
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide-regulated growth of electrocatalytic metal nanoparticles directly onto a carbon paper electrode†
Yaovi
Holade
,
David P.
Hickey
and
Shelley D.
Minteer
*
Departments of Chemistry and Materials Science and Engineering, University of Utah, 315 S 1400 E Rm 2020, Salt Lake City, Utah 84112, USA. E-mail: minteer@chem.utah.edu
First published on 14th October 2016
Abstract
Direct growth of hierarchical micro/nanostructured metal arrays on a 3D substrate is a powerful tool to enhance the catalytic efficiency of metal particles towards a wide range of substrates. In this contribution, we demonstrate a novel and versatile method for growing anisotropic microstructures directly onto a 3D carbon paper electrode, using Au, Pd and Pt nanoparticles as elementary building blocks. It was determined that halides play a crucial role in the morphology of synthesized Au particles, leading to either complex flower-like rough surfaces (exhibiting high catalytic activity) or featureless smooth surfaces (exhibiting low catalytic activity). Of the three metal materials studied, the Pt decorated carbon paper (Pt@carbon) material exhibits a high electrocatalytic activity toward the oxygen reduction reaction in an alkaline medium. We demonstrate that this new Pt material can operate as a cathode in an alkaline glucose fuel cell exhibiting outstanding peak power density of 2–3 mW cm−2 (using an anode of Au@carbon or Pt@carbon at room temperature and low metal loading without any fuel circulation). This newly described fabrication approach allows for the rational control of metallic particle growth, paving a new way towards materials with regulated surface roughness allowing for tuneable physical, optical and catalytic properties.
Introduction
The development of electrochemical energy conversion devices, such as solid alkaline membrane fuel cells (SAMFCs) that utilize alcohols as a fuel source, is of paramount importance. Their effectiveness requires the design of innovative procedures for synthesizing advanced electrode materials that help to optimize the catalytic efficiency while reducing the metal catalyst loading. Therefore, seeking highly active and durable catalysts has become a prerequisite for achieving efficiency in both the oxygen reduction reaction (ORR) and alcohol oxidation. An ideal catalytic material should have a high dispersion of the active sites, controllable surface structure and morphology, and minimal metal loading that allow for optimization of activity, selectivity, and durability. For platinum group metals (PGMs), the most widely used approaches rely on engineering materials with tuneable surface structure and composition, such as core–shell materials and multimetallic nanoalloys.1–7 Another common approach for increasing the efficiency of metallic nanoparticles (NPs) is to partially or completely remove a sacrificial component in alloys, so that the removal of the more reactive metal leads to nanocages8–10 or nanoframes11–13 with well-defined and controllable facets. The obtained materials exhibit nanoscale defects in their channels and on their pore walls, which are of particular interest for most catalytic applications because these sites are often a source of catalytic activity. The majority of existing synthetic methodologies require materials to be stabilized in a liquid or dispersed in a powder before using a binder, such as Nafion, to immobilize the NPs on a substrate (i.e. glassy carbon) prior to use. While the use of a Nafion ionomer contributes to the proton transport within the electrode materials, the use of such binders adds a layer of complexity to the fabrication process, and degradation of binders results in the undesired loss of NPs and subsequent decrease in catalytic performance.14–16 Thus far, only a few attempts have been devoted to direct synthesis of NPs onto an electrode surface such as carbon paper to further reduce these issues. One of the solutions was the self-assembled 3D metallic nano-architectures, which have received tremendous interest in catalysis.17–20 Hence, direct syntheses of NPs onto carbon papers or glassy carbon have been initiated from methods of electrodeposition,19–23 electrospinning,24–26 and wall-jet configuration.27 However, these methods result in high loading of precious metals and loss of catalytic activity due to the packing of NPs inside the electrode fibers. Furthermore, many attempts have been made to use the halide anion, Br−, as a capping agent to regulate the aqueous synthesis of Pd and Ag NPs in a polyol-based system.28,29 The effect of halide ions on the growth rate of NPs in solution has been addressed during the last ten years and the obtained particles have a smooth surface, mostly from pre-made small seeds.30 However, it is still unknown how other halides, such as Cl−, Br− and I−, could impact the direct growth of metallic particles on a 3D substrate and subsequently the electrochemical performances.To address all these challenges, we report herein a versatile chemical method to grow 3D metallic microstructures at the surface of carbon paper. The success of this approach for in situ growth relies on a control of the reaction time together with the use of halide anions, Br− and I−, as effective tools. We discovered that the incorporation of Br− into the reaction mixture is critical for tailoring both highly rough surfaces and intrinsic catalytic activity of Au decorated carbon paper (Au@carbon paper), thus contributing to high electrocatalytic activity of the resulting particles towards glucose oxidation and excellent stability. Since the particles are directly deposited onto the carbon paper, there is no need to prepare a catalytic ink, which enables (i) bypassing the use of binders, (ii) avoiding active site-obstruction, (iii) minimizing metal detachment from the electrode, and (iv) preventing particle agglomeration.
