
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of the direct effects of poly(dopamine) on the in vitro response of human osteoblastic cells
Alexander J.
Steeves†
a,
Aman
Atwal†
b,
Sarah C.
Schock
cd and
Fabio
Variola
*ade
aFaculty of Engineering, Department of Mechanical Engineering, University of Ottawa, Canada. E-mail: fabio.variola@uottawa.ca
bFaculty of Science, Department of Biopharmaceutical Sciences, University of Ottawa, Canada
cThe Children's Hospital of Eastern Ontario (CHEO) Research Institute, Canada
dFaculty of Medicine, Department of Cellular and Molecular Medicine, University of Ottawa, Canada
eFaculty of Science, Department of Physics, University of Ottawa, Canada
First published on 13th April 2016
Abstract
Functional polymeric coatings have rapidly become one of the most efficient strategies to endow biomaterials with enhanced bioactive properties. Among the bio-inspired polymers used for biomedical applications, mussel-derived poly(dopamine) (PDA) has increasingly attracted considerable interest because of its unique characteristics. In this work, we carried out detailed physicochemical characterization of a PDA film deposited on nanoporous titanium. In particular, we employed spectroscopic techniques (Raman and ATR-FTIR) and Digital Pulsed Force Mode Atomic Force microscopy (DPFM-AFM) to probe the chemical makeup and the nanomechanical properties of PDA-coated surfaces. In addition, we investigated protein adsorption by ATR-FTIR and quantified it with ten different serum proteins by Liquid Chromatography Mass spectroscopy (LC-MS), aiming at elucidating their potential contribution to the subsequent cell colonization. Successively, we assessed the response of MG-63 human osteoblastic cells to PDA-coated titanium both the multiple- and single-cell levels. Results for this study demonstrate that, compared to bare and nanoporous titanium, the PDA coating positively influences the adhesion and proliferation of MG-63 cells. In addition, we focus on how the three different substrates influence cell morphology (i.e. aspect ratio and form factor), the establishment of focal adhesions and the expression of RhoA, a protein involved in cell contractility. In conclusion, our work provides a deeper insight on the in vitro response of human osteoblastic cells to poly(dopamine) by closing in on specific aspects of cell–PDA interactions, ultimately reaffirming the potential of this bio-inspired polymer as a functional coating for bone tissue engineering applications.
Introduction
The modern history of biomedical research and regenerative medicine has been characterized by a concerted effort aimed at developing strategies to precisely control the interactions between biomaterials and the surrounding biological milieu to ultimately guide cellular events along predetermined pathways.1–3 The motivation that has been propelling such collective endeavour is the evidence that the physicochemical environment of surfaces at the micro- and nanoscale not only controls cell behaviour in vitro,4 but also influences the in vivo outcome of implanted devices.5,6 The knowledge of how cells sense and respond to surfaces has rapidly progressed in the last two decades, thanks also to the concomitant development of nanotechnology-based approaches which provided researchers with novel strategies to nanoengineer functional biomaterials,4 and increasingly more sophisticated toolboxes to evaluate the biological response at the molecular and cellular level (e.g. Atomic Force Microscopy).7 Methods currently used to nanostructure biomaterials are numerous, but they can all be classified into two main categories, used either independently or in combination: “top-down” techniques incorporate smaller details in a bulk material (e.g. nanotubular structures, organized nanopatterns),3,8 and “bottom-up” fabrication rearranges smaller components into a larger or more complex assembly with unique biomolecular functionalities (e.g. layer-by-layer assemblies).3Among the methods that permit to engineer the surface of synthetic biomaterials,4 the use of biopolymer coatings has yielded a remarkable enhancement of the bioactivity of biomaterials by providing advantageous physicochemical cueing to adhering cells.9,10 While these techniques will keep evolving to further enhance their in vivo and in vitro impact, the search is still on for valuable alternatives to provide increasingly more effective biological functionalities.
In the quest for the next generation of functional biomaterials, researchers have sought inspiration from nature by focusing on biological materials (e.g. collagen, keratin, chitin) and on approaches that mimic naturally occurring phenomena.11 In this context, biologically inspired adhesive interfaces have attracted much attention because of their potentially beneficial applications in medicine, technology and industry as intermediate linkers to immobilize bioactive agents onto surfaces.12 In particular, the investigation of the composition of adhesive proteins in mussels permitted to identify dopamine, a small molecule that contains functional groups (catechol and amine) believed to be crucial for achieving adhesive properties onto inorganic and organic materials.13,14 Multifunctional films resulting from the self-polymerization of dopamine have been successfully used to covalently graft proteins and peptides, as well as other bioactive agents onto a wide variety of surfaces.13,15–18 Poly(dopamine) (PDA) has also been exploited in tissue engineering applications as a bioactive coating for direct cueing to cells.19,20 Results from these studies showed that adherent PDA films facilitate the crystallization of hydroxyapatite and mineralization,21,22 and promote specific cellular functions, such as endothelial cell adhesion and growth.19,23–25 In addition, a poly(dopamine) coating showed to improve osteoblastic (MC3T3-E1 murine) cell adhesion and viability on non-wetting polymers (i.e. PE, PTFE, silicone and PDMS).26 This study showed that the enhancement of cell viability on PDA-coated samples was significant when using as controls surfaces which are highly resistant to cell adhesion. Conversely, when compared to substrates that support cell adhesion such as glass or titanium, poly(dopamine) coating impacted MC3T3-E1 cell viability to a lesser extent.19,26
Despite such previous fundamental work, the precise mechanisms that govern how cells respond to poly(dopamine) still need to be fully elucidated. In addition, the comparison with surfaces that not only support but also enhance cell functions (i.e. bioactive surfaces) will put the evaluation of cellular response on PDA coating to a more stringent test, ultimately assessing its potential to become a valid alternative for bone tissue engineering applications. To address these challenges, we closed in on the physicochemical characteristics of a PDA coating of titanium and carried out a comprehensive investigation of its in vitro bioactive effects on human MG-63 osteoblastic cells at both the multiple- (i.e. viability, expression of RhoA) and single-cell (i.e. spreading, morphology and focal adhesions) levels. Distinctively from previous studies, we employed Digital Pulse Force Mode Atomic Force microscopy (DPFM-AFM) to visualize the nanoscale surface morphology of the PDA film, one of the main parameters known to affect cell response,27 as well as its surface mechanical properties (i.e. stiffness), a factor that influences cell contractility, motility, and spreading.28 Successively, we employed Raman and Attenuated Total Reflectance Fourier-Transform Infrared (ATR-FTIR) spectroscopy to probe the chemical characteristics of the PDA coating. Infrared analysis was also used in combination with Liquid Chromatography Mass (LC-MS) spectroscopy to single out the potential contribution of preferential adsorption of serum proteins onto surfaces, a factor which may affect the subsequent cellular colonization.26,29 We successively evaluated the in vitro bioactivity of PDA-coated surfaces by investigating various aspects of the response of MG-63 osteosarcoma human osteogenic cells. In particular, we quantified cellular adhesion, proliferation and morphology, and quantitatively investigated the establishment of focal adhesions (FAs) and the expression of the RhoA, a protein which has been shown to have its activity modulated through mechanotransduction of cell adhesion molecules.30 Cellular results on PDA-coated samples were compared with both bare and nanoporous titanium31–33 (i.e. surfaces that, respectively, support and enhance osteoblastic cell adhesion and proliferation), thereby generating a more stringent context to investigate cellular behaviour on poly(dopamine).
Our results provide new evidences of the direct effects of poly(dopamine) on osteoblastic cell activity, ultimately casting new light on cell–PDA interactions and reaffirming the potential of such bio-inspired polymer as a functional bioactive coating for bone engineering applications.
Experimental
Sample preparation
Grade 2 titanium disks (12 mm in diameter and 1 mm in thickness) were mechanically polished with a 2-step process by the supplier (Firmetal Co., Ltd, Shanghai, China). Before treatment, disks were first cleaned in toluene in an ultrasonic bath for 20 min and successively rinsed in deionized water and ethanol. Nanoporous surfaces were generated by immersing titanium disks in a freshly prepared 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of H2SO4 (37N, Fisher Scientific)/H2O2 (30%, Fisher Scientific) at room temperature for 2 hours, according to a previously established protocol.34 Prior to the deposition of the poly(dopamine) coating, disks were thoroughly rinsed in distilled water to remove any acid remnants.34 This oxidative nanopatterning treatment was shown to increase surface area as well as the availability of surface binding sites,34 and therefore was chosen not only to provide a bioactive reference for the cellular assays but also to enhance the adhesion of the poly(dopamine) film onto the titanium substrate. Disks were successively coated with a poly(dopamine) film by immersion in a solution of 2 mg of 3-hydroxytyramine–HCl (Sigma Aldrich) dissolved in 10 mM Tris buffer (Sigma Aldrich) at pH 8.5 for 24 hours,35 ultimately creating three different experimental groups: mechanically polished (Ti), nanoporous (NPTi) and PDA-coated nanoporous (NPTi + PDA) titanium (Fig. 1).
:50 mixture of H2SO4 (37N, Fisher Scientific)/H2O2 (30%, Fisher Scientific) at room temperature for 2 hours, according to a previously established protocol.34 Prior to the deposition of the poly(dopamine) coating, disks were thoroughly rinsed in distilled water to remove any acid remnants.34 This oxidative nanopatterning treatment was shown to increase surface area as well as the availability of surface binding sites,34 and therefore was chosen not only to provide a bioactive reference for the cellular assays but also to enhance the adhesion of the poly(dopamine) film onto the titanium substrate. Disks were successively coated with a poly(dopamine) film by immersion in a solution of 2 mg of 3-hydroxytyramine–HCl (Sigma Aldrich) dissolved in 10 mM Tris buffer (Sigma Aldrich) at pH 8.5 for 24 hours,35 ultimately creating three different experimental groups: mechanically polished (Ti), nanoporous (NPTi) and PDA-coated nanoporous (NPTi + PDA) titanium (Fig. 1).
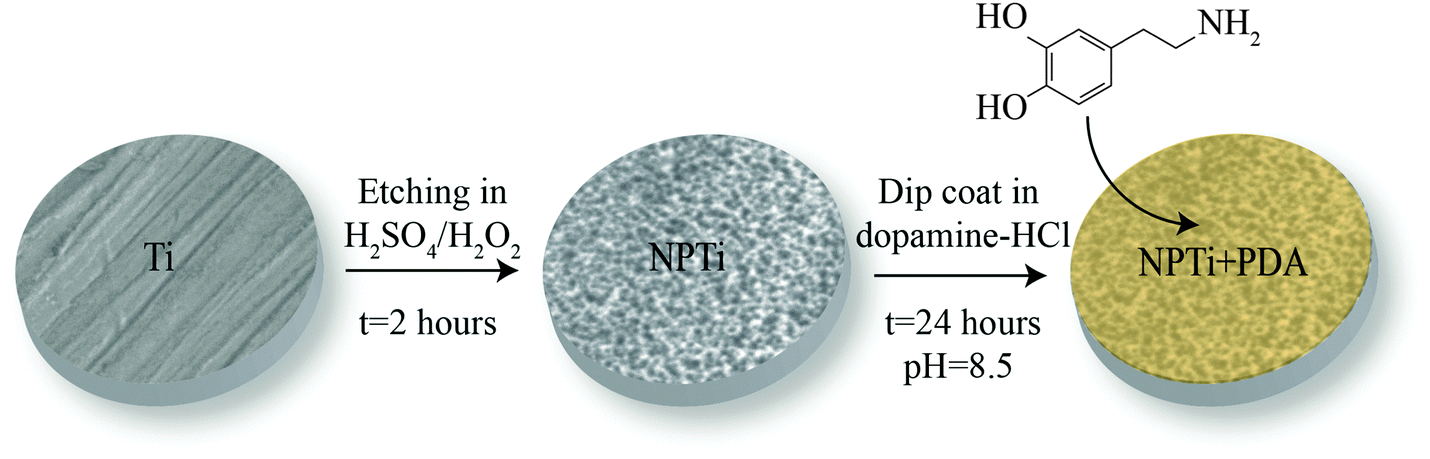 | ||
| Fig. 1 Sample preparation. Grade 2 titanium disks (Ti) were etched in a 50:50 mixture of H2SO4 and H2O2 (oxidative nanopatterning) to create nanoporous surfaces (NPTi).34 Treated disks were successively immersed in a solution of 2 mg of 3-hydroxytyramine–HCl dissolved in 10 mM Tris buffer at pH 8.5 for 24 hours to create the poly(dopamine) coating (NPTi + PDA). | ||
Scanning electron (SEM) and atomic force (AFM) microscopy
A JSM-7500F Field Emission Scanning Electron Microscope (FESEM, JEOL, Japan) was used to image treated samples in order to ensure the presence of the nanoporous structure on NPTi samples before the deposition of the poly(dopamine) coating and to visualize the nanoscale morphology of the PDA films.A WITec Alpha 300 integrated microscope (WITec, Germany) was used to collect AFM images by Digital Pulsed Force (DPFM) mode. This technique not only permitted to characterize the nanoscale surface morphology of the PDA films, but also to probe their nanomechanical properties, such as stiffness and Young's modulus. Images were obtained with a cantilever characterized by a tetrahedral tip (tip radius less than 10 nm) and a nominal spring constant of 0.2 N m−1 (Arrow™ FM Nanoworld, Switzerland). Each DPFM image contains 2.5 × 105 force–distance curves, which were converted into force [N] VS indentation depth [nm] according to a previously published procedure.36 From these data, stiffness values were calculated by measuring the slope of 40 randomly selected curves after indentation on different samples.37 In order to extract the Young's modulus and compare it to previous literature,38,39 these curves were successively fitted with the Hertz model.40
Raman and attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy
The WITec Alpha 300 was also used to map out the chemical composition of samples and ensure the homogeneity of the PDA film across the nanoporous surface. High resolution (150 points × 150 lines) 20 × 20 μm Raman maps were collected in randomly selected areas of three samples/condition by using a 100× objective and a 524 nm frequency doubled Nd:YAG laser (power of 9.5 mW at the focal plane, acquisition time of 0.1 seconds) as excitation source. Raman data were processed by OriginPro (OriginLab corporation, United States) and WITec Project 4 PLUS. The assignment of Raman bands in the 250–3000 cm−1 interval was carried according to previous literature.41,42Infrared analysis was carried out by exploiting a Bruker Tensor 37 FTIR spectrometer equipped with (RT) DLaTGS detector (Bruker, USA) and a platinum ATR accessory with a 2 mm diameter single reflection diamond crystal. Three samples per conditions were analyzed, and three spectra were collected in randomly selected areas of each sample to ensure statistical significance. Spectroscopic information was collected in the 1100–1800 cm−1 range, with a 4 cm−1 resolution and 256 acquisitions per spectrum, using a Ti sample for reference. The choice of the spectral range was based on the data interpretation and infrared peak assignment found in previous literature.43–46 Infrared data were processed by using the OriginPro software. Spectra were smoothed with a Savitzky–Golay filter after linear baseline subtraction and fitted with Gaussian functions to resolve secondary vibrational components. Infrared analysis was also used to investigate the adsorption of serum proteins onto the three surfaces.
Serum proteins adsorption
Ti, NPTi and NPTi + PDA disks (three samples per condition) were immersed in the complete medium used for cell cultures, a solution of α-MEM (GIBCO, USA) and 10% Fetal Bovine Serum (FBS) (GIBCO, USA). After 6 hours and 3 days, samples were removed from the solution, rinsed thoroughly in deionized water, air-dried and analyzed by ATR-FTIR, using dry Ti, NPTi and NPTi + PDA disks, respectively, as reference in order to highlight the contribution of adsorbed species. Three spectra were collected in randomly selected areas of each sample submersed in the α-MEM + 10% FBS solution.In-solution digestion
In parallel, four samples per condition were immersed in the complete medium for LC-MS spectroscopy. After 6 hours, samples were removed from the solution, rinsed thoroughly in deionized water and placed in a sterile 24-well plate. 200 μl of 8 M urea (UltraPure™, Invitrogen, USA) were added in each well and incubated for 15 min with moderate shaking. Successively, 200 μl of DTT (dithiothreitol, electrophoresis grade, Fisher Scientific, USA) in 100 mM ABC (Ammonium Bicarbonate, BioUltra, from Fluka, USA) were added into each well to bring the final concentration of DTT at 10 mM, and incubated at room temperature for 1 hour. 200 μl of IA (iodoacetamide, GE Healthcare, UK) in 100 mM ABC were then added into each well to make the final concentration of IA at 55 mM, and incubated at room temperature in the dark for 1 hour. The alkylation reaction was quenched with 5 mM dithiothreitol in 100 mM ABC solution.200 μl of MS grade trypsin (Pierce, USA)/100 mM ABC solution with concentration of 1 ng μl−1 were added into each well and incubated at 37 °C overnight. Digestion was stopped by adding 5 μl of 5% formic acid (Optima LC/MS, Fisher Scientific, USA) into each well. The content of each well wan transferred into a 1.5 mL Eppendorf tube, and the volume of liquid in each vile was reduced to 20 mL by using the SpeedVac. Five samples per condition were combined into 1 tube. Experiments were carried out in quadruplicates to ensure statistical significance. Samples were finally cleaned by 10 μl C18 tips according to manufacturer's protocol (Pierce, USA), lyophilized with SpeedVac and re-suspended in 10 μl 0.1% formic acid for LCMS/MS analysis.
Liquid chromatography mass spectroscopy (LC-MS)
LC-MS measurements were carried out by using an Orbitrap Fusion mass spectrometer (Thermo Scientific, USA) equipped with an Ultimate 3000 UHPLC (Thermo Scientific, USA), a PepSwift monolithic pre-concentration column (Thermo Scientific, USA) and an Acclaim PepMap C18 analytical column (Thermo Scientific, USA).The mobile phase for HPLC separation was composed of 2 components: buffer A and B. Buffer A was 0.1% formic acid in water and buffer B was composed of 0.1% formic acid, 90% acetonitrile and 9.9% water. 5 μl of sample was loaded onto the pre-concentration column with the loading pump flow rate set at 3 μl min−1. The nano pump flow rate was set at 0.3 μl min−1. Mobile phase B started at 2% at the beginning of the run and increased to 4% at 7 min. Then it increased to 13.5% at 32 min, 24.5% at 60 min, 33.5% at 70 min, followed by 5 min wash at 80% B. The column was re-equilibrated for 20 min at 2% B.
The peptides eluted from the analytical column were ionized by electrospray by a Thermo Scientific Nanospray Flex ion source and analyzed by the Thermo Orbitrap Fusion mass spectrometer. The full MS scan was performed by the Orbitrap and the resolution was set at 60 K and AGC target set at 2 × 105 with maximum injection time 50 ms. The MS/MS were done in the ion trap with the following filters in effect: (1) monoisotopic precursor selection was turned on; (2) intensity threshold was set at 5 × 103; (3) charge states 2–6; (4) dynamic exclusion 35 s. The MS/MS was performed at data dependent mode under “Top Speed” setting, meaning the instrument will maximize the MS/MS scans based on the cycle time and the frequency of the full scans. Peptide fragmentation activation mode was CID and the collision energy was set at 35%. The AGC target was set at 1 × 104 and maximum injection time was 35 ms.
Cell cultures
MG-63 human osteosarcoma (ATCC, USA) cells were grown in Dulbecco's Modified Eagle Medium (DMEM, GIBCO) containing 10% fetal bovine serum (FBS), 20 U mL−1 penicillin and 20 U mL−1 streptomycin (ThermoFisher Scientific, USA) in a 5% CO2 incubator at 37 °C. Following trypsinization with 0.25% trypsin–EDTA (ThermoFisher Scientific, USA), cells were washed by centrifugation at 190g for 8 min to give a pellet that was resuspended in 10 mL of fresh supplemented medium to a concentration of 2 × 104 cells per mL. Disks were sterilized in ethanol prior to use in cell culture experiments. They were placed in 24-well plates and 500 μl of the cell suspension was added to each well. The plated cells were cultured for 1, 4 and 8 h for cell spreading and adhesion, and 1, 2, and 3 days for viability experiments. Each experiment was performed in triplicate with at least three samples/condition.Immunofluorescence microscopy
The medium was removed and the samples were washed three times in phosphate-buffered saline (PBS, GIBCO). Cells were fixed by adding 600 μl per well of 4%-paraformaldehyde (pH 7.2) for 10 min at room temperature. They were subsequently permeabilized in 0.5% Triton X-100 and blocked in 5% normal goat serum. Nuclei were visualized with NucBlue® Fixed Cell ReadyProbes® Reagent (ThermoFisher Scientific, USA) while Actin was labelled by Alexa Fluor® 555 conjugated with Phalloidin (ThermoFisher Scientific, USA). Samples were then mounted on #1.5 coverslides using ProLong Diamond Antifade Hardset Mountant (ThermoFisher Scientific, USA).For viability and morphological assessment, cells were imaged using an AxioObserver.D1 inverted microscope (Carl Zeiss, Germany) and driven by AxioVision 4.8 (Carl Zeiss, Germany). For viability assessment, a total of 16 images per disc were captured in a 4 × 4 grid pattern across the disc using a 10× A-Plan 0.24 Ph1 objective (Carl Zeiss, Germany). For cell spreading, aspect ratio and form factor assessment, images were taken with a 20× Plan-Apochromat 0.8 and 40× Plan-NeoFluar 1.3 Oil Ph3 objectives (Carl Zeiss, Germany) as 12 images per disc in a 4 × 3 grid.
For visualization of focal adhesions, samples were further immunolabeled with 1:400 primary mouse monoclonal anti-Vinculin, clone hVIN-1 (Sigma Aldrich, USA) and 1:400 secondary goat anti-mouse IgG (H + L) Alexa Fluor® 488 conjugate (ThermoFisher Scientific, USA) for one hour each in blocking buffer. Images were captured on a Leica BMI16000B inverted microscope (Leica Microsystems, Germany), configured with a Quorum Spinning-disk Confocal (Quorum Technologies, Canada), using a 40× PL APO 1.3 Oil objective, driven by MetaMorph Software (Molecular Devices, USA).
Image analysis
Images for nuclei count, cell–substrate contact, aspect ratio and form factor were processed by using custom pipelines in CellProfiler (Broad Institute).47 Digital area and aspect ratio (i.e. minimum diameter/maximum diameter) are indicators of cellular spreading and elongation along a preferential direction, respectively. Form factor (calculated as 4π × area/perimeter2)48,49 is associated to the regularity of cell shape: a form factor of 1 describes a perfectly round cell, while lower values represent a polygonal and more elongated cell.48 In this context, form factor has been indicated to be representative of jagged cellular edge, such as during membrane ruffling.50Images processed for focal adhesion assessment were captured in a z-series on a motorized stage at 0.2 μm steps. They were then focus stacked using an algorithm included in the MetaMorph Software (Molecular Devices, USA). Quantification was achieved using a custom macro in ImageJ that performed background subtraction, thresholding and particle analysis.
Reverse transcription and polymerase chain reaction (PCR) assay
After MG-63 cells were incubated on Ti, NPTi and NPTi + PDA for 24 hours, 1 mL of Trizol Reagent (Gibco, USA) was added and the solution was transferred to microcentrifuge tubes. After the addition of chloroform, samples were centrifuged (12000g, 10 min, 4 °C). The aqueous phase was then mixed with isopropyl alcohol, incubated at room temperature for 10 minutes and centrifuged again. The pellet was dissolved in DNAse/RNAse free water and the protein concentration was measured. Total RNA (2 μg) was subjected to RT for 60 min at 42 °C using SuperScript™ II (200 units) and First Strand Buffer (Invitrogen, USA), in the presence of 0.5 μg μl−1 oligo dT, 10 mM DTT and 1 mM dNTP mix. Amplifications of cDNA (1 μl) obtained from RT were then performed in ThermoPol Buffer (NEB, USA), containing 1 mM dNTP mix, in the presence of 10 μM of each specific primer and 2.5 units of Taq DNA polymerase (NEB, USA). Amplifications were performed in an Eppendorf thermocycler (Brinkmann, USA). Each sample was run in triplicate. cDNA was also amplified with RhoA primers (sense 5′-CCA GTT CCC AGA GGT GTA TGT-3′; antisense 5′-CCT GTT TGC CAT ATC TCT GCC-3′) and GAPDH primers (sense 5′-CAT GGC CTT CCG TGT TCC TAC CC-3′; antisense 5′-CCT CGG CCG CCT GCT TAC-3′) as a positive control. Ten microliter aliquots of the PCR products were size-separated by electrophoresis on a 1–2% agarose gel. Experiments were repeated in triplicates.
Results and discussion
In agreement with previous work, while polished controls did not exhibit any reproducible features at the nanoscale, nanoporous surfaces showed the characteristic 3-dimensional network of nanometric pits (image not shown, the reader can refer to ref. 34, 51 and 52 for details). In the case of PDA-coated nanoporous titanium, SEM and AFM analysis revealed the distinctive granular appearance that typifies deposited films of PDA (Fig. 2A and B).46 High resolution (5 × 5 μm2) AFM imaging and line scans (Fig. 2C) permitted to close in on the 3-dimensional structure of the granular features, which resulted to be 95 ± 11 nm in width and 22 ± 6 nm in height. The average dimension of these elements is significantly larger than that previously reported for PDA films deposited via dip coating process on gold (in the 25–30 nm range in diameter).46 However, the granular features we report are similar in size to those resulting from a multiple dip cycles deposition (>100 nm).53 This may be ascribed to the longer deposition used for this study (24 hours), a factor that could have permitted the growth of the granular features with time.46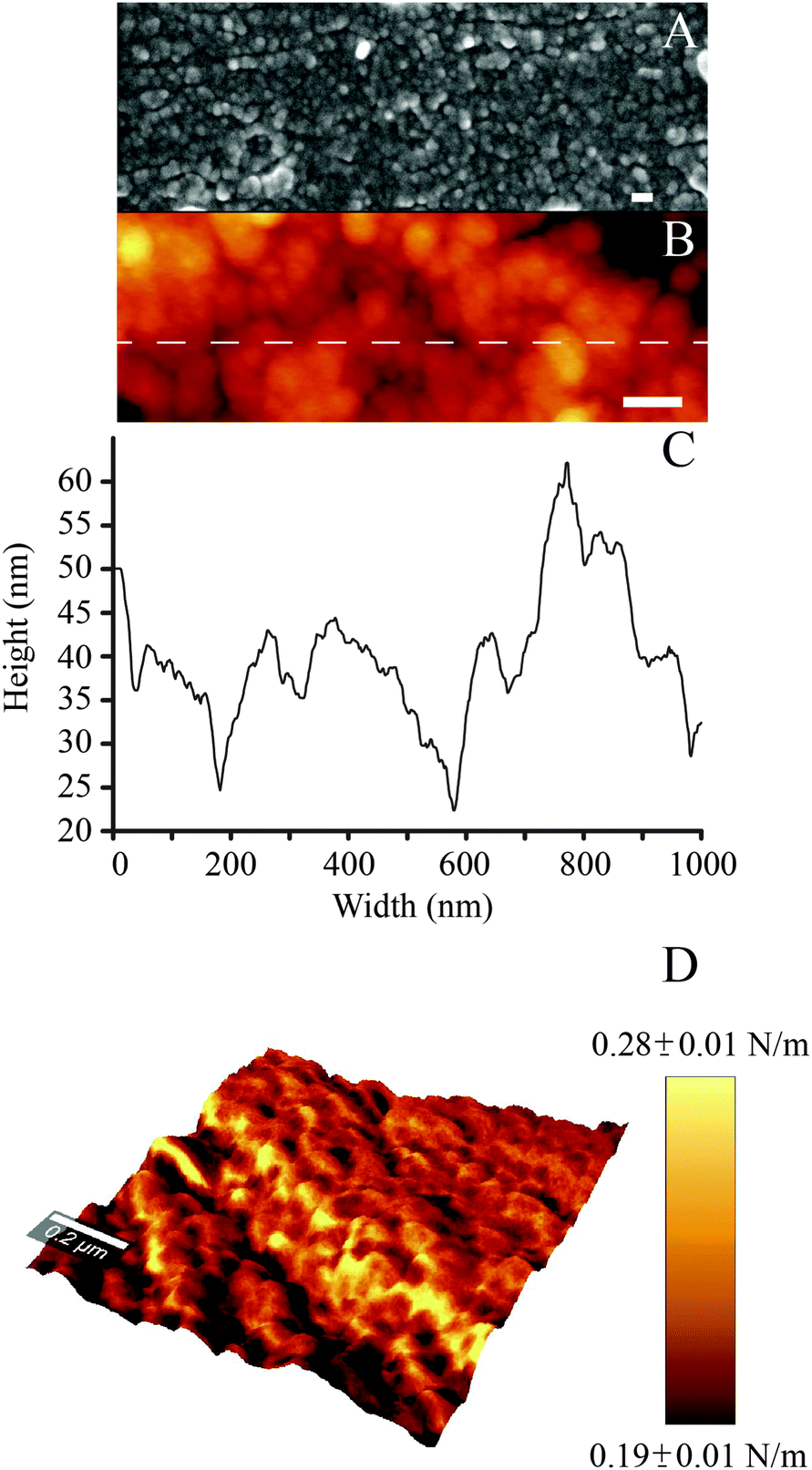 | ||
| Fig. 2 (A) SEM and (B) AFM images of the PDA coating (scale bar = 100 nm); (C) AFM line scan; (D) DPFM-AFM image showing the stiffness + topography overlay for the PDA coating. | ||
By exploiting the DPFM capabilities of the AFM, we generated stiffness maps by assigning a color code to the cantilever's deflection when indenting the sample's surface (Fig. 2D). The average stiffness of the poly(dopamine) coating resulted to be 0.24 ± 0.06 N m−1. This value is similar to the stiffness of rubbery polymeric films, such as styrene–butadiene copolymers (0.23 N m−1).54 DPFM data also permitted to quantify the Young's modulus. Every point of a DPFM map can in fact be converted into a force–distance curve from which the value of the elastic modulus can be extracted. In particular, the PDA coating exhibited an average Young's modulus of 92 ± 17 MPa. The comparison of our results with the existing literature was not unequivocal because of the variability in the published results. For example, a previous study which employed the AFM to determine the elastic modulus of poly(dopamine) films deposited on a gold substrate reported a value of 12.1 ± 1.6 MPa.38 Conversely, a PDA coating of mica was shown to exhibit an elastic modulus of 870 MPa.39 The inconsistencies between our results and previous literature may originate from different assumptions used in the contact mechanics models, the adhesion strength between the substrate and the polymeric layer,55 as well as from the different parameters used to deposit the PDA coating (which, in turn, may affect its physical properties), all factors which may have influenced the AFM-based nanoindentation measurements.
In order to probe the physicochemical characteristics of the PDA coating and its ability to retain serum proteins, we exploited Raman (Fig. 3) and infrared (Fig. 4) spectroscopy.
