis of these PEGylated Au NPs evidenced a C–O–C/O–CO ratio of 5.7.16 In contrast, the same ratio for the PEGylated Au NPs described in this work is 17. A higher C–O–C/O–CO suggested that the relative ratio of PEG is higher in the Au NPs produced by PLAL, which is in agreement with the presence of a ligand-free surface which facilitates the grafting of PEG molecules even using only a slight excess of these molecules.
Neither the Au NPs@PEG-Mn2+ dispersed in de-ionized water, nor that dispersed in saline, showed any change in HD (Fig. 5A and B). Therefore, the Au NPs@PEG-Mn2+ may be kept at room temperature in the saline medium necessary for the injection during three days without risk of flocculation or precipitation. Also, the small amount of agglomerates present in the saline solution after seven days do not compromise the application of Au NPs@PEG-Mn2+ as an intravenous CA, as they can be easily removed by centrifugation before injection of the colloid. On the other hand, the formation of protein corona and subsequent consequences observed when the Au NPs@PEG-Mn2+ are dispersed in complete cell medium are a well-known limitation, and research has been on-going to circumvent this phenomenon.63 Nevertheless, in the present work, the percentage of these aggregates was rather low (only detectable in intensity-weighted DLS measurements due to the very high light scattering power of the larger particles). In addition, the size these agglomerates is not large enough to cause vascular occlusion, hence Au NPs@PEG-Mn2+ are safe for vascular injection.64
The toxicity assays for this study were performed using long incubation times (e.g. 48 hours and 72 hours) and high concentrations of Au. This is a conservative strategy that is very challenging for cells and places clear emphasis on possible toxicity risks. This approach was necessary at this step, as several types of cells may be in contact with Au NPs for prolonged periods of time. In fact, blood half-lives of 11 to 30 hours may be achieved with PEG-coated Au NPs.17,37 Consequently, the endothelial cells in contact with circulating Au NPs are expected to remain in contact with high particle concentrations, although not for more than 24–48 hours.
The present study revealed a clear difference between Au NPs@PEG and Au NPs@PEG-Mn2+. The higher toxicity of the latter colloid is most likely related to an excess of DMSA-DTPA, and not to the presence of Mn2+.65,66 In fact, both Mn2+ and Mn-DTPA are well tolerated by cells at the Mn2+ concentrations necessary to enhance MR image contrast (e.g. human lymphocytes can tolerate up to 1 mM of MnCl2 during 24 hours incubation).65 However, the presence of a high concentration of DTPA functionalities, at the surface of ultra-small NPs showing a very high specific surface, could affect the cells (membranes and organelles) and their division during the 48 hours and 72 hours incubation time. Therefore, the concentration of DMSA-DTPA attached at the surface of Au NPs, must be lowered in subsequent studies. On the other hand, the metabolism of PC3 cells was not significantly affected in the presence of relatively high concentrations of Au NPs@PEG. This appears to be a very promising result, as most CT contrast media studies have reported cell viability studies of PEGylated Au NPs performed with much lower Au concentrations.67,68 However, as for any Au NP CT CA, further assays should be performed to clarify whether such concentrations alter signaling pathways and gene expression in the cells.
To confer MRI-visibility, Au NPs@PEG were functionalized with DTPA, which is a chelating agent commonly used in clinically-approved CAs. This aminopolycarboxylic acid forms a relatively stable complex with Mn2+.69 Au NPs@PEG-Mn2+ provided a clear signal enhancement at 1.0 T in T1-weighted images, and NMR characterization revealed a r1 higher than that of free Mn-chelates.70 This increase in r1 could be partially related to the lower tumbling rates of larger hydrodynamic-size nanoconstructs as compared with small chelates, as well as to an increase in the viscosity of the medium.71,72 By comparison, for Mn-containing NPs designed as “positive” MRI CAs, the most promising r1 and relaxometric ratios obtained were 5.5 mM−1 s−1 (r2/r1 = 1.8, at 1.5 T), 3.4 mM−1 s−1 (r2/r1 = 3.2, at 1.41 T), 7.0 mM−1 s−1 (r2/r1 = 6.8, at 1.5 T), and 2.06 (r2/r1 = 4.7, at 3 T), for MnO nanoplates (8 nm),73 USPMnO (8.4 nm),74 MnO NPs (2–3 nm)75 and Mn3O4 nanoplates (10 nm)76 respectively.69 Therefore, the insertion of Mn-chelates within the Au NPs-PEG layer provided a r1 and relaxometric ratio equivalent to other Mn-containing NP-based CAs, and allowed the design of a CA that is also useful for CT.