Experimental
Chemicals and materials
Untreated Toray carbon paper TGP-H-060 was purchased from Fuel Cell Earth (see Fig. S1†). Trihydrate tetrachloroauric(III) acid (HAuCl4·3H2O, 99.9%), potassium tetrachloropalladate(II) (K2PdCl4, 99.99%), hexahydrate hexachloroplatinic(IV) acid (H2PtCl6·6H2O, 37.50% Pt basis), lead(II) nitrate (Pb(NO3)2, ≥99.0%) and D-(+)-glucose (99.5%) were obtained from Sigma-Aldrich. Potassium bromide (KBr, 99.6%) and potassium chloride (KCl, 100%) were purchased from Fisher Scientific. Potassium iodide (KI) was purchased from Amresco® (USA). Potassium nitrate (KNO3) and sodium hydroxide (NaOH, 97%) were purchased from MACRON Fine Chemicals™. Potassium sulphate (K2SO4, 100%) was supplied from J. T. Baker (USA). L-(+) ascorbic acid (AA, >99%) was purchased from Research Products International Corp. (USA). All chemicals were used as received. All name ultra-pure water is provided from Milli-Q Millipore source (MQ: 18.2 MΩ cm at 20 °C).Synthesis of anisotropic particles on Toray carbon paper
The synthesis was performed using a square of 5 cm sides as exposed areas, (2 × 25 cm2 available for both sides) of TGP-060-H Toray carbon paper (bulk density: 0.4514 ± 0.003 g cm−3).Results and discussion
Novel method for particles growth at carbon substrate
Compared to the traditional method for particle preparation (Fig. 1a, pathway 1), the proposed approach (pathway 2) simplifies the process and post-synthetic steps into one overall route by assembling the metallic particles directly onto a carbon support. An image of the support before and after the reaction where the red colour indicates the deposition of Au particles is illustrated in Fig. 1a. In order to regulate the growth of Au particles, potassium halide precursors, KX (X = Cl, Br, I) were added so that the ratio of KX to Au (φ) follows φ = n(KX)/n(HAuCl4). Fig. 1b–d highlight the colour change of HAuCl4 aqueous solutions as a function of the ratio of added KX to Au. This is further illustrated by UV-Vis spectra (Fig. 1e), indicating the replacement of Cl− by other halides Br− or I− to form [AuCl4−xXx]−. Further chemical reduction of the complex in the presence of halide ions yields the nucleation and growth of NPs directly onto carbon paper. | ||
| Fig. 1 Schematic illustration of the two general approaches for the modification of substrates by metallic (nano)particles. (a) Pathway 1: the traditional method that leads to particles (solution or powder), requires the preparation of an ink before the incorporation on the support; pathway 2: simplified method for the direct growth of metallic particles onto a carbon paper by a chemical method in presence of halide ions. AA: L-ascorbic acid. (b–d) From top to down: photo showing the colour change of HAuCl4 aqueous solution (fixed at 2.5 mM) after the addition of KCl (b), KBr (c) and KI (d) for different amount of φ = n(KX)/n(HAuCl4). (e) UV-Vis spectra of HAuCl4 aqueous solution (diluted 25-times) in presence of different halides to regulate the Au particles growth. | ||
Physical structure and morphological characterization of the metal particles decorated carbon fiber paper
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was fixed. Indeed, considering its larger ionic radius and preferential adsorption on crystallographic facets (hkl),30–32 we hypothesized that Br− would have a greater effect on the anisotropic growth rate than Cl−. Upon addition of KBr in the aqueous solution of HAuCl4, the mixture instantaneously undergoes a colour change from yellow to brown, Fig. 1b–d (extended data are reported within Fig. S2a†). This transition was studied by UV-Vis measurements (Fig. 1e), which showed that the main absorption peak at 200–220 nm and 290–320 nm (assigned to the ligand-to-metal charge transfer transition phenomenon in metal complex ions33) disappeared while two new characteristic features appear at 253 and 380 nm, indicating that [AuCl4]− was successfully replaced by [AuBr4]− (extended UV-Vis data are reported within Fig. S2 and S3†). Upon addition of AA, the mixture turned progressively from brown to colourless, indicating that Au(III) was being reduced into the metallic state, i.e., Au0 and the previous peaks disappeared from the UV-Vis spectrum as reported in Fig. S2a and b.†
:1 was fixed. Indeed, considering its larger ionic radius and preferential adsorption on crystallographic facets (hkl),30–32 we hypothesized that Br− would have a greater effect on the anisotropic growth rate than Cl−. Upon addition of KBr in the aqueous solution of HAuCl4, the mixture instantaneously undergoes a colour change from yellow to brown, Fig. 1b–d (extended data are reported within Fig. S2a†). This transition was studied by UV-Vis measurements (Fig. 1e), which showed that the main absorption peak at 200–220 nm and 290–320 nm (assigned to the ligand-to-metal charge transfer transition phenomenon in metal complex ions33) disappeared while two new characteristic features appear at 253 and 380 nm, indicating that [AuCl4]− was successfully replaced by [AuBr4]− (extended UV-Vis data are reported within Fig. S2 and S3†). Upon addition of AA, the mixture turned progressively from brown to colourless, indicating that Au(III) was being reduced into the metallic state, i.e., Au0 and the previous peaks disappeared from the UV-Vis spectrum as reported in Fig. S2a and b.†
To evaluate the influence of reaction time on the growth and subsequently the morphology of Au particles, we quenched the reaction at 0.5, 1, 2 and 4 h after the addition of AA. Fig. 2 depicts SEM and HRSEM micrographs for the different reaction times. After 30 minutes, AuNPs were observed by HRSEM assembling into 3D sponges-like structure with a diameter of 500–800 nm composed of 40–60 nm ultrathin 3D particles. However, longer reaction times resulted in the loss of surface features on the nanostructures. After four hours, the initial morphology deteriorated towards aggregated featureless NPs of 30–70 nm in diameter. The initial morphology results from the simultaneous adsorption of Br− on various crystallographic (hkl) facets. Indeed, the growth rate of low-indexed Au surfaces (100), (110) and (111) is similar in the presence of Br−.30–32 The continuous diffusion and reduction of Au(III) will lead to a rough surface. Previous reports have suggested that a combination of molecular oxygen and halide ions can result in oxidative etching of NPs that induces anisotropic growth and leads to the disappearance of twinned nanostructures.17,34,35 Therefore, considering the abundance of Br− in the solution as well as O2 from the air, we attributed the observed degradation of the grown AuNPs to the etching capability of Br−/O2 as well as Cl−/O2. To prevent this morphology deformation, we quench the reaction at 30 min, unless specified elsewhere.
 | ||
| Fig. 2 Au particles growth on Toray carbon paper (in the presence of Br−) with n(KBr)/n(HAuCl4) = 12 and 4 wt% Au. SEM/HRSEM images for different reaction time: (a and e) 0.5 h, (b and f) 1 h, (c and g) 2 h, (d and h) 4 h. | ||
 | ||
| Fig. 3 (a–d) SEM and (e–h) HRSEM micrographs of metallic particles growth on Toray carbon paper. Au for n(KBr)/n(HAuCl4) = 100:4 wt% (a and e) and 8 wt% (b and f) at room temperature. (c and g) Pd for n(KBr)/n(K2PdCl4) = 100 and 8 wt% at 40 °C. (d and h) Pt for 8 wt% at 40 °C. | ||
To elucidate the role of larger halide ions X− (X = Br, I) on AuNP growth, various amounts of each halide were introduced in the form of KX, where the relative amount of X− to Au is described as φ = n(KX)/n(HAuCl4). The standard redox potentials of the involved Au(I) and Au(III) halides, the dissociation constants of Au(III) halide at 25 °C, and the standard redox potentials of the involved metal complexes with halides within Tables S1–S3† demonstrate that the addition of X−, will result in the conversion of [AuCl4]− into mixed-ion complexes that evolve towards the more stable [AuX4]− in the presence of ionic ligand X−. Otherwise, the mixed ion-complex [AuCl4−xXx]−, with 0 < x ≤ 4, is obtained according to eqn (1).
| [AuCl4](aq)− + xKX(aq) → [AuCl4−xXx](aq)− + xKCl(aq) | (1) |
Since the ionic radii (r) of I− and Br− (r(I−) = 220 pm and r(Br−) = 195 pm) are larger than their counterpart chloride (r(Cl−) = 181 pm), the partial or total substitution of Cl− by X− is expected to accentuate the steric hindrance around the metal cation, which will further control the seed growth, similar to that observed in the presence of carbon black.36 Fig. S5–S7† show (HR)SEM images for NPs grown using Br− or I− solutions where φ = 0, 2, 4, 24 and 96. These results indicate that anisotropic growth and control of particle distribution on the support depend on excess amounts of X− in the solution. Indeed, according to eqn (1), φ = 4 corresponds to an equivalent replacement of Cl− by X−. However, SEM images of NPs prepared with solutions where φ = 4 (Fig. S5c, S5g, S6a and b†) show few of the expected morphological features, suggesting that an excess of halide ions is required. Indeed, HRSEM images of NPs prepared from solutions of Br− where φ = 96 (Fig. 3e and f) reveal intricate 3D microstructures composed of hundreds of NPs. The slats protruding from these structures are 5–10 nm thick and tens of square micrometers across each face. However, HRSEM images of NPs grown in solutions containing a large excess of I− (Fig. S5i–k and S7†), reveal the self-assembly of AuNPs to form leaf crystals. Taken together, these results suggest that the rapid anisotropic growth of Au particles results from preferential adsorption of X− at primary NPs, and that these adsorption sites act as “seeds” for growth in different directions. We further performed control syntheses to determine whether the anisotropy can be influenced by the nature of other transition metals, Pd and Pt. Pd enables obtaining microstructures exhibiting higher exposed surface area because of the presence of many defects as can be seen in Fig. 3c and g. Pt material is composed of microspheres ≈200–600 nm diameter decorating homogeneously the entire external surface of each fiber (Fig. 3d and h). To better support the role of halides, we performed control experiments at 8 wt% using halides (HRSEM images within Fig. S8†) and other ions, i.e., NO3− and SO42− (HRSEM images within Fig. S9†). Both non-halide ions lead to 200–400 nm microspheres even if NO3− leads to rougher surface. The control syntheses of Au at 40 °C in absence of X− (HRSEM images within Fig. S10†) and its presence (HRSEM images within Fig. S11–S13†) substantiate the main role of the halides. These results underpin completely the role of the halides on the morphological control of the gold crystals growth.
 | ||
| Fig. 4 HRSEM micrographs for the Au particles morphology evolution for initial solution containing: (a) Br− for φ = 12 and 4 wt%, (b) Br− for φ = 96 and 4 wt%; (c) I− for φ = 96 and 4 wt%; (d) Br− for φ = 100 and 8 wt%; (e) Cl− for φ = 100 and 8 wt%; (f) I− for φ = 100 and 8 wt%; (g) control in absence of halides. The reaction time is 30 minutes at room temperature. | ||
 | ||
| Fig. 5 Compositional, structural and surface characterizations of metallic particles growth on Toray carbon paper with metal loading of 8 wt%. (a) EDX, (b) XRD patterns of Au (synthesis in presence of I−, Br−, Cl−): inset shows the patterns for Au (111) crystallographic plane and (c) high-resolution XPS spectra of the Au 4f core level: inset shows the surface atomic (at%: left y-axis) and weight (wt%, right y-axis) compositions of Au. (d) UPD of lead of Au electrodes in 0.1 M NaOH + 1 mM Pb(NO3)2 at 20 mV s−1. | ||
Electrochemical performance
Next we aimed to study the newly described NP materials as electrode materials. We investigated the electrocatalytic properties by considering two model reactions in alkaline medium. Prior to that, we first compared the responses of the electrodes in CV using a model of PbUPD, which has proved its efficiency for characterizing the surface of metallic nanostructures, as well as for the assessment of the crystallographic structure.Fig. S14c† depicts the recorded CVs in 0.1 M NaOH showing well defined electrochemical characteristics of Au. Fig. 5d and S14d and e† depict the PbUPD for different electrodes exhibiting the same overall behaviour. Starting at 0.8 V vs. RHE and sweeping the potential down, two reduction peaks appear at 0.55 and 0.41 V vs. RHE, which are typically attributed to the deposition of lead on (110) and (111) Au facets, respectively.38 Similarly, the two stripping peaks at 0.45 and 0.58 V vs. RHE recorded during the positive scan are basically assigned to a reversible desorption of the previous Pb layer on (111) and (110) Au facets, respectively. It is worth noting that the slight shift of the above peaks compared to common observations38 and the duplication of (111) could be assigned to the presence of high-index (hkl) facets and/or defects. As shown within Fig. S14e,† a close examination reveal the presence of (100) facets, which are known to display high activity towards glucose oxidation.38,39 The presence of these low-coordination number (100), (110) and (111) is expected to enhance the catalytic properties of Au materials. Importantly, the obtained results greatly support the above hypothesis of Au particles growth at different (hkl) directions leading to various facets.
We next evaluated the electrocatalytic properties of Au, Pd and Pd materials for ORR in 0.1 M NaOH (Fig. 6a) while the steady-state blank CVs are reported in Fig. S15.† Note that the convention used to plot the current–potential curves corresponds to a positive reduction current and a negative oxidation current. As shown within the inset, the results reveal that ORR occurs at Pt electrodes at ca. 1.1 V vs. RHE, ultimately approaching the theoretical equilibrium potential of 1.18 V vs. RHE. Pd electrode shows better ORR performances for E ≥ 0.8 V vs. RHE that comes from the availability of hydroxyl species, the main ORR performance descriptors. Overall, the outcomes indicate that Au material is not an efficient cathode electrode while Pt and Pd aspire to be potential active cathodic electrocatalysts for ORR.
 | ||
| Fig. 6 Electrochemical characterization of the as-synthesized materials for the metal loading of 8 wt% (i.e. 0.4 mgmetal cm−2). (a) Positive-going ORR polarization curves (insets show the dashed region) recorded at Au, Pd and Pt materials at 5 mV s−1 in O2-saturated 0.1 M NaOH aqueous solution. (b) Voltammograms for glucose (10 mM) oxidation at Au materials in 0.1 M NaOH at 50 mV s−1: inset shows the chronoamperometry curves recorded at 0.65 V vs. RHE. (c) Fuel cell polarization curves (iR-uncorrected): the Pt cathode (1 cm2) operated in 0.5 M NaOH + O2; the Au anode (1 cm2 for FC-1), Pt anode (1 cm2 for FC-2) and Pt anode (2 cm2 for FC-3) operated in 0.5 M NaOH + 0.3 M glucose; a Fumatech AEM separated both compartments. (d) Voltammetry polarization curves recorded in 0.5 M NaOH for O2 reduction (upper) and glucose (0.3 M) electrooxidation (lower). | ||
The electrocatalytic activity of the as-prepared Au-based materials was then studied for glucose (10 mM) electrooxidation in 0.1 M NaOH. Among our investigated parameters during the synthesis of Au materials using the halide Br−, a ratio φ = n(KBr)/n(HAuCl4) of 96 gives the best performances in terms of activity and stability (CV within Fig. S16a†). This outcome substantiates the SEM results (Fig. S5†), the rough surface provides more active sites for the reaction. Fig. 6b and S16b† highlight high catalytic activity of the material synthesized in the presence of Br− compared to Cl− and I−. For 0.1 M glucose (Fig. S16c†), an enhanced peak current density jpeak of 14 mA cmgeo−2 is reached. From Fig. 6b and S16d–f,† the chronoamperometry curves recorded at 0.65 V vs. RHE show that Br− enables obtaining a stable electrocatalyst that is 2- and 5-fold higher than Cl− and I−, respectively. The high activity of the material obtained with Br− is rationally ascribed to its structure, especially the presence of different defects that lead to high surface area. Otherwise, Fig. 6b shows that Au electrodes exhibits improved kinetics (the reaction starts at E ≤ 0.3 V vs. RHE) with ca. 100 mV shift towards lower electrode potentials compared to their counterparts in literature,20,24 which is a good indicator for future application as anode material. Importantly, the peak current density is at least 2-fold higher while the present Au weight is only 4 wt% compared to 30 wt% for electrospinning24 and low loading of 0.4 mgmetal cm−2 compared to 3 mgmetal cm−2 for electrodeposition.20 In other words, the present carbon paper based Au materials exhibit highly enhanced activity with very low Au amount that is 7-fold lower than the literature. Furthermore, Fig. S15a† demonstrates that the achieved peak current density of 9 mA cmAu−2 is higher than the conventional chemically synthesized AuNPs by an order of magnitude.38 Thus, using the as-synthesized bromide-regulated particles growth, for instance Au (having rough surface) as anode for glucose oxidation should enable designing an efficient DGFC.
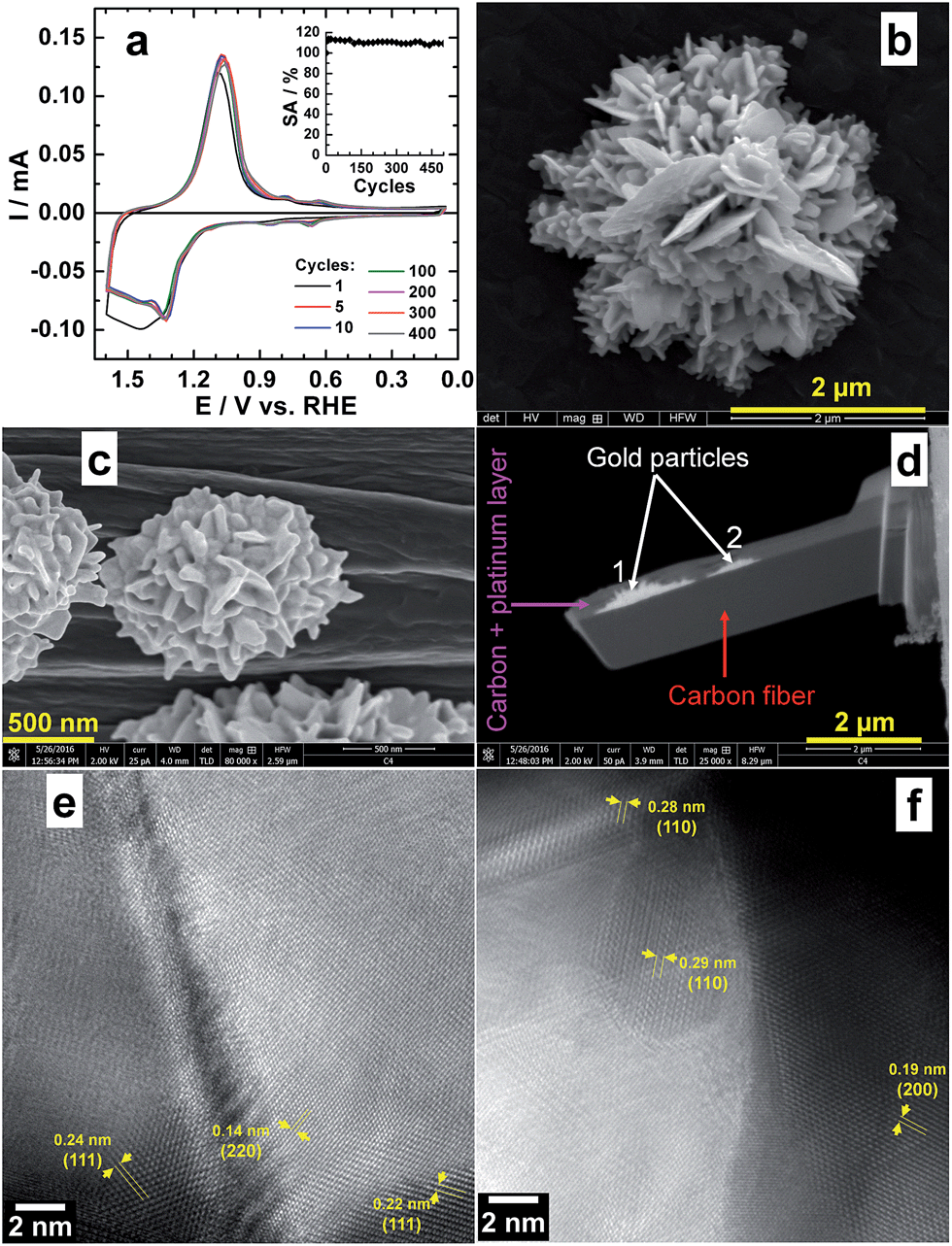 | ||
| Fig. 7 (a and b) Electrochemical durability tests of Au particles growth on Toray carbon paper for n(KBr)/n(HAuCl4) = 100 and 8 wt% Au: (a) CVs between 0.05–1.6 V vs. RHE at 50 mV s−1 in 0.1 M NaOH (inset: SA decay) and (b) HRSEM micrograph of aged material after 500 cycles. (c) HRSEM micrograph taken during the sample preparation by dbFIB, (d) SEM image of the as-prepared sample by dbFIB and (e and f) atomic-resolution HRTEM images showing the different atom arrangements. | ||
Finally, we tested the cell performance employing the as-synthesized Pt as cathode and Au (from KBr) or Pt as anode electrodes, respectively. The polarization and power density curves are presented in Fig. 6c. A high OCV of 1.01 ± 0.02 V indicates that both electrodes have good kinetics, which is higher than OCV of 0.64 V for electrodes obtained from electrodeposition method.20 Using Au as anode, the present DGFC exhibits a peak power density of 1.7 ± 0.1 mW cm−2, which is higher than relevant reported values that range from 0.6 to 1.5 mW cm−2 (see Table S5† for relevant DGFCs from literature). By increasing the Pt anode surface area from 1 to 2 cm2, a record value of 3.1 ± 0.1 mW cm−2 is obtained at 0.7 V cell voltage without any fuel circulation, which is higher than the reported power densities of previously reported Pt/C and Au/C materials.40 In 1 M KOH, a DGFC using the state-of-the-art commercial PtRu/C (60 wt%, Johnson Matthey) anode material (3 mgmetal cm−2) yields 1.38 mW cm−2 at 0.51 V (OCV = 0.91 V) and 1.08 mW cm−2 at 0.50 V (OCV = 0.90 V) using 0.2 and 0.3 M glucose at 30 °C, respectively.41 The superior performance of the materials obtained here is explained by the absence of any organic contaminants or protective ligands on the surface of the metal, the good interaction between metal and support, and the particular morphology of the particles. To explain the difference between Pt and Au anodes, voltammetric curves were recorded in 0.5 M NaOH (Fig. 6d). For the same electrode size (anode and cathode), the results indicate that the DGFC performances are limited by the glucose activation at lower current densities for Au anode; at higher current densities, both anode and cathode are limiting in the case of Pt anode. The estimated ohmic resistance from the fuel cell polarization curves (fitting the ohmic loss region by a linear model, Fig. S17†) highlights a relatively small resistance (40–80 Ω cm2), i.e., good conductivity. For comparison, the value of 40 Ω cm2 (Au = anode and Pt = cathode) is smaller than the value of 45 Ω cm2 that can be estimated from the conventional Pt/Vulcan and Au/Vulcan materials used in direct glucose fuel cell.40
Electrochemical durability and textural analysis of Au particles
We also evaluated the long-term electrochemical stability of the Br− regulated Au particles growth on Toray carbon paper through an accelerated durability test. The goal was to evaluate the impact of the electrochemical treatment (potential cycling from 0.05 to 1.6 V vs. RHE at 50 mV s−1 in 0.1 M NaOH for a total of 1000 segments) of the evolution of the obtained structure. Fig. 7a shows the recorded voltammograms and depicts the general behaviour of Au and the active surface area (SA) decay. The electrode exhibits good electrochemical stability, fully supported by the post-reaction HRSEM analyses (Fig. 7b) which do not show any morphology modification.To gain deeper insights about the structural and textural features of the as-prepared materials, a representative sample is prepared by focused ion beam dual beam (dbFIB). Fig. 7c depicts the HRSEM micrograph taken during the sample preparation by dbFIB. Protective thin layers of carbon and platinum were deposited before cutting the sample. SEM image of as-prepared sample by dbFIB is displayed within Fig. 7d showing two Au particles. Fig. 7e and f shows HRTEM images taken at different places. The presence of low and high indexes (110), (111), (200) and (220) as well as various defects and twins confirms that the sample have several NPs growing a different directions. This atomic-resolution analysis fully substantiates the previous electrochemical characterizations by PbUPD.
Next, electron diffraction studies were undertaken to confirm the crystallinity of the sample already revealed by XRD. Fig. 8a shows the NBD pattern, mainly composed of small spots making up rings and testifies the polycrystalline structure of Au, containing various facets, (111), (200), (200), (311), etc. However, the bright spots in the center originate from the textured grains that had preferential (100) and (110) orientations during the analysis. Furthermore, the measured interplanar space for all lattice fringes roughly corresponded to those of XRD and inter-atomic distance values obtained from HRTEM. These NBD results endorse the multi-facet and multi-twin nature of the Au material synthesized in presence of Br−.
 | ||
| Fig. 8 Structural and textural characterizations of Au particle growth on Toray carbon paper (in the presence of Br−) for n(KBr)/n(HAuCl4) = 100 and 8 wt%: (a) NBD patterns, (b and c) SE images, (e) bright-field STEM images, (d and f) HAADF-STEM images. | ||
We finally combined various tomography techniques to establish the topological structure of the different areas of the sample. SE images within Fig. 8b and c show the landforms with different defects and holes (dark spots). Dark-field (HAADF) STEM tomography (Fig. 8d and f) and high resolution bright-field STEM (Fig. 8e) of selected regions confirms the evidence of stress, strain and hole within Au nanocrystallites. Deep comparison by complementary bright-field STEM (dark regions represent the heavier element, that is Au) and HAAD-STEM (the bright represent Au) images reveal that each particles has holes and the density of gold atoms is heterogeneous confirming the formation of particles with a rough surface. All these results support the high catalytic performances of the materials obtained in presence of Br− as regulator for the morphology control.
Conclusions
In summary, a novel class of electrode materials composed of carbon paper decorated by metallic particles was fabricated for both electrocatalytic oxygen reduction (ORR) and glucose oxidation reactions in alkaline medium. In this contribution, we demonstrated that the direct formation of anisotropic gold and palladium at the surface of carbon paper is feasible and can be regulated by the introduction of different halides (Cl−, Br− and I−) during nanoparticle (NP) growth. This enables bypassing the use of any Nafion binder during the electrode preparation and minimized the particle detachment during the reaction, which was fully supported by the post-reaction analysis by scanning electron microscopy. Since the metal particles are directly assembled onto the carbon paper, there is no need to prepare a catalytic ink and the developed approach allows simplifying the membrane-electrode-assembly (MEA). Then, we thoroughly determined the main parameters that enable tuning the particles growth kinetics. By fine-tuning the experimental conditions (ions, concentrations, temperature), we were able to generate different materials of Au, Pd and Pt structures having various roughness and distinguished electrocatalytic properties. The carbon paper decorated by Pt particles shows distinguished electrochemical kinetic towards ORR in an alkaline medium, starting at 1.1 V vs. RHE, ultimately approaching the theoretical equilibrium potential of 1.18 V vs. RHE. For glucose electrooxidation, the use of Br− as regulator enables obtaining Au materials with rough surface leading to a record peak current density close to 10 mA cmAu−2, which is higher than the conventional chemically synthesized AuNPs by an order of magnitude. All these high performances result from the particular morphology of the particles and the simplicity of the developed method. Moreover, the constructed glucose fuel cell has an outstanding open-circuit voltage (OCV) of 1.0 V and delivers a maximum power of 3.1 mW cm−2 (at high cell voltage of 0.7 V without any fuel circulation at room temperature with metal loading of 0.35 mg cm−2), which surpasses the relevant reported data as well as the commercial 60 wt% PtRu/C anode41 (1.38 mW cm−2 at 0.51 V, OCV of 0.91 V using a high metal loading of 3 mg cm−2). To date, this work constitutes the first proof that metallic particles can be directly assembled onto large surface (i.e. 5 cm × 5 cm) of conducting carbon paper electrode (currently used in fuel cell technology) by means of a chemical reduction method to used as effective electrocatalysts for ORR, organic molecule oxidation and high-performance fuel cell construction with low metal content. Owing to these unique outcomes, it is believed that this work will offer a new strategic approach for simplifying the design of electrocatalysts, and pave a pathway for the breakthroughs in the development of alcohol-based fuel cell technology to achieve superior cell performances.Acknowledgements
This work was supported by the US Army Research Office MURI (#W911NF1410263). For the materials characterization by Scanning Transmission Electron Microscope (S/TEM: JEOL JEM-2800) and Focused Ion Beam Dual Beam Microscopy (dbFIB, FEI Helios NanoLab 650), these works made use of University of Utah USTAR shared facilities support, in part, by the MRSEC Program of NSF under Award No. DMR-1121252. We thank Prof. Boniface K. Kokoh (University of Poitiers, France) for providing the anion exchange membrane (AEM).References
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
- V. Mazumder, M. Chi, K. L. More and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 9368–9372 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, Y. Chen, E. Zhu, M. Li, X. Duan and Y. Huang, Energy Environ. Sci., 2014, 7, 2957–2962 CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- H. A. Miller, M. Bevilacqua, J. Filippi, A. Lavacchi, A. Marchionni, M. Marelli, S. Moneti, W. Oberhauser, E. Vesselli, M. Innocenti and F. Vizza, J. Mater. Chem. A, 2013, 1, 13337–13347 CAS.
- B. Jiang, C. Li, J. Henzie, T. Takei, Y. Bando and Y. Yamauchi, J. Mater. Chem. A, 2016, 4, 6465–6471 CAS.
- J. Chen, M. Yang, Q. Zhang, E. C. Cho, C. M. Cobley, C. Kim, C. Glaus, L. V. Wang, M. J. Welch and Y. Xia, Adv. Funct. Mater., 2010, 20, 3684–3694 CrossRef CAS.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- D. S. He, D. He, J. Wang, Y. Lin, P. Yin, X. Hong, Y. Wu and Y. Li, J. Am. Chem. Soc., 2016, 138, 1494–1497 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- X. Lu, L. Au, J. McLellan, Z.-Y. Li, M. Marquez and Y. Xia, Nano Lett., 2007, 7, 1764–1769 CrossRef CAS PubMed.
- S. Chen, H. Su, Y. Wang, W. Wu and J. Zeng, Angew. Chem., Int. Ed., 2015, 54, 108–113 CrossRef CAS PubMed.
- A. P. Yadav, T. Okayasu, Y. Sugawara, A. Nishikata and T. Tsuru, J. Electrochem. Soc., 2012, 159, C190–C194 CrossRef CAS.
- S. Chen, H. A. Gasteiger, K. Hayakawa, T. Tada and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A82–A97 CrossRef CAS.
- Y. Sugawara, T. Okayasu, A. P. Yadav, A. Nishikata and T. Tsuru, J. Electrochem. Soc., 2012, 159, F779–F786 CrossRef CAS.
- L. Zhang, W. Niu and G. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- W. Luo, C. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. Fan, ACS Nano, 2010, 4, 7451–7458 CrossRef CAS PubMed.
- M. S. El-Deab, T. Sotomura and T. Ohsaka, J. Electrochem. Soc., 2005, 152, C730–C737 CrossRef.
- S.-P. Tung, T.-K. Huang, C.-Y. Lee and H.-T. Chiu, RSC Adv., 2012, 2, 1068–1073 RSC.
- T.-M. Cheng, T.-K. Huang, H.-K. Lin, S.-P. Tung, Y.-L. Chen, C.-Y. Lee and H.-T. Chiu, ACS Appl. Mater. Interfaces, 2010, 2, 2773–2780 CAS.
- X. Li, K. Yao, W. Lu and J. Wang, CrystEngComm, 2013, 15, 1289–1292 RSC.
- T. X. H. Le, R. Esmilaire, M. Drobek, M. Bechelany, C. Vallicari, d.-l. nguyen, A. Julbe, S. Tingry and M. Cretin, J. Mater. Chem. A, 2016 10.1039/c6ta05443a.
- A. Both-Engel, M. Bechelany, O. Fontaine, A. Cherifi, D. Cornu and S. Tingry, ChemElectroChem, 2016, 3, 629–637 CrossRef CAS.
- C. D. Saquing, J. L. Manasco and S. A. Khan, Small, 2009, 5, 944–951 CrossRef CAS PubMed.
- D. Selloum, A. A. Chaaya, M. Bechelany, V. Rouessac, P. Miele and S. Tingry, J. Mater. Chem. A, 2014, 2, 2794–2800 CAS.
- L. M. Alencar, G. L. Caneppele and C. A. Martins, Electroanalysis, 2016 DOI:10.1002/elan.201600472.
- A. Ruditskiy and Y. Xia, J. Am. Chem. Soc., 2016, 138, 3161–3167 CrossRef CAS PubMed.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef CAS.
- M. Grzelczak, A. Sánchez-Iglesias, B. Rodríguez-González, R. Alvarez-Puebla, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Funct. Mater., 2008, 18, 3780–3786 CrossRef CAS.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679–726 CrossRef CAS PubMed.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- A. Usher, D. C. McPhail and J. Brugger, Geochim. Cosmochim. Acta, 2009, 73, 3359–3380 CrossRef CAS.
- N. Zettsu, J. M. McLellan, B. Wiley, Y. Yin, Z.-Y. Li and Y. Xia, Angew. Chem., Int. Ed., 2006, 45, 1288–1292 CrossRef CAS PubMed.
- Y. Xiong, H. Cai, B. J. Wiley, J. Wang, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2007, 129, 3665–3675 CrossRef CAS PubMed.
- Y. Holade, K. Servat, T. W. Napporn and K. B. Kokoh, Electrochim. Acta, 2015, 162, 205–214 CrossRef CAS.
- Y. Xiong, J. M. McLellan, J. Chen, Y. Yin, Z.-Y. Li and Y. Xia, J. Am. Chem. Soc., 2005, 127, 17118–17127 CrossRef CAS PubMed.
- S. Hebié, L. Cornu, T. W. Napporn, J. Rousseau and B. K. Kokoh, J. Phys. Chem. C, 2013, 117, 9872–9880 Search PubMed.
- J. Wang, J. Gong, Y. Xiong, J. Yang, Y. Gao, Y. Liu, X. Lu and Z. Tang, Chem. Commun., 2011, 47, 6894–6896 RSC.
- Y. Holade, K. Servat, T. W. Napporn, C. Morais, J.-M. Berjeaud and K. B. Kokoh, ChemSusChem, 2016, 9, 252–263 CrossRef CAS PubMed.
- D. Basu and S. Basu, Electrochim. Acta, 2010, 55, 5775–5779 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis spectra, additional tables, additional scheme, additional SEM images, additional CVs. See DOI: 10.1039/c6ta08288b |
| This journal is © The Royal Society of Chemistry 2016 |