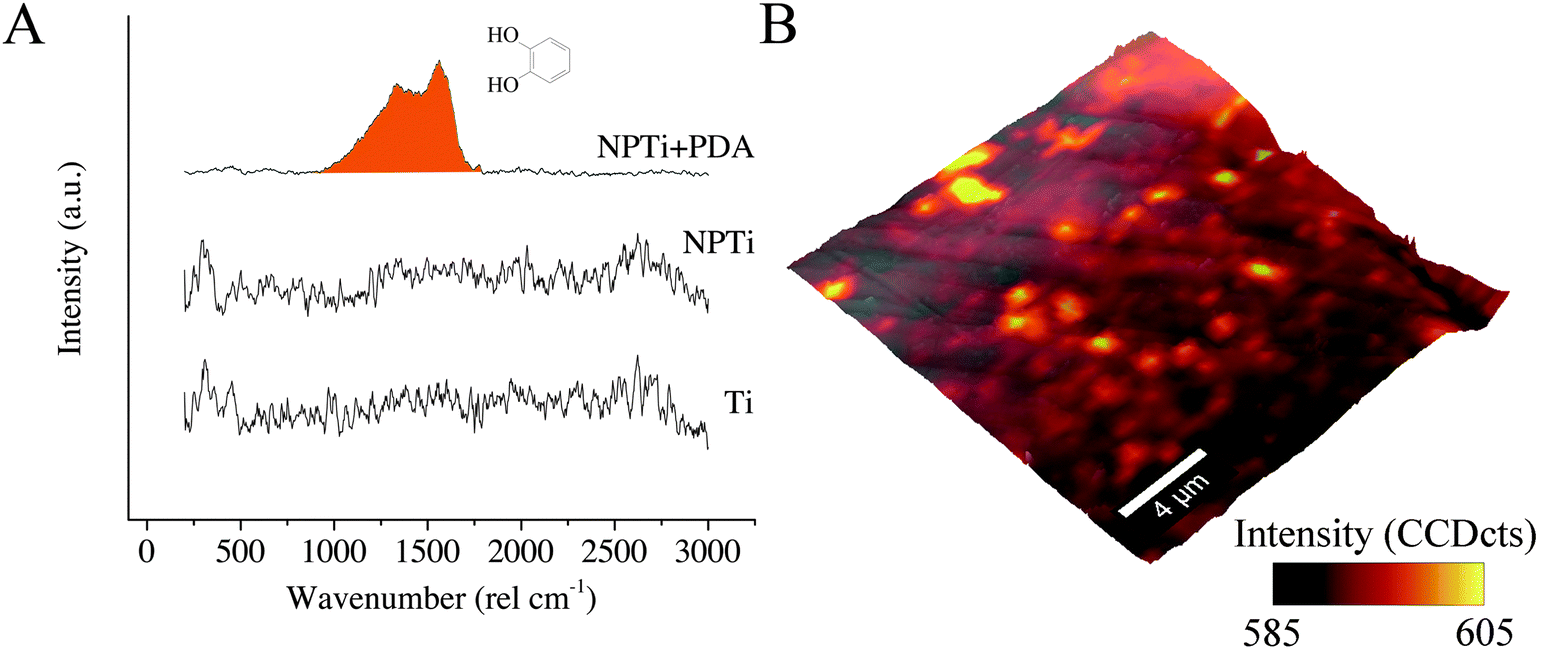 | ||
Fig. 3 (A) Representative Raman spectrum of Ti, NPTi and NPTi + PDA; (B) representative Raman + AFM overlay showing the intensity distribution of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O catechol stretching vibrations band for NPTi + PDA samples. O catechol stretching vibrations band for NPTi + PDA samples. | ||
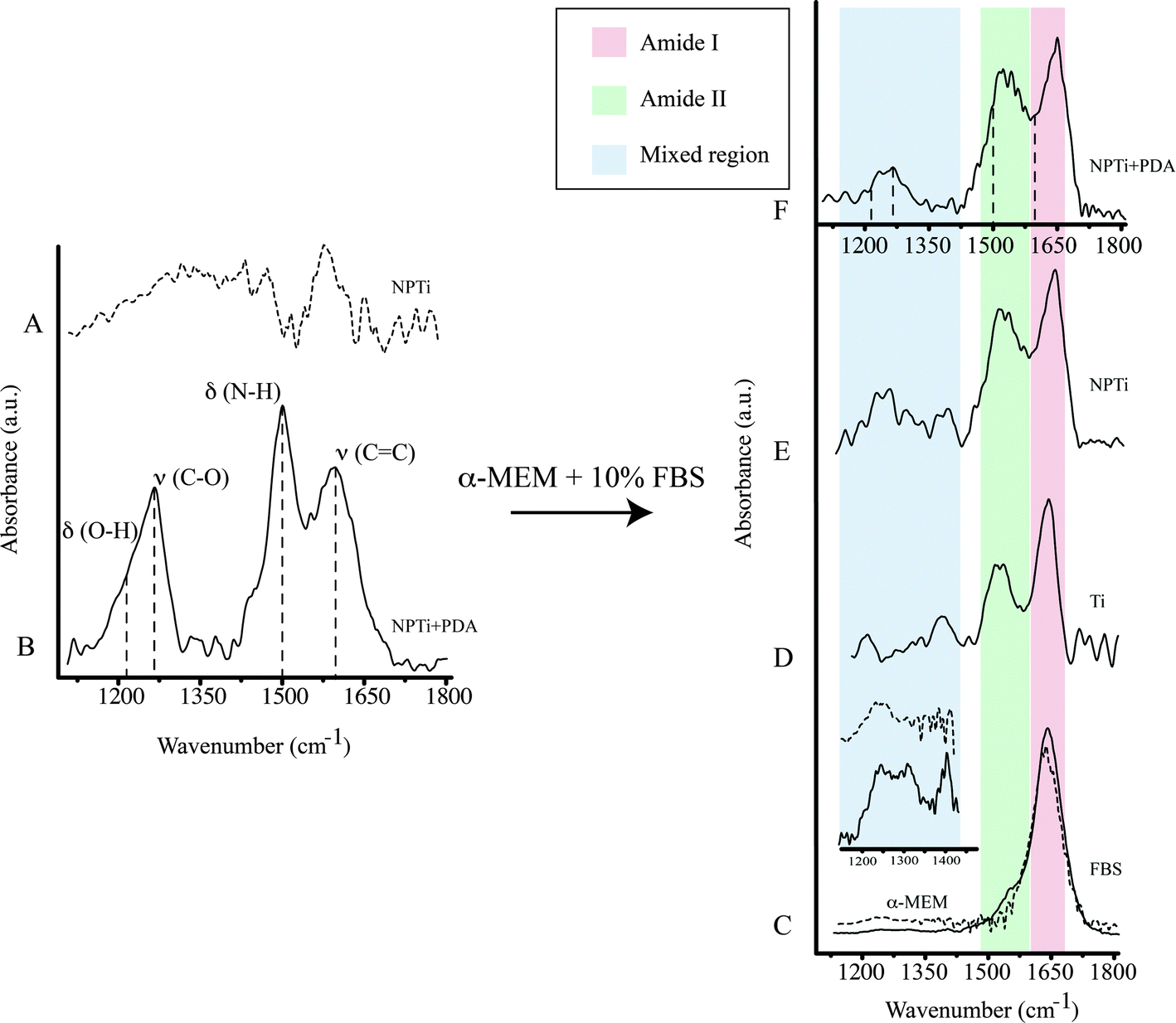 | ||
| Fig. 4 Representative ATR-FTIR spectrum of (A) NPTi, (B) NPTi + PDA, (C) 10% FBS (solid line) and α-MEM (dashed line). Enhancement of the mixed region in 10% FBS (solid line) and α-MEM (dashed line) (inset). Representative ATR-FTIR spectrum of (D) Ti, (E) NPTi and (F) NPTi + PDA (dashed lines show the center maxima of PDA bands) after immersion in α-MEM + 10% FBS for 6 hours. | ||
In the case of Raman analysis, while the Ti and NPTi disks did not show any significant band in the 250–3000 cm−1 range (indicative of a mainly amorphous outer TiO2 layer),34,52 spectra collected on NPTi + PDA samples were characterized by two peaks at 1350 and 1580 cm−1, attributed to CO catechol stretching vibrations (Fig. 3A).41,42 The Raman signature of the dopamine molecule was successively used to map out the chemical composition of NPTi + PDA samples to ensure that the deposited films were homogeneously distributed on the surface of the disks. Fig. 3B displays a representative Raman map (superimposed to the corresponding AFM topographical image) which demonstrates the presence of a continuous PDA surface film. However, localized variations in the band intensity were detected, and although minimal (Fig. 3B, intensity scale), they may nonetheless suggest the presence of distinct domains in the PDA film in the 600–2000 nm range in diameter, therefore larger than the individual granular features detected by AFM. Because of the different spatial resolution of AFM (∼1 nm)7 and Raman imaging (diffraction-limited spatial resolution of about 300 nm for a 524 nm laser with a 100× objective),56 we were not able to unequivocally correlate these domains to the surface distribution of the granular features resolved by the AFM. However, we can infer that such contrast in Raman images may derive from localized variations in the film's thickness, most likely resulting from microtopographical alterations (e.g. polishing marks) of the underlying titanium surface and/or from the deposition of PDA aggregates from solution.57
A previous study determined that FTIR spectroscopy of native (Ti) and modified (NPTi) titanium permits to probe the characteristics (i.e. thickness and crystallinity) of the surface oxide layer by considering the characteristics IR bands in the 600–1000 cm−1 interval which result from the Ti–O stretching vibrations.58 At larger wavenumbers, Ti (data not shown, the reader can refer to ref. 34) and NPTi samples did not display distinctive absorption bands (Fig. 4A). Conversely, NPTi + PDA disks revealed the characteristic infrared signature of poly(dopamine), featuring absorption bands at 1210 cm−1 (O–H bending in the catechol group), 1260 cm−1 (C–O stretching in the catechol group), 1510 cm−1 (N–H scissoring) and 1600 cm−1 (CC stretching in the aromatic ring) (Fig. 4B).43–46 Together with the Raman analysis, these results confirm that the PDA film was successfully deposited across the surface of nanoporous titanium samples.
ATR-FTIR was successively employed to investigate protein adsorption on surfaces. Cellular processes on surfaces are believed to be affected by an adlayer of proteins adsorbed from bodily fluids prior to cell colonization.59–62 Therefore, investigating the distinctive capacity of surfaces to retain proteins may contribute to understand the differential cellular response observed on the three conditions tested. In order to reproduce the in vitro conditions of cell cultures, we immersed Ti, NPTi and NPTi + PDA samples in a solution of 10% FBS in α-MEM for 6 hours and 3 days, thereby considering a large number of proteins at once without any biological bias. Noteworthy, although the selected intervals considerably exceed the time required for proteins to adsorb (in a timeframe of seconds to minutes),61,63 such a choice permitted us to obtain a snapshot of the surface adlayer presented to cells between the adhesion and the growth phase.
The infrared spectrum of α-MEM (Fig. 4C, dotted line) and FBS (Fig. 4C, solid line) as well as that of Ti (Fig. 4D), NPTi (Fig. 4E) and NPTi + PDA (Fig. 4F) samples after 6 hours of immersion in α-MEM + 10% FBS are shown in Fig. 4. Infrared analysis of α-MEM revealed a main absorption band at 1640 cm−1 (H–O–H bending in water)64 and minor peaks in the “mixed region” (1450–1220 cm−1) (Fig. 4D inset, dotted line) which includes contributions from proteins, amino acids and glucose in the medium.65 Conversely, FBS displayed two main absorption bands at 1650 cm−1 (amide I) and at 1550 cm−1 (amide II),64 associated to the secondary structure of proteins,66 as well as other minor peaks in the mixed region, most likely associated to additional vibrational modes of serum proteins (Fig. 4D inset, solid line).65
After immersion for 6 hours and thorough rinse in deionized water, all samples displayed the amide I and II bands (Fig. 4D–F, red and green shadowing) as well as bands in the mixed region (Fig. 4D–F, blue shadowing). In this case, the precise assignment of the bands in the mixed region was not possible since they can derive from constituents of both the medium and the FBS. In the case of NPTi + PDA samples, the amide I and II bands overlaid with those of poly(dopamine), represented with dotted vertical lines in Fig. 4F. As one could expect, the same result was also observed after immersion for a longer interval (i.e. 3 days).
Taken together, our infrared data allow us to infer that proteins are retained by the three surfaces, regardless of their physicochemical characteristics. The amide I and II bands, evidence of the presence of adsorbed proteins, were in fact detected on Ti, NPTi and NPTi + PDA samples after exposure to complete medium for 6 hours. Therefore, we can conclude that during the growth phase, cells adhered and proliferated on surfaces all capable of retaining a protein adlayer. However, because of the multi-component nature of the system under investigation, we cannot draw definite conclusions about the precise identity, amount, conformational characteristics and/or surface-specific affinity of the adsorbed proteins. We can nonetheless hypothesize that the retention mechanisms most likely vary from one surface to the other, encompassing a combination of specific and non-specific interactions as well as physical entrapment effects exerted by the nanometric pores in the case of NPTi samples.51
To provide a better insight on whether protein adsorption was affected by a differential affinity with surfaces, we carried out liquid chromatography mass spectroscopic (LC-MS) analysis. Fig. 5 shows the unique spectral count for the 10 most abundant proteins retrieved from NPTi and NPTi + PDA samples after immersion in complete medium, normalized against the counts obtained on bare titanium. It can be readily observed that Ti, NPTi and NPTi + PDA samples are characterized by similar counts for 8 different proteins, suggesting that their adsorption occurs irrespective of the surface treatment. Only two proteins among the ones we tested (i.e. #5 and #10) showed a statistically significant difference when compared to bare titanium. Noteworthy, despite these differences, NPTi and NPTi + PDA samples exhibited similar values. In this context, it should be noted that the same trend was obtained by considering the total ion current (TIC) values (data not shown).
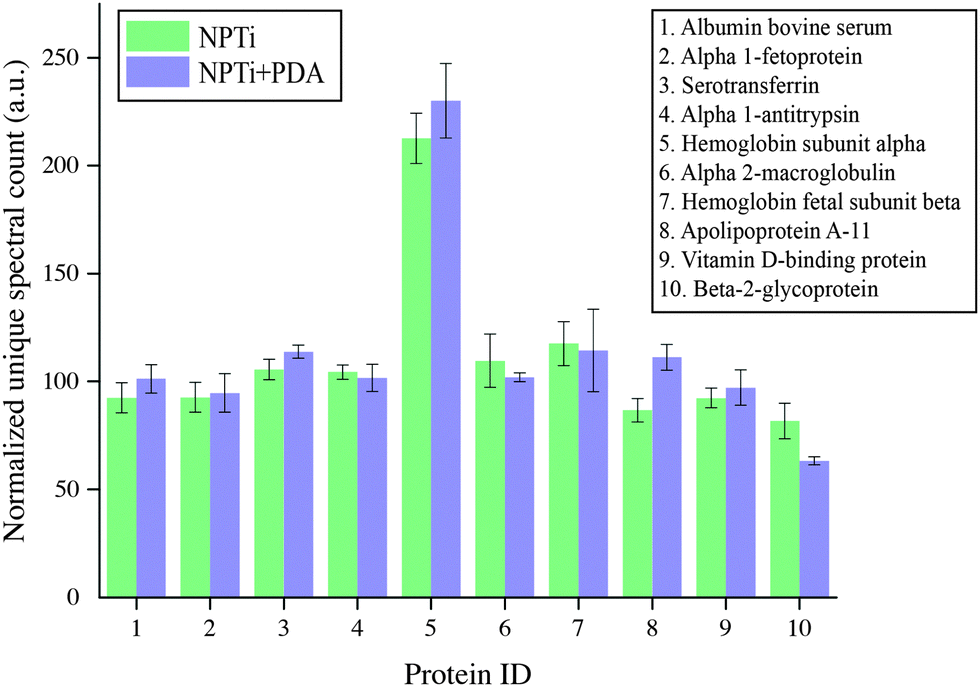 | ||
| Fig. 5 Liquid chromatography mass spectroscopic (LC-MS) analysis of serum proteins adsorbed on NPTi and NPTi + PDA surfaces. Results show the unique spectral count normalized against the values determined on bare titanium controls. | ||
Taken together, the FT-IR and LC-MS results indicate that the PDA coating does not affect the amount of adsorbed serum proteins when compared to nanoporous surfaces as well as bare titanium. Noteworthy, a potential substrate-induced protein denaturation could still occur on surfaces and affect cell adhesion and proliferation, an aspect that, however, we could not address by these techniques.
In order to assess the biological response to PDA-coated titanium, we carried out cell culture studies with MG-63 human osteosarcoma cells. The choice of this cell line was motivated by the fact that among osteoblastic cell models used for in vitro studies (e.g. MC3T3-E1, Saos-2), MG-63 may provide a better alternative for studies interested in initial attachment to various materials.67 This cell line also manifests a behavior closer to that of human osteoblasts while still providing the beneficial aspects of a cell line, such as the ease of maintenance, unlimited number of cells without the need for isolation.67 In addition, MG-63 cells permit to avoid the interspecies and genomic differences that may occur with MC3T3-E1 cells, a factor which can make extrapolation to human clinical disease and treatment outcomes challenging.67
In this study, we first evaluated MG-63 cell viability at short (at 1, 4 and 8 hours) and longer (1, 2 and 3 days) intervals to validate the beneficial effects of PDA on cell adhesion and proliferation previously reported with different cell lines. Fig. 6 displays the relative variations in cell number determined by employing the CellProfiler software68 to count DAPI-stained nuclei on the three surfaces. Our results demonstrate that in the shorter interval (1–4 hours), cell number did not significantly vary in relation to the substrate. While significant cell growth is not expected to occur in such relatively short interval, we can however consider this finding an indicator of a consistent seeding efficiency across different samples. Therefore, any difference in cell number that may result at longer intervals will be most likely associated to a substrate-dependent differential behavior during the late adhesion/proliferation phase. Noteworthy, from 8 hours onwards, proliferation of cells grown on NPTi + PDA samples experienced a statistical significant increase (p < 0.05) of 15–25% in respect to Ti samples, an evidence of the bioactive effects exerted by poly(dopamine). This result demonstrates that cells grown on the coating still maintain a higher viability at 72 hours when compared to uncoated controls, unlike what was previously reported for PDA-coated silicon oxide chips.57
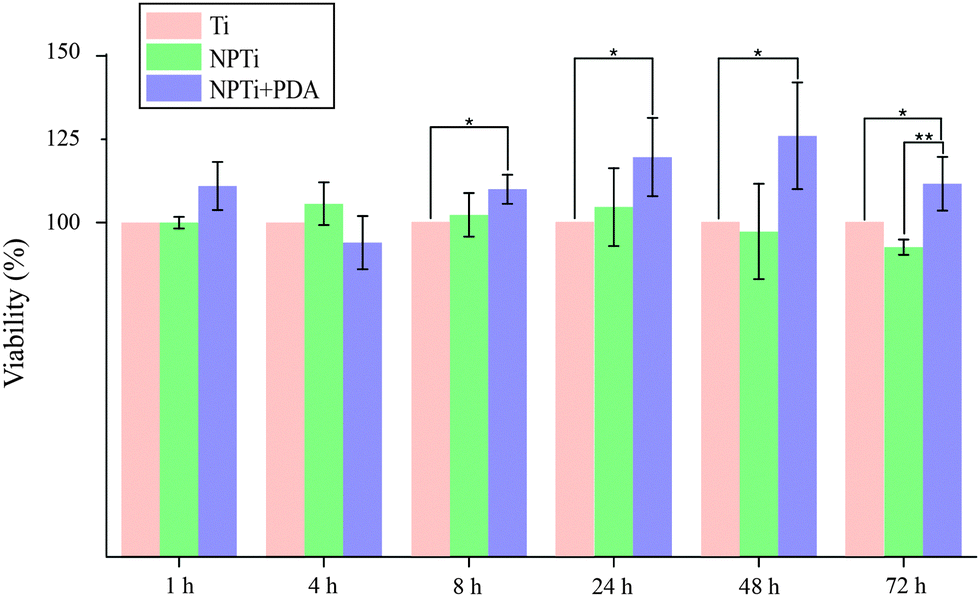 | ||
| Fig. 6 Nuclei counting at different intervals (ANOVA test: * = p < 0.05, ** = p < 0.01). Results are expressed as percentage increase/decrease in respect to bare titanium (Ti). | ||
However, when compared to nanoporous titanium, a marked increase in cell number was only detected at 3 days. It should be noted that nanoporous surfaces generated by oxidative nanopatterning inherently posses bioactive capacities, in particular the ability to enhance the proliferation of UMR-106 and primary calvaria-derived osteoblastic cells.31–33 Therefore, while the PDA coating offers a distinctive advantage over bare titanium in terms of cell proliferation from early intervals onwards, its bioactive effects are similar to that of the bioactive nanoporous surface, and only become more evident in the longer term. Interestingly, our ATR-FTIR and LC-MS results demonstrate that despite the differential cellular behavior, serum proteins were equally adsorbed onto the three surfaces, a factor which was expected to yield similar biological outcome. The differential behavior in cellular colonization reported herein could be attributed to a denaturation of proteins adsorbed onto Ti and NPTi samples26 and/or their nanoscale distribution.69
In order to close in on the effects exerted by poly(dopamine) at the single-cell level, we quantified substrate-dependent variations in the cellular morphology in terms of area (spreading), aspect ratio and form factor.70,71 From the representative fluorescence images of cells shown in Fig. 7A, it readily transpires that the morphology of cells adhering onto the poly(dopamine) after 1 hour of culture is distinctive, exhibiting a larger and more elongated structure with membrane projections. At 4 hours, cells on bare and nanoporous surfaces begin to assume a similar morphology, until it is no longer possible to qualitatively discern evident morphological differences at 8 hours. To further our investigation of cell morphology, we carried out image analysis to quantify parameters such as area, aspect ratio and form factor. Fig. 7B shows that the surface area of cells in contact with the PDA layer is significantly larger (20–25%) when compared to that of cells adhering on both Ti and NPTi samples, demonstrating an enhanced cellular spreading at early intervals. The aspect ratio and the form factor were also affected by the substrate. In fact, the boxplots in Fig. 7C confirm the qualitative observations: a more elongated shape characterizes cells on the PDA coating at 1 hour, but after 4–8 hours such morphology becomes predominant also in cells adhering on Ti and NPTi samples. Fig. 7D provides information about the complexity of cellular shape by comparing the form factor, an indicator of a jagged edge. Cells on the PDA coating increasingly assume a more complex geometry as a function of time, which could be related to a membrane ruffling and/or the emission of membrane protrusions. The same trend was exhibited by cells adhering on the other two substrates, but their morphological complexity consistently resulted less pronounced than that of cells on poly(dopamine). A different trend was observed at later intervals. Cell area was greater on PDA-coated samples until 3 days of culture, when compared to that of cells proliferating on Ti and NP-Ti samples (Fig. 8A and B). However, both the aspect ratio (Fig. 8C) and the form factor (Fig. 8D) were similar on the three surfaces, indicating that an elongated and well-spread morphology was achieved regardless of the substrate. Noteworthy, the form factor at 3 days for cells adhering onto NPTi and NPTi + PDA samples decreased, suggesting the onset of cellular events (e.g. differentiation) that could contribute to modify their membrane edge.