As expected in CT, Au NPs@PEG-Mn2+ provided contrast enhancement at significantly higher doses of Au compared to the Mn2+ doses required to increase the MRI contrast (e.g. 66.4 mM of Au and 1.5 mM of Mn2+, Au/Mn = 44). In the CAM studies, injection sites could be clearly visualized by both imaging modalities. CT enabled a more accurate follow-up of Au NPs@PEG-Mn2+ diffusion than did MRI. This effect was probably related to the fact that the diffusion of the NPs within the tumor matrix decreased the local concentration of Mn-chelates below MRI detection thresholds. On the contrary, the low attenuation of X-ray photons by soft tissues, and the very strong attenuation potential of Au at 40 kVp facilitated the visualization of Au NPs@PEG-Mn2+.
Finally, the dynamic contrast-enhanced studies performed in the mouse model demonstrated a steady contrast enhancement in the bloodstream, liver and spleen for time-windows characteristic of angiographic procedures (e.g. 90 minutes). No evidence of NP agglomeration or of liver/spleen uptake (except for the blood concentration in these organs) was found. These long blood retention times without evidence of opsonization of the NPs by the mononuclear phagocyte system (i.e. liver and spleen uptake)49 confirmed the quality of PEG grafting at the surface of PLAL-synthesized Au NPs. The biodistribution profiles of Au NPs@PEG-Mn2+ were similar to those obtained with commercial Au NPs CT agents (Aurovist™), and to others observed in studies using PEGylated Au NPs for blood pool imaging with CT.16,17
4. Experimental
4.1 Synthesis of gold nanoparticles
Pure Au NPs were synthesized by PLAL using a pre-established methodology.56,58 A high-purity gold target (99.99%, Allgemeine Gold und Silberscheideanstalt AG, Pforzheim) was fixed inside a self-constructed batch ablation chamber.77 The chamber was filled with 30 mL of 3.3 mM NaOH (0.1 M solution VWR, Germany) (pH 11.1) dissolved in de-ionized water (Milli-Q water). The ablation was performed with a Rofin PowerLine E20 ns laser (8 ns pulse width) with a wavelength of 1064 nm. The laser beam was directed horizontally at the surface of the metal target by a laser scanner (SCANcube® 10, SCANLAB AG, Munich, Germany) and focused with a lens at a focal length of 100 mm. The laser beam passed through a 3 mm-thick liquid layer before striking the target for five minutes, while describing a spiral scanning pattern (6 mm external diameter). Prior to passing through the scan head, the laser's energy output was 367 μJ per pulse and the repetition rate was 15 kHz. After laser ablation, the Au NPs colloidal suspension was centrifuged (4400g for 20 min) and stored at 4 °C between the time of synthesis and the functionalization.4.2 PEG functionalization of Au NPs synthesized by PLAL
The particles were sterically stabilized with thiolated polyethylene glycol (NH2-PEG-SH, MW 5000 Da, Laysan BIO, Arab, AL, USA). First, the concentration of Au NPs was estimated using UV-visible spectroscopy, assuming that all NPs are spheres of 8 nm (methodology details described in Section 4.4). PEG was dissolved in de-ionized water (18.2 MΩ cm, Barnstead ultrapure system) at 2.1 mM and added to the Au NPs (20 mL, 2.5–3.4 × 1013 NPs mL−1) at a molecular ratio of 1:1000 (AuNP:PEG molecules). This mixture was vortexed for 30 seconds and gently agitated overnight at room temperature (RT). Next, the particles were concentrated using centrifuge filtration at a centrifugal force of 550g (Amicon Ultra – 15 centrifugal filters, 30K NMWL, Millipore, Fisher Scientific, CA), up to 9.7 × 1013–1.2 × 1014 NPs mL−1, and dialyzed for six hours in de-ionized water (10 kDa membrane, Spectra/Por 6 dialysis tubing, Rancho Dominguez, CA, USA) to remove the remaining excess PEG and NaOH. The water was changed twice and the sample-to-dialysis water volume ratio was kept at 1:150. PEG-grafted Au NPs (Au NPs@PEG) were stored overnight at 4 °C.
4.3 Functionalization of Au NPs@PEG with manganese (Mn2+) chelates
2,3-meso-Dimercaptosuccinic acid (DMSA, Sigma-Aldrich, Oakville, ON, CA) was dissolved in 0.031 M of NaOH (∼pH 8). Maleimido-mono-amide-DTPA (mal-DTPA) (Macrocyclics Inc., Dallas, TX, USA) was dissolved in PBS (1×) (10×, ultrapure grade, Amresco, Solon, Ohio, USA) at a concentration of 1.8 mM. DMSA and mal-DTPA were reacted (maleimide groups with sulfhydryl groups) in a 1:1 molecular ratio at pH 6.4 (30 seconds vortex; then moderate agitation for 90 min at RT). A volume of DMSA-DTPA solution was added to Au NPs@PEG (4.89 mL, 1.63 × 1014 NPs mL−1) to reach a final Au NP:DMSA-DTPA number ratio of 1:2500. The solution was vortexed for 30 seconds, then kept at RT under gentle agitation for three hours, and finally dialyzed against de-ionized water (1:100 volume ratio; water changed twice; fourteen hours total time; 10 kDa membrane). The Au NPs@DMSA-DTPA (7.5 mL, 1.06 × 1014 NPs mL−1) recovered from dialysis were incubated with approximately 0.37 mM MnCl2·4H2O (99.999% purity, Puratronic, Alfa Aesar, Ward Hill, MA, USA), in order to reach a final Au NP:Mn2+ number ratio of 1:2000. The solution was vortexed for 30 seconds and gently agitated at RT for 90 minutes. Finally, the particles were dialyzed against de-ionized water (1:100 volume ratio; water changed twice; five hours total time; 10 kDa membrane) and then stored at 4 °C. This suspension of Mn2+ functionalized and PEG-stabilized Au NPs will be referred to as Au NPs@PEG-Mn2+ in the following text and figures.
4.4 UV-visible spectroscopy
UV-visible spectroscopy was used to estimate Au concentration throughout all experimental procedures. This concentration was calculated based on the absorption at 380 nm, which is mainly influenced by the inter-band transition of Au atoms;78 these calculations were made using a correlation curve obtained with commercially available Au NPs (reactant-free 15 nm Au NPs, Cytodiagnostics, Sigma-Aldrich, Oakville, ON, CA). Moreover, UV-visible spectroscopy allowed for rapidly assessing the stability of the Au NPs after each functionalization step by monitoring the red shift of the SPR band as well as the level of aggregation detectable by the scattering of agglomerates and large particles in the near infrared region.For all measurements, pure Au NPs were diluted in 3.3 mM NaOH, while functionalized Au NPs (i.e. Au NPs@PEG, Au NPs@DMSA-DTPA and Au NPs@PEG-Mn2+) were diluted in de-ionized water, to ensure the maximum absorption at the SPR band was equal to or less than 1. For measurement purposes, the Au NPs solutions were dispensed into a quartz cell and the absorption spectra acquired with a UV-1600 spectrophotometer (Shimadzu, Kyoto, Japan) from 200 to 800 nm.
4.5 Attenuated total reflectance Fourier transform infrared spectroscopy (A TR-FTIR)
The colloids (30 μL) were drop-cast on a Si crystal and dried under vacuum in an oven at 40 °C. The spectra were acquired with an ATR-FTIR (Agilent Cary 660 FTIR, Agilent Technologies, USA), equipped with a deuterated L-alanine-doped triglycine sulfate (DLa-TGS) detector, a germanium-coated KBr beam splitter, and a DTGS/KBr detector. The data was obtained in absorbance mode and 64 scans were recorded with a spectral resolution of 4 cm−1.4.6 X-ray photoelectron spectroscopy (XPS)
Si wafers (Cemat Silicon S.A., Poland) were cleaned first by TL2 solution (de-ionized water, 30% hydrogen peroxide (H2O2), (Fluka, ON, Canada) and concentrated hydrochloric acid (HCl) (Fisher, Canada) mixed with a 6:1:1 volume ratio; wafers left in solution at 80 °C for 10 minutes), thoroughly rinsed with de-ionized water, and then cleaned with TL1 solution (de-ionized water, 30% H2O2 and 25% ammonia (Fisher Scientific, IL, USA) mixed with a 5:1:1 volume ratio; wafers left in solution at 80 °C for 10 minutes). Subsequently, the substrates were again extensively rinsed with de-ionized water, anhydrous ethanol (Commercial Alcohols, Brampton, ON, Canada) and dried with pressurized air. Pure Au NPs, Au NPs@PEG, Au NPs@DMSA-DTPA or Au NPs@PEG-Mn2+ were deposited on these Si substrates (4 × 15 μL = 60 μL) and dried under vacuum at 40 °C. XPS analysis were performed with a non-monochromatic aluminum X-ray source for survey spectra (0–1400 eV; PHI 5600-ci spectrometer, Physical Electronics U.S.A., Eden Prairies, MN, USA). The high-resolution (HR) spectra of C 1s and O 1s were acquired with a non-monochromatic magnesium X-ray source (1253.6 eV). The analysis was performed on a spot size of 0.005 cm2 and the photoelectrons were detected at 45° relative to the surface normal (no charge neutralization applied). All HR spectra were corrected so that the peak corresponding to the adventitious C 1s had a binding energy of 285.0 eV. The HR spectra were analyzed with CasaXPS software (version 2.3.16 PR 1.6, 1999-2011 Casa Software Ltd). A Shirley-type background and Gaussian-Lorentz functions were used. A 70% Gaussian – 30% Lorentz ratio was employed for C 1s and O 1s. The full width at half-maximum was kept constant for all peaks fitted in the HR spectrum of each element. The best quality fitting was achieved by obtaining a residual standard deviation close to the unit.
4.7 Dynamic light scattering (DLS) and zetapotential measurements
The hydrodynamic diameter (HD) of the Au NPs was measured at the end of each functionalization step to assess the colloidal stability in different solutions. All measurements were performed at 25 °C by DLS using a Nano S Zetasizer system (Malvern Instruments, Worcestershire, UK) including a HeNe laser with a wavelength of 633 nm, and the scattered light detection was performed at 173°. Each measured sample had a NP concentration in the order of 100 to 150 μg mL−1 of Au as assessed by UV-visible absorption spectroscopy. Water was selected as the dispersant for all samples, therefore the viscosity and refractive index were 0.8872 cP and 1.33, respectively; the refractive index for Au NPs was 0.408 and the absorption was 3.32. The HD was calculated from the average of three measurements and the polydispersity index (PdI) was calculated using the Malvern Zetasizer software (version 7.11). All results met the quality criteria of Malvern Zetasizer software. The colloidal stability of Au NPs@PEG-Mn2+ was assessed for seven days in de-ionized water and in 154 mM of NaCl (Sigma-Aldrich, Oakville, ON, Canada) as well as for three days in complete RPMI cell culture medium (RPMI 1640 medium + 1× L-glutamine (Corning, Fisher Scientific) + 10% FBS (Life Technologies Inc., Burlington, ON, CA)).The zetapotential was measured at 25 °C by electrophoretic light scattering using the Nano zetasizer and a folded capillary cell (DTS1070). The refractive index and viscosity were the same selected for the HD measurements and the zetapotential values were calculated using the Smoluchowski equation. The pure Au NPs were prepared by diluting in 3.3 mM NaOH to approximately 118 μg mL−1, whereas the Au NPs@PEG and Au NPs@PEG-Mn2+ were diluted in deionized water to 72 μg mL−1 and 96 μg mL−1 respectively. The final pH of both suspensions was approximately 7. The results are the average of three measurements and met the Malvern Zetasizer software's quality criteria for phase plot and distribution data.
4.8 Transmission electron microscopy (TEM)
For TEM observation, drops of Au NPs or Au NPs@PEG-Mn2+ were dried on carbon-coated copper grids (Electron Microscopy Sciences, Hatfield, PA, USA). The samples were observed by a JEM-2100F (Jeol, USA) operating at an accelerating voltage of 200 kV. The Feret diameter (i.e. the largest distance between two parallel tangential lines which restrict the particles perpendicularly to that direction) of the Au NPs was measured using the ImageJ software (version 1.47v, Wayne Rasband, National Institutes of Health, USA) and used for size distribution graphs.4.9 Relaxometric analysis
Au NPs@PEG-Mn2+ were concentrated to 15.94 Au/0.55 Mn mM and several dilutions (12.75/0.44; 9.56/0.33; 7.97/0.28; 6.38/0.22) of this stock solution were prepared with de-ionized water. 300 μL of each sample were dispensed in a 7.0 mm o.d. NMR tube. The longitudinal and transversal relaxation times (T1 and T2) were measured by a time-domain Nuclear Magnetic Resonance (NMR) benchtop system (Bruker Minispec 60 mq, 60 MHz – 1.41 T, 37 °C, Bruker, Billerica, MA, USA). The relaxivities (r1, r2) were extracted from the slope of a linear regression obtained by plotting the relaxation rates (1/T1, 1/T2) in function of the Mn concentration of each Au NPs@PEG-Mn2+ sample. The concentration of Au and Mn in the stock suspension was measured by ICP-MS (described in Section 4.10 – Elemental analysis).4.10 Elemental analysis
Au and Mn concentrations in suspensions were confirmed by ICP-MS (Perkin Elmer Elan 6000, Waltham, MA, USA) or MP-AES (Agilent 4200 Microwave Plasma-Atomic Emission Spectrometer, Agilent Technologies, USA). Before elemental analysis, the Au NPs@PEG-Mn2+ and Au NPs@PEG were digested in aqua regia (HCl and nitric acid; 3:1 volume ratio; Sigma-Aldrich, Oakville, ON, Canada) and 30% H2O2 at 115 °C, until the suspension turned light yellow or completely transparent.
4.11 Cell viability assays
PC3 human prostate cancer cells (CRL-1435, American Type Culture Collection, Manassas, USA) were seeded in 96-well plates (5000 cells per well in 100 μL) and incubated for 24 hours at 37 °C under humidified atmosphere of 5% CO2 (standard cell incubation conditions used in this study). Complete cell culture medium was used: RPMI 1640 medium with L-glutamine (Corning Cellgro, Manassas, VA, USA) supplemented with 10% FBS (Gibco, NY, USA) and 1% of penicillin streptomycin (Gibco, NY, USA). The cells were incubated in quadruplicate in the cell culture medium with concentrations of Au NPs@PEG or Au NPs@PEG-Mn2+ ranging from 0.3–17.46 mM Au. The NPs solution was administrated by removing up to 10 μL from the 100 μL cell medium in the well. This volume of cell medium was replaced by an equivalent volume of NPs suspension.Untreated cells and cells incubated with 10% (10 μL) of de-ionized water were used as controls. The cells were left in incubation for 48 and 72 hours, followed by removal of the cell medium (100 μL), and two cycles of gentle rinsing (200 μL of PBS (1×), Gibco, NY, USA). Then, 100 μL of complete RPMI 1640 medium containing resazurin (25 μg mL−1) were added to each well and incubated with the cells for three hours. The fluorescence measurements were performed with a Bio-Tek FL600 96-well microtiter plate fluorescence reader (Bio-Tek Instrument Inc., VT, USA). The excitation was set to 530 nm and the emission detection to 590 nm. The background fluorescence of complete cell medium, including resazurin, was subtracted from the fluorescence values obtained in the controls and NP-treated wells. The percentage of viable cells relative to controls was calculated by dividing the average fluorescence (n = 4) of the treated wells by the average fluorescence of the control wells (n = 4, cells not treated with either NPs or de-ionized water). Statistical analysis was performed using Prism 5 (GraphPad Software, La Jolla, CA, USA). All cell viability data are presented as mean ± SEM of quadruplicates. Statistical significance for cell viability was determined using one-way ANOVA followed by the post hoc Bonferroni multiple comparison test. A P < 0.05 was considered statistically significant. Significance between samples is indicated in the figures as follows: ***P < 0.001. Statistical significance analysis was performed between the control and the different concentrations of Au NPs@PEG and Au NPs@PEG-Mn2+.
4.12 In vitro dual imaging by CT and MR
For in vitro imaging, 80 μL of Au NPs@PEG-Mn2+ (Au/Mn ratio: 44) were dispensed in small polymer tubes (0.2 mL) at Au concentrations of 10.0, 17.4, 22.2, 44.3, 66.4 (mM). The tubes were immersed in a vial of nanopure water, and first imaged with a preclinical CT (eXplore Locus 80, GE Healthcare Technologies, Milwaukee, WI, USA) using the following parameters: 40 kVp, 450 μA, 90 ms detector exposure time, 4 × 4 detector binning; 89 μm voxel resolution. Image reconstruction and standard Hounsfield unit (HU) calibration were performed using the Parallax Innovations Reconstruction tool (Ilderton, ON, Canada). Next, the tubes were inserted into a 60 mm coil for imaging at RT with a 1.0 T MRI pre-clinical scanner (M2M, Aspect Imaging, Netanya, Israel). A T1-weighted echo sequence was performed with the following parameters: echo time (TE) 10.8 ms, repetition time (TR) 400 ms, dwell time 16 μs, field of view 70 mm, flip angle 90°, slice thickness 1.4 mm, interslice 0.1 mm, matrix 200 × 200, number of excitations 3, acquisition time 4 minutes.PC3 chick embryo chorioallantoic (CAM) tumor models were used to assess the potential of Au NPs@PEG-Mn2+ as a dual MRI and CT CA. The details are described in the ESI.†
4.13 In vivo vascular CT contrast performance of Au NPs@PEG-Mn2+
The performance of Au NPs as high blood-retention CT CAs was assessed in the mouse model. Au NPs@PEG-Mn2+ were synthesized according to the previously-described protocol, except purification was done by centrifugal filters instead of dialysis. In brief, 20 mL aliquots of pure Au NPs, and 10 mL aliquots of Au NPs@DMSA-DTPA and Au NPs@PEG-Mn2+ were concentrated down to 5 mL each, followed by three wash/centrifugation cycles (+6 mL de-ionized water; 500 g; 15 minutes per cycle). Before the animal injections, the Au NPs@PEG-Mn2+ were concentrated up to 140 mg mL−1 of Au and re-suspended in 154 mM NaCl through buffer exchange using centrifugal filters.All animal experiments were performed according to the guidelines issued by the Animal Ethical Committees of Université Laval and CHU de Québec. Four (4) eleven-week-old CD-1 female mice (approximately 25 g) (Charles River, Montreal, Canada) were anesthetized with 1% isoflurane (induction chamber, then using a bed-integrated nose cone). The animals were cannulated in the caudal tail vein (30G, winged needle) and connected to a catheter (280 μm i.d. intramedic polyethylene tubing PE-10, 60 cm, total volume 60 μL) prewashed with heparin (25 units mL−1) in 0.9% NaCl solution (9 mg mL−1, Hospira, Montreal, CA). Animals were CT-scanned (same parameters as described in Section 4.12) before the contrast media was injected. Next, the catheter was connected to a glass syringe (Hamilton 1710–100 μL syringe) containing the CA. Mice were injected with Au NPs@PEG-Mn2+ (6.8 ± 1.1 mg Au per mouse) in triplicate. After injection, the catheter was flushed with 100 μL of heparin/0.9% NaCl solution.
The animals were scanned approximately every six minutes for a period of 90 minutes, followed by euthanasia by CO2. The images were reconstructed and calibrated for the standard Hounsfield Unit (HU) scale with the Parallax Innovations Reconstruction tool (Ilderton, ON, Canada). For dynamic contrast enhancement analysis, 3D regions of interest (ROIs) were positioned over the abdominal aorta, inferior vena cava, liver, spleen, kidney and muscle. The average HU of each ROI was measured with Microview software (Parallax Innovations, Ilderton, ON, Canada). The contrast enhancement was calculated from the ratio between the average HU of each region after the injection at different time points, and the average HU in the same region before the injection.
5. Conclusions
PLAL was used to synthetize Au NPs with highly pure surfaces. This is an important advantage in the development of nanosized CT CAs, as it minimizes chemical surface contamination of these products that must be administered at concentrations typically higher than 1 g Au kg−1 to reach detection thresholds.For the imaging studies, the surface of pure Au NPs was densely covered with PEG-SH, using a low ligand excess, and further functionalized with Mn-chelates to obtain a CA also suitable for MRI. Detailed characterization of Au NPs@PEG-Mn2+ confirmed the production of colloids with (1) a surface composition only related with the ligands used for functionalization, (2) small sizes, (3) low polydispersity, and (4) stability in various media. The Au NPs@PEG-Mn2+ enhanced the image contrast in both CT and MRI after intratumoral injection in CAM prostate cancer models, and showed stable and long-lasting vascular contrast enhancement in CT using mouse models.
Therefore, PLAL proved to be an advantageous method for synthesizing Au NPs for use as CAs. The pure surface of PLAL-synthesized Au NPs bypasses concerns about the impact of chemical contaminants in vivo, and enables the use of lower amounts of ligands necessary for effective functionalization, a key step in the production of CAs with strong blood retention and multimodal imaging potential.
Acknowledgements
This work was supported by grants from NSERC (Discovery Grants 05821-2015 – D. Guay; 355524-2012 – M.-A. Fortin) and the Deutsche Forschungsgemeinschaft BA 3580/10-1 (S. Barcikwoski). T. Simão is grateful to FRQNT/CQMF for an International Training Program Fellowship and to NSERC-CREATE for the Integrated Sensor Systems (ISS) Fellowship and Travelling Award. The authors would like to acknowledge the valuable contributions of Mr Jean-Philippe Masse (Polytechnique Montréal; TEM images), Ms Vicky Dodier (U. Laval; elemental analysis) and Ms Myriam Laprise-Pelletier (U. Laval; assistance in imaging). All CT and MRI studies were performed at the Small Animal Imaging Platform (CR-CHU de Québec).Notes and references
- A. S. Thakor, J. Jokerst, C. Zavaleta, T. F. Massoud and S. S. Gambhir, Nano Lett., 2011, 11, 4029–4036 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- P. A. Jackson, W. N. W. A. Rahman, C. J. Wong, T. Ackerly and M. Geso, Eur. J. Radiol., 2010, 75, 104–109 CrossRef PubMed.
- M. W. Galper, M. T. Saung, V. Fuster, E. Roessl, A. Thran, R. Proksa, Z. A. Fayad and D. P. Cormode, Invest. Radiol., 2012, 47, 475–481 CrossRef CAS PubMed.
- L. Nebuloni, G. A. Kuhn and R. Muller, Acad. Radiol., 2013, 20, 1247–1255 CrossRef PubMed.
- D. P. Clark, K. Ghaghada, E. J. Moding, D. G. Kirsch and C. T. Badea, Phys. Med. Biol., 2013, 58, 1683–1704 CrossRef PubMed.
- C. Schürmann, F. Gremse, H. Jo, F. Kiessling and R. P. Brandes, PLoS One, 2015, 10, e0130374 Search PubMed.
- M. F. Kircher and J. K. Willmann, RY, 2012, 263, 633–643 CrossRef PubMed.
- M. Magnoni, E. Ammirati and P. G. Camici, J. Cardiol., 2015, 65, 261–269 CrossRef PubMed.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- A. Heidenreich, J. Bellmunt, M. Bolla, S. Joniau, M. Mason, V. Matveev, N. Mottet, H.-P. Schmid, T. van der Kwast, T. Wiegel and F. Zattoni, Eur. Urol., 2011, 59, 61–71 CrossRef PubMed.
- C. H. Lee, D. D. Dershaw, D. Kopans, P. Evans, B. Monsees, D. Monticciolo, R. J. Brenner, L. Bassett, W. Berg, S. Feig, E. Hendrick, E. Mendelson, C. D'Orsi, E. Sickles and L. W. Burhenne, J. Am. Coll. Radiol., 2010, 7, 18–27 CrossRef PubMed.
- D. Djoumessi, M. Laprise-Pelletier, P. Chevallier, J. Lagueux, M. F. Côté and M. A. Fortin, J. Mater. Chem. B, 2015, 3, 2192–2205 RSC.
- K. T. Butterworth, S. J. McMahon, F. J. Currell and K. M. Prise, Nanoscale, 2012, 4, 4830–4838 RSC.
- V. Patel, R. V. L. Papineni, S. Gupta, R. Stoyanova and M. M. Ahmed, Radiat. Res., 2012, 177, 483–495 CrossRef CAS PubMed.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS PubMed.
- Q. Y. Cai, S. H. Kim, K. S. Choi, S. Y. Kim, S. J. Byun, K. W. Kim, S. H. Park, S. K. Juhng and K. H. Yoon, Invest. Radiol., 2007, 42, 797–806 CrossRef CAS PubMed.
- H. Chen, H. Paholak, M. Ito, K. Sansanaphongpricha, W. Qian, Y. Che and D. Sun, Nanotechnology, 2013, 24, 355101 CrossRef PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- J. W. Park and J. S. Shumaker-Parry, ACS Nano, 2015, 9, 1665–1682 CrossRef CAS PubMed.
- R. Naccache, P. Chevallier, J. Lagueux, Y. Gossuin, S. Laurent, L. Vander Elst, C. Chilian, J. A. Capobianco and M. A. Fortin, Adv. Healthcare Mater., 2013, 2, 1478–1488 CrossRef CAS PubMed.
- D. P. Cormode, T. Skajaa, M. M. van Schooneveld, R. Koole, P. Jarzyna, M. E. Lobatto, C. Calcagno, A. Barazza, R. E. Gordon, P. Zanzonico, E. A. Fisher, Z. A. Fayad and W. J. M. Mulder, Nano Lett., 2008, 8, 3715–3723 CrossRef CAS PubMed.
- S. J. Soenen, P. Rivera-Gil, J.-M. Montenegro, W. J. Parak, S. C. De Smedt and K. Braeckmans, Nano Today, 2011, 6, 446–465 CrossRef CAS.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS PubMed.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS PubMed.
- A. M. Smith, L. E. Marbella, K. A. Johnston, M. J. Hartmann, S. E. Crawford, L. M. Kozycz, D. S. Seferos and J. E. Millstone, Anal. Chem., 2015, 87, 2771–2778 CrossRef CAS PubMed.
- W. E. Ghann, O. Aras, T. Fleiter and M. C. Daniel, Langmuir, 2012, 28, 10398–10408 CrossRef CAS PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- T. Skotland, T.-G. Iversen and K. Sandvig, Nanomedicine, 2010, 6, 730–737 CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Barcikowski and G. Compagnini, Phys. Chem. Chem. Phys., 2013, 15, 3022–3026 RSC.
- S. Petersen and S. Barcikowski, J. Phys. Chem. C, 2009, 113, 19830–19835 CAS.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- M. Laprise-Pelletier, M. Bouchoucha, J. Lagueux, P. Chevallier, R. Lecomte, Y. Gossuin, F. Kleitz and M.-A. Fortin, J. Mater. Chem. B, 2015, 3, 748–758 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Theory and Applications in Inorganic Chemistry, Wiley, 2008 Search PubMed.
- A. Shchukarev and D. Korolkov, Open Chem., 2004, 2, 347–362 CAS.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley, 1992 Search PubMed.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- C. Rehbock, V. Merk, L. Gamrad, R. Streubel and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3057–3067 RSC.
- K. Rahme, L. Chen, R. G. Hobbs, M. A. Morris, C. O'Driscoll and J. D. Holmes, RSC Adv., 2013, 3, 6085–6094 RSC.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- W. P. Wuelfing, S. M. Gross, D. T. Miles and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 12696–12697 CrossRef CAS.
- A. Albanese, P. S. Tang and W. C. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- D. Pan, S. D. Caruthers, A. Senpan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 162–173 CrossRef CAS PubMed.
- L. E. Cole, T. Vargo-Gogola and R. K. Roeder, ACS Nano, 2014, 8, 7486–7496 CrossRef CAS PubMed.
- D. Kim, Y. Y. Jeong and S. Jon, ACS Nano, 2010, 4, 3689–3696 CrossRef CAS PubMed.
- D. H. Tsai, F. W. DelRio, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2010, 26, 10325–10333 CrossRef CAS PubMed.
- U. Taylor, C. Rehbock, C. Streich, D. Rath and S. Barcikowski, Nanomedicine, 2014, 9, 1971–1989 CrossRef CAS PubMed.
- J. P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2009, 11, 3805–3821 RSC.
- C. Pfeiffer, C. Rehbock, D. Huhn, C. Carrillo-Carrion, D. J. de Aberasturi, V. Merk, S. Barcikowski and W. J. Parak, J. R. Soc., Interface, 2014, 11, 20130931 CrossRef PubMed.
- P. Leena, H. Jouni and L. Timo, Handbook of Nanobiomedical Research, World Scientific, 2013, vol. 3, pp. 169–197 Search PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- K. Larson-Smith and D. C. Pozzo, Langmuir, 2012, 28, 13157–13165 CrossRef CAS PubMed.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- A. K. Murthy, R. J. Stover, W. G. Hardin, R. Schramm, G. D. Nie, S. Gourisankar, T. M. Truskett, K. V. Sokolov and K. P. Johnston, J. Am. Chem. Soc., 2013, 135, 7799–7802 CrossRef CAS PubMed.
- D. A. Bakan, M. A. Longino, J. P. Weichert and R. E. Counsell, J. Pharm. Sci., 1996, 85, 908–914 CrossRef CAS PubMed.
- I. Aoki, Y. Takahashi, K.-H. Chuang, A. C. Silva, T. Igarashi, C. Tanaka, R. W. Childs and A. P. Koretsky, NMR Biomed., 2006, 19, 50–59 CrossRef CAS PubMed.
- D. Pan, S. D. Caruthers, A. Senpan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 3, 162–173 CrossRef PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- M. Kueny-Stotz, A. Garofalo and D. Felder-Flesch, Eur. J. Inorg. Chem., 2012, 1987–2005 CrossRef CAS.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- G. Wegner, M. Auger, D. Bélanger, P. Bénard, J. Claverie, M. A. Fortin, J. Greener, F. Kleitz, G. Laroche and J. F. Morin, Functional Materials: For Energy, Sustainable Development and Biomedical Sciences, De Gruyter, 2014 Search PubMed.
- A. J. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921–2959 CrossRef CAS PubMed.
- M. Park, N. Lee, S. H. Choi, K. An, S.-H. Yu, J. H. Kim, S.-H. Kwon, D. Kim, H. Kim, S.-I. Baek, T.-Y. Ahn, O. K. Park, J. S. Son, Y.-E. Sung, Y.-W. Kim, Z. Wang, N. Pinna and T. Hyeon, Chem. Mater., 2011, 23, 3318–3324 CrossRef CAS.
- M. Letourneau, M. Tremblay, L. Faucher, D. Rojas, P. Chevallier, Y. Gossuin, J. Lagueux and M. A. Fortin, J. Phys. Chem. B, 2012, 116, 13228–13238 CrossRef CAS PubMed.
- M. J. Baek, J. Y. Park, W. Xu, K. Kattel, H. G. Kim, E. J. Lee, A. K. Patel, J. J. Lee, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, ACS Appl. Mater. Interfaces, 2010, 2, 2949–2955 CAS.
- C. C. Huang, N. H. Khu and C. S. Yeh, Biomaterials, 2010, 31, 4073–4078 CrossRef CAS PubMed.
- L. Gamrad, C. Rehbock, J. Krawinkel, B. Tumursukh, A. Heisterkamp and S. Barcikowski, J. Phys. Chem. C, 2014, 118, 10302–10313 CAS.
- H. Muto, K. Yamada, K. Miyajima and F. Mafune, J. Phys. Chem. C, 2007, 111, 17221–17226 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: In vivo dual imaging CT/MR studies, hydrodynamic diameter results of Au NPs during the functionalization procedure, proton NMR relaxation data of the functionalized colloid and CT dynamic contrast enhancement study using AurovistTM. See DOI: 10.1039/c6tb01162d |
| This journal is © The Royal Society of Chemistry 2016 |