
Gallium-containing mesoporous bioactive glass with potent hemostatic activity and antibacterial efficacy
Sara
Pourshahrestani
a,
Ehsan
Zeimaran
a,
Nahrizul
Adib Kadri
a,
Nicola
Gargiulo
 b,
Shani
Samuel
c,
Sangeetha Vasudevaraj
Naveen
c,
Tunku
Kamarul
c and
Mark R.
Towler
*ad
b,
Shani
Samuel
c,
Sangeetha Vasudevaraj
Naveen
c,
Tunku
Kamarul
c and
Mark R.
Towler
*ad
aDepartment of Biomedical Engineering, Faculty of Engineering, University of Malaya, Kuala Lumpur 50603, Malaysia
bLaboratori di Chimica Applicata, Dipartimento di Ingegneria Chimica, dei Materiali e della Produzione Industriale, Università Federico II, P.le Tecchio 80, 80125 Napoli, Italy
cTissue Engineering Group (TEG), Department of Orthopedic Surgery, NOCERAL, Faculty of Medicine, University of Malaya, Kuala Lumpur 50603, Malaysia
dDepartment of Mechanical & Industrial Engineering, Ryerson University, Faculty of Engineering and Architectural Science, 350 Victoria Street, Toronto M5B 2K3, Ontario, Canada. E-mail: mtowler@ryerson.ca
First published on 18th November 2015
Abstract
Haemorrhage remains the leading cause of potentially survivable death in both military and civilian populations. Although a large variety of hemostatic agents have been developed, many of them have an inadequate capacity to induce hemostasis and are not effective in killing bacteria. In recent years, mesoporous bioactive glasses (MBGs) were found to be effective in inducing hemostasis. However, the materials may not be considered as ideal hemostats since they do not offer antimicrobial activity. The gallium ion (Ga+3) not only exhibits antibacterial properties but also accelerates the blood coagulation cascade. The aim of this study was to develop MBGs containing various concentrations of Ga2O3 (1, 2 & 3 mol%) via the evaporation-induced self-assembly (EISA) process and investigate whether the addition of Ga3+ would induce both hemostatic and antibacterial effects. The results indicated that the incorporation of lower Ga2O3 content (1 mol%) into the MBG system improved structural properties including the specific surface area, mesopore size and pore volume as well as the release of silicon and calcium ions. The bioactive glass was found to stimulate blood coagulation, platelet adhesion and thrombus generation and exerted an antibacterial effect against both Escherichia coli and Staphylococcus aureus. Likewise, Ga-doped MBGs showed excellent cytocompatibility even after 3 days, with the 1% Ga2O3-containing MBG attaining the best biocompatibility that render them safe hemostatic agents for stopping bleeding. This study demonstrated that the lowest Ga2O3-substituted MBG can be a potent candidate for controlling haemorrhage and wound infection.
Introduction
Based on the recent combat mortality analysis, hemorrhage was the main cause in almost 91% of all pre-hospital deaths which were deemed potentially survivable.1 In the last decade, a number of hemostatic agents have been used for the rapid control of bleeding. Among those, inorganic hemostats have demonstrated good efficacy because the materials are free of animal or human derived proteins that can cause allergic reactions.2 QuickClot (QC, zeolite-based hemostat) and QuikClot® Combat Gauze™ (QCG, kaolin-based hemostat) are inorganic coagulation accelerators that have attracted the scientific community because of their ability to staunch bleeding.3–7 However, animal studies and case reports revealed that the application of QC can result in thermal injuries and abnormal foreign-body reaction because of its highly exothermic reaction and poor biodegradability.5,8,9 Some studies also found that QCG does not often provide immediate hemostasis when applied to the bleeding site, resulting in larger blood loss compared with other agents.10 Furthermore, the dressing may not be reliable for controlling hemorrhage in patients with coagulopathy since its hemostatic abilities depend solely on the blood-clotting activity of the host.10,11 Hence, the development of a promising hemostatic agent is still a challenge. Infection of wounds is a further serious complication that can delay or impair healing. In this sense, an ideal hemostat should be bacteriostatic and/or bactericidal when applied to the wound.12 Although many antibiotics have been developed to reduce bacteria at the wound site, the most important challenge facing caregivers is the increasing rate of antibiotic-resistant bacterial infections.13,14 To address this problem, alternatives to antibiotics need to be considered for controlling infection and accelerating healing.A new generation of bioactive glasses, referred to as MBGs, were first prepared by Yan et al. in 2004 using the EISA process.15 The bioactive glasses in the 80% SiO2–15% CaO–5% P2O5 system are distinct from conventional non-mesoporous bioactive glasses developed by Hench and co-workers16 since they possess highly ordered mesoporous channel structures and significantly greater specific surface area.15 These MBGs also offer other unique characteristics including a greater pore volume and pore sizes ranging from 2 to 10 nm that render them suitable for a variety of drug delivery and musculoskeletal applications.15,17,18 In the recent years, it is demonstrated that incorporation of some trace elements in the MBG matrix such as titanium (Ti+4), cerium (Ce3+), zinc (Zn2+), silver (Ag+) or Ga3+ as well as surface modification with some polymers can improve the biological behavior of MBG materials.19–23 Ag-containing MBG with efficient antibacterial properties, has recently shown promise as hemostatic agent for staunching bleeding.24–27 Dai et al. found that mesoporous silica spheres doped with calcium (Ca+2) and Ag+ can exhibit good hemostatic efficacy and antibacterial activity.25 They reported that high surface area as well as the presence of Ca2+ play a ubiquitous role in the coagulation cascade, contributing to the hemostatic effect. The antimicrobial activity of the material was also a result of the presence of Ag+.25 Similar to the study performed by Dai et al., Hu et al. demonstrated the superiority of the Ag-doped nanoporous bioactive glass (n-BGS) over the non-porous bioactive glass (BGS) in terms of procoagulant and antibacterial activity.24 According to the authors, the higher specific surface area of n-BGS compared with BGS was responsible for its hemostatic ability. Additionally, another already published work demonstrated that macroporous chitosan-coated mesoporous silica xerogel beads (CSSX) can also accelerate the intrinsic pathway of the coagulation cascade and achieve a desirable hemostasis.26 The thrombotic effect of the hybrid material was ascribed to the interaction of negatively charged blood cells with positively charged chitosan, which resulted in a more pronounced adhesion of blood cells onto CSSX beads, as well as to a negatively charged surface of mesoporous silica xerogel beads, which led to activation of the intrinsic pathway.26
Ga3+ is a chemotherapeutic ion whose biological function has already been approved by US Food and Drug Administration (FDA).28 Ga3+ has shown to be effective for various disorders such as bone resorption and autoimmune diseases, alongside cancer.29 Ga3+ was found to be beneficial in treating localized infection,30 inhibiting biofilm formation and imparting bactericidal activity against both free living bacteria and biofilm cells.30,31 Additionally, Ga3+ ions share antibacterial activity against both Gram-positive (Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, and Clostridium difficile) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacteria. Thus, the delivery of the ion from Ga-containing biomaterials could be of special interest in biomedical applications. Recently, in vitro bioactivity and controlled drug-release ability of Ga-substituted MBG have been investigated by Maria Vallet-Regi's group.20,21 Their studies indicated that incorporation of Ga3+ into the network of MBGs made them capable of exhibiting high bioactive response and facilitating drug loading and release while maintaining mesostructural order. Accordingly, it was proposed that Ga2O3-containing glasses can be suitable for bone tissue engineering applications and drug delivery.
It was also found that some other Ga-containing compounds, such as aqueous solution of gallium nitrate Ga(NO3)3, have appeared to be efficient for the immediate treatment of wounds and for the enhancement of the early stage of hemostasis (coagulation, platelet activation, or clot formation) when directly applied to an open wound.32–34 Although the exact mechanism of action for the ability of Ga(NO3)3 to facilitate hemostasis is not fully understood, the main principle behind this function may be dependent upon the presence of the Ga3+ in solution.32,33 Goodley and Rogosnitzky studied the hemostatic activity of Ga(NO3)3 on ceasing blood flow from an open wound and concluded that the solution has the potential to considerably reduce clotting time in both a warfarin-treated (S1) and non-warfarin-treated (S2) subjects compared to the control that was not treated with Ga(NO3)3. It was also found that the blood-stemming property of Ga(NO3)3 was not influenced by warfarin in S1.33 This behavior of Ga(NO3)3 in individuals with an inhibited extrinsic pathway can lead to some support for the theory that Ga-containing compounds may achieve desirable hemostasis via another coagulation pathway, probably involving the activation of the intrinsic pathway or platelet activation.32 To date, there are no studies that investigate the effects of the incorporation of Ga3+ into MBG compositions for hemostasis.
The present study aims to evaluate the in vitro acceleratory effect on the blood coagulation cascade and the antibacterial function of Ga-containing MBGs (Ga-MBGs), in comparison with a Ga-free MBG. Accordingly, a series of MBGs containing three different concentrations of Ga2O3 (1, 2 & 3 mol%) were prepared by the EISA process, and their physiochemical properties, in vitro hemostatic activity, biocompatibility and antibacterial activity against Echerichia coli (E. coli) and Staphylococcus aureus (S. aureus) were investigated.
Materials and methods
Materials
Tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich), triethyl phosphate (TEP, ≥99.8%, Sigma-Aldrich), calcium nitrate tetrahydrate [Ca(NO3)2·4H2O, ≥99%, Sigma-Aldrich], gallium(III) nitrate hydrate [Ga(NO3)3·xH2O, 99.9%, Sigma-Aldrich], triblock copolymer EO20PO70EO20 (P123, Sigma-Aldrich), ethyl alcohol (EtOH, Sigma-Aldrich) and nitric acid (HNO3, Sigma-Aldrich) were of reagent grade and used without any further purification.Synthesis of MBGs
Ordered MBGs (80 − x)% SiO2–15% CaO–5% P2O5–xGa2O3, doped with different contents of Ga2O3, were synthesized by using P123 as a structure directing agent. In the present work, we kept CaO and P2O5 content constant and varied SiO2 and Ga2O3 content. The nominal chemical composition of the synthesized samples are reported in Table 1.| Components (mol%) | ||||
|---|---|---|---|---|
| Glass code | SiO2 | CaO | P2O5 | Ga2O3 |
| MBG | 80 | 15 | 5 | 0 |
| 1% Ga-MBG | 79 | 15 | 5 | 1 |
| 2% Ga-MBG | 78 | 15 | 5 | 2 |
| 3% Ga-MBG | 77 | 15 | 5 | 3 |
Typically, 4 g of P123 was dissolved in 60 g of EtOH, followed by the addition of TEOS, TEP, Ca(NO3)2·4H2O, Ga(NO3)3·xH2O (the latter only in the case of Ga-MBGs) and 1.0 mL HNO3 (0.5 M). The mixture was vigorously stirred overnight at room temperature, and then the as-derived clear sol was transferred into a Petri dish and kept for several days at room temperature for the EISA process. The dried gels were calcined at 600 °C for 5 h to remove the redundant organic template and to obtain the final glass powder. The as-synthesized MBG and Ga-MBGs were then ground into two particle sizes ranging in the 32–75 and 250–300 μm intervals and stored in a desiccator until usage.
Characterization of MBG and Ga-MBGs
Small and wide angle X-ray diffractions
Small and wide angle X-ray diffraction (SAXRD and WAXRD, respectively) patterns were obtained using a X-ray diffractometer (PANalytical Empyrean) with Cu Kα (λ = 0.154 nm) radiation (40 kV, 40 mA). The SAXRD patterns were collected in the 2θ range between 0.6° and 7° with a step size of 0.02° and a counting time of 5 s per step. In addition, WAXRD patterns were collected in the 2θ range between 10° and 60° with a step size of 0.02° and a counting time of 5 s per step.Transmission electron microscopy (TEM)
The inner microstructure of the samples was examined by transmission electron microscopy (TEM, JEOL JEM-2100F microscope) operating at 200 kV.Scanning electron microscopy and energy-dispersive X-ray spectroscopic analysis (SEM-EDS)
An energy-dispersive X-ray spectrometer (EDS: 20 mm X-Max, Oxford Instruments, Oxford, UK) and the INCA software connected to a scanning electron microscope (SEM: Quanta™ 250 FEG-FEI, USA) at 20 kV were used for the assessment of the surface morphology and composition of the glasses.Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra of the synthesized samples were collected using a Fourier transform infrared spectrometer (ATR-FTIR400, Perkin Elmer instruments, USA) within the 400–1400 cm−1 wavelength range at room temperature.Zeta potential measurement
Zeta potential determination was carried out in a Zetasizer Nanoseries (Malvern Instruments) via suspending MBG and Ga-MBG particles in phosphate buffered solution (PBS) at pH 7.4.In vitro ion release and degradation measurements
Ion release measurements of MBG and Ga-MBGs were performed using a microwave plasma-atomic emission spectrometer (MP-AES) Agilent 4100 (Agilent Technologies, Inc., Santa Clara, CA, USA) to investigate the concentration of silicate (SiO44−), phosphate (PO43−), Ca2+ and Ga3+ ions in the soaking solution. For this purpose, 20 mg of each dried sample was immersed in a polypropylene vial containing 10 mL of 0.05 M Tris(hydroxymethyl)aminomethane–HCl (Tris–HCl, Sigma-Aldrich) buffer solution (pH = 7.4), and incubated upto 3 days at 37 °C with continuous shaking at 120 rpm (n = 3).37 At specified time points (0.5, 1, 2, 3, 6, 12, 24, 48 and, 72 h), the powders were collected by filtration, and the extract was used for ion release studies.The degradation properties of MBG and Ga-MBGs were also evaluated at 37 °C in Tris–HCl solution (pH 7.4). The samples were immersed in vials containing 10 mL of the solution following incubation upto 14 days. At the scheduled time, the samples were separated from the solution by filtration and rinsed with acetone followed by drying in an oven at 37 °C to a constant weight. For measuring the weight loss, the following equation was used: weight loss (%) = [(W0 − Wt)/W0] × 100%, where W0 is the initial weight of the samples and Wt is the weight of samples after immersion in the solution.
In vitro blood plasma coagulation assay
To evaluate coagulation behaviors of MBG and Ga-MBGs, the activated partial thromboplastin time (APTT) and prothrombin time (PT) were performed. Fresh human blood anticoagulated with 3.2% sodium citrate was collected from healthy volunteers with approval from the Medical Ethics Committee of University of Malaya, Kuala Lumpur, Malaysia (UMMC reference number 967.10). The platelet poor plasma (PPP) was obtained via centrifugation of whole blood at 3500 rpm for 10 min at 25 °C. For each assay, the MBG and Ga-MBGs were placed into test tubes containing 100 μL PPP.For the APTT assay, the above test tubes with selected samples (MBG or Ga-MBGs) and 100 μL APTT reagent were incubated at 37 °C in a water bath for 2 minutes. Then, 100 μL of pre-warmed CaCl2 was added and the clotting time at which the fibrous substance appeared was recorded.
For the PT assay, the pre-warmed plasma was incubated with the samples for 2 min at 37 °C. The PT was measured simultaneously after the addition of 100 μL of pre-warmed PT reagent. The tests for negative control were performed without MBG and Ga-MBGs.
PBS absorption efficiency in vitro
The in vitro absorption efficiency of MBG and Ga-MBGs was assessed in PBS (pH = 7.4). The test was performed according to Dai's method as already described.25 Before doing the test, the samples were dried at 50 °C in a vacuum overnight to completely eliminate the residual water. Briefly, the pre-weighed dried samples (having mass Wdry) were placed at the center of a folded filter paper in a funnel so that fluid (PBS) can be uniformly absorbed by the samples. After that, the fluid was added dropwise at a rate of 10 mL min−1 until the absorption by the samples reached its saturation state (when the first drop of fluid trickled from the funnel). The mass of the wet sample was then recorded as Wwet after removing the filter paper. The absorption ratio of samples with PBS was calculated by using the following equation: (%) = [(Wwet − Wdry)/Wdry] × 100%, where Wdry and Wwet are the weight of the samples before and after the PBS immersion, respectively. For each absorption study, three samples were examined.In vitro thrombus formation
The in vitro thrombogenic activity of the samples was evaluated with citrated human blood using a method adapted from Imai.38 Thirty milligrams of MBG and Ga-MBGs were introduced into the wells of 24-well culture plates containing 1 mL of citrated human blood to start the thrombus formation followed by incubation at 37 °C for three different time points (15, 30 and 60 min). The reactions were stopped at appropriated time intervals through adding 10 mL of deionized water. The formed thrombus was fixed in 37% formaldehyde solution for 10 min at room temperature. The fixed samples were then placed in an oven overnight at 50 °C until constant weight was obtained. The degree of thrombogenicity (DT) of the samples at a given time was defined as follows: DT = [(Wt − W0)/W0] × 100%, where W0 and Wt are the weights of the samples before and after in contact with blood, respectively. The assay was done at least three times for each sample. The thrombus formed on the surfaces of the samples was photographed by a digital camera. The interaction of whole blood cells with the synthesized samples was also visualized by a field emission scanning electron microscope (FESEM: Quanta™ 250 FEG-FEI, USA). Prior to fixing as described above, the soaked samples were washed with PBS to remove the blood cells not fully adhered and then were fixed using 2.5% glutaraldehyde for 2 h, immediately after the rinsing step. Afterwards, samples underwent a graded dehydration series of ethanol and HMDS (hexamethyldisilazane) drying for 10 min. The particles were then left drying for 2 days in a fume hood at room temperature. The dried samples were then sputtered with a thin layer of gold and the photomicrographs were collected with an accelerating voltage of 15 kV.In vitro evaluation of platelet adhesion
A lactate dehydrogenase (LDH) assay was used to measure the number of adherent platelets. Human blood samples were freshly obtained from volunteers and anticoagulated with 3.2% sodium citrate. The citrated human blood samples were centrifuged at 1500 rpm for 10 min to remove leukocytes and erythrocytes. Then the supernatant, platelet rich plasma (PRP), was again centrifuged at 3000 rpm for 10 min to yield a platelet pellet with platelet poor plasma as the supernatant. The platelet pellet was re-suspended in PBS to a concentration of 4 × 108 platelets per mL. The MBG and Ga-MBGs were then exposed to the platelets and incubated at 37 °C for three different time intervals (15, 30, and 60 min). At the end of each time interval, the samples were removed and dip rinsed ten times in PBS to remove the platelets that are not attached. The samples were then placed into PBS containing 1% Triton X-100 for 1 h at 37 °C to lyse the adherent platelets. The number of platelets adhered on the surface of the materials was quantified by the LDH assay kit (Sigma-Aldrich, USA). A platelet calibration curve was obtained using serial dilutions of a known number of platelets by measuring optical density (OD) at 450 nm by an Epoch microplate spectrophotometer (BioTek; Winooski, VT, USA). Each measurement was performed at least thrice. After the measurement of platelet adhesion, the interaction of platelets with MBG and Ga-MBGs was observed by FESEM (Quanta™ 250 FEG-FEI, USA) at 15 kV. In this regard, samples were fixed in 2.5% glutaraldehyde followed by dehydration in an ascending series of ethanol upto 100%. The samples were then dried in HMDS and coated with gold in a sputter coater for FESEM studies.Antibacterial efficacy
Killing time assays were used to evaluate antibacterial efficacy of MBGs doped with various concentrations of Ga2O3 against E. coli and S. aureus.39,40 Briefly, a suspension of E. coli or S. aureus was obtained from freshly grown colonies on tryptic soy agar after incubation at 37 °C overnight. 10 μL of the bacteria suspension (4 × 108 CFU mL−1) was added to each vial containing 3 mL of tryptic soy broth. Ga-MBGs that were previously sterilized under ultraviolet (UV) light for 12 h were then added and incubated at a temperature of 37 °C for 0, 1, 3, 6 and 12 h. After that, aliquots were taken at specified time intervals and, subsequently, serial dilutions of each bacterial culture were plated on agar plates to count the number of living bacterial colonies. Each evaluation was carried out in triplicate. The antibacterial activity was quantitatively estimated by the following relationship: R(%) = 100 × [(B − C)/B], where R is the antibacterial activity (%), B is the mean number of survived bacteria on the control samples (CFU mL−1), and C is the mean number of bacteria on the MBG and Ga-MBGs (CFU mL−1). The MBG and pure LB broth culture were used as positive control and negative control, respectively. After 12 h incubation at 37 °C, digital images of the plates were captured.In vitro cytotoxicity assays
MBG and Ga-MBG samples, which were previously sterilized under UV light for 12 h, were soaked in 100 mL of DMEM, preheated to 37 °C. After 24 h under sterile conditions, DMEM was filtered to separate MBG and Ga-MBGs, and these extracts were used as culture medium. To evaluate the effect of the bioactive glasses on cell viability, human dermal fibroblast (HDF) cells were seeded (1 × 104 cells per well) in a 24-well plate and allowed to attach overnight. Then the cells were treated in the presence of different extracts obtained from MBG and Ga-MBGs. HDF cells without extract (treated with normal medium) were used as control. Each condition was set up in triplicate. Plates were assayed 1 and 3 days after the addition of extracts. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used in this study to measure cell vitality and proliferation. The MTT agent (Biotium, Inc., Hayward, CA) reacts with its tetrazolium ring to produce blue formazan crystals in viable cells. At 1 and 3 days, the supernatant was removed from the wells, and the cells were rinsed three times with PBS to eliminate nonviable cells. 500 μL of fresh media were added to each well and then fifty microliters of the MTT solution (5 mg mL−1 in PBS) were added in each well, and the cells were incubated at 37 °C for 4 h to allow the formation of formazan crystals. After incubation, the supernatant was removed, and 1 mL of dimethyl sulfoxide (DMSO) was then added to each well to dissolve the formazan crystals under continuous pipetting. The optical density was measured at a wavelength of 570 nm using a microplate reader. The results from three individual experiments were averaged and normalized to the control.Statistical analysis
All data were expressed as mean ± standard deviations of a representative of three experiments carried out in triplicate. The data were analyzed using statistical software (IBM SPSS Statistics for Windows, Version 22). In all the statistical evaluations, a value of p < 0.05 was considered statistically significant.Results
Structural and morphological characterization of MBGs
MBGs with different concentrations of Ga2O3 were successfully synthesized via EISA. The N2 adsorption–desorption isotherms at 77 K of the synthesized samples were measured and are shown in Fig. 1 together with the corresponding pore size distribution. The main textural parameters of the prepared samples are shown in Table 2.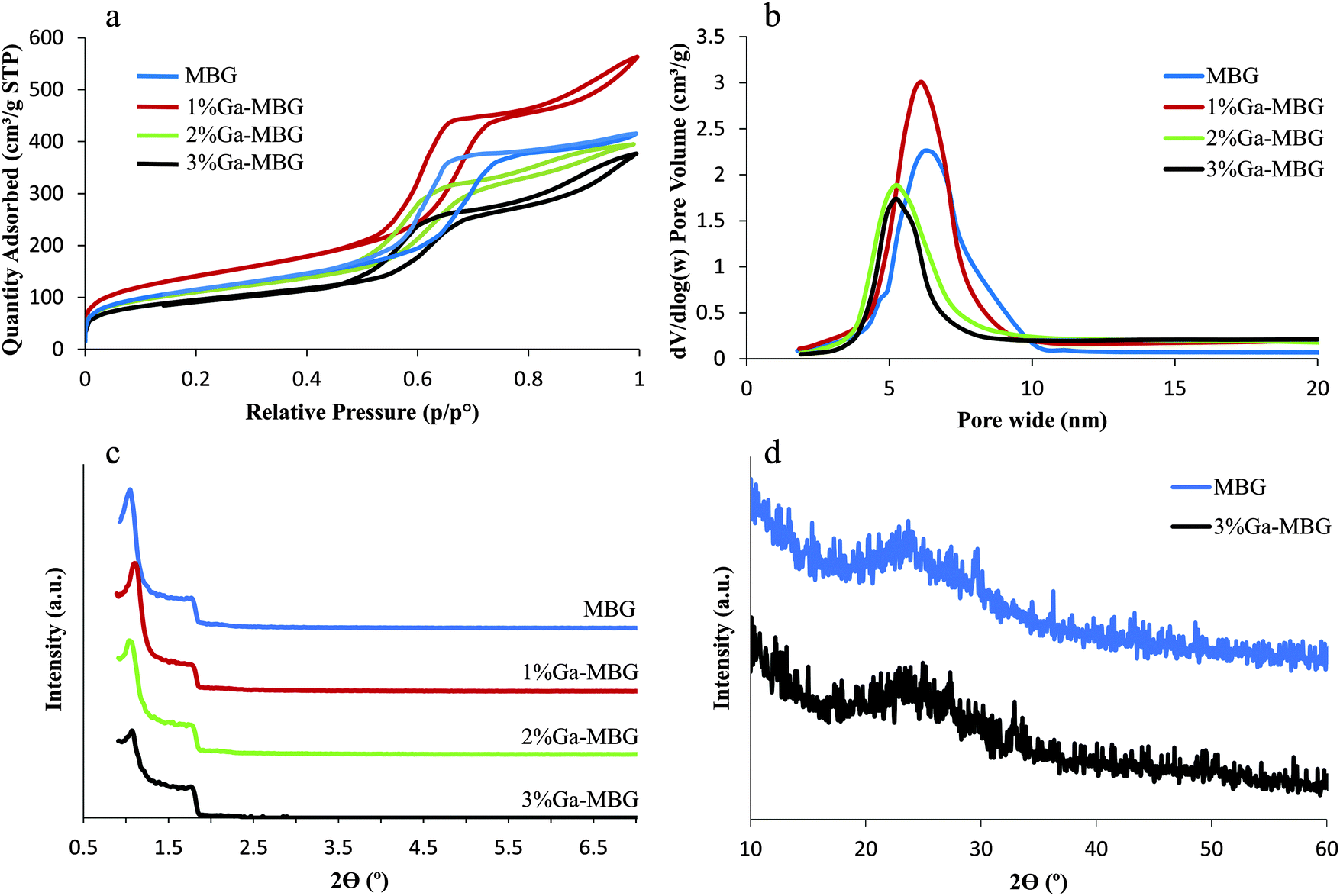 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms and (b) the corresponding pore size distributions of MBG and Ga-MBGs. (c) SAXRD of MBG and Ga-MBGs. (d) WXRD analysis of MBG and 3% Ga-MBG glasses. | ||
| Sample code | Ga2O3 content (mol%) | Surface area (m2 g−1) | Average pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| MBG | 0 | 415 | 5.75 | 0.55 |
| 1% Ga-MBG | 1 | 509 | 6.34 | 0.78 |
| 2% Ga-MBG | 2 | 396 | 5.74 | 0.54 |
| 3% Ga-MBG | 3 | 340 | 6.65 | 0.52 |
All the glasses revealed type IV isotherms along with H1 hysteresis loops, which are characteristic of a mesoporous structure and non-defective cylindrical pores, respectively (Fig. 1a).41Fig. 1b illustrates the pore size distribution curves as calculated from the adsorption branches by the BJH model. Apparently, all the samples exhibited very narrow pore size distributions centered between 5 and 7 nm. As shown in Table 2, 1% Ga-MBG showed a higher surface area and pore volume compared with MBG and higher substituted Ga-MBGs. These results are in good agreement with those presented by Salinas et al.20 The authors claimed that the inclusion of higher Ga2O3 content in the MBG framework result in a decrease of the textural properties (pore diameter, surface area and pore volume). The pore structure of the samples with different Ga2O3 contents was investigated by SAXRD (Fig. 1c). All glasses exhibited two characteristic peaks at around 2θ = 1.1° and 1.8° that are associated with the ordered p6mm hexagonal space group. The presence of these diffraction peaks indicated that the synthesized glasses have a high degree of hexagonal mesoscopic organization. However, 2 and 3% Ga-MBGs showed a weaker diffraction peak at around 2θ = 1.1° compared with MBG and 1% Ga-MBG. No evidence of residual crystallinity was observed in the WXRD pattern of the samples indicating that each glass is completely amorphous (Fig. 1d).
The presence of ordered mesoporous channels in the prepared glasses was also confirmed by TEM images. All synthesized glasses showed a typical 2D-hexagonal ordered mesoporous arrangement. The TEM images of the MBG and 1% Ga-MBG are presented in Fig. 2. Well-ordered pore arrays can be clearly observed in MBG (Fig. 2a) and 1% Ga-MBG (Fig. 2b) samples. However, the TEM images indicated a loss of order as the Ga2O3 content in the pores was increased (data not shown), confirming the results of SAXRD. The EDS analysis of the synthesized samples confirmed the presence of Si, Ca, P and Ga. The SEM morphologies and EDS spectra of the MBG and 3% Ga-MBG are also shown in Fig. 2. Based on the EDS results, the composition of the glasses were approximately similar to the theoretical value.
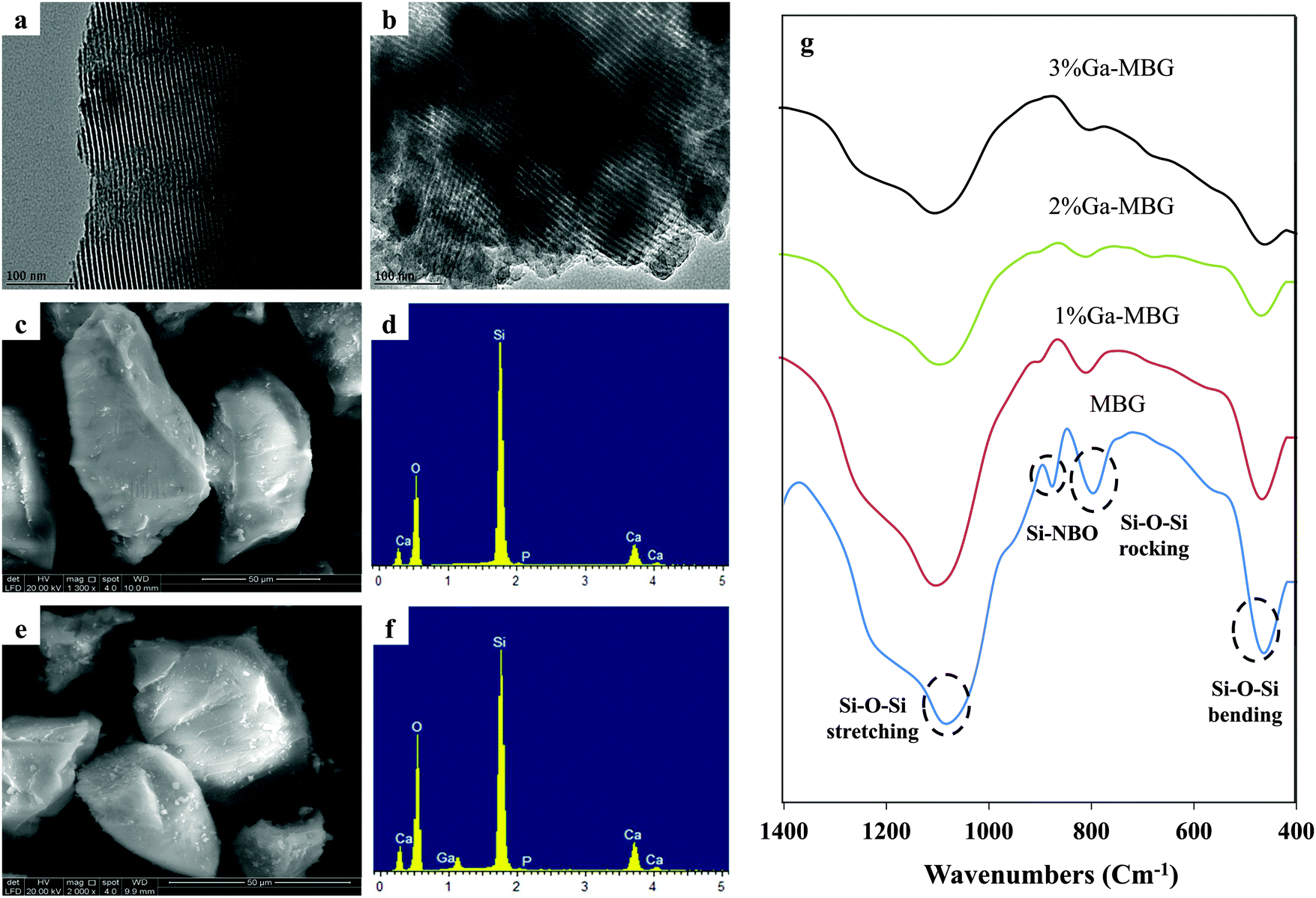 | ||
| Fig. 2 TEM images of the (a) MBG and (b) 1% Ga-MBG samples presenting their highly ordered mesoporous channel structure. SEM images and the EDS pattern of the (c and d) MBG and (e and f) 3% Ga-MBG samples. (g) FT-IR spectra of the MBG; 1% Ga-MBG; 2% Ga-MBG; and 3% Ga-MBG. | ||
Evidence for the local structural changes with addition of different concentrations of Ga2O3 can be obtained from the FT-IR spectroscopy. FT-IR spectra of the MBG and Ga-MBGs are given in Fig. 2g. All glasses represented two absorption peaks centered at around 1070 and 800 cm−1 corresponding to the Si–O–Si asymmetric stretching mode and symmetric Si–O–Si stretching or vibration modes of the silica ring structures, respectively.42,43 The peak at around 460 cm−1 is also assigned to the Si–O–Si bending mode.42 As can be seen in Fig. 2g, the intensity of the 460, 800 and 1070 cm−1 bands decreased with increasing Ga2O3 content upto 3 mol% indicating the depolymerizing role of Ga3+ on the silicate network. Therefore incorporation of Ga3+ into the MBG framework led to a decrement in the intensity of 460, 800 and 1070 cm−1 absorption bands, thus confirming the formation of more non-bridging oxygen NBOs (corresponding to the 970 cm−1 vibrational band). However, this band is not observed in 2% Ga-MBG and 3% Ga-MBGs due to the overlap with the broad Si–O–Si rocking peak.44
The zeta potentials of MBG and Ga-MBGs in PBS solution were measured in order to characterize their surface properties (Table 3). The surface of MBG was highly negative; the zeta potential was −30.4 mV, as a result of negatively charged silanol groups. In contrast to MBG, Ga-MBGs negligibly increased the zeta potential which can be attributed to the incorporation of Ga3+.
| Sample | Zeta potential (mV) in PBS at pH 7.4 |
|---|---|
| MBG | −30.4 |
| 1% Ga-MBG | −27.3 |
| 2% Ga-MBG | −26.2 |
| 3% Ga-MBG | −25.5 |
Ion release and degradability of MBG and Ga-MBGs in Tris–HCl solution
Changes in SiO44−, Ca2+, PO43− and Ga3+ concentrations in Tris–HCl buffer solution after various soaking times are presented in Fig. 3. As can be observed in Fig. 3a, the SiO44− concentration drastically increased in all samples during the first 48 h and then reached a steady state ranging from 60–70 ppm. A sharp increase in the Ca2+ concentration was also detected during the first 72 h of the test for all samples (Fig. 3b). Nevertheless, the concentration of Ca2+ for the glasses with higher Ga2O3 content (2% Ga-MBG and 3% Ga-MBG) was lower than that of MBG and 1% Ga-MBG. PO43− ion release profiles (Fig. 3c) of all the glasses also followed the same trend and increased up to 72 h, reaching values close to 6–8 ppm. Surprisingly, the concentration of the PO43− ions were higher for 1% Ga-MBG as compared with others. However, a different trend was found for Ga3+ ion release in Ga-substituted MBGs (Fig. 3d). The Ga3+ concentration increased after 3 h in the three Ga-containing samples and then experienced a steady state, reaching a value of 0.32 ppm in 3% Ga-MBG, that was approximately triple the amount found in 1% Ga-MBG. The reason why leaching of Ga3+ is low with respect to other ions is because of its high electric charge, which make it difficult to release from the glass network into the solution.45 It is worth noting that the concentration of Ga3+ ions arrived to a steady state earlier than that of SiO44−, Ca2+, and PO43− concentration in the glasses. The in vitro degradation properties of the samples were also determined by testing the weight loss ratio of the samples after soaking in Tris–HCl solution for various times. As can be inferred from Fig. 3, the weight loss increased upon prolonged immersion upto 14 days for all the materials, and then negligibly increased or remained almost constant. However, the degradation rates of the MBGs with higher amounts of Ga2O3 were found to be lower with respect to MBG and 1% Ga-MBG, probably due to their slow dissolution. The low dissolution of 3% Ga-MBG can be attributed to the acidic nature of its surface as a result of the presence of a high amount of Ga3+.46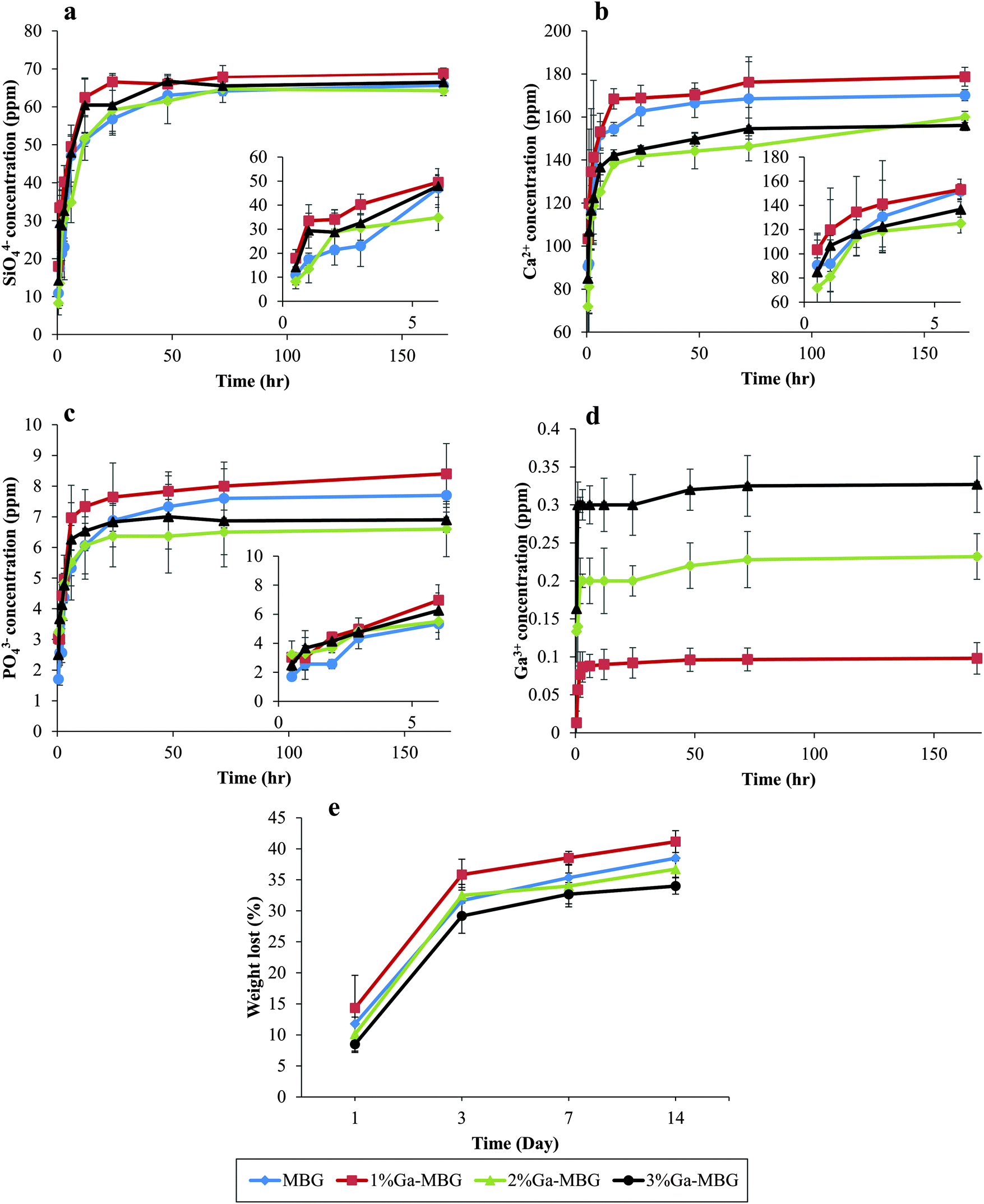 | ||
| Fig. 3 Concentration variation of (a) SiO44−, (b) Ca2+, (c) PO43− and (d) Ga3+ with the soaking time in Tris–HCl solution. The inset shows the ion release profile of the ions during the first 6 h. (e) Change of weight loss of MBG, 1% Ga-MBG, 2% Ga-MBG and 3% Ga-MBG samples in Tris–HCl solution. | ||
Blood coagulation experiments
To study the effect of the MBG and Ga-MBGs on the blood coagulation process, the PT and APTT of the glasses at two different amounts (5 and 10 mg) were measured. The APTT test is used to investigate the intrinsic pathway of blood coagulation and PT is a test performed to investigate extrinsic pathway of blood coagulation. The PT and APTT results are indicated in Fig. 4. The APTT results (Fig. 4a) revealed that all of the samples resulted in a significant activation of intrinsic pathways of the coagulation cascade since the values obtained for APTT were significantly diminished with respect to the negative control (blood without the sample). Notably, 1% Ga-MBG induced somewhat more pronounced effect on the APTT value than that observed with other glasses. However, no significant dosage-dependent trends were observed in APTT values since increasing the amount of samples from 5 to 10 mg did not significantly decline the values of APTT (Fig. 4a). Additionally, at 5 mg there was no significant difference in the PT values of the glasses (Fig. 4b). All the glasses showed approximately the same effect on PT values of that of the negative control. However, in the case of the higher glass amount (10 mg), PT was prolonged.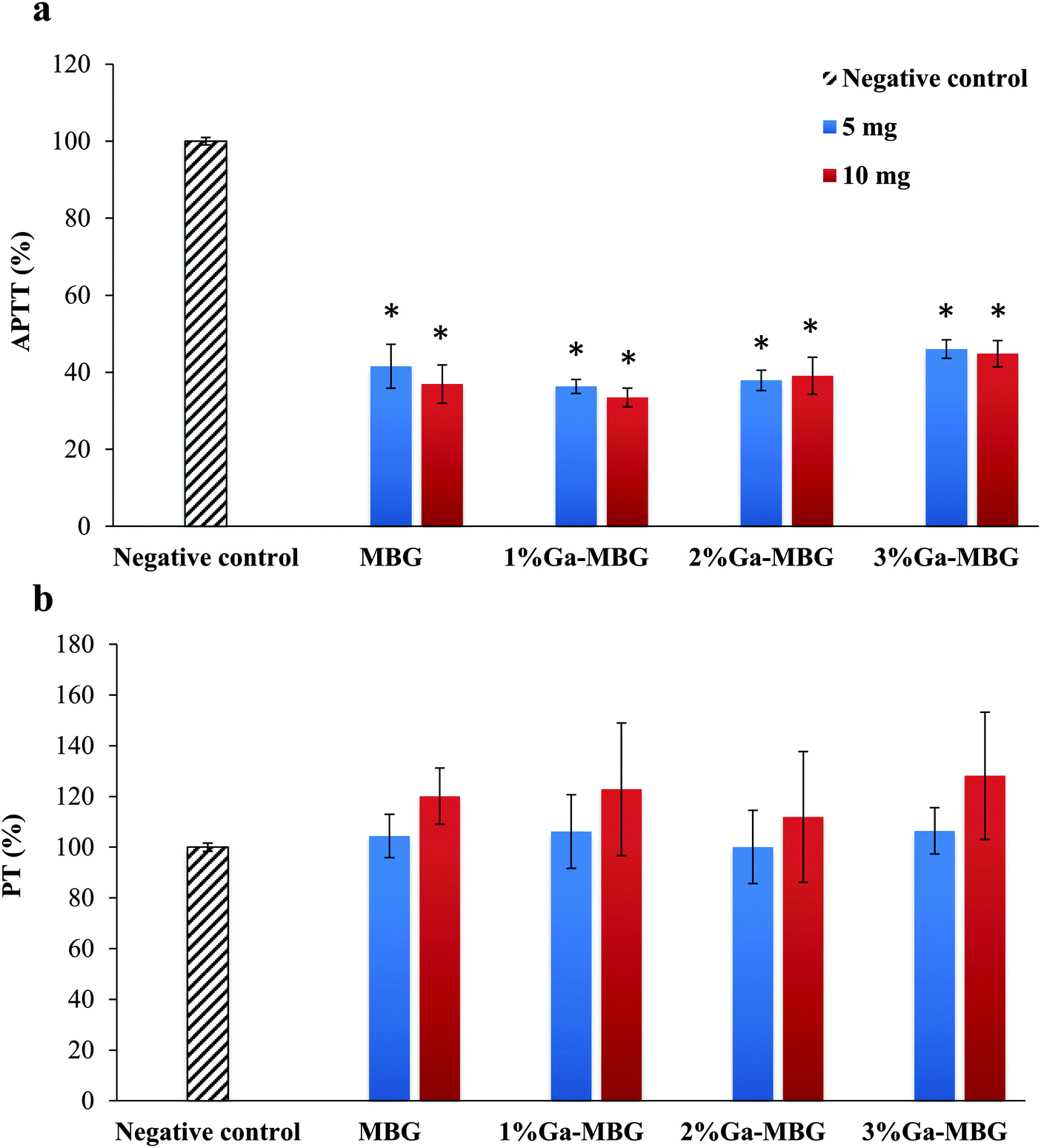 | ||
| Fig. 4 Influence of MBG and Ga-MBGs on (a) APTT and (b) PT of human plasma. * Represents a significant difference with respect to negative control at p < 0.05. | ||
PBS absorption study in vitro
The absorption capacity of the prepared samples was evaluated in vitro using PBS. The absorption ratios are presented in Fig. 5. The results revealed that the absorption coefficient of MBG with lowest Ga2O3 content (1% Ga-MBG) increased as compared with other glasses, which may be due to its outstanding textural properties (high surface area and pore volume).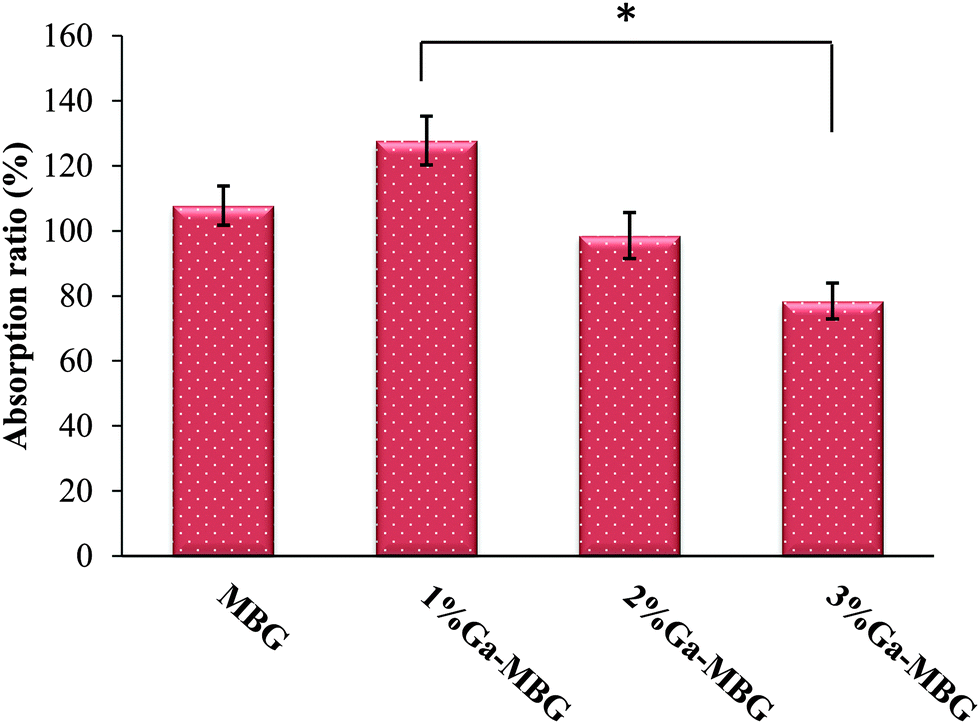 | ||
| Fig. 5 PBS absorption ratio of MBG and Ga-MBG samples. PBS absorption for 3% Ga-MBG indicated a tendency of lower absorption ratio. The difference between 1% Ga-MBG and 3% Ga-MBG was statistically significant (*p < 0.05). | ||
In vitro thrombus formation
The effect of Ga-substituted MBGs in comparison with MBG on thrombus formation is described in Fig. 6a. In all cases, thrombus formation increased with incubation time. As observed from Fig. 6a, the amount of thrombus formed was not significantly different between the groups during the initial 15 min. With increasing incubation periods upto 60 min, 1% Ga-MBG produced much more thrombus in contact with blood than MBG, indicating its higher thrombogenic activity, while 3% Ga-MBG exhibited lower thrombus formation. These results revealed the same trends as the blood coagulation experiments and PBS absorption studies. The digital image of MBG (Fig. 6b) and 1% Ga-MBG (Fig. 6c) also confirmed the results of the thrombus formation test. More red blood cells (RBCs) surrounded by a fibrin meshwork were aggregated on the 1% Ga-MBG surface with respect to MBG. The interaction of whole blood with MBG and Ga-MBGs was also seen by FESEM. FESEM images of both MBG (Fig. 6d and e) and 1% Ga-MBG samples (Fig. 6f and g) revealed that RBCs adhered onto their surface were dehydrated and aggregated, causing a change in cellular morphology.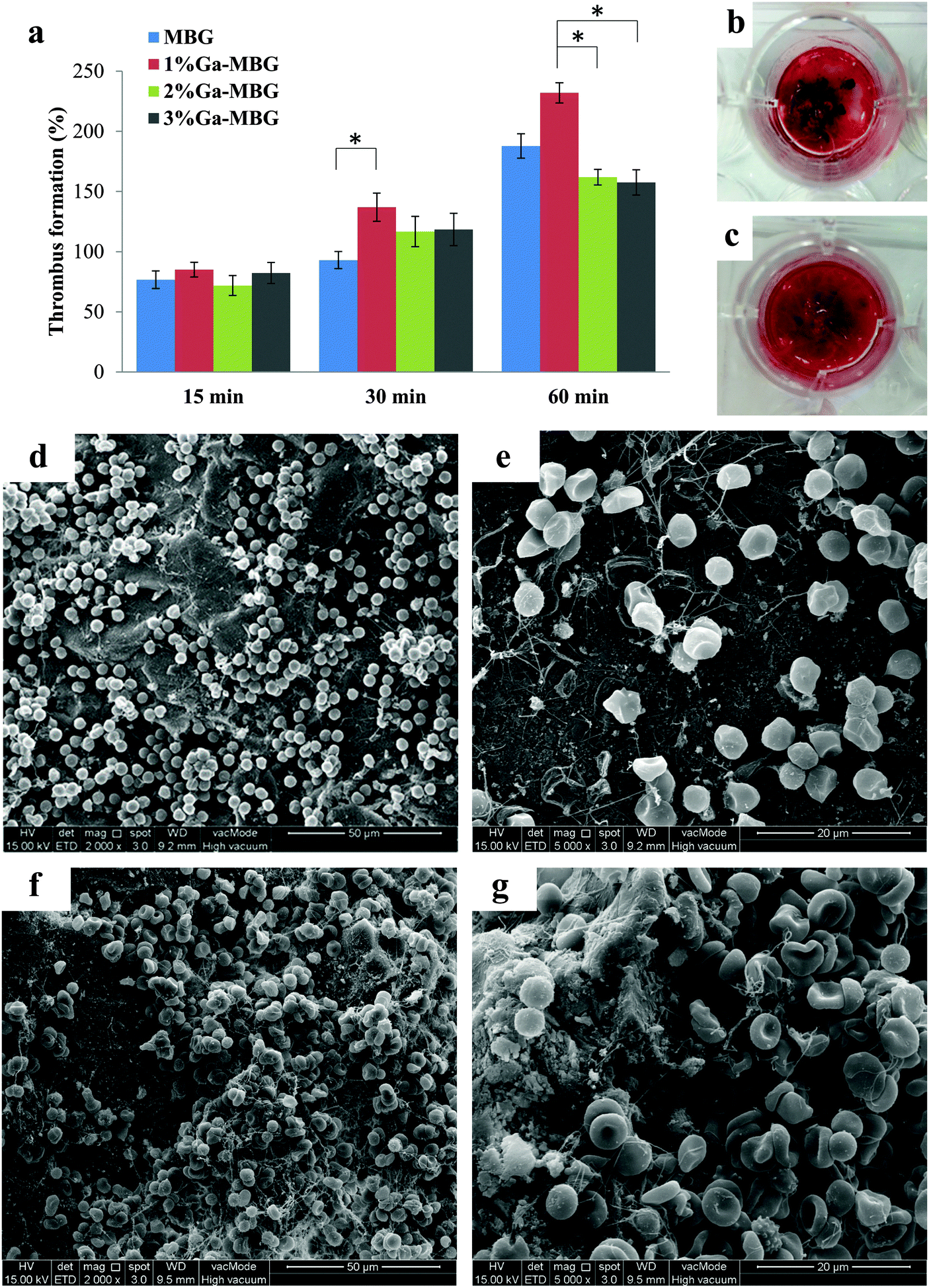 | ||
| Fig. 6 (a) Quantitative results of thrombus formation to the material surface after different incubation times. The blood clot formed on (b) MBG and (c) 1% Ga-MBG surfaces captured by a digital camera. FESEM micrographs of RBC agglutination and adhesion on the surfaces of (d) MBG, and (f) 1% Ga-MBG glasses after 30 min of incubation. Accumulated RBCs on (e) MBG and (g) 1% Ga-MBG surfaces under higher magnification (5000×). Many RBCs are trapped in a mesh of fibrin fibers and coalesced into thrombus on the 1% Ga-MBG surface. * Indicated a significant difference, p < 0.05. | ||
More specifically, the RBCs on the 1% Ga-MBG surface (Fig. 6f and g) seemed to form larger aggregates and coalesced into an erythrocyte plug trapped in the more extensive fibrin protein mesh with respect to MBG (Fig. 6d and e), for which much fewer aggregates were observed on its surface. The images were in agreement with the results of thrombus formation measurements.
Platelet adhesion
To assess the ability of Ga-substituted MBGs in enhancing platelet adhesion and aggregation in vitro, the synthesized materials were mixed with platelet suspensions for three time intervals (15, 30 and 60 min), and platelet adhesion was quantitatively measured using an LDH assay kit. Fig. 7 illustrates the qualitative and quantitative results of platelet adhesion on MBG and Ga-MBGs. It can be deduced from the results (Fig. 7d) that all the synthesized materials were able to significantly increase the number of adherent platelets at different time intervals. After 15 min incubation, no significant differences were found in the numbers of adherent platelets to the surfaces of the four types of glass studied. Most changes in the platelet number occurred during the subsequent 30 and 60 min. As compared with non-Ga-containing MBG, 2% Ga-MBG and 3% Ga-MBG, numerous platelets adhered on the surface of 1% Ga-MBG after 30 and 60 min incubation with PRP. However, the results showed an almost negligible increment in the number of platelets adhered onto the surface of other glasses with increasing incubation time. The FESEM also confirmed the significant ability of the 1% Ga-MBG to initiate platelet aggregation (Fig. 7b and c) after 30 min of incubation with PRP. The images show that the 1% Ga-MBG surface was covered with more platelets alongside silky fibrin than MBG (Fig. 7a), and also many pseudopodia were observed on the surface of 1% Ga-MBG, implying higher activation and aggregation of platelets. By contrast, less platelet adhesion and fibrin formation occurred on the surface of non-Ga-containing MBG (Fig. 7a).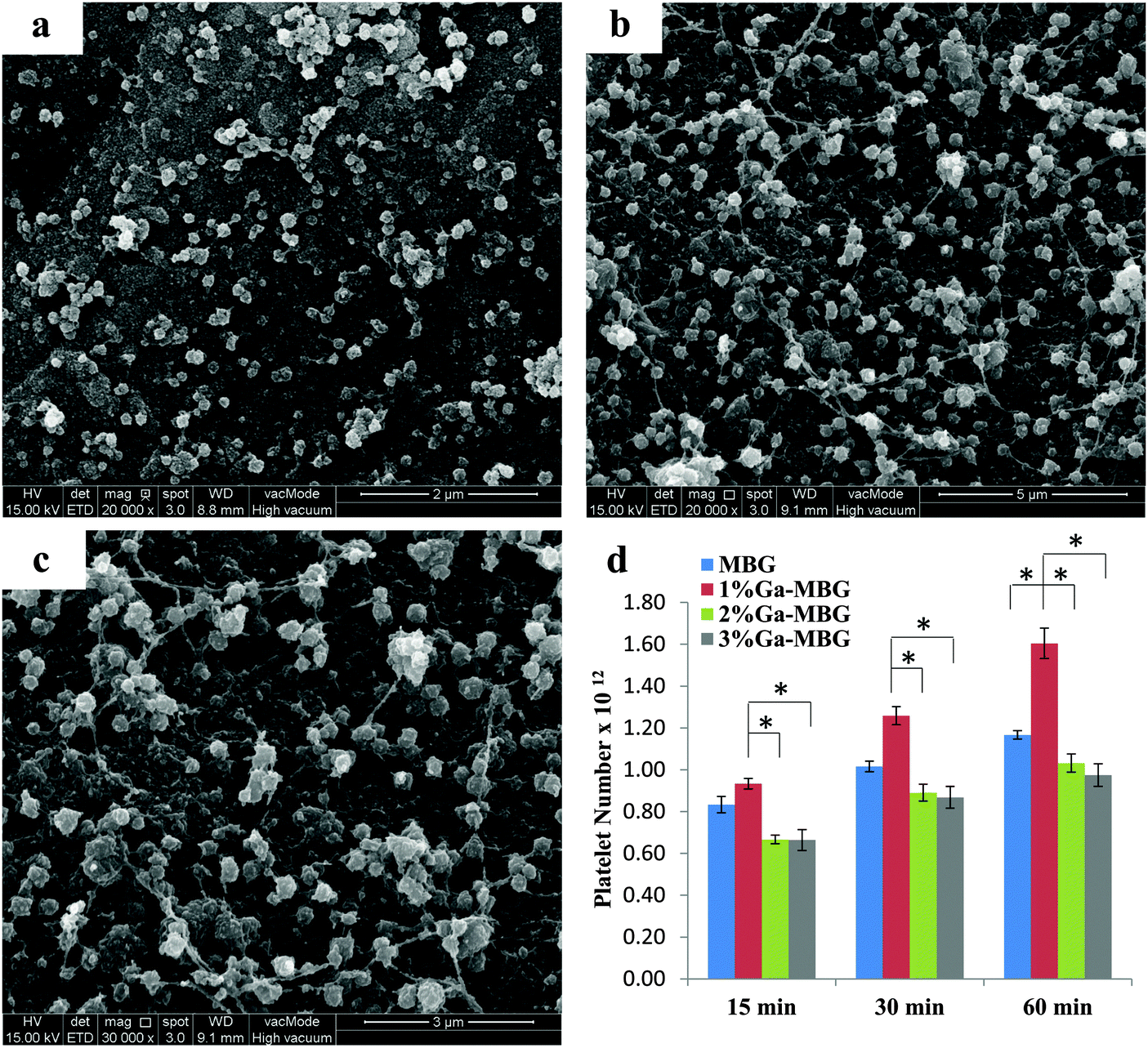 | ||
| Fig. 7 SEM images of platelets adhered on the glasses: (a) MBG, (b) 1% Ga-MBG, and (c) higher magnification image of (b). (d) Quantification of platelet adhesion on the MBG and Ga-MBGs. * Represented a significant difference, p < 0.05. | ||
Antibacterial activity
The antibacterial activity of Ga-MBGs was compared with that of non-Ga-containing MBG against both E. coli and S. aureus over 12 h as shown in Fig. 8a and b. Ga-MBGs demonstrated a potent antibacterial effect against both pathogens which can be attributed to the presence of Ga3+, since MBG did not impart a significant antibacterial effect. The increase in pH resulting from ion release in the solution is considered a possible antibacterial mechanism of MBG. Of note, the antibacterial efficacy of Ga-MBGs against both E. coli and S. aureus increased with Ga2O3 content over time. Fig. 8a indicated that 3% Ga-MBG exerted higher antibacterial activity against E. coli compared to MBGs with lower Ga2O3 content (1% Ga-MBG and 2% Ga-MBG) and achieved significant reduction in the number of viable cells within 12 h (Fig. 8a). However, differences between the antibacterial effect of 1% Ga-MBG and 2% Ga-MBG were not significant. In the case of S. aureus (Fig. 8b), 3% Ga-MBG had slightly more inhibitory effect compared to 1% Ga-MBG and 2% Ga-MBG, reaching its antibacterial rate of 99% at 12 h. Fig. 8 also presents images of colonies of E. coli (c and e) and S. aureus (d and f) incubated on agar plates obtained from cultured suspensions with negative control and 3% Ga-MBG. Obvious reduction was detected in the population of the E. coli colonies after exposure to the 3% Ga-MBG for 12 h (Fig. 8e) compared with negative control (Fig. 8c). A more pronounced reduction in S. aureus colonies was also seen upon exposure to 3% Ga-MBG (Fig. 8f) with respect to the negative control (Fig. 8d). It should be noted that Ga-MBGs displayed a more significant antibacterial impact against S. aureus than against E. coli.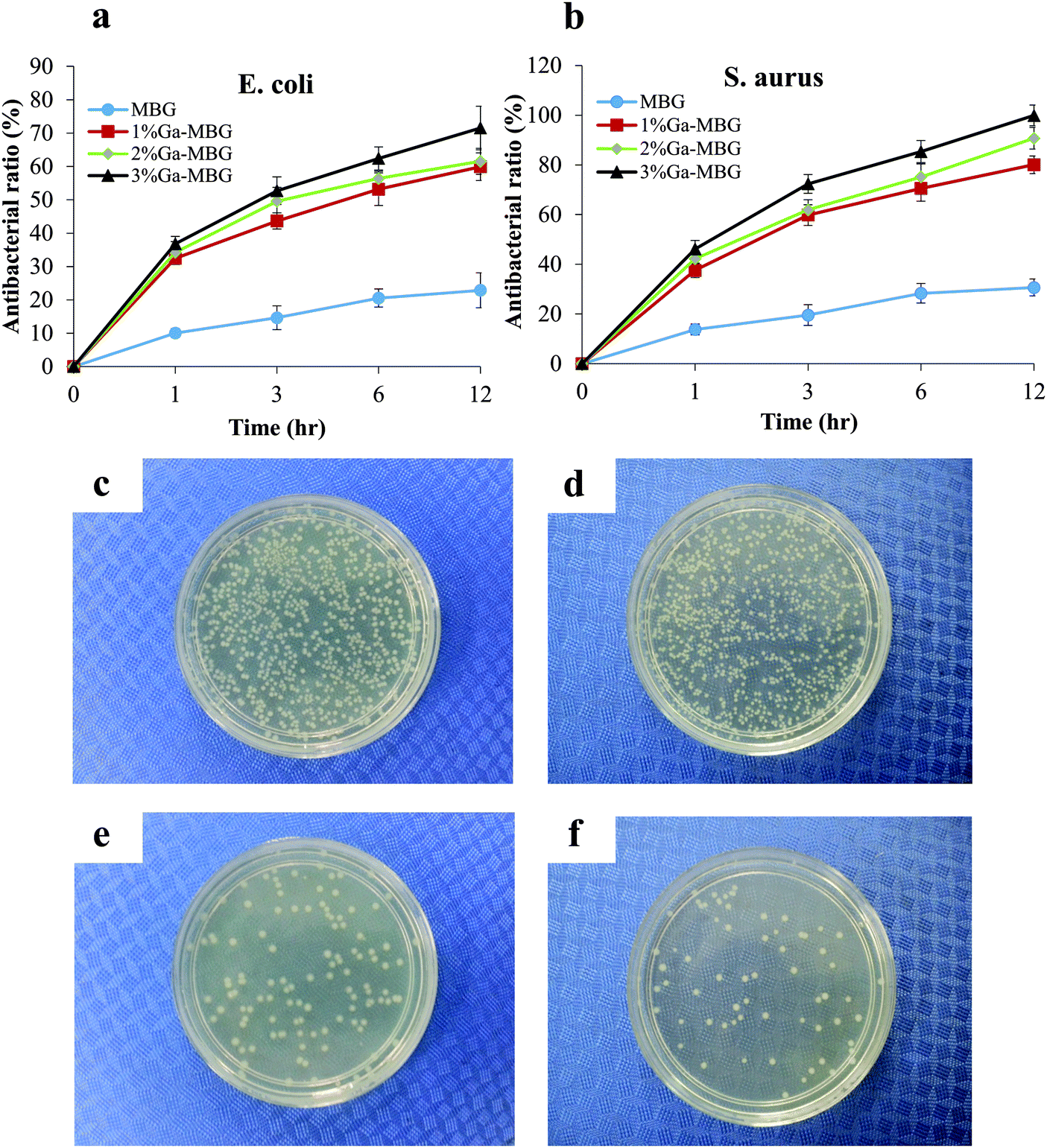 | ||
| Fig. 8 Bacteriostatic efficacies of Ga-MBGs against (a) E. coli and (b) S. aureus. Digital images representing the bacterial culture plates of E. coli upon exposure to (c) the negative control and (e) 3% Ga-MBGs and the bacterial culture plates of S. aureus treated with (d) the negative control and (f) 3% Ga-MBG. | ||
Cytotoxicity effects
To study the biocompatibility of the MBG and Ga-MBGs, cell viability of HDF cells was evaluated after they were treated with extracts of the bioactive glasses for 1 and 3 days. The MTT assay (Fig. 9) indicated that the cell viability of HDF cells in the presence of the MBG and Ga-MBG extracts increases with culture time, suggesting that all glasses were non-cytotoxic to HDF cells. In comparison to the control, a slightly higher cell viability was noted for all MBG materials at day 1. However, a significant increase in cell numbers for all the materials were observed after 3 days when compared to the control. Interestingly, the increment in the cell viability was more pronounced in the presence of 1% Ga-MBG.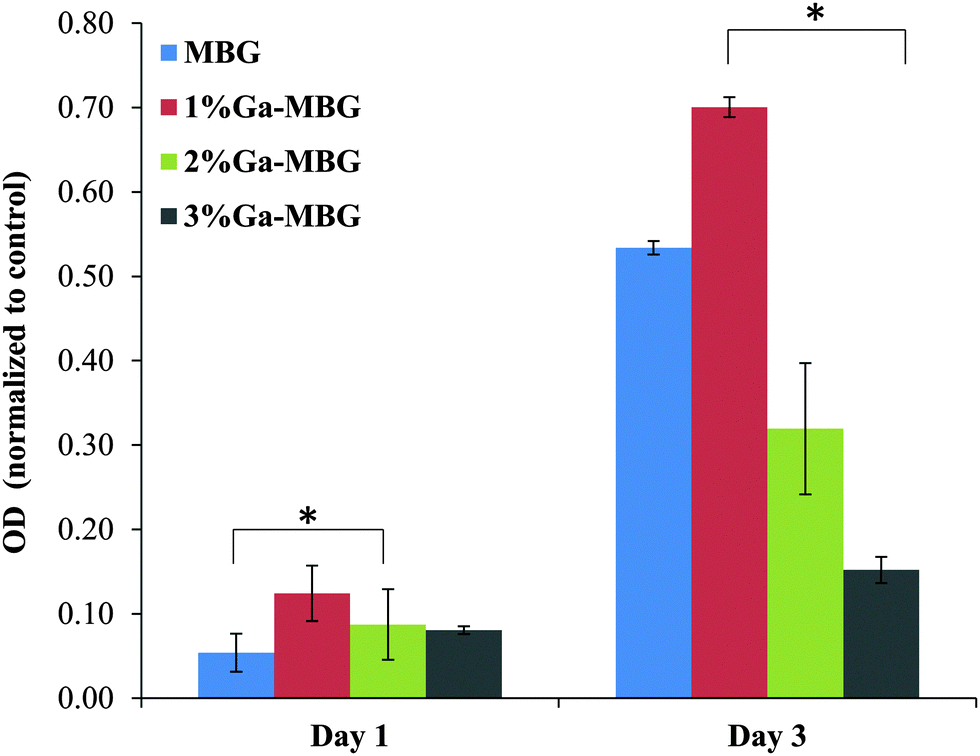 | ||
| Fig. 9 Viability of fibroblast cells after 1 day and 3 day exposure to the extracts obtained from MBG and Ga-MBGs. Data were obtained using MTT assay. * Represents a significant difference, p < 0.05. | ||
Discussion
Much attention has been focused on the fabrication of inorganic mesoporous materials as a new generation of inorganic hemostats that may be able to overcome any shortcoming associated with commercial inorganic hemostats such as QC and QCG. Whilst some reports highlight the hemostatic performance and antibacterial properties of MBGs,25,26,40 more studies are needed to clarify the hemostatic properties and antibacterial activity of these materials. Chemical modification of MBGs through incorporation of therapeutic elements into their frameworks may be one way to improve their hemostatic capacity and antibacterial activity. As discussed earlier, Ga(NO3)3 has the ability to induce the earliest stages of hemostasis. Considering this, in the present study we prepared Ga-MBGs that not only demonstrated an accelerating effect on the blood coagulation cascade but, also impart an antibacterial effect against both E. coli and S. aureus.Hemostasis is a complex physiological process that prevents blood loss and proceeds via the synchronized action of three mechanisms including vasoconstriction, formation of a platelet plug and blood coagulation.2 So, the hemostatic agents that perform their task by accelerating one or more of the processes above can be of help in achieving hemostasis. There are multiple mechanisms underlying MBG and Ga-MBGs' hemostatic properties. Based on the obtained results in the coagulation assays, it was observed that, unlike the extrinsic pathway that was insignificantly affected by the bioactive glasses, the intrinsic coagulation pathway was considerably accelerated in the presence of the synthesized materials with respect to the negative control (Fig. 4). The accelerated coagulation response induced by the synthesized samples can depend on multiple factors. In detail, both MBG and Ga-MBGs, due to possessing mesoporous interconnected structures with large specific surface area, can act as factor concentrators and share a mechanism of action similar to QC. Such materials capture and store large volumes of water within their pores resulting from the electrostatic interaction with Ca2+ ions residing in their pores. These interactions lead to the hyperconcentration of coagulation factors and other protein and cellular components of the plasma that in turn would accelerate the coagulation cascade and the subsequent fibrin clot formation. However, a prolonged APTT was observed with increasing Ga2O3 content in the MBG network (e.g., for 3% Ga-MBG) in comparison with MBG (Fig. 4). We may speculate that this prolonged effect of MBG with higher Ga2O3 content on APTT values can be related to deterioration of the hexagonal ordered structures of the MBG, which decreases its textural properties (specific surface area and pore volume) and subsequently reduces its water absorption coefficient. In contrast to 3% Ga-MBG and other prepared bioactive glasses, a shortening of the APTT was occurred as a result of adding MBG with lowest Ga2O3 content (1% Ga-MBG), which can be attributed to its outstanding textural properties. The 1% Ga-MBG surface, due to its larger specific surface area (509 m2 g−1) and pore volume (0.78 cm3 g−1), serve plenty of spaces resulting in greater permeability of the water molecules to their pores and subsequent blood clot formation. Accordingly, the mesoporous structure of the MBG materials plays a pronounced role in the activation of the intrinsic pathway. These results were consistent with the PBS absorption results: indeed, the 1% Ga-MBG sample was superior to all other synthesized glasses in water absorption ability (Fig. 5).
Another favorable factor that contributes to the hemostatic function of the glasses can be related to their potent procoagulant activity resulting from their negative surface charge. The presence of the negative charge on the surface of all synthesized materials were confirmed by zeta potential measurements (Table 3). The synthesized materials exhibited significant negative zeta potential in PBS at pH 7.4. The accepted explanation is that, when the bioactive glasses come in contact with body fluids, a unique surface reaction occurs including rapid ion exchange reactions between the glass network modifiers with H+ (or H3O+) ions from the solution.47,48 This reaction leads to hydrolysis of the silica groups, subsequent formation of silanol (Si–OH) groups on the glass surface and, eventually, the production of a negatively charged surface with functional groups (Si–O−). As previously proved, negatively charged materials such as kaolin can accelerate the hemostasis of the blood via activation of a coagulation cascade.25,49–51 Due to this, such materials are expected to have a hemostatic mode of action similar to that of QCG, which is a kaolin-based hemostat.52,53 When the MBG and Ga-MBGs are exposed to blood, their negative surface charge trigger the intrinsic blood coagulation cascade. The basic principle of such phenomena relies on the fact that coagulation factor XII (Hageman factor) binds to negatively charged surfaces through positively charged amino acids in its heavy chain.54,55 The binding facilitates subtle conformational changes in FXII, leading to the formation of active FXIIa through auto-activation. It is evident that this activated factor directly contributes to fibrin formation.56 Therefore, negatively charged surfaces of Ga-MBGs are considered as a key contributor to the activation of the intrinsic pathway of blood coagulation. However, the zeta potential became less negative when a higher amount of Ga2O3 was incorporated in the MBG framework as compared with Ga-free MBG (Table 3). The less negative charge imparted on the surface of highest substituted Ga-MBG can be correlated with the presence of a high amount of acidic Ga3+ species at the surface of the glass as previously demonstrated by Aina et al.46 The authors demonstrated that the presence of abundant surface Ga3+-species acting as strong Lewis acid sites is responsible for the enhanced surface acidity of the Ga-modified glasses compared with Ga-free glasses. Therefore, this could be another acceptable reason for the prolongation of the APTT in the presence of 3% Ga-MBG.