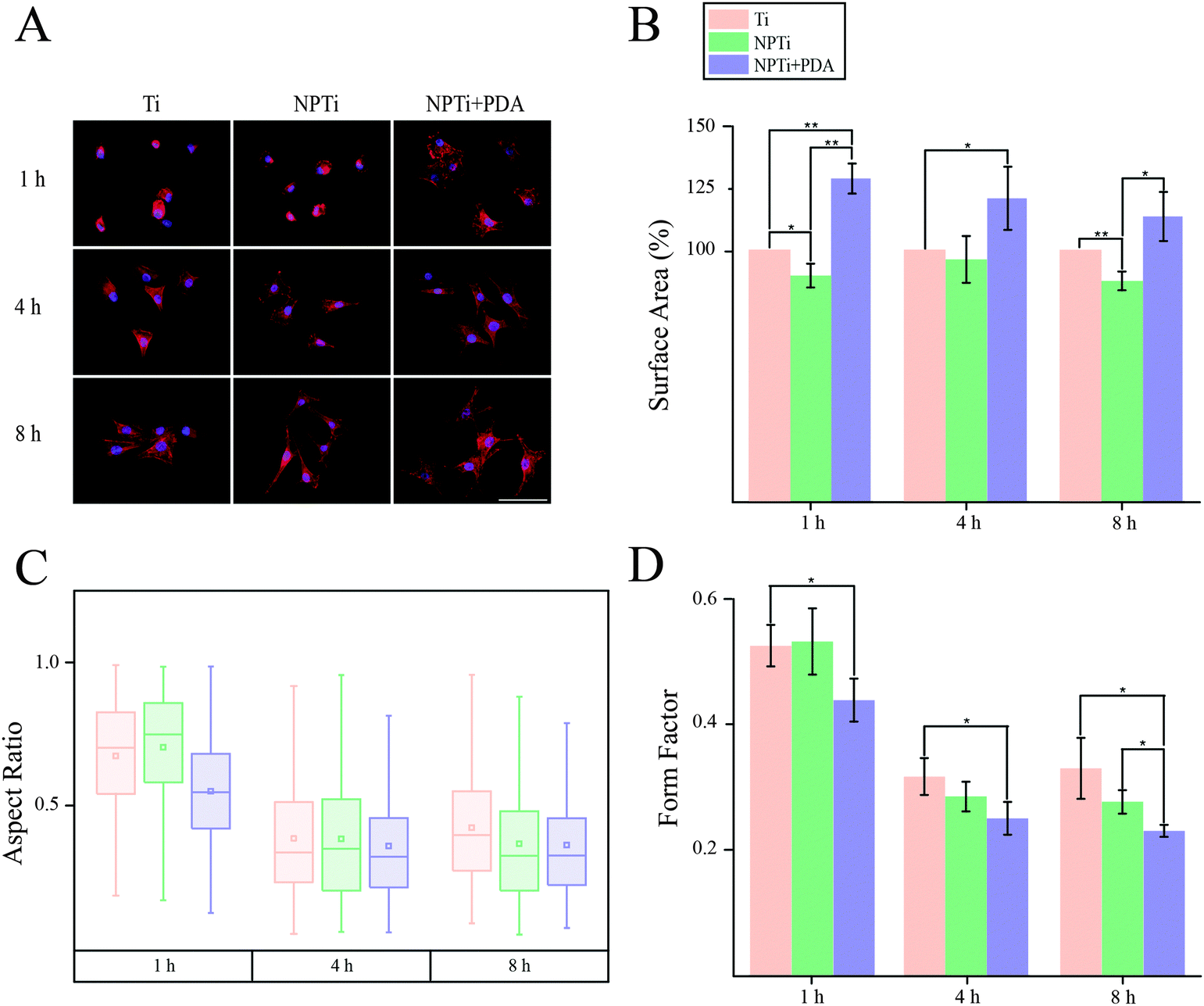 | ||
| Fig. 7 (A) Fluorescence images (scale bar = 200 μm), (B) area (expressed as percentage increase/decrease in respect to bare titanium), (C) aspect ratio and (D) form factor of MG-63 osteoblastic cells adhering on Ti, NPTi and NPTi + PDA surfaces at 1, 4 and 8 hours. ANOVA test: * = p < 0.05, ** = p < 0.01. | ||
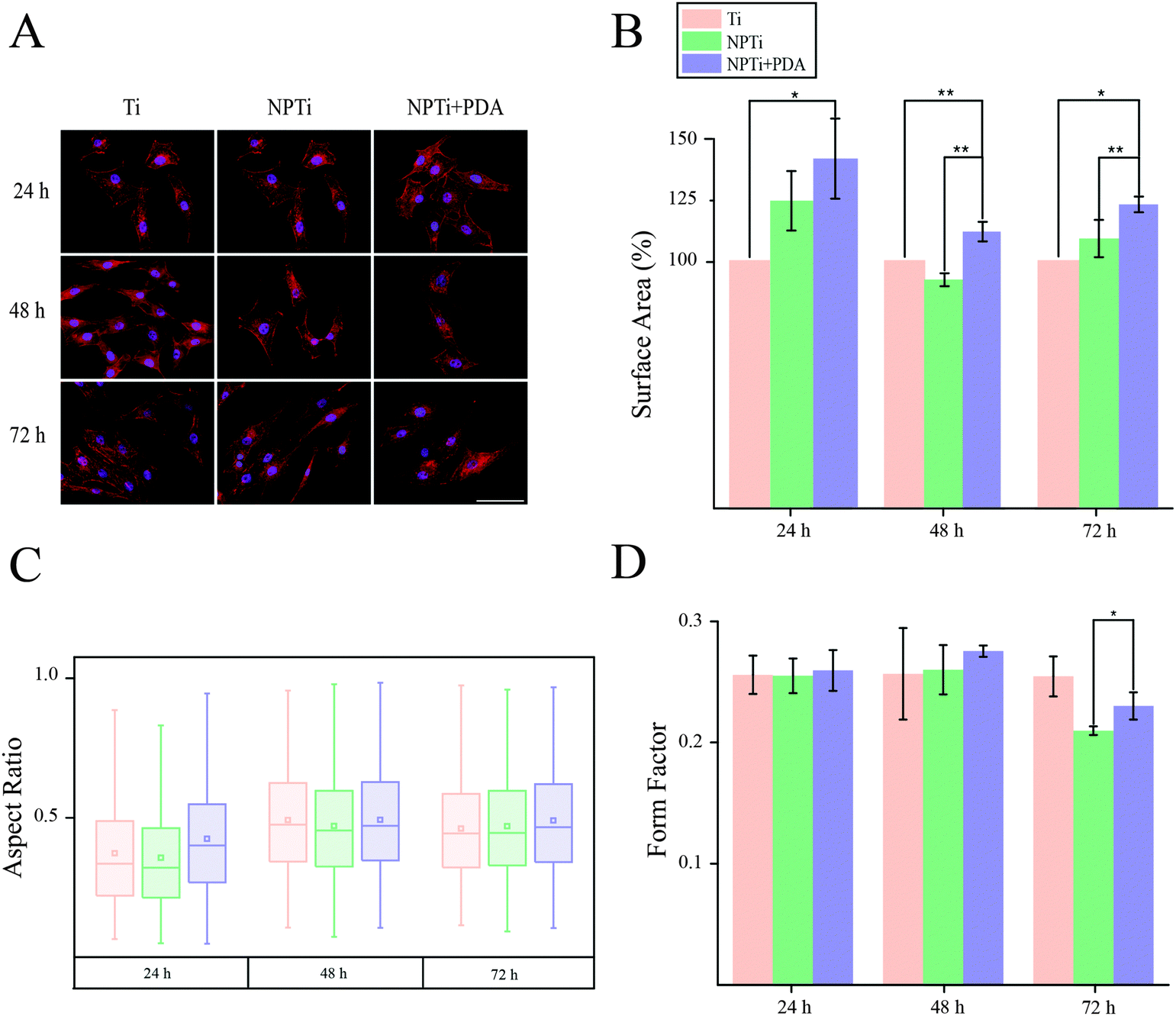 | ||
| Fig. 8 (A) Fluorescence images (scale bar = 200 μm), (B) area (expressed as percentage increase/decrease in respect to bare titanium), (C) aspect ratio and (D) form factor of MG-63 osteoblastic cells adhering on Ti, NPTi and NPTi + PDA surfaces at 1, 2 and 3 days. ANOVA test: * = p < 0.05, ** = p < 0.01. | ||
To better investigate potential molecular mechanisms for the variations in cell morphology on PDA-coated substrates, we investigated the development of focal adhesions (FAs) at earlier time points, where differences were most pronounced in both cell–substrate contact (Fig. 7B) and aspect ratio (Fig. 7D). FAs are dynamic membrane-associated protein complexes act as the interface between the extracellular matrix (ECM) and actin cytoskeleton through sensing of biochemical and/or physical cues. These cues, such as ECM proteins, interact with integrin transmembrane proteins leading to clustering and formation of early focal complexes, dot-like structures of ∼0.5 μm in length, that can mature into focal adhesions, ∼1–5 μm in length, in response to force transduction through myosin-II mediated contractile force.72 Proper cell–ECM tethering and FA development has shown to be important for proper cell function including proliferation, motility and osteospecific differentiation of stem cells.73 Qualitative assessment to investigate detectable differences in FA maturity indicated that presence and maturation of these complexes varied across all three substrates (Fig. 9A). In particular, we observed that cells adhered on NPTi + PDA showed abundant and mature, dash-like, FAs as early as 1 hour after seeding whereas Ti and NPTi tended to show smaller round-like aggregations of vinculin that resembled early focal complexes. Wide spread mature focal adhesions were then seen on the NPTi and Ti conditions at 4 hours and 8 hours, respectively. Although accurate analysis of FA size was excluded because of the difficulty to measure it by confocal microscopy, assessing the quantity of FAs (focal adhesion and complexes together) during initial adhesion was achieved as displayed in Fig. 9B. At 1 hour, there is a noticeable difference in detected FAs between all three substrates. When normalized to Ti, NPTi showed a substantial ∼25% increase whereas NPTi + PDA had the most dramatic increase of nearly 50%. After 4 hours, the effect of the substrate seemed to widen between Ti and the other two conditions. Although NPTi and NPTi + PDA conditions were approximately the same, they presented an almost 70% increase over Ti. Following 8 hours, all three conditioned presented cells with similar numbers of detected FAs. Later time point results were similar to that of 8 hours (data not shown). Quantitative confirmation of increased FA assembly and qualitative maturation help to explain the accelerated cell spreading on PDA-coated substrate seen in Fig. 7B, although the mechanisms, whether physical or biochemical cued, to which this is achieved is still undetermined. Noteworthy, while the FAs presence normalizes across all substrate after 8 hours, cells continue to present a more spread morphology and viability extending up to 3 days later. This indicates that there is a prolonged mechanotransductional effect between the substrate and intracellular signaling.
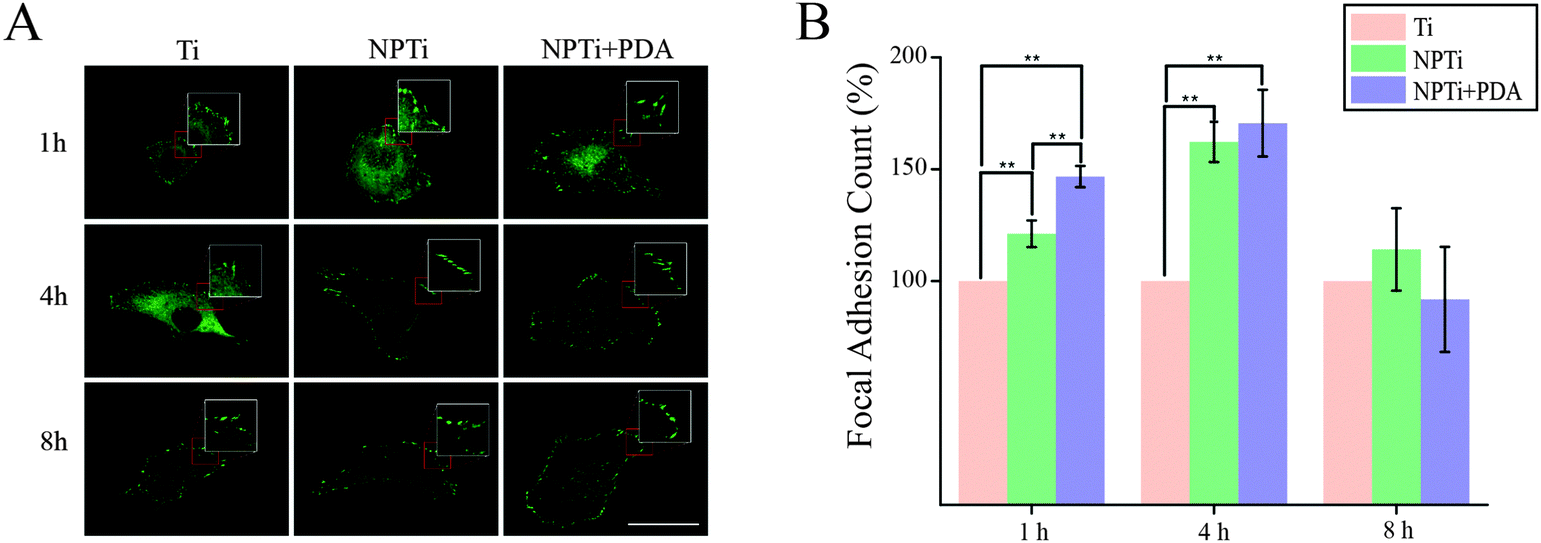 | ||
| Fig. 9 (A) Confocal fluorescence images of focal adhesion complexes at short intervals (scale bar = 50 μm). Insets were magnified 2×. (B) Number of focal adhesion complexes at short intervals. ANOVA test: **=p < 0.01. | ||
Of interest in mechanotransductional signaling pathways is the Rho family of GTPases, including Rac1, Cdc42 and RhoA, which act as molecular switches that influence cell processes such as cell cycle regulation, adhesion and actin cytoskeleton reorganization. These proteins switch between their active GTP-bound and inactive GDP-bound conformations. Integrin mediated Rac1 and Cdc42 activation has been shown to promote cell spreading through associated extension of lamellipodia and filopodia, respectively.74 RhoA which plays an important role in actomyosin contractility and cell cycle maintenance, has been shown to have its activity modulated through mechanotransduction of cell adhesion molecules.30 PCR assay suggested a trend towards lower expression of RhoA after 24 hours in NPTi and NPTi + PDA conditions when compared to bare titanium (67 ± 23% and 77 ± 22%, respectively), indicating lowered relative RhoA activity. In turn, lowered activity in NPTi and NPTi + PDA conditions could help to explain cell–substrate morphology as integrin-mediated lowering of RhoA though p190RhoGAP activity has been shown to promote cell spreading in addition to motility through regulation of cell protrusion and polarity.75 Conversely, enhanced RhoA activity through use of constitutively active RhoA-V14 promotes commitment of hMSCs towards osteogenic lineages, as seen previously on PDA coated substrate, whereas dominant-negative RhoA-N19 led to commitment to an adipogenic fate.76 This implies that there is an intricate mechanotransductional relationship between PDA and MG-63 cells that is not fully understood. Future work should have an emphasis on understanding the dynamics of Rho GTPase expression and activity, at both early and late time points, to elucidate how the accelerated cell adhesion, prolonged spreading, enhanced viability and amplified osteogenic differentiation is regulated and achieved.
Conclusion
In conclusion, our results reaffirm the potential of poly(dopamine) for biomedical applications, not only as an intermediate linker to immobilize bioactive agents, but also as a functional coating capable of beneficially affecting adhering cells. In this study, we have carried out detailed physicochemical characterization of a poly(dopamine) film deposited on nanoporous titanium and demonstrated its effects on MG-63 cell activity at short and longer intervals in terms of adhesion, viability and morphological changes. These may be associated to the granular nanotopography of deposited PDA, its nanomechanical characteristics and chemistry, a differential influence on proteins adsorption (in terms of conformational arrangement) or, most likely, a synergistic combination of these factors. In addition, our results open the door new fundamental studies aimed at understanding additional aspects of the mechanisms that control cell spreading mechanics and motility on poly(dopamine).Acknowledgements
A. A. acknowledges financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Undergraduate Student Research Award (USRA). This work was supported by NSERC through the Discovery grant, by the Canada Foundation for Innovation (CFI) and the Ontario Ministry of Research and Innovation (MRI) through the Leaders of Opportunity (LOF) fund. We thank Drs Li Zhang and Maxim V. Berezovski for LC-MS measurements, data analysis and interpretation.References
- S. M. Oliveira, R. L. Reis and J. F. Mano, Biotechnol. Adv., 2015, 33, 842–855 CrossRef CAS PubMed.
- P. Koegler, A. Clayton, H. Thissen, G. N. C. Santos and P. Kingshott, Adv. Drug Delivery Rev., 2012, 64, 1820–1839 CrossRef CAS PubMed.
- D. Guduru, M. Niepel, J. r. Vogel and T. Groth, Int. J. Artif. Organs, 2011, 34, 963–985 CrossRef CAS PubMed.
- Y. Shao and J. Fu, Adv. Mater., 2013, 26, 1494–1533 CrossRef PubMed.
- A. P. Tomsia, J. S. Lee, U. G. Wegst and E. Saiz, Int. J. Oral Maxillofac. Implants, 2013, 28, e535–e546 CrossRef PubMed.
- F. Variola, J. B. Brunski, G. Orsini, P. T. d. Oliveira, R. Wazen and A. Nanci, Nanoscale, 2011, 3, 335–353 RSC.
- F. Variola, Phys. Chem. Chem. Phys., 2015, 17, 2950–2959 RSC.
- K. S. Brammer, C. J. Frandsen and S. Jin, Trends Biotechnol., 2012, 30, 315–322 CrossRef CAS PubMed.
- R. R. Costa and J. F. Mano, Chem. Soc. Rev., 2014, 43, 3453–3479 RSC.
- C. M. Kirschner and K. S. Anseth, Acta Biomater., 2013, 61, 931–944 CrossRef CAS PubMed.
- M. A. Meyers and P.-Y. Chen, Biological Materials Science, Cambridge University Press, 2014 Search PubMed.
- C. Brubaker and P. Messersmith, Langmuir, 2012, 28, 2200–2205 CrossRef CAS PubMed.
- H. Lee, S. Dellatore, W. Miller and P. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. Lynge, R. van der Westen, A. Postma and B. Städler, Nanoscale, 2011, 3, 4916–4928 RSC.
- J. Kang, S. Tada, T. Kitajima, T. I. Son, T. Aigaki and Y. Ito, BioMed Res. Int., 2013, 2013, 265980 Search PubMed.
- E. Ko, K. Yang, J. Shin and S.-W. Cho, Biomacromolecules, 2013, 14, 3202–3213 CrossRef CAS PubMed.
- Q. Liu, N. Wang, J. r. Caro and A. Huang, J. Am. Chem. Soc., 2013, 135, 17679–17682 CrossRef CAS PubMed.
- C.-Y. Chien and W.-B. Tsai, ACS Appl. Mater. Interfaces, 2013, 5, 6975–6983 CAS.
- J.-J. Lee, I.-S. Park, G.-S. Shin, S.-K. Lyu, S.-G. Ahn, T.-S. Bae and M.-H. Lee, Int. J. Precis. Eng. Man., 2014, 15, 1647–1655 CrossRef.
- S. Zhong, R. Luo, X. Wang, L. Tang, J. Wu, J. Wang, R. Huang, H. Sun and N. Huang, Colloids Surf., B, 2014, 116, 553–560 CrossRef CAS PubMed.
- J. Ryu, S. Ku, H. Lee and C. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- Y.-Z. Zhou, Y. Cao, W. Liu, C. Chu and Q.-L. Li, ACS Appl. Mater. Interfaces, 2012, 4, 6901–6910 CAS.
- J. L. Wang, K. F. Ren, H. Chang, F. Jia, B. C. Li, Y. Ji and J. Ji, Macromol. Biosci., 2013, 13, 483–493 CrossRef CAS PubMed.
- S. Ku, J. Lee and C. Park, Langmuir, 2010, 26, 15104–15108 CrossRef CAS PubMed.
- M. Lynge, R. Ogaki, A. Laursen, J. Lovmand, D. Sutherland and B. Stadler, ACS Appl. Mater. Interfaces, 2011, 3, 2142–2147 CAS.
- S. Ku, J. Ryu, S. Hong, H. Lee and C. Park, Biomaterials, 2010, 31, 2535–2541 CrossRef CAS PubMed.
- M. J. Dalby, N. Gadegaard and R. O. C. Oreffo, Nat. Mater., 2014, 13, 558–569 CrossRef CAS PubMed.
- J. Li, D. Han and Y.-P. Zhao, Sci. Rep., 2014, 4, 1–10 Search PubMed.
- V. Nedovic and R. Willaert, Fundamentals of Cell Immobilisation Biotechnology, Springer, 2004 Search PubMed.
- R. J. Marjoram, E. C. Lessey and K. Burridge, Curr. Mol. Med., 2014, 14, 199–208 CrossRef CAS PubMed.
- L. Richert, F. Vetrone, J.-H. Yi, S. F. Zalzal, J. D. Wuest, F. Rosei and A. Nanci, Adv. Mater., 2008, 20, 1488–1492 CrossRef CAS.
- F. Variola, J.-H. Yi, L. Richert, J. D. Wuest, F. Rosei and A. Nanci, Biomaterials, 2008, 25, 1285–1298 CrossRef PubMed.
- F. Vetrone, F. Variola, P. T. d. Oliveira, S. F. Zalzal, J.-H. Yi, J. Sam, K. F. Bombonato-Prado, A. Sarkissian, D. F. Perepichka, J. D. Wuest, F. Rosei and A. Nanci, Nano Lett., 2009, 9, 659–665 CrossRef CAS PubMed.
- J. H. Yi, C. Bernard, F. Variola, S. F. Zalzal, J. D. Wuest, F. Rosei and A. Nanci, Surf. Sci., 2006, 600, 4613–4621 CrossRef CAS.
- H. Lee, J. Rho and P. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- C. A. Rezende, L.-T. Lee and F. Galembeck, Langmuir, 2009, 25, 9938–9946 CrossRef CAS PubMed.
- J. Domke and M. Radmacher, Langmuir, 1998, 14, 3320–3325 CrossRef CAS.
- B. Stöckle, D. Y. W. Ng, C. Meier, T. Paust, F. Bischoff, T. Diemant, R. J. r. Behm, K.-E. Gottschalk, U. Ziener and T. Weil, Macromol. Symp., 2014, 346, 73–81 CrossRef.
- S. Nirasay, A. Badia, G. g. Leclair, J. P. Claverie and I. Marcotte, Materials, 2012, 5, 2621–2636 CrossRef CAS.
- D. C. Lin, E. K. Dimitriadis and F. Horkay, J. Biomech. Eng., 2007, 129, 430–440 CrossRef PubMed.
- S. Ku and C. Park, Biomaterials, 2010, 31, 9431–9437 CrossRef CAS PubMed.
- T. Shalev, A. Gopin, M. Bauer, R. Stark and S. Rahimipour, J. Mater. Chem., 2012, 22, 2026–2032 RSC.
- M. Baginska, B. Blaiszik, T. Rajh, N. Sottos and S. White, J. Power Sources, 2014, 269, 735–739 CrossRef CAS.
- R. Luo, L. Tang, S. Zhong, Z. Yang, J. Wang, Y. Weng, Q. Tu, C. Jiang and N. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1704–1714 CAS.
- H. Wei, J. Ren, B. Han, L. Xu, L. Han and L. Jia, Colloids Surf., B, 2013, 110, 22–28 CrossRef CAS PubMed.
- R. Zangmeister, T. Morris and M. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- A. E. Carpenter, T. R. Jones, M. R. Lamprecht, C. Clarke, I. H. Kang, O. Friman, D. A. Guertin, J. H. Chang, R. A. Lindquist, J. Moffat, P. Golland and D. M. Sabatini, Genome Biol., 2006, 7, R100 CrossRef PubMed.
- A. Kasten, T. Naser, K. Brullhoff, J. Fiedler, P. Muller, M. Moller, J. Rychly, J. Groll and R. E. Brenner, PLoS One, 2014, 9, e109411 Search PubMed.
- C. Barnes, L. Speroni, K. P. Quinn, M. Montevil, K. Saetzler, G. Bode-Animashaun, G. McKerr, I. Georgakoudi, C. S. Downes, C. Sonnenschein, C. V. Howard and A. M. Soto, PLoS One, 2014, 9, e93325 Search PubMed.
- H. Z. Sailem, J. E. Sero and C. Bakal, Nat. Commun., 2015, 6, 5825 CrossRef CAS PubMed.
- A. Ketabchi, K. Komm, M. Miles-Rossouw, D. A. D. Cassani and F. Variola, PLoS One, 2014, 9, e92080 Search PubMed.
- F. Variola, S. F. Zalzal, A. Leduc, J. Barbeau and A. Nanci, Int. J. Nanomed., 2014, 9, 2319–2325 CrossRef PubMed.
- F. Bernsmann, O. Ersen, J. C. Voegel, E. Jan, N. Kotov and V. Ball, ChemPhysChem, 2010, 11, 3299–3305 CrossRef CAS PubMed.
- U. Schmidt, S. Hild, W. Ibach and O. Hollricher, Macromol. Symp., 2005, 230, 133–143 CrossRef CAS.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kachar and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS PubMed.
- S. Ayas, G. Cinar, A. D. Ozkan, Z. Soran, O. Ekiz, D. Kocaay, A. Tomak, P. Toren, Y. Kaya, I. Tunc, H. Zareie, T. Tekinay, A. B. Tekinay, M. O. Guler and A. Dana, Sci. Rep., 2013, 3, 2624–2632 Search PubMed.
- P. Zhou, Y. Deng, B. Lyu, R. Zhang, H. Zhang, H. Ma, Y. Lyu and S. Wei, PLoS One, 2014, 9, e113087 Search PubMed.
- F. Variola, A. Nanci and F. Rosei, Appl. Spectrosc., 2009, 63, 1187–1190 CrossRef CAS PubMed.
- C. J. Wilson, R. E. Clegg, D. I. Leavesley and M. J. Pearcy, Tissue Eng., 2005, 11, 1–18 CrossRef CAS PubMed.
- P. Roach, D. Eglin, K. Rohde and C. C. Perry, J. Mater. Sci.: Mater. Med., 2007, 18, 1263–1277 CrossRef CAS PubMed.
- B. Kasemo, Surf. Sci., 2002, 500, 656–677 CrossRef CAS.
- M. Salmeron-Sanchez and G. Altankov, in Tissue Eng., ed. D. Eberli, InTech, 2010 Search PubMed.
- R. A. Latour, in Encyclopedia of Biomaterials and Biomedical Engineering, ed. G. E. Wnek and G. L. Bowlin, Taylor & Francis, 2004 Search PubMed.
- M. Malmsten, Biopolymers at interfaces, Marcel Dekker, Inc., 2003 Search PubMed.
- S. Ede, L. Hafner, R. Frost and G. Will, Hydrol.: Curr. Res., 2012, 3, 1–4 Search PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- E. M. Czekanska, M. J. Stoddart, R. G. Richards and J. S. Hayes, Eur. Cells Mater., 2012, 24, 1–17 CAS.
- L. Kamentsky, T. R. Jones, A. Fraser, M. Bray, D. Logan, K. Madden, V. Ljosa, C. Rueden, G. B. Harris, K. Eliceiri and A. E. Carpenter, Bioinformatics, 2011, 27, 1179–1180 CrossRef CAS PubMed.
- Y. Hiraguchi, K. Nagahashi, T. Shibayama, T. Hayashi, T. A. Yano, K. Kushiro and M. Takai, Acta Biomater., 2014, 10, 2988–2995 CrossRef CAS PubMed.
- Z.-X. Ren, H.-B. Yu, J.-S. Li, J.-L. Shen and W.-S. Du, Biosci. Rep., 2015, 35, e00202 Search PubMed.
- J. R. Eidet, L. Pasovic, R. Maria, C. J. Jackson and T. P. Utheim, Diagn. Pathol., 2014, 9, 92 CrossRef PubMed.
- J.-C. Kuo, Prog. Mol. Biol. Transl. Sci., 2014, 126, 55–73 Search PubMed.
- M. J. P. Biggs and M. J. Dalby, Proc. Inst. Mech. Eng., Part H, 2010, 224, 1441–1453 CrossRef CAS.
- L. S. Price, J. Leng, M. A. Schwartz and G. M. Bokoch, Mol. Biol. Cell, 1998, 9, 1863–1871 CrossRef CAS PubMed.
- W. T. Arthur and K. Burridge, Mol. Biol. Cell, 2001, 12, 2711–2720 CrossRef CAS PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |