
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica core–shell particles for the dual delivery of gentamicin and rifamycin antibiotics†
Andrea M.
Mebert
ab,
Carole
Aimé
c,
Gisela S.
Alvarez
ab,
Yupeng
Shi
c,
Sabrina A.
Flor
d,
Silvia E.
Lucangioli
d,
Martin F.
Desimone
*ab and
Thibaud
Coradin
*c
aUniversidad de Buenos Aires, Facultad de Farmacia y Bioquimica, Junin 956 Piso 3, (1113) Ciudad Autónoma de Buenos Aires, Argentina. E-mail: desimone@ffyb.uba.ar
bIQUIMEFA-CONICET, Ciudad Autónoma de Buenos Aires, Argentina
cSorbonne Universités, UPMC Univ Paris 06, CNRS, Collège de France, UMR 7574, Laboratoire de Chimie de la Matière Condensée de Paris, 11 Place Marcelin Berthelot, F-75005 Paris, France
dCONICET – Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Departamento de Tecnología Farmacéutica, Ciudad Autónoma de Buenos Aires, Argentina
First published on 30th March 2016
Abstract
Increasing bacterial resistance calls for the simultaneous delivery of multiple antibiotics. One strategy is to design a unique pharmaceutical carrier that is able to incorporate several drugs with different physico-chemical properties. This is highly challenging as it may require the development of compartmentalization approaches. Here we have prepared core–shell silica particles allowing for the dual delivery of gentamicin and rifamycin. The effect of silica particle surface functionalization on antibiotic sorption was first studied, enlightening the role of electrostatic and hydrophobic interactions. This in turn dictates the chemical conditions for shell deposition and further sorption of these antibiotics. In particular, the silica shell deposition was favored by the positively charged layer of gentamicin coating on the core particle surface. Shell modification by thiol groups finally allowed for rifamycin sorption. The antibacterial activity of the core–shell particles against Staphylococcus aureus and Pseudomonas aeruginosa demonstrated the dual release and action of the two antibiotics.
1. Introduction
Drug carriers for local antibiotic delivery have gained attention in the last few years due to the need to deliver effective antibiotic doses, and avoid the high toxicity associated with systemic administration.1 Those delivery systems can be applied in situations such as periodontal disease,2 orthopedic trauma3–5 or in skin wound healing.6 However, due to the presence of multiple pathogens, and the increasing drug resistance, the co-delivery of different antibiotics or other compounds is a promising alternative for increasing treatment efficacy.7When the antimicrobial treatment is associated with the implantation of a biomaterial, the relevant drugs are ideally loaded within the biomaterial scaffold or at its surface.8 However, for soft tissue repair where hydrogel-based dressings are used, the kinetics of drug release is usually too fast due to the large porosity of the scaffold compared to antibiotic dimensions.9 Another strategy is to introduce strong chemical bonds between the material and the drug but this can become complex if multiple drug delivery is targeted.
As an alternative, the use of nanocomposite materials where the drugs are associated with nanoparticles embedded in the scaffold has attracted increasing attention.10,11 The intrinsic properties of these nanoparticles, as well as their interactions with the host hydrogel, can be tuned so as to optimize the drug release profiles. Among possible antibiotic carriers, silica nanoparticles have been extensively studied. They can be prepared in a wide range of size, porosity and chemical composition. Whereas a full assessment of their biocompatibility is still to be achieved,12 numerous studies have demonstrated that silica nanoparticles with size ranging from 10 nm to 200 nm exhibit limited cytotoxicity.13–16 Moreover, grafting sulfonate, amine and especially thiol moieties further decreases their detrimental effect.17
To address the issues related to co-delivery of antibiotics, two options can be envisioned. The first one would rely on the incorporation of different populations of particles, each loaded with a unique drug. Although each drug-carrier can be individually optimized, this approach requires the use of a high total concentration of silica particles. Alternatively, the incorporation of multiple drugs within a single type of particle can be envisioned. However, because the different drugs exhibit sometimes diverging physico-chemical properties (charge and hydrophobicity), a chemical compartmentalization is required for the design of the carrier particle. A rather simple option is to use core–shell nanoparticles, consisting of a core, or inner material surrounded by a shell, or outer layer.18 Core–shell nanoparticles are widely used for bioimaging19 and biosensing,20 as well as for other biomedical applications including the co-delivery of drugs.21,22 In particular, silica-based core–shell particles are commonly formed from a metal,23,24 metal oxide25,26 or micellar core covered with a silica shell.27 This outer layer gives the particle the same properties as silica particles, such as lower reactivity, enhanced stability in suspension and slower drug release.28 Core–shell particles entirely formed by silica have also been reported exhibiting different characteristics such as bimodal pore structures29 or selective functionalization of the inner and outer regions of the particles using hybrid sols.30
Inspired by the latter approach, we report here a new type of silica core–shell particle having the capability to simultaneously deliver two common topical antibiotics, gentamicin and rifamycin. For this purpose, we have developed a strategy similar to layer-by-layer deposition routes where the positively charged coating of the drugs on the core surface can act as the reactive interface for silica shell deposition. The role of electrostatic and hydrophobic interactions between silanes and antibiotics in the successful synthesis of these core–shell particles is discussed, enlightening the foreseeable versatility of this approach.
2. Materials and methods
2.1 Materials
Tetraethyl orthosilicate (TEOS, 98%), (3-aminopropyl)triethoxysilane (APTES, ≥98%) and (3-mercaptopropyl)trimethoxysilane (MPTMOS, ≥95%) were purchased from Sigma-Aldrich, as well as the antibiotics sodium rifamycin and gentamicin sulfate. Ammonium hydroxide (30%) was obtained from Carlo Erba Reagents. All other reagents were of analytical grade and commercially available.2.2. Synthesis of silica particles
2.3. Particle loading
Antibiotic-loaded particles were prepared in deionized water by mixing the particles and the corresponding antibiotic, in a ratio of 375 mg of particles![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mg of antibiotic:50 mL of deionized water. Suspensions were stirred overnight at room temperature and protected from light. The resulting particles were recovered by centrifugation and washed three times with deionized water. Sodium rifamycin and gentamicin sulfate were used as antibiotics.
:20 mg of antibiotic:50 mL of deionized water. Suspensions were stirred overnight at room temperature and protected from light. The resulting particles were recovered by centrifugation and washed three times with deionized water. Sodium rifamycin and gentamicin sulfate were used as antibiotics.
2.4. Preparation of core–shell particles
The core was synthesized by the Stöber method, modified with thiol or sulfonate for the loading of rifamycin or gentamicin, respectively as described before. The silica shell layer synthesis was carried out with the drug in the ethanol–water medium, in a ratio 500 mg of particles:27 mg of antibiotics:0.4 mL of TEOS:0.5 mL of ammonium hydroxide:1.5 mL of deionized water:50 mL of ethanol. This shell was modified as described before to achieve core sulfonate-modified and shell thiol-modified particles (SiSO3@SiSH) or core thiol-modified and shell sulfonate-modified particles (SiSH@SiSO3). The second layer was loaded with the second antibiotic (i.e. that was not used in the first step) in the same drug:particle:deionized water ratio. The loaded core–shell particles were then washed and resuspended as described before.
2.5. Transmission electron microscopy (TEM)
The size and the shape of the particles were investigated by TEM. Briefly a drop of the sample in aqueous solution was deposited on 400-mesh carbon-coated copper grids. After one minute, the liquid was blotted with filter paper (Whatman no. 4). TEM was performed at room temperature using a JEOL 1011 electron microscope operating at 100 kV.2.6. Zeta potential
The zeta potential of the particles was measured using Zeta Plus Brookhaven equipment. For this purpose a 5 mg mL−1 suspension of particles was prepared in 10 mM KCl aqueous solution. In another experiment, 0.5 mg mL−1 of particles were confronted with increasing concentrations of antibiotics. After each step of antibiotic addition, particles were recovered by centrifugation before zeta-potential measurements.2.7. Microbiological assay (diffusion method)
The microbiological assay was carried out using the disk diffusion method as described in the United States Pharmacopeia. Petri dishes with 2 to 5 mm thick Luria Bertani (LB) agar medium were inoculated and homogeneously scattered with 100 μL of 1 × 107 cfu mL−1 in PBS of the bacterial suspension sensitive to the antibiotic of interest. Sterile absorbent paper disks were impregnated with 10 μL of the particle suspension and placed on the surface of the agar. Staphylococcus aureus ATCC 29213 (sensitive to rifamycin and gentamicin) and Pseudomonas aeruginosa ATCC 27853 (sensitive to gentamicin) were used. The inhibition zone was measured after incubation at 37 °C for 24 hours, and evaluated using the corresponding calibration curve of antibiotics selected according to the known sensitivity of the microorganisms to the antibiotic of interest (i.e.: 20–60 μg mL−1 for gentamicin and 0.3–1.2 μg mL−1 for rifamycin). In all cases the results are expressed as mean ± SD from triplicate experiments.2.8. Quantification of gentamicin and rifamycin
The kinetics of the dual drug release behaviour of core–shell particles was determined by Mass Spectrometry. Nanoparticles were suspended at a 30 mg mL−1 concentration in deionized water at 37 °C. At regular intervals of time, they were recovered by centrifugation and the amount of drug released was measured by MS. For this purpose, quantitative and qualitative mass analysis was carried out on a TSQ Quantum Access™ Max (Thermo Fisher Scientific, San Jose, CA, USA) triple stage quadrupole mass spectrometer with an electrospray ionization (ESI) source and a Rheodyne™ 7750E-185 divert/inject valve. Sample injection was performed via the Rheodyne injection valve with a 10 μl loop. The linearity of the detector response to sample concentration was assessed by measuring five calibration points ranging from 5 to 600 ng mL−1 for gentamicin and rifamycin. Each calibration point was injected five times. Regression coefficients were obtained by plotting the average peak area versus concentration, using the least squares method (ESI-1†). Three ions were selected for the quantification of gentamicin (m/z 464, 322 and 160) and rifamycin (720, 660 and 574).2.9. Statistical analysis
In all cases data are mean ± SD of triplicate experiments. The differences were analyzed using two-way ANOVA, followed by the Tukey post-test; p < 0.05 was considered significant.3. Results and discussion
3.1 Characterization of surface-modified silica particles
Monodisperse spherical silica particles were obtained by the Stöber method.31,32 The mean diameters for each type of particle were 277 ± 12 nm (SiOH), 273 ± 5 nm (SiNH2), 269 ± 7 nm (SiSH) and 277 ± 2 nm (SiSO3) as determined by TEM (Fig. 1A and B), indicating that the particle mean diameter was not significantly affected by the surface modification (ESI-2†). The surface modification was also analyzed by measuring the zeta potential (ζ) of the particles at different pH values (Fig. 1C). Starting from pH 2, the bare silica particles (SiOH) are slightly positively charged and undergo a rapid surface charge reversal between pH 3 and 4 before reaching a constant value of ca. −65 mV. For thiol-bearing particles (SiSH), the ζ value is almost zero in acidic media and reaches ca. −30 mV at pH 8. After sulfonation, the ζ value is negative over the whole range of pH, with a slight decay between pH 3 and 5. Finally, the ζ value of amino-modified particles (SiNH2) follows a similar trend except that values go from a highly positive value (+65 mV) to zero. | ||
| Fig. 1 (A and B) TEM photo of unmodified SiOH particles. (C) Zeta-potential of sulfonate-modified (SiSO3), unmodified (SiOH), amino-modified (SiNH2) and thiol-modified (SiSH) silica particles at different pH values (10 mM KCl). | ||
The measured evolution for bare silica nanoparticles is in good agreement with the literature, indicating that silanols can exist in three different forms on the silica surface: SiOH2+, SiOH and SiO−. The positively charged species are present in a significant amount up to pH ca. 3. This is in agreement with the point of zero charge of 3 for ca. 270 nm silica particles as reported elsewhere.33 Then it was suggested that further deprotonation involves two populations of silanol groups, 20% being acidic (pKa SiOH/SiO− = 4.5) and 80% basic (pKa SiOH/SiO− = 8.5). In the case of thiol groups, with a pKa (SH/S−) of ca. 10.5, the organic function should not contribute to the surface charge of the particle in the investigated pH range. Therefore the observed ζ evolution can be attributed to the decrease of available silanol groups on the particle surface after grafting, leading to a lower density of SiOH2+ in acidic media and SiO− in basic solution, therefore narrowing the range of ζ variation. After sulfonation, the strongly acidic SO3− groups (pKa SO3H/SO3− < 1) should contribute to a constant negative charge that can be evidenced at low pH. However, in basic medium, the absolute value of ζ for sulfonated particles is smaller than that for bare silica particles. This indicates that, from a surface charge point of view, the decrease in the number of free silanol groups on the silica surface that results from the grafting reaction is not compensated by the presence of sulfonate groups on the organic chain of the silane. Accordingly, since pKa of primary amines is ca. 10.5, their protonation degree should be constant over the 2–8 pH range of this study and should therefore contribute in a constant manner to the surface charge of SiNH2 particles. Comparison of the ζ evolution of SiNH2 and SiOH shows that this is indeed the case except in the 3–4 pH range. This probably reflects the existence of direct acid–base reactions between surface silanolate and ammonium groups from APTES, as reported in the literature.34
3.2 Characterization of antibiotic-coated silica particles
Silica particles were loaded with two oppositely charged antibiotics, gentamicin being positively charged, while rifamycin is negatively charged. TEM analysis revealed that the particle mean diameter was not significantly affected by the antibiotic adsorption (ESI-3†). Based on preliminary experiments, particle loading was performed in a large excess of antibiotics (20 mg of antibiotics for 375 mg of particles in water). After washing, the gentamicin and rifamycin loadings were evaluated on P. aeruginosa and S. aureus, respectively. Antibiograms were obtained using the disk diffusion method, and inhibition zones were measured after incubation for 24 h at 37 °C. After this period, impregnated paper discs were transferred to new agar media seeded with bacteria and incubated for an additional 24 h period. However, no antibacterial activity could be observed after this delay, suggesting that a major fraction of the antibiotics was released over the first 24 h. Unloaded particles showed no antibacterial activity in all cases indicating that the measured bactericidal effects originated from the drugs only.In the case of gentamicin, the largest antibiotic amount was found for the most negatively charged particles SiOH and SiSO3− compared to thiol-(SiSH) and amino-(SiNH2) modified silica particles (Fig. 2). This suggests that attractive electrostatic interactions are the main driving force for the sorption process. Indeed, the amounts of gentamicin in SiOH and SiSO3− are 1509 μg g−1 and 1793 μg g−1, respectively, which are 6 and 7 times higher than the amount found in SiSH (Table 1).
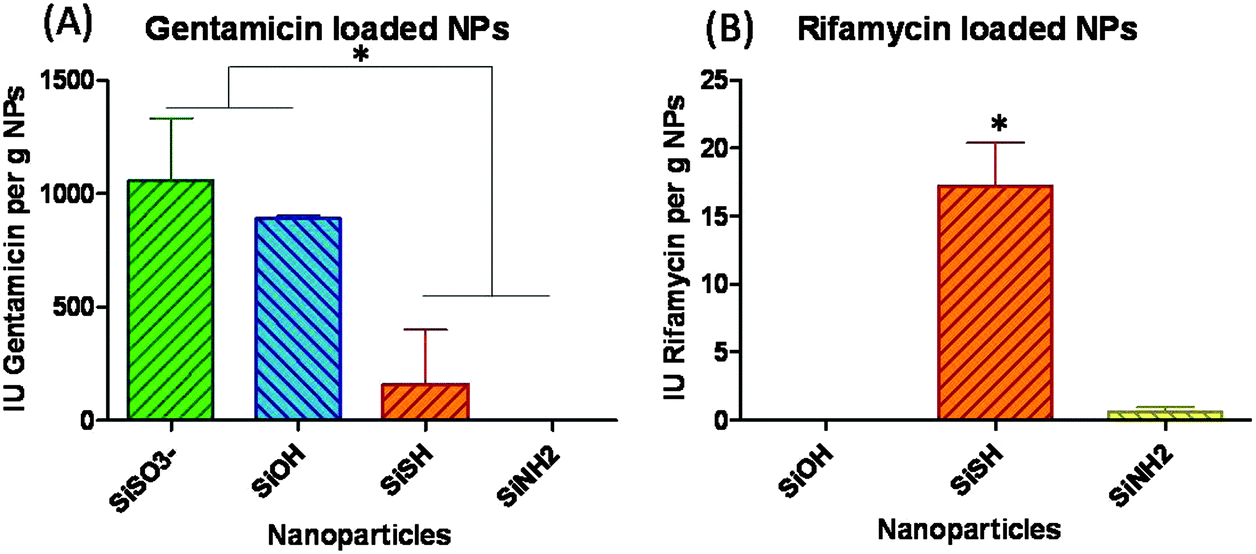 | ||
| Fig. 2 Disk diffusion method evaluation of the antibacterial activity. Activities expressed as International Units (IUs) of the drug per gram of particles: (A) gentamicin and (B) rifamycin. The results are expressed as mean ± SD from at least triplicate experiments. * indicates the statistical significant difference (p < 0.0001). | ||
| IU Drug per g of NPs | μg of drug per g of NPs | |
|---|---|---|
| (A) Gentamicin | ||
| SiSO3− | 1058 ± 256 | 1793 ± 467 |
| SiOH | 890 ± 11 | 1509 ± 20 |
| SiSH | 160 ± 240 | 271 ± 407 |
| SiNH2 | 0 ± 0 | 0 ± 0 |
| (B) Rifamycin | ||
| SiSH | 17.2 ± 3.2 | 19.2 ± 3.5 |
| SiNH2 | 0.6 ± 0.3 | 0.7 ± 0.4 |
| SiOH | 0.0 ± 0.0 | 0.0 ± 0.0 |
On the other hand, in the case of the negatively charged rifamycin, the situation is more complex. Despite their positively charged surface, SiNH2 particles do not adsorb significantly more antibiotic than bare particles. Moreover, the highest rifamycin loading was obtained for slightly negatively charged SiSH particles with 19.2 μg g−1 (Table 1). This can be explained by considering that rifamycin is classified as an hydrophobic molecule, with an octanol–water partition coefficient of 2.77,35 while thiol groups have the capability to confer non-polar-properties to modified surfaces.36,37 Therefore it is very likely that hydrophobic interactions are involved in the rifamycin sorption on SiSH. Sulfonated particles were not evaluated as their negative surface charge was expected to repel anionic rifamycin molecules.
To clarify the sorption process, the evolution of zeta potential with antibiotic concentration was studied for the antibiotic-loaded particles that exhibited even a low antimicrobial activity, namely SiSO3, SiOH, and SiSH for gentamicin, and SiNH2, and SiSH for rifamycin. In the case of gentamicin adsorption, a gradual increase in the ζ value with antibiotic concentration was observed for SiOH, SiSO3− and SiSH up to slightly positive values (+5/+15 mV, Fig. 3). The difference in the ζ value between uncoated and saturated surfaces for each type of particle nicely correlates with the drug loading as determined by the disk method. Altogether, this supports the previous hypothesis that the sorption process is driven by attractive electrostatic interactions and that saturation occurs after neutralization of the negative charge of the particle surface by positively charged gentamicin. Nevertheless, it must be noticed that the final ζ value is slightly positive. This suggests that a fraction of the antibiotic is adsorbed via other interactions either with the silica surface or with already-deposited molecules. In particular, the presence of three hydroxyl groups on the gentamicin backbone should favor hydrogen bond formation.
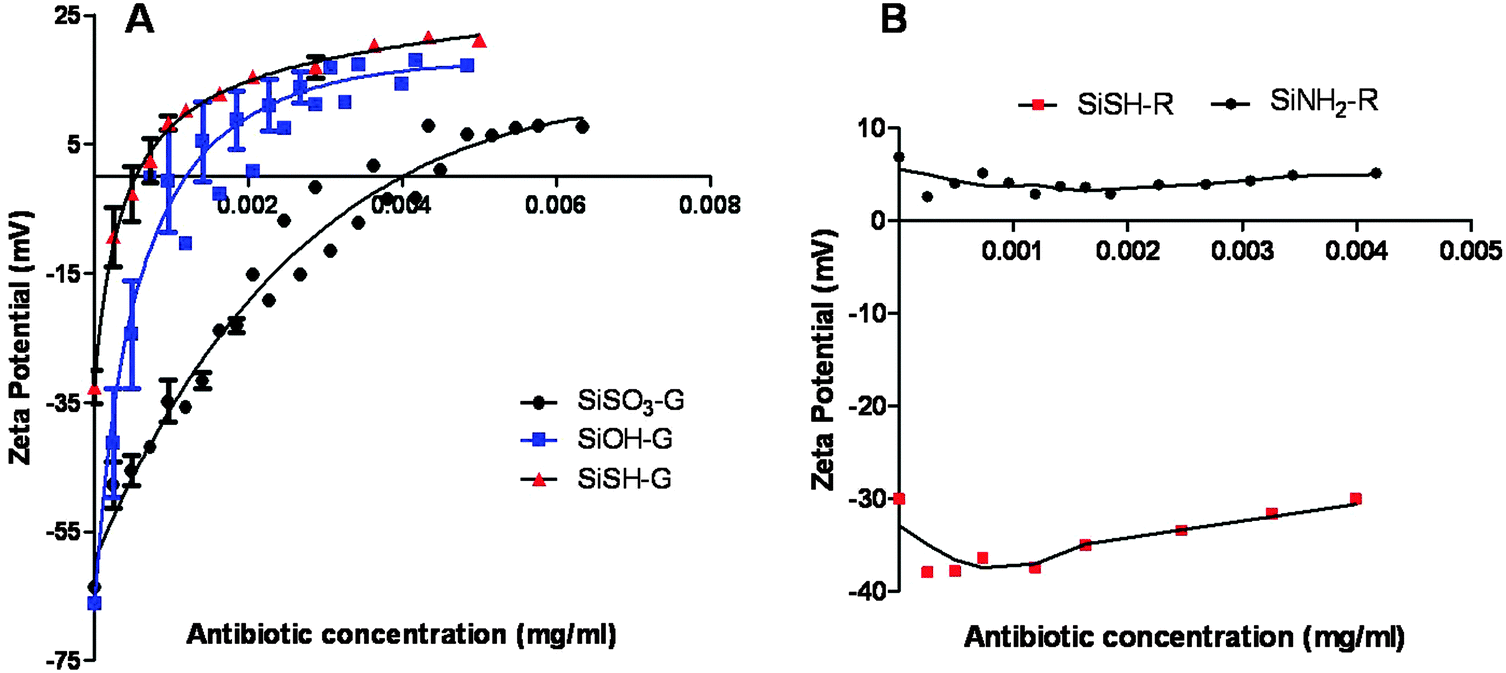 | ||
| Fig. 3 Zeta-potential of silica particles as a function of antibiotic concentration. (A) Gentamicin-loaded sulfonate (SiSO3-G), unmodified (SiOH-G), and thiol (SiSH-G) modified particles. (B) Rifamycin loaded amino (SiNH2-R) and thiol (SiSH-R) modified particles. | ||
In the case of rifamycin, the relationship between zeta-potential and drug adsorption is not straightforward. The addition of a small amount of antibiotic leads to a decrease of ζ for both SiSH and SiNH2. This event is more pronounced for the former than for the latter, suggesting that it corresponds to the adsorption of the negatively charged antibiotics. Increasing further the antibiotic concentration does not significantly modify the ζ value, but a slight continuous increase is obtained for SiSH. Since the rifamycin loading capacity of these two systems is about hundred times lower than for gentamicin (see Table 1), it can be expected that surface saturation is reached at such low concentrations. Moreover, near neutral pH, gentamicin bears five positive charges per molecule, whereas rifamycin has only one negatively charged group so that the former should have more influence on the overall particle surface charge than the latter at a similar surface concentration (Fig. 4). As a matter of fact, the two drugs also differ from the point of view of the accessibility of the ionized groups. In the gentamicin structure, the ammonium functions point out of the glycosidic rings, whereas the OH group of rifamycin belongs to a naphthalene ring inducing a sterical barrier and conferring a strong hydrophobic character to the molecule. This can explain its low sorption on SiNH2 particles despite their high positive charge. It also strengthens our hypothesis about the key role of hydrophobic interactions in rifamycin sorption.
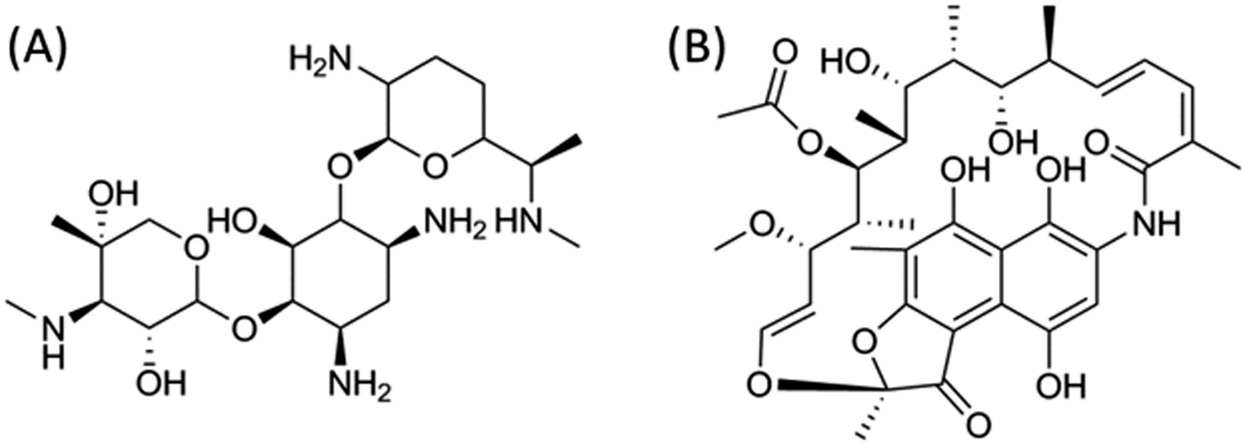 | ||
| Fig. 4 Chemical structures of (A) gentamicin and (B) rifamycin. | ||
3.3. Core–shell nanoparticles and their dual antibiotic drug release properties
In a step forward, particles capable of releasing two antibiotics were designed. Our strategy involves the synthesis and functionalization of a Stöber silica core and its loading with the first antibiotic (drug A), followed by the deposition of the shell that is further functionalized and loaded with the second antibiotic (drug B) (Fig. 5). The evolution of the particle size, as determined by DLS, and of their surface charge, as obtained by ζ measurements, are provided in Table 2 at each step of the preparation process.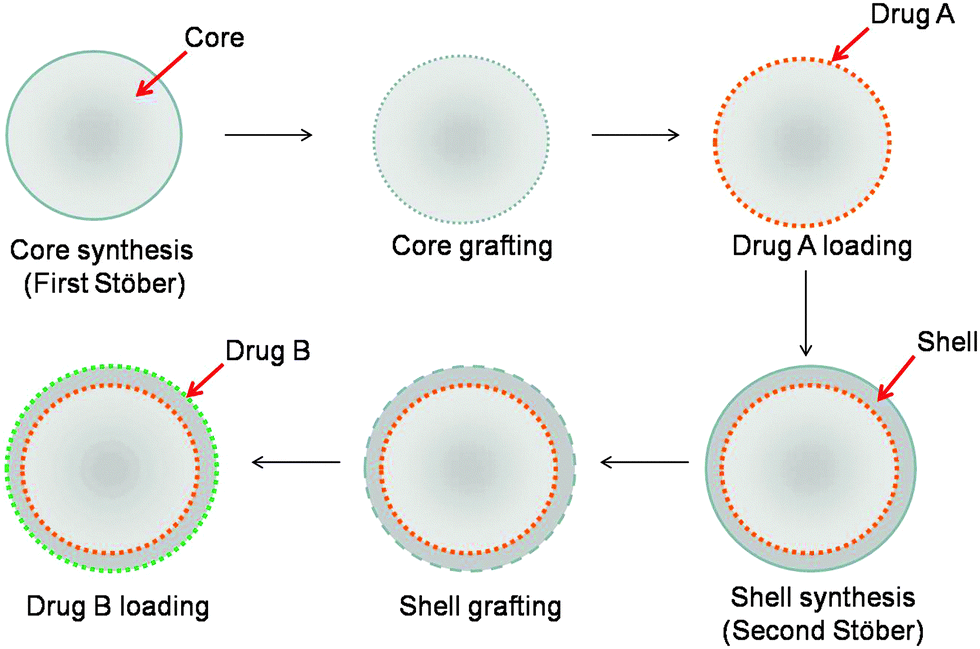 | ||
| Fig. 5 Scheme of the synthesis of double drug-loaded core–shell particles based on a two step Stöber process. | ||
| Sample | ζ (mV) | d H (nm) | Sample | ζ (mV) | d H (nm) |
|---|---|---|---|---|---|
| SiSO3 | −50 ± 3 | 257 ± 16 | SiSH | −38 ± 2 | 258 ± 7 |
| SiSO3-G | −18 ± 1 | 325 ± 42 | SiSH-R | −32 ± 2 | 253 ± 17 |
| SiSO3-G@SiOH | −59 ± 2 | 358 ± 17 | SiSH-R@SiOH | −55 ± 2 | 267 ± 5 |
| SiSO3-G@SiSH | −32 ± 1 | 377 ± 13 | SiSH-R@SiSO3 | −46 ± 2 | 275 ± 13 |
| SiSO3-G@SiSH-R | −13 ± 2 | 1240 ± 157 | SiSH-R@SiSO3-G | −6 ± 1 | 988 ± 130 |
A first configuration was studied using the sulfonated core loaded with gentamicin, as it exhibited the higher drug loading, further coated with a silica shell and functionalized with thiol groups followed by rifamycin sorption (SiSO3-G@SiSH-R). ζ data show that the adsorption of gentamicin on SiSO3 leads to a decrease in negative charge of the particles, as expected for the deposition of the cationic antibiotic. DLS also indicates a slight increase in the particle diameter. Note that these data were obtained after rinsing the particles so that, compared to the values obtained in the presence of an excess of antibiotics and representative of the synthesis process (Fig. 3), partial gentamicin desorption has occurred leading to a negative value of ζ. Subsequently, reaction of SiSO3-G with TEOS leads to an increase of the absolute value of ζ, reaching ca. −60 mV and therefore close to the value of bare Si–OH particles, strongly supporting the formation of a silica shell. In parallel, DLS data also suggest an increase in particle diameter but the relatively high value of the standard deviation for SiSO3-G (40 nm) does not allow the calculation of the shell thickness. After outer grafting with MPTMOS, the ζ decreases in absolute value, similar to our previous observation of MPTMOS reaction on core silica particles. The successful formation and functionalization of the shell were also supported by analyzing the pH dependence of the ζ value of the particles after synthesis (Fig. 6). The evolution of the zeta potential in the pH range of 2 to 8 closely follows that of the thiolated core particles, indicating that the resulting surface chemistry is similar. Finally, further contact with rifamycin decreases again the absolute value of ζ. In parallel, DLS data suggest an important aggregation of the particles.
 | ||
| Fig. 6 Zeta-potential of silica particles and core–shell particles as a function of pH: SiSH-R@SiSO3 as compared with SiSO3 and SiSO3-G@SH as compared with SiSH. | ||
The mirror situation using the thiolated core loaded with rifamycin coated with a silica shell and functionalized with sulfonated groups, followed by gentamicin sorption, (SiSH-R@SiSO3-G) was studied. As seen in Table 2, after contact of TEOS with the rifamycin-coated thiolated core particles, only a slight variation of ζ is measured for the expected (SiSH-R@SiOH) system and this value is not significantly modified after MPTMOS grafting and sulfonation (SiSH-R@SiSO3). In parallel all particle sizes obtained from DLS were within the standard deviation range, except for the final gentamicin deposition. A more detailed study of the pH-dependence of the ζ for SiSH-R@SiSO3 systems shows that it follows that of sulfonated cores but with a significantly more negative value under acidic conditions (Fig. 6). This suggests that the oxidation reaction required for sulfonation of thiol groups of the shell also impacts the thiol groups present on the core particle. In addition, the experimental conditions for the sulfonation of the shell are particularly harsh when considering the shell formation of preloaded cores, involving a multistep process with hydrogen peroxide, sulfuric acid and successive washings. This suggests that such a combination is not suitable to obtain bi-functional core–shell particles so that only gentamicin loading of the core will be considered for the rest of the manuscript.
The structure of the particles obtained with and without antibiotics was then analyzed by TEM. As can be seen in Fig. 7, for antibiotic-free core particles, no significant modification of the surface of the final colloids was observed, even after the sorption of the second drug. In contrast, when gentamicin was initially deposited on the core particle, an additional thin layer was observed on the particle surface. This layer could still be observed after further shell surface sorption of rifamycin.
 | ||
| Fig. 7 TEM images of: (A) SiOH@SiSH, (B) SiSH@SiSH and (C) SiSO3@SiSH core–shell particles, with or without gentamicin (G)-loaded cores and with or without rifamycin (R)-loaded shells (scale bar 50 nm). | ||
The deposition of a silica shell was confirmed by SEM imaging (Fig. 8 and ESI-4†). The surface of the silica shell grown on the surface of gentamicin-coated SiSO3 particles shows a granular aspect that is also strikingly observed after rifamycin sorption on these core–shell systems.
 | ||
| Fig. 8 SEM images of (A) SiSO3@SiSH and (B) SiSO3-G@SiSH-R particles (scale bar 50 nm). | ||
At this point, it is important to consider the shell process formation. The possibility to form SiO2 shells on silica particles under the Stöber conditions has been widely described.38 Although electrostatic interactions are unfavorable for silica deposition on bare silica particles, it may proceed thanks to the promotion of the condensation reaction of silanols and silonates under basic conditions. Surface modification by cationic coating introduces favorable electrostatic interactions promoting silica deposition.39 In contrast, the introduction of anionic groups on the particle surface should limit shell formation. The effect of grafting hydrophobic moieties should also make the coating process of hydrophilic silica less favorable than for bare particles. Coming back to our samples, the efficiency of the shell layer deposition of silica particles should vary as SiOH > SiSH > SiSO3. Noticeably, TEM images could not provide any evidence that such a reaction occurred under these conditions. However, this situation was changed after gentamicin sorption as the surface turned positive, favoring the formation of an observable silica shell. On this basis, only the SiSO3@SiSH configuration was further studied.
Antimicrobial activity against the two bacterial strains (gentamicin-sensitive P. aeruginosa and gentamicin–rifamycin-sensitive S. aureus) was achieved when the two drugs were present within the core–shell particles. The diameters of the corresponding inhibition zones were taken as 100% to compare the efficiency of these dual systems with particles containing a single antibiotic (Fig. 9). For both bacteria, drug-free core–shell particles showed no significant antibacterial activity. For P. aeruginosa, the gentamicin-free particles were ineffective whereas the gentamicin-loaded particles showed an antibacterial activity similar to the particles with both antibiotics. This is in good agreement with the fact that rifamycin is poorly effective towards Gram-negative bacteria. It is important to point out that gentamicin-coated core particles and core–shell systems have a similar antibacterial efficiency, indicating that no important leaching of gentamicin occurred from the particle core during rifamycin sorption on the shell. For S. aureus, the core–shell particles containing both drugs presented an antimicrobial activity 1.5 times higher than the two systems containing only one antibiotic. These results indicate that these core–shell particles can efficiently deliver both antibiotics.
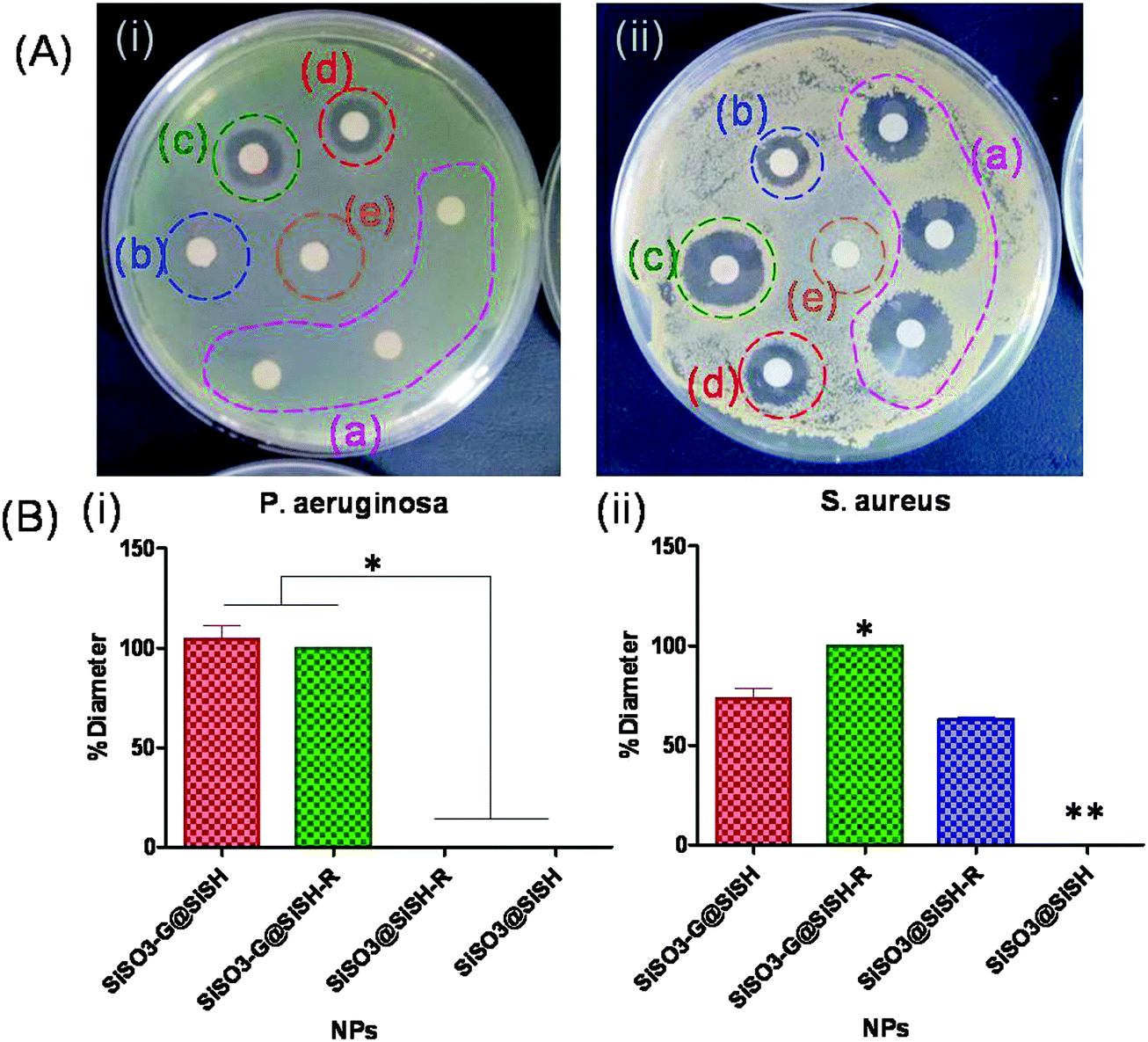 | ||
| Fig. 9 Disk diffusion method evaluation of the antibacterial activity of core–shell particles: (A) plates obtained with (a) rifamycin calibration curve (0.3–0.6–1.2 μg mL−1), (b) SiSO3@SiSH-R, (c) SiSO3-G@SiSH-R, (d) SiSO3-G@SiSH and (e) SiSO3@SiSH on (i) P. aeruginosa and (ii) S. aureus. (B) Corresponding diameter size relative to SiSO3-G@SiSH-R (100%) for (i) P. aeruginosa and (ii) S. aureus. * and ** indicate the statistical significant difference (p < 0.0001). | ||
To clarify this point, mass spectrometry analysis of a particle suspension supernatant allowed us to identify the presence of gentamicin and rifamycin40,41 (ESI-5†) and further confirmed the release of both antibiotics from the core–shell particles. The kinetics profiles revealed that 43% of the rifamycin is released during the first 30 minutes while the release of 50% of the gentamicin requires a four time longer incubation time. Moreover, rifamycin is completely released from the particles within 3 hours while only 71% of the gentamicin is released over the same period (Fig. 10). This is in agreement with the proposed mechanism where the outer rifamycin coating is rapidly desorbed whereas gentamicin release requires the progressive diffusion through the silica shell or dissolution of the silica shell.
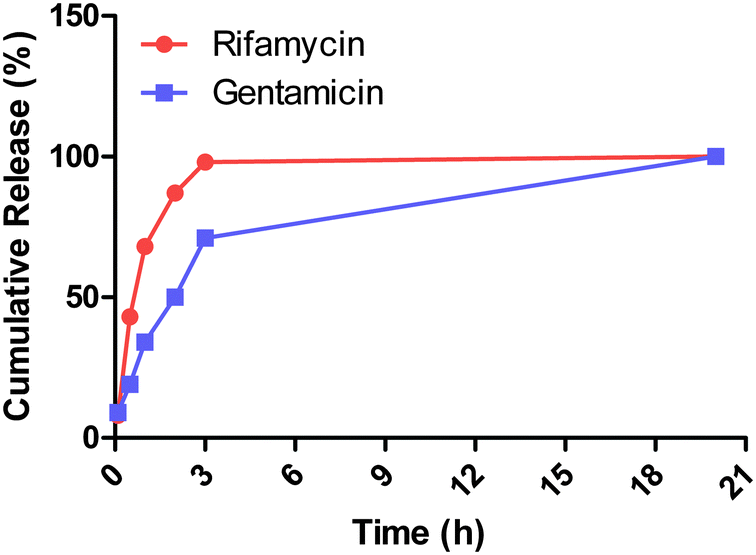 | ||
| Fig. 10 Dual release profiles of rifamycin and gentamicin from the core–shell particles measured by mass spectrometry. | ||
Our procedure has some similarities with layer-by-layer routes, where the core–shell structure is built up from the alternation of coatings with opposite charges. Whereas a close strategy was previously described to build up bi-functional core–shell mesoporous particles using hybrid sols,30 we have here taken advantage of the antibiotic layer itself as a charged interface for shell deposition, limiting its leaching during silica formation and further chemical modification. This strategy should therefore be applicable to any positively charged drug as a core component, whereas the shell coating may be of various natures. Importantly, although the mirror situation, i.e. direct shell coating from TEOS on a negatively charged core, does not appear to be possible as such, the use of hybrid sols containing cationic silanes, such as TEOS/APTES mixtures, should allow for further silica deposition.42 Accordingly, the use of hydrophobic sols can favor shell formation on the surface of core particles coated with lipophilic drugs.43
This strategy can also be extended to mesoporous particles that may offer the possibility for the loading of up to four drugs (inside core, outside core, inside shell, and outside shell) whose sequential release may be additionally tuned by gate-keeping approaches.44–46 Such a strategy may be of particular interest in so-called combination therapy that has become a key strategy in cancer treatment.47
4. Conclusions
The co-encapsulation of drugs with different and sometimes divergent, physico-chemical properties within a single carrier requires the development of compartmentalization strategies. The core–shell approach appears to be particularly suitable for this purpose but faces possible interference in the presence of drugs associated with the core particle with further deposition of the shell. In contrast, we show here that it is possible to benefit from the presence of a positively charged antibiotic coating to promote further silica deposition. This approach also has the advantage to avoid premature leaching of the core drug during shell formation. As a step further, it is now important to extend this layer-by-layer strategy to anionic and hydrophobic drugs. In addition, further control of the shell structure should allow for a temporally differentiated control of the release kinetics of the two molecules.Acknowledgements
A. M. Mebert is grateful for her doctoral fellowship granted by the National Research Council (CONICET). Y. Shi is funded by the China Scholarship Council. The authors would like to acknowledge the support of grants from the Universidad de Buenos Aires UBACYT 20020110100081, from CONICET PIP 11220120100657CO and from Agencia Nacional de Investigaciones Cientificas y Técnicas PICT 2012-1441 (to M. F. D). M. F. D. and T. C. thank the Argentina-France MINCYT-ECOS-Sud (project A12S01) and CONICET-CNRS programs for financial support of their collaboration. The authors would like to thank J. Nesterzak for his technical assistance.References
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey, P. Pandey and S. K. Bannerjee, Int. J. Pharm. Invest., 2012, 2, 2–11 CrossRef PubMed.
- N. J. Medlicott, M. J. Rathbone, I. G. Tucker and D. W. Holborow, Adv. Drug Delivery Rev., 1994, 13, 181–203 CrossRef CAS.
- I. Hornyxak, E. Madacsi, P. Kalugyer, G. Vacz, D. B. Horvathy, M. Szendroi, W. Han and Z. Lacza, BioMed Res. Int., 2014, 2014, 8 Search PubMed.
- S. J. McConoughey, R. P. Howlin, J. Wiseman, P. Stoodley and J. H. Calhoun, J. Biomed. Mater. Res., Part B, 2015, 103, 870–877 CrossRef PubMed.
- A. R. Short, D. Koralla, A. Deshmukh, B. Wissel, B. Stocker, M. Calhoun, D. Dean and J. O. Winter, J. Mater. Chem. B, 2015, 3, 7818–7830 RSC.
- A. Saito, H. Miyazaki, T. Fujie, S. Ohtsubo, M. Kinoshita, D. Saitoh and S. Takeoka, Acta Biomater., 2012, 8, 2932–2940 CrossRef CAS PubMed.
- T. Bollenbach, Curr. Opin. Microbiol., 2015, 27, 1–9 CrossRef CAS PubMed.
- A. Santos, M. Sinn Aw, M. Bariana, T. Kumeria, Y. Wang and D. Losic, J. Mater. Chem. B, 2014, 2, 6157–6182 RSC.
- M. Zilberman, D. Egozi, M. Shemesh, A. Keren, E. Mazor, M. Baranes-Zeevi, N. Goldstein, I. Berdicevsky, A. Gilhar and Y. Ullmann, Acta Biomater., 2015, 22, 155–163 CrossRef CAS PubMed.
- G. S. Alvarez, C. Helary, A. M. Mebert, X. Wang, T. Coradin and M. F. Desimone, J. Mater. Chem. B, 2014, 2, 4660–4670 RSC.
- B. Song, C. Wu and J. Chang, Acta Biomater., 2012, 8, 1901–1907 CrossRef CAS PubMed.
- Y. Wang, A. Santos, A. Evdokiou and D. Losic, J. Mater. Chem. B, 2015, 3, 7153–7172 RSC.
- H. Zhang, D. R. Dunphy, X. Jiang, H. Meng, B. Sun, D. Tarn, M. Xue, X. Wang, S. Lin, Z. Ji, R. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- D. Lison, L. C. J. Thomassen, V. Rabolli, L. Gonzalez, D. Napierska, J. W. Seo, M. Kirsch-Volders, P. Hoet, C. E. A. Kirschhock and J. A. Martens, Toxicol. Sci., 2008, 104, 155–162 CrossRef CAS PubMed.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846–853 CrossRef CAS PubMed.
- S. Quignard, G. Mosser, M. Boissière and T. Coradin, Biomaterials, 2012, 33, 4431–4442 CrossRef CAS PubMed.
- C. G. Gonzalez, G. S. Álvarez, D. E. Camporotondi, M. L. Foglia, C. Aimé, L. E. Diaz, T. Coradin and M. F. Desimone, Silicon, 2014 DOI:10.1007/s12633-12014-19203-12635.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- T.-J. Yoon, K. N. Yu, E. Kim, J. S. Kim, B. G. Kim, S.-H. Yun, B.-H. Sohn, M.-H. Cho, J.-K. Lee and S. B. Park, Small, 2006, 2, 209–215 CrossRef CAS PubMed.
- I. Al-Ogaidi, H. Gou, A. K. A. Al-kazaz, Z. P. Aguilar, A. K. Melconian, P. Zheng and N. Wu, Anal. Chim. Acta, 2014, 811, 76–80 CrossRef CAS PubMed.
- Y. Wang, S. Gao, W.-H. Ye, H. S. Yoon and Y.-Y. Yang, Nat. Mater., 2006, 5, 791–796 CrossRef CAS PubMed.
- G.-F. Luo, W.-H. Chen, Y. Liu, Q. Lei, R.-X. Zhuo and X.-Z. Zhang, Sci. Rep., 2014, 4, 6064 CrossRef CAS PubMed.
- H. U. Lee, C. Park and S. W. Kim, Process Biochem., 2012, 47, 1282–1286 CrossRef CAS.
- S. C. Larsen, S. E. Lehman, A. S. Morris, P. S. Mueller, A. Salem and V. Grassian, Environ. Sci.: Nano, 2016, 3, 56–66 RSC.
- A. A. Ansari and M. Alam, J. Lumin., 2015, 157, 257–263 CrossRef CAS.
- F. Jiang, Y. Fu, Y. Zhu, Z. Tang and P. Sheng, J. Alloys Compd., 2012, 543, 43–48 CrossRef CAS.
- J. Liu, C. Detrembleur, S. Mornet, C. Jérôme and E. Duguet, J. Mater. Chem. B, 2015, 3, 6117–6147 RSC.
- Q. Huo, J. Liu, L.-Q. Wang, Y. Jiang, T. N. Lambert and E. Fang, J. Am. Chem. Soc., 2006, 128, 6447–6453 CrossRef CAS PubMed.
- H. Ishii, T. Ikuno, A. Shimojima and T. Okubo, J. Colloid Interface Sci., 2015, 448, 57–64 CrossRef CAS PubMed.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- L. C. J. Thomassen, A. Aerts, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, D. Napierska, P. H. Hoet, C. E. A. Kirschhock and J. A. Martens, Langmuir, 2010, 26, 328–335 CrossRef CAS PubMed.
- C. Aimé, G. Mosser, G. Pembouong, L. Bouteiller and T. Coradin, Nanoscale, 2012, 4, 7127–7134 RSC.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244–6256 CrossRef CAS PubMed.
- R. G. Acres, A. V. Ellis, J. Alvino, C. E. Lenahan, D. A. Khodakov, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2012, 116, 6289–6297 CAS.
- W. H. Cover and S. C. Rittenberg, J. Bacteriol., 1984, 157, 391–397 CAS.
- H. Niu, S. Wang, T. Zeng, Y. Wang, X. Zhang, Z. Meng and Y. Cai, J. Mater. Chem., 2012, 22, 15644–15653 RSC.
- M. H. Marchena, M. Granada, A. V. Bordoni, M. Joselevich, H. Troiani, F. J. Williams and A. Wolosiuk, J. Solid State Chem., 2012, 187, 97–102 CrossRef CAS.
- A. van Blaaderen and A. Vrij, Langmuir, 1992, 8, 2921–2931 CrossRef CAS.
- T. Coradin, E. Mercey, L. Lisnard and J. Livage, Chem. Commun., 2001, 2496–2497 RSC.
- N. Isoherranen and S. Soback, Analyst, 2000, 125, 1573–1576 RSC.
- B. Prasad and S. Singh, J. Pharm. Biomed. Anal., 2010, 52, 377–383 CrossRef CAS PubMed.
- S. Sakai, T. Ono, H. Ijima and K. Kawakami, Biomaterials, 2001, 22, 2827–2834 CrossRef CAS PubMed.
- S. Li, X. Jiao and H. Yang, Langmuir, 2013, 29, 1228–1237 CrossRef CAS PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- L. Palanikumar, E. S. Choi, J. Y. Cheon, S. H. Joo and J. H. Ryu, Adv. Funct. Mater., 2015, 25, 957–965 CrossRef CAS.
- T. Chen, W. Wu, H. Xiao, Y. Chen, M. Chen and J. Li, ACS Macro Lett., 2016, 5, 55–58 CrossRef CAS.
- S. Gadde, MedChemComm, 2015, 6, 1916–1929 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM and SEM images of nanoparticles and MS data of antibiotics. See DOI: 10.1039/c6tb00281a |
| This journal is © The Royal Society of Chemistry 2016 |