Another possible component that dominates the procoagulant trend of the investigated samples is ascribed to their polar framework resulting from the silanol groups resident on their surface, which have very strong polar interactivity. Previous reports have suggested that blood coagulates faster on polar glass surfaces than nonpolar plastic surfaces; this phenomenon is referred to as the “glass effect”.57,58 The underlying principle for this observation is that polar surfaces are involved in the autocatalytic activation of clotting Factors XII and XI along with prekallikrein and high-molecular-weight-kininogen (HMWK), and in the subsequent activation of the multiple mechanisms responsible for the association of the thrombin enzyme and, eventually, of fibrin production.55,59 The polar framework of the MBG materials that is generated by the presence of surface silanol groups can thus act as a desirable surface for the contact activation of the intrinsic pathway. More specifically, FTIR spectra of the MBG and Ga-MBGs confirmed the presence of non-bridging oxygen (NBOs) in the glass network, which in turn facilitates the formation of Si–OH functional groups on the surface of glasses. We assume that increasing the Ga2O3 content in the MBG framework shifts its role from that of a network former to a network modifier, since a significant decrease in the intensity of 460, 800 and 1070 cm−1 bands was observed in higher substituted Ga-MBG, thus confirming the formation of more NBOs (corresponding to 970 cm−1 vibrational band). These results prove the discussion presented by Franchini et al. who investigated the structural role of different concentrations of Ga3+ in bioactive phospho-silicate glasses.45 The authors explained that the structural behavior of this ion relies on its amount in the glass network so that the increasing Ga2O3 content changes its role from that of a network former to a network modifier. Although we expect that shortening of APTT becomes more pronounced in the presence of 3% Ga-MBG due to a more pronounced formation of NBOs and, subsequently, of silanol groups, it was much shorter in with the presence of 1% Ga-MBG, which has the lowest network modifier content. This indicates that the role of surface area can be more noticeable in the formation of nucleation sites (Si–OH) rather than the presence of a network modifier. Indeed, 1% Ga-MBG with a high surface area released ions quickly, inducing a large number of OH− groups to deposit on its surface, and further activation of the intrinsic pathway.
Similarly to zeolite-based hemostats, the presence of Ca2+ in the glass network also plays an important role in its procoagulant action. The synthesized materials have ability to present Ca2+ to blood, which speeds up the intrinsic pathway of blood coagulation. Ca2+ (known as clotting factor IV) is a cofactor that plays a ubiquitous role in the coagulation cascade. It is involved in the immobilization and orientation of clotting enzymes on cellular surfaces, serving as the ionic bridge between two negatively charged residues (such as the cellular surface and clotting factors). It also serves as a required co-factor for several enzymatic reactions, including conversion of prothrombin to thrombin, the enzyme that aids in the generation of insoluble fibrin from the soluble fibrinogen. Ca2+ ions are also consumed during splitting fibrinogen into fibrin monomers and polymerization of monomers into a fibrin strand and, eventually, lead to crosslinking and formation of ticker fibrin strands. Indeed, they act as an adhesive to hold the fibrin monomers to each other to form the polymeric fibrin fibers. Hence, Ca2+ availability in the MBG framework is another factor favorable to the activation of the intrinsic pathway and subsequent shortened APTT. Interestingly, comparing the results from the ion release study and coagulation assay indicated a very close correlation between shortened APTT and Ca2+ release profiles so that 1% Ga-MBG, having a higher capability to release Ca+2 than other glass formulations, led to the further reduction of APTT. Contrary to what occurs in conventional glasses (melt and sol–gel bioactive glasses), where a higher amount of network modifier leads to lower network connectivity, a rapid ion exchange and a subsequent rapid ion release, mesoporous bioactive glasses do not apparently exhibit the same trend. Here, we observed that an increase in the Ga2O3 content provoked a decrease in the release rate of the ions, which is evidenced by MP-AES results. Such results may support the idea that the incorporation of a higher amount of Ga+3 into MBG disrupts the order of the mesostructure, resulting in a decrement of the textural properties, which are essential parameters to produce a unique ion release profile in the mesoporous bioactive glasses. Therefore, the lower Ca2+ release rate of the 3% Ga-MBG system was considered to be another leading cause of APTT prolongation. On the contrary, in the case of 1% Ga-MBG, with the lowest Ga2O3 content and outstanding textural properties (higher specific surface area and pore volume), the Ca2+ release profile evidenced that, just after 0.5 h immersion in Tris–HCl solutions, a substantial amount of Ca2+ (∼103 ppm) was released. These results could be explained as owing to the synergy of both the low Ga2O3 content (1 mol%) and high specific surface area (502 m2 g−1), which provoked a larger Ca2+ release, necessary for the activation of the intrinsic pathway.
It is reported that polyvalent cations such as trivalent ions can also promote the natural blood clotting cascade.60,61 Ferric (Fe3+) ions are examples of the ions that can generate the most biologically reactive hydroxyl radicals (HRs) through reaction with hydroxyl groups of water.60,62–64 These radicals, in turn, lead to polymerization of fibrinogen into strong fibrin clots and subsequently stimulate the blood coagulation cascade.65 It is interesting to note that Ga3+ not only is a polyvalent cation, but also has an ionic radius nearly to that of Fe3+. There is some speculation that Ga3+ may share with Fe3+ a similar mechanism of action to accelerate coagulation and blood clotting. Therefore, another reason why 1% Ga-MBG despite lower Ga2O3 content is a stronger activator of the coagulation cascade compared to the other samples can be attributed to the synergy of both its excellent intrinsic textural and structural features alongside the presence of Ga3+.
Besides the role of the glasses in the activation of the intrinsic pathway, they were also found to be effective in thrombus formation and platelet aggregation. From the results of thrombus formation assay in Fig. 6, distinct amounts of thrombus were noted on four MBGs, in particular on the 1% Ga-MBG surface. It is encouraging that, unlike typical clotting which is composed of a platelet plug and a mesh of cross-linked fibrin protein, the unique blood clot formed by MBG materials also consisted of a homogenous layer of RBCs that are stuck in the fibrin mesh to form a clot. It is expected that two main factors, including the silanol-rich surface of MBGs that bind with the phosphatidyl choline-rich RBC membrane66 and the large surface area and pore volume of the glasses that provide more lattice to trap cellular and blood plasma components, are involved in this process. Hence, a possible explanation for the higher thrombogenic potential of 1% Ga-MBG is attributed to its higher surface area that increases the accessibility of silanol groups to the blood cells, leading to the production of a stable hemostatic clot.
These results were consistent with those of the platelet adhesion assay. 1% Ga-MBG dramatically enhanced the platelet adhesion and activation compared to other glasses, since a large number of platelets were detected on its surface with spread morphology and protruding pseudopodia. Our hypothesis is that a larger surface area in 1% Ga-MBG served as a favourable surface for interaction with platelets and fibrinogen, causing clumping and activation of platelets. Likewise, the Ca2+ ions that are released from the synthesized materials can contribute to platelet activation, since they accelerate the production of the proteolytic enzyme thrombin. Thrombin is a serine protease that stimulates platelet activation and aggregation through activation of protease-activated receptors on the platelet cell membrane.67
The major concern with inorganic hemostats such as QC and Woundstat (WS, a Smectite clay) is related to their non-biodegradability, which can lead to thrombosis in distal organs such as brain and lungs.52 For this reason, the degradability properties of hemostatic agents should be evaluated. Here, we assessed the degradation ability of MBG and Ga-MBGs after immersion in Tris–HCl solution over time. 1% Ga-MBG showed a more pronounced degradation over 14 days compared with other glasses and lost about 32% of its initial weight after soaking for 14 days. The faster degradation rate of 1% Ga-MBG is probably due to its quick dissolution.
In addition to assessing the hemostatic functions of the prepared bioactive glasses, their antibacterial properties and biocompatibility were investigated, since infection and cellular toxicity at the site of bleeding are other challenges faced by hemostatic agents. Based on the results of the antibacterial activity, incorporation of Ga3+ into the MBG framework implied a faster and more potent bacteriostatic action than non-Ga-containing MBG against both pathogens that were tested. As compared to both pure LB broth culture and MBG, 3% Ga-MBG had a significant inhibitory effect on the growth of both E. coli and S. aureus in time-kill assays, in particular at 12 h. Moreover, 1% Ga-MBG and 2% Ga-MBG samples exhibited more bacteriostatic activity with respect to either pure LB broth culture or MBG. These results were in good agreement with the Ga3+ release profile from the glass network (Fig. 3). Although Ga3+ had a slower release rate than other ions, it was good enough to achieve a significant antibacterial effect. Interestingly, MBG demonstrated slight antibacterial activity against the two bacterial strains considered in this work. This could be due to the change in pH (data not shown) during the glass degradation, as observed in Tris–HCl. One possible explanation of the antibacterial effect of Ga3+ may be that, differently from another well-known antibacterial ion such as Ag+,22 this ion has an ionic radius nearly identical to that of Fe+3, which is critical to the pathogenesis of infections.68 It is believed that Ga3+ may compete with the Fe3+ ions in various biochemical reactions and function as a Trojan horse since many biological systems are unable to distinguish it from Fe3+.69 Notably, supplementation of Ga3+ can result in disruption or inhibition of those biomedical reactions requiring Fe3+. However, unlike Fe3+, Ga3+ cannot be reduced under the same conditions and can lead to inhibition of a number of essential biological reactions such as those responsible for DNA and protein synthesis.68,70 Accordingly, Ga3+ has the potential to serve as a Fe3+ analog and is an effective antibacterial. However, compared with S. aureus that was found to be the most susceptible organism to Ga3+, E. coli developed resistance to Ga3+, which can be ascribed to structural differences in the cell walls of these two pathogens. S. aureus is surrounded by a peptidoglycan layer with a loosely packed network structure, which allows Ga3+ to pass through the cell wall into the interior without much difficulty, while E. coli have a thicker peptidoglycan cell wall, which protects the cell from penetrating Ga3+.71 For these reasons, Ga-MBG samples exerted a better antibacterial effect against S. aureus than E. coli. Furthermore, the MTT assay illustrated that HDF cells maintained high levels of viability in the presence of both non-Ga-containing MBG and Ga-MBGs, indicating that the synthesized materials have good cytocompatibility. The higher O.D. values in MBG and Ga-MBGs, which show more live cells, demonstrated an enhanced HDF cells' proliferation in the samples as compared with control in a short culture time period such as 1 day. For a longer culture time, such as 3 days, we found that cells can grow on 1% Ga-MBG dramatically better than those on the other glass samples. It is supposed that both the high surface area and high rate of ion release may stimulate cell responses and eventually affect the viability of cells. Therefore, the mentioned factors could be possible reasons for higher cell viability of HDF cells in the presence of 1% Ga-MBG extract. It is interesting to find that Ga3+ does not appear cytotoxic to HDF cells as its release was maintained well below the toxicity level (14 ppm).45
In short, the synergistic effect of all the factors presented above, including chemical activation and physical absorption, promote the blood coagulation cascade and render Ga-MBG materials very suitable hemostatic materials. Among the samples, 1% Ga-MBG exhibited the best overall performance.
Conclusions
In the present study, Ga-MBG materials have been successfully developed for hemostatic application and their antihemorrhagic effect alongside biocompatibility and antibacterial properties were evaluated. Our results demonstrated that the proportion of Ga2O3 in MBG was a critical parameter affecting hemostatic events. While the 1% Ga-MBG accelerated intrinsic coagulation cascade, thrombus formation and platelet adhesion compared to MBG, the benefits of Ga2O3 were negated at higher concentrations (3 mol%). Therefore, the lowest Ga-substituted MBG sample, with its combination of improved hemostatic performance, high degradability, high cytocompatibility and antibacterial activity against both E. coli and S. aureus, may be a candidate for consideration as a hemostat for controlling hemorrhage and infection. To the best of our knowledge, these results are the first instance of a Ga-substituted MBG for hemostatic application being discussed in the literature. Further investigation will be required to more precisely delineate the principle behind the improved hemostatic efficacy of Ga-MBGs, and validate the findings in vivo.Acknowledgements
Financial support from the Ministry of Higher Education (MOHE), Government of Malaysia, under the High Impact Research (UM.C/625/1/HIR/MoE/ENG/58) scheme and the University of Malaya Research Grants scheme (UMRG, RG156-12AET) is gratefully acknowledged.References
- B. J. Eastridge, R. L. Mabry, P. Seguin, J. Cantrell, T. Tops, P. Uribe, O. Mallett, T. Zubko, L. Oetjen-Gerdes and T. E. Rasmussen, J. Trauma Acute Care Surg., 2012, 73, S431–S437 CrossRef PubMed.
- S. Pourshahrestani, E. Zeimaran, I. Djordjevic, N. A. Kadri and M. R. Towler, Mater. Sci. Eng., C, 2016, 58, 1255–1268 CrossRef CAS PubMed.
- F. L. Wright, H. T. Hua, G. Velmahos, D. Thoman, D. Demitriades and P. M. Rhee, J. Trauma Acute Care Surg., 2004, 56, 205–208 CrossRef PubMed.
- V. Shanmugam and M. Robinson, Colorectal Dis., 2009, 11, 221–222 CrossRef CAS PubMed.
- P. Rhee, C. Brown, M. Martin, A. Salim, D. Plurad, D. Green, L. Chambers, D. Demetriades, G. Velmahos and H. Alam, J. Trauma Acute Care Surg., 2008, 64, 1093–1099 CrossRef PubMed.
- Y. Ran, E. Hadad, S. Daher, O. Ganor, J. Kohn, Y. Yegorov, C. Bartal, N. Ash and G. Hirschhorn, Prehosp. Disaster Med., 2010, 25, 584–588 Search PubMed.
- M. E. Chávez-Delgado, C. V. Kishi-Sutto, X. N. A. de la-Riva, M. Rosales-Cortes and P. Gamboa-Sánchez, J. Surg. Res., 2014, 192, 678–685 CrossRef PubMed.
- D. S. Kauvar, R. Lefering and C. E. Wade, J. Trauma Acute Care Surg., 2006, 60, S3–S11 CrossRef PubMed.
- J. K. Wright, J. Kalns, E. A. Wolf, F. Traweek, S. Schwarz, C. K. Loeffler, W. Snyder, L. D. Yantis Jr and J. Eggers, J. Trauma Acute Care Surg., 2004, 57, 224–230 CrossRef.
- B. Kheirabadi, US Army Med. Dep. J., 2011, 2, 25–37 Search PubMed.
- D. R. King, J. Trauma Acute Care Surg., 2011, 71, 1775–1778 CrossRef PubMed.
- Ç. Çinar, M. E. Odabaş, G. Akca and B. Işik, J. Clin. Exp. Dent., 2012, 4, e151 CrossRef PubMed.
- C. K. Murray, S. A. Roop, D. R. Hospenthal, D. P. Dooley, K. Wenner, J. Hammock, N. Taufen and E. Gourdine, Bacteriology of war wounds at the time of injury, DTIC Document, 2006 Search PubMed.
- N. E. Aronson, J. W. Sanders and K. A. Moran, Clin. Infect. Dis., 2006, 43, 1045–1051 CrossRef PubMed.
- X. Yan, C. Yu, X. Zhou, J. Tang and D. Zhao, Angew. Chem., Int. Ed., 2004, 43, 5980–5984 CrossRef CAS PubMed.
- L. L. Hench, J. Am. Ceram. Soc., 1998, 81, 1705–1728 CrossRef CAS.
- Y. Li, Y.-Z. Liu, T. Long, X.-B. Yu, T. T. Tang, K.-R. Dai, B. Tian, Y.-P. Guo and Z.-A. Zhu, J. Mater. Sci.: Mater. Med., 2013, 24, 1951–1961 CrossRef CAS PubMed.
- Y. Zhu, C. Wu, Y. Ramaswamy, E. Kockrick, P. Simon, S. Kaskel and H. Zreiqat, Microporous Mesoporous Mater., 2008, 112, 494–503 CrossRef CAS.
- P. Jiang, F. Qu, H. Lin, X. Wu, R. Xing and J. Zhang, IET Nanobiotechnol., 2012, 6, 93–101 CrossRef CAS PubMed.
- A. Salinas, S. Shruti, G. Malavasi, L. Menabue and M. Vallet-Regi, Acta Biomater., 2011, 7, 3452–3458 CrossRef CAS PubMed.
- S. Shruti, A. J. Salinas, E. Ferrari, G. Malavasi, G. Lusvardi, A. L. Doadrio, L. Menabue and M. Vallet-Regi, Micropor Mesopor Mat, 2013, 180, 92–101 CrossRef CAS.
- N. Gargiulo, A. M. Cusano, F. Causa, D. Caputo and P. A. Netti, J. Mater. Sci.: Mater. Med., 2013, 24, 2129–2135 CrossRef CAS PubMed.
- C. Wu and J. Chang, Interface Focus, 2012, rsfs20110121 Search PubMed.
- G. Hu, L. Xiao, P. Tong, D. Bi, H. Wang, H. Ma, G. Zhu and H. Liu, Int. J. Nanomed., 2012, 7, 2613 CrossRef CAS PubMed.
- C. Dai, Y. Yuan, C. Liu, J. Wei, H. Hong, X. Li and X. Pan, Biomaterials, 2009, 30, 5364–5375 CrossRef CAS PubMed.
- C. Dai, C. Liu, J. Wei, H. Hong and Q. Zhao, Biomaterials, 2010, 31, 7620–7630 CrossRef CAS PubMed.
- X. Wu, J. Wei, X. Lu, Y. Lv, F. Chen, Y. Zhang and C. Liu, Biomed. Mater., 2010, 5, 035006 CrossRef PubMed.
- E. Zeimaran, S. Pourshahrestani, B. Pingguan-Murphy, N. A. Kadri, H. A. Rothan, R. Yusof, M. R. Towler and I. Djordjevic, J. Mater. Sci., 2015, 50, 2189–2201 CrossRef CAS.
- L. R. Bernstein, Pharmacol. Rev., 1998, 50, 665–682 CAS.
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877 CrossRef CAS PubMed.
- E. Zeimaran, S. Pourshahrestani, I. Djordjevic, B. Pingguan-Murphy, N. A. Kadri and M. R. Towler, Mater. Sci. Eng., C, 2015, 53, 175–188 CrossRef CAS PubMed.
- M. Rogosnitzky, US Pat., US8652509 B2, 2014 Search PubMed.
- P. H. Goodley and M. Rogosnitzky, J. Case Rep. Med., 2011 DOI:10.1155/2011/819710.
- J. Goncalves, N. Wasif, D. Esposito, J. M. Coico, B. Schwartz, P. J. Higgins, R. S. Bockman and L. Staiano-Coico, J. Surg. Res., 2002, 103, 134–140 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- W. Fan, D. Wu, T. Ma and B. Fan, Dent. Mater. J., 2015, 34, 54–60 CrossRef CAS PubMed.
- Y. Imai and Y. Nose, J. Biomed. Mater. Res., 1972, 6, 165–172 CrossRef CAS PubMed.
- S.-Y. Ong, J. Wu, S. M. Moochhala, M.-H. Tan and J. Lu, Biomaterials, 2008, 29, 4323–4332 CrossRef CAS PubMed.
- M. Ip, S. L. Lui, V. K. Poon, I. Lung and A. Burd, J. Med. Microbiol., 2006, 55, 59–63 CrossRef CAS PubMed.
- A. López-Noriega, D. Arcos, I. Izquierdo-Barba, Y. Sakamoto, O. Terasaki and M. Vallet-Regí, Chem. Mater., 2006, 18, 3137–3144 CrossRef.
- D. Arcos, M. Vila, A. López-Noriega, F. Rossignol, E. Champion, F. Oliveira and M. Vallet-Regí, Acta Biomater., 2011, 7, 2952–2959 CrossRef CAS PubMed.
- C. Vaid, S. Murugavel, R. Kashayap and R. P. Tandon, Micropor Mesopor Mat, 2012, 159, 17–23 CrossRef CAS.
- X. Yan, X. Huang, C. Yu, H. Deng, Y. Wang, Z. Zhang, S. Qiao, G. Lu and D. Zhao, Biomaterials, 2006, 27, 3396–3403 CrossRef CAS PubMed.
- M. Franchini, G. Lusvardi, G. Malavasi and L. Menabue, Mater. Sci. Eng., C, 2012, 32, 1401–1406 CrossRef CAS PubMed.
- V. Aina, C. Morterra, G. Lusvardi, G. Malavasi, L. Menabue, S. Shruti, C. L. Bianchi and V. Bolis, J. Phys. Chem. C, 2011, 115, 22461–22474 CAS.
- M. N. Rahaman, D. E. Day, B. S. Bal, Q. Fu, S. B. Jung, L. F. Bonewald and A. P. Tomsia, Acta Biomater., 2011, 7, 2355–2373 CrossRef CAS PubMed.
- V. Grover, A. Kapoor, R. Malhotra and R. S. Uppal, J. Indian Soc. Periodontol., 2013, 17, 104 CrossRef PubMed.
- H. B. Alam, Z. Chen, A. Jaskille, R. I. L. C. Querol, E. Koustova, R. Inocencio, R. Conran, A. Seufert, N. Ariaban and K. Toruno, J. Trauma Acute Care Surg., 2004, 56, 974–983 CrossRef CAS.
- T. A. Ostomel, Q. Shi and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 8384–8385 CrossRef CAS PubMed.
- P. Walsh, Br. J. Haematol., 1972, 22, 393–405 CrossRef CAS PubMed.
- B. S. Kheirabadi, J. W. Edens, I. B. Terrazas, J. S. Estep, H. G. Klemcke, M. A. Dubick and J. B. Holcomb, J. Trauma Acute Care Surg., 2009, 66, 316–328 CrossRef PubMed.
- J. W. Carraway, D. Kent, K. Young, A. Cole, R. Friedman and K. R. Ward, Resuscitation, 2008, 78, 230–235 CrossRef CAS PubMed.
- R. W. Colman and A. H. Schmaier, Blood, 1997, 90, 3819–3843 CAS.
- C. Sperling, M. Fischer, M. F. Maitz and C. Werner, Biomaterials, 2009, 30, 4447–4456 CrossRef CAS PubMed.
- T. Renné, A. H. Schmaier, K. F. Nickel, M. Blombäck and C. Maas, Blood, 2012, 120, 4296–4303 CrossRef PubMed.
- T. A. Ostomel, Q. Shi, P. K. Stoimenov and G. D. Stucky, Langmuir, 2007, 23, 11233–11238 CrossRef CAS PubMed.
- T. A. Ostomel, P. K. Stoimenov, P. A. Holden, H. B. Alam and G. D. Stucky, J. Thromb. Thrombolysis, 2006, 22, 55–67 CrossRef CAS PubMed.
- G. D. Stucky, T. A. Ostomel, Q. Shi, P. K. Stoimenov and P. A. Holden, US Pat., US7858123 B2, 2010 Search PubMed.
- T. K. Keene, J. A. Patterson, J. W. Reding and J. A. Thompson, WO Pat., WO2001082896 A1, 2001 Search PubMed.
- J. A. Patterson, US Pat., US6521265 B1, 2003 Search PubMed.
- B. Lipinski and E. Pretorius, Pol. Arch. Med. Wewn., 2012, 122, 115–122 CAS.
- B. Lipinski, E. Pretorius, H. M. Oberholzer and W. J. Van Der Spuy, Microsc. Res. Tech., 2012, 75, 1185–1190 CrossRef CAS PubMed.
- E. Pretorius, J. Bester, N. Vermeulen and B. Lipinski, Curr. Drug Targets, 2013, 14, 13 CrossRef CAS PubMed.
- B. Lipinski and E. Pretorius, Front. Hum. Neurosci., 2013 DOI:10.3389/fnhum.2013.00735.
- Y. Zhao, X. Sun, G. Zhang, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, ACS Nano, 2011, 5, 1366–1375 CrossRef CAS PubMed.
- S. R. Coughlin, Nature, 2000, 407, 258–264 CrossRef CAS PubMed.
- S. P. Valappil, D. Ready, E. A. A. Neel, D. M. Pickup, W. Chrzanowski, L. A. O'Dell, R. J. Newport, M. E. Smith, M. Wilson and J. C. Knowles, Adv. Funct. Mater., 2008, 18, 732–741 CrossRef CAS.
- C. Chitambar and J. Narasimhan, Pathobiology, 1991, 59, 3–10 CrossRef CAS PubMed.
- C. R. Chitambar, Int. J. Environ. Res. Public Health, 2010, 7, 2337–2361 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |