
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dots with tunable concentrations of trapped anti-oxidant as an efficient metal-free catalyst for electrochemical water oxidation†
Abheek
Datta
,
Sutanu
Kapri
and
Sayan
Bhattacharyya
*
Department of Chemical Sciences and Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur – 741246, India. E-mail: sayanb@iiserkol.ac.in; Fax: +91-33-25873020; Tel: +91-9051167666
First published on 24th August 2016
Abstract
The challenging water oxidation reaction to generate molecular oxygen requires low-cost efficient catalysts for its application in renewable energy technologies. Carbon dot (C-dot) catalysts synthesized by microwave irradiation can trap an anti-oxidant, 5-hydroxymethyl-2-furaldehyde (5-HMF) inside the carbon framework. The C-dot with the highest concentration of 5-HMF acts as a stable metal-free oxygen evolution reaction (OER) catalyst which operates at a decently low 0.21(±0.03) V overpotential and can generate current density up to 33.6(±2.3) mA cm−2. With increased microwave reaction time, the concentration of 5-HMF inside the C-dots decreases at the cost of different furan derivatives which decreases the OER activity. The 5-HMF molecules in close vicinity to the catalytically active sites containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups can extract the ˙OH/˙OOH radicals and can increase the in situ H2O concentration to facilitate the forward reaction of O2 evolution. During continuous electrolysis beyond 10 min, 5-HMF gets converted to 2,5-diformylfuran entities, which increases the catalytically active sites and thereby maintains the OER activity of the C-dots for at least 4 h. The ability of microwave irradiated sucrose derived C-dots to electro-oxidize water is generalized with C-dots and graphene dots (G-dots) prepared from different precursors.
O groups can extract the ˙OH/˙OOH radicals and can increase the in situ H2O concentration to facilitate the forward reaction of O2 evolution. During continuous electrolysis beyond 10 min, 5-HMF gets converted to 2,5-diformylfuran entities, which increases the catalytically active sites and thereby maintains the OER activity of the C-dots for at least 4 h. The ability of microwave irradiated sucrose derived C-dots to electro-oxidize water is generalized with C-dots and graphene dots (G-dots) prepared from different precursors.
1. Introduction
The impending energy crisis in today's world is driving the search for mature, clean and renewable energy in the form of fuels and water oxidation to molecular oxygen has attracted immense attention in this respect.1–3 However, water electrolysis is thermodynamically unfavorable since the standard reduction potential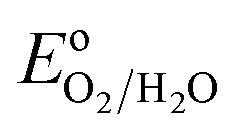 per electron transferred is 1.23 V versus reversible hydrogen electrode (RHE) and the associated free energy (ΔG) is 237.2 kJ mol−1.4,5 The OER in the natural enzyme photosystem II is catalyzed by cubane-like Mn4CaOx active sites, in a physiological pH environment with a moderate overpotential (η) of 0.3–0.4 V.5,6 Developing a nature mimicking, stable OER catalyst for artificial photosynthesis is a formidable challenge and a body of recent work is available in the search for efficient, robust and inexpensive electro-catalysts.2,7,8 A majority of the OER catalysts known to date suffer from limitations such as low current density (J) and overpotential of several hundred millivolts. Although precious metals such as Ru and Ir metals are known to demonstrate reliable OER activities,9 the nanoparticles of these metals are prone to corrosion at applied OER potentials and under non-physiological pH conditions. Moreover, IrOx, RuOx and earth abundant transition metal oxides are some of the well-known OER catalysts which work at 0.32–0.5 V.10,11 In addition, homogeneous molecular complexes based on Ru, Ir and 3d transition metals have demonstrated decent OER activities.2,5,12–14 All the metal based catalysts have issues such as scarcity, toxic nature, difficulties in disposal and secondary pollution. Bioinspired catalysts based on the naturally occurring metalloenzymes have also gained attention.15 In recent years, metal-free heterogeneous catalysts have emerged as the new alternatives to the metal based catalysts.16 The development of an earth abundant, metal-free, easy to synthesize and stable OER catalyst that can operate at low η close to pH 7 remains a fundamental challenge.17
per electron transferred is 1.23 V versus reversible hydrogen electrode (RHE) and the associated free energy (ΔG) is 237.2 kJ mol−1.4,5 The OER in the natural enzyme photosystem II is catalyzed by cubane-like Mn4CaOx active sites, in a physiological pH environment with a moderate overpotential (η) of 0.3–0.4 V.5,6 Developing a nature mimicking, stable OER catalyst for artificial photosynthesis is a formidable challenge and a body of recent work is available in the search for efficient, robust and inexpensive electro-catalysts.2,7,8 A majority of the OER catalysts known to date suffer from limitations such as low current density (J) and overpotential of several hundred millivolts. Although precious metals such as Ru and Ir metals are known to demonstrate reliable OER activities,9 the nanoparticles of these metals are prone to corrosion at applied OER potentials and under non-physiological pH conditions. Moreover, IrOx, RuOx and earth abundant transition metal oxides are some of the well-known OER catalysts which work at 0.32–0.5 V.10,11 In addition, homogeneous molecular complexes based on Ru, Ir and 3d transition metals have demonstrated decent OER activities.2,5,12–14 All the metal based catalysts have issues such as scarcity, toxic nature, difficulties in disposal and secondary pollution. Bioinspired catalysts based on the naturally occurring metalloenzymes have also gained attention.15 In recent years, metal-free heterogeneous catalysts have emerged as the new alternatives to the metal based catalysts.16 The development of an earth abundant, metal-free, easy to synthesize and stable OER catalyst that can operate at low η close to pH 7 remains a fundamental challenge.17
The applications of carbon containing nanomaterials are abundant in photoelectrochemical water splitting,18,19 photocatalysis,20 fuel cells,21,22 solar cells,23 spintronic devices,24,25 drug delivery,26–28 and greener approaches to materials design.29 Carbon based catalysts have also been used for oxygen reduction and other miscellaneous reactions.16,30–33 However, reports on metal-free OER electro-catalysts are rather scarce except the OER activities shown by N-doped 30–40 nm carbon nanoparticles,7 g-C3N4 coupled N-doped graphene,8 and surface modified carbon nanotubes.3 Also, most of the carbon catalysts require heteroatom modification especially doping with nitrogen atoms.7 The OER involves a large variety of possible pathways and irrespective of the absence of an unambiguous mechanism, it is argued that in alkaline medium, the overall OER activity according to the half-cell reaction: 4OH− → O2 + 2H2O + 4e− is determined by the rates of adsorption and desorption of hydroxyl radicals on the catalyst surface.34 In this regard, encasing a ˙OH scavenger inside a metal-free nanoparticle such as C-dots is highly lucrative to test the OER activity. On the other hand, the multifaceted C-dots containing encapsulated aromatic molecules within a carbon scaffold are usually employed in optoelectronics, bio-imaging and sensing applications (ESI Table S1†),35 but were rarely used as catalysts,36–39 and particularly no report is available on their use as OER catalysts. We show that the water-dispersible pristine C-dots are excellent OER catalysts providing one of the highest J and lowest η to date. Although microwave irradiation is known to be a successful technique for synthesizing C-dots from various precursors,40–44 the novelty of our approach lies in microwaving a precursor mixture of carbohydrate (sucrose), weak acid (oxalic acid) and a binder (polyvinylpyrrolidone, PVP) for a limited duration to arrest the anti-oxidant inside the C-dots. The catalytic activity of the synthesized C-dots depends on the microwave irradiation time. The radical scavenger anti-oxidant 5-HMF encased C-dots C5, C10 and C15 were prepared by altering the microwave reaction time as 5, 10 and 15 min, respectively. Increasing the microwave reaction time to 20 min (C20), 25 min (C25) and 30 min (C30) cannot retain 5-HMF which subsequently undergoes complete polymerization into aromatic compounds with CO linkages. C-dots were also synthesized from other precursors such as citric acid and ethylenediamine (C-A), sucrose, oxalic acid and cetyl trimethyl ammonium bromide (CTAB) with a microwave time of 5 and 10 min (C-B and C-C), glucose and NaOH in the presence and absence of PVP (C-D and C-E). The G-dots G5 and G10 were synthesized from citric acid and ethanolamine for 5 and 10 min, respectively.
2. Experimental
2.1 Materials
Sucrose, glucose (pure grade, Merck India), citric acid (Sigma Aldrich, ACS reagent, >99.5%), oxalic acid dehydrate (Purified, Merck India), ethylenediamine (synthesis grade, Merck India), ethanolamine (Sigma Aldrich, >99.5%), potassium hydroxide and sodium hydroxide pellets (EMPLURA, Merck India), PVP (Loba Chemie, mol. wt. 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), CTAB (Sigma Aldrich, 98%), 5-HMF (Alfa Aesar, 97%) and Nafion perfluorinated resin solution (5 wt%, Sigma Aldrich) were used without further purification.
000), CTAB (Sigma Aldrich, 98%), 5-HMF (Alfa Aesar, 97%) and Nafion perfluorinated resin solution (5 wt%, Sigma Aldrich) were used without further purification.
2.2 Synthesis of C-dots and G-dots
To synthesize the C-dots (C5 to C30, C-A to C-E) microwave reactions were performed in a Sineo MASII-1000W commercial microwave oven, with a well-equipped refluxing system under constant stirring at 900 W. The microwave heating was constantly monitored with an automated on/off program to avoid the risk of overheating. G5 and G10 were synthesized with an Anton Paar Monowave 300 microwave reactor, where the reactions can be controlled up to 30 bar and 300 °C.:3 and the solution was treated by microwave irradiation at 180 °C. 5 and 10 min reactions gave greenish-yellow G5 and yellow G10 colloidal solutions.
2.3 Separation and purification of C-dots and G-dots
The uniform sized particles were separated and the samples were purified to remove the impurities and by-products. The crude colloidal suspensions were half-diluted with a 2:1 ethanol water mixture and centrifuged for 15 min at 13000 rpm followed by collection of a clear supernatant. The whole exercise was repeated 3 times for each sample. For purification, the dispersions were introduced on a Fisher scientific dialysis tubing membrane (with a molecular weight cut-off of 3500 Da) and extensively dialyzed against de-ionized water for 36 h. The as-received suspensions were solidified using a rotary evaporator. All the samples were kept under high vacuum to remove the moisture.
2.4 Characterization methods
The ICP-MS measurements were performed with a Thermo Scientific X-series using Plasma lab software. TEM images were recorded using a JEOL JEM 2100 HR model using a 200 kV electron source. UV-visible absorption spectra were collected using a JASCO V-670 spectrophotometer. PL spectra were measured with a Horiba ScientificFluoromax-4 spectrofluorometer using a Xe lamp as the excitation source and excitation at 309 nm. LC-MS studies were performed with an Agilent Q-TOF LC-MS 6520. 1H NMR and 13C NMR spectra were recorded on a JEOL ECS 400 MHz spectrometer and on a Bruker Avance III 500 MHz spectrometer. Chemical shifts (δ) downfield from the reference standard were assigned positive values. FTIR studies were performed with a Perkin Elmer spectrum RX1 with KBr pellets. pH of the solutions was measured using a standard pH meter (pH 510, Eutech Instruments). Electron paramagnetic resonance (EPR) measurements were performed with a Bruker A-300 instrument. The amount of evolved oxygen was measured using a Fischer Scientific dissolved oxygen meter. The amount of dissolved oxygen before and after one cycle was obtained from the difference between the two readings. The impedance measurements were carried out using a CHI Electrochemical workstation. The plots were analyzed with inbuilt software in the electrochemical workstation.2.5 Electrochemical measurements
The electrocatalytic performance of the C-dots was measured with a conventional three electrode cell in a CHI604D electrochemical workstation. The working electrode was designed as follows: 1 mg of dried C-dot was dispersed in a mixed solution containing 250 μL distilled water, 250 μL of ethanol and 25 μL of 5% aqueous Nafion solution. 5 μL of the resulting suspension was carefully drop cast onto the surface of the GC disk followed by drying at 25 °C for 0.5 h. The scanning rate was maintained constant at 100 mV s−1. Since the catalyst shows a near steady state behavior at the electrode surface over 4 h, the scan rate was not lowered according to a generic protocol.103. Results and discussion
3.1 Characterization of the C-dots and G-dots
The OER-active C-dots and G-dots were found to be “pure” by inductively coupled plasma – mass spectroscopy (ICP-MS) and all metal impurities were at the ppb level (Table S2†). X-ray diffractograms (XRD) with a lone (002) broad reflection of sp2 hybridized carbon show the primarily amorphous nature of the C-dots (Fig. S1†). The broadening of the XRD peaks from C10 to C30 is due to the increase in the diameter of the C-dots. All the C-dots show optical absorption in the UV region with π–π* and n–π* transitions of the CC and CO bonds, respectively (Fig. S2†). The photoluminescence emission is dependent on the excitation wavelength (Fig. S3†), a property attributed to differently sized C-dots and various surface emissive traps.48 The evidence of dominant surface trap states which can serve as catalytically active sites was obtained from the extremely low fluorescence quantum yield (QY) of the samples (Fig. S4†).35,48 QY shows a steady increase from 1.3 for C5 to 2.6 for C30, varies between 1.6 and 5.3 for C-A to C-E and is highest for the G-dots, 15.1 for G5 and 18.5 for G10.
O linkages. PVP plays the role of a surfactant to stabilize the C-dots by encapsulating 5-HMF and other intermediates present due to incomplete carbonization, within an elemental carbon scaffold.51 In fact, 5-HMF was observed to form within 2.5 min of the microwave reaction between sucrose, oxalic acid and PVP, as shown by 13C NMR spectroscopy (Fig. S6†). 13C NMR of a commercial 5-HMF sample confirms this peak (Fig. S7†). 2.5 min is however not enough to encapsulate the 5-HMF molecules within a sp2 hybridized carbon scaffold, and because of the water-soluble nature of 5-HMF, the yield of this sample is pretty low, not enough for OER experiments.
The 13C NMR spectra in Fig. 1a–f show the descending concentration of 5-HMF from C5 to C30. A single peak at 177.9 ppm characteristic of C1 in 5-HMF (Fig. 1g) is observed in C5 and C10,52 which is shifted to 178.6 for C15 as a result of increased carbohydrate decomposition.53,54 In C20 and C25, the signals at 162.9–182.9 ppm are due to the furan derivative intermediates formed by decomposition of 5-HMF. In fact, 160–200 ppm peaks in C20–C30 can correlate with CO groups of these furan derivatives. Likewise, the population of aromatic sp2 carbon at 100–120 ppm starts increasing from C5 up to C30. The final condensation to polyaromatic rings is evident from the 120–140 ppm signals in C30. For C5–C30, multiple peaks in the 20–80 ppm region correspond to sp3 hybridized carbon and the C–OH groups such as C6 in 5-HMF. The 80–100 ppm signals are from the carbon attached to ether linkages in unreacted sucrose or its byproduct glucose trapped inside the C-dots.53
 | ||
| Fig. 1
13C NMR spectra of (a) C5, (b) C10, (c) C15, (d) C20, (e) C25, and (f) C30. (i) sp3 hybridized carbon and the C–OH groups, (ii) carbon attached to ether linkages in leftover carbohydrates, (iii) aromatic sp2 carbon, and (iv) CO groups of furan derivatives. (g) Structure of 5-HMF. (h) FT-IR spectra of different C-dots where subscripts s and b correspond to stretching and bending vibrations, respectively. | ||
Liquid chromatography-mass spectroscopy (LC-MS) studies also support the trapping of 5-HMF in the C-dots. LC-MS spectra of C5, C10 and C15 show the presence of a prominent peak at m/z 127.4 which confirms the presence of 5-HMF (Fig. S8†).55 This peak is absent in C20 which indicates the absence of 5-HMF due to complete degradation to the furan derivatives with increased microwave time. Moreover, 5-HMF could be quantified by LC-MS analysis and the abundance of 5-HMF in C5, C10 and C15 is 93, 89 and 9%, respectively.
The C-dots prepared from other precursors and surfactants fail to meet the reaction conditions required for the formation of 5-HMF (Fig. S5†). For example, for the formation of C-A, citric acid was used as the carbon precursor instead of sucrose. In the case of C-B and C-C, CTAB was used as the surfactant instead of PVP and could not trap the 5-HMF molecules. C-D and C-E were formed by basic hydrolysis of glucose instead of acid hydrolysis. G5 and G10 were prepared from citric acid instead of sucrose. The 13C NMR spectra of C-A to C-E (Fig. S9†) likewise show the absence of the 5-HMF signal and the presence of sp3 hybridized carbon, C–OH groups and ether linkages in leftover sucrose/glucose.
Anti-oxidants are presumably unstable and soluble in water. Since all the samples post-microwave irradiation were dialyzed against de-ionized water for 36 h, the left-over 5-HMF is not a physical mixture with the C-dots, rather the molecules are loaded inside the C-dots. The stability of the anti-oxidant is thus enhanced by its impregnation inside the C-dots, beneficial to long term stability during electrocatalysis experiments. Moreover, the trapping of 5-HMF within a scaffold helps preventing them getting soluble in water medium which further benefits the OER activity of the C-dots by increasing the reaction kinetics through free-radical scavenging.56 Representative Fourier transform infrared (FT-IR) spectra in Fig. 1h show the carbonyl bands corresponding to either the carboxylic acid attached to the C-dot surface and/or ketonic CO groups of the furan derivatives which have been demonstrated earlier to alter the electronic distribution of the adjacent carbon atoms assisting the adsorption of OER intermediates.3 All the indexed functional groups were observed in C5–C15, and their origin is likely due to the 5-HMF molecules (Fig. 1g) trapped in the C-dots along with the furan molecules. In addition, the topological defects can promote the formation of charged sites through the formation of localized orbitals favourable for the adsorption of intermediates.57
3.2 Electrochemical activities of the C-dots and G-dots
| Catalyst | J (mA cm−2) | η@10 mA cm−2 (V) | Tafel slope (mV dec−1) | ECSA (cm2) | Reference |
|---|---|---|---|---|---|
| C5 | 33.6(±2.3) | 0.21(±0.03) | 68(±4) | 17.5(±2) | This work |
| C10 | 29.2(±1.5) | 0.36(±0.06) | 90(±8) | 12.8(±2) | This work |
| C15 | 23.6(±1.1) | 0.34(±0.03) | 78 (±3) | 11.6(±1.5) | This work |
| C20 | 18.9(±1.6) | 0.4(±0.08) | 112(±2) | 10.7(±0.9) | This work |
| C25 | 17.6(±2.1) | 0.41(±0.02) | 104(±3) | 8.96(±0.9) | This work |
| C30 | 16.3(±1.4) | 0.43(±0.05) | 124(±8) | 8.2(±0.6) | This work |
| C-A | 22.5(±2.1) | 0.39(±0.03) | 98(±5) | 10.56(±1) | This work |
| C-B | 19.0(±0.9) | 0.4 (±0.04) | 110(±6) | 7.25(±1) | This work |
| C-C | 19.05(±0.9) | 0.39(±0.04) | 97(±5) | 8.65(±0.8) | This work |
| C-D | 15.87(±1.1) | 0.41(±0.05) | 115(±4) | 6.9(±0.5) | This work |
| C-E | 24.8(±2.5) | 0.3(±0.01) | 74(±4) | 11.3(±0.5) | This work |
| G5 | 16.4(±1.9) | 0.4 (±0.05) | 111(±6) | 2.3(±0.2) | This work |
| G10 | 20.1(±1.7) | 0.4 (±0.03) | 125(±10) | 1.8(±0.2) | This work |
| Surface modified CNT | 8 | 0.30 | 72 | 0.14 | 3 |
| N-Doped carbon | 55 | 0.38 | — | 18.2 | 7 |
| IrOx | 42 | 0.32 | — | 21 | 10 |
| Ni–Co | 60 | 0.325 | 39 | — | 62 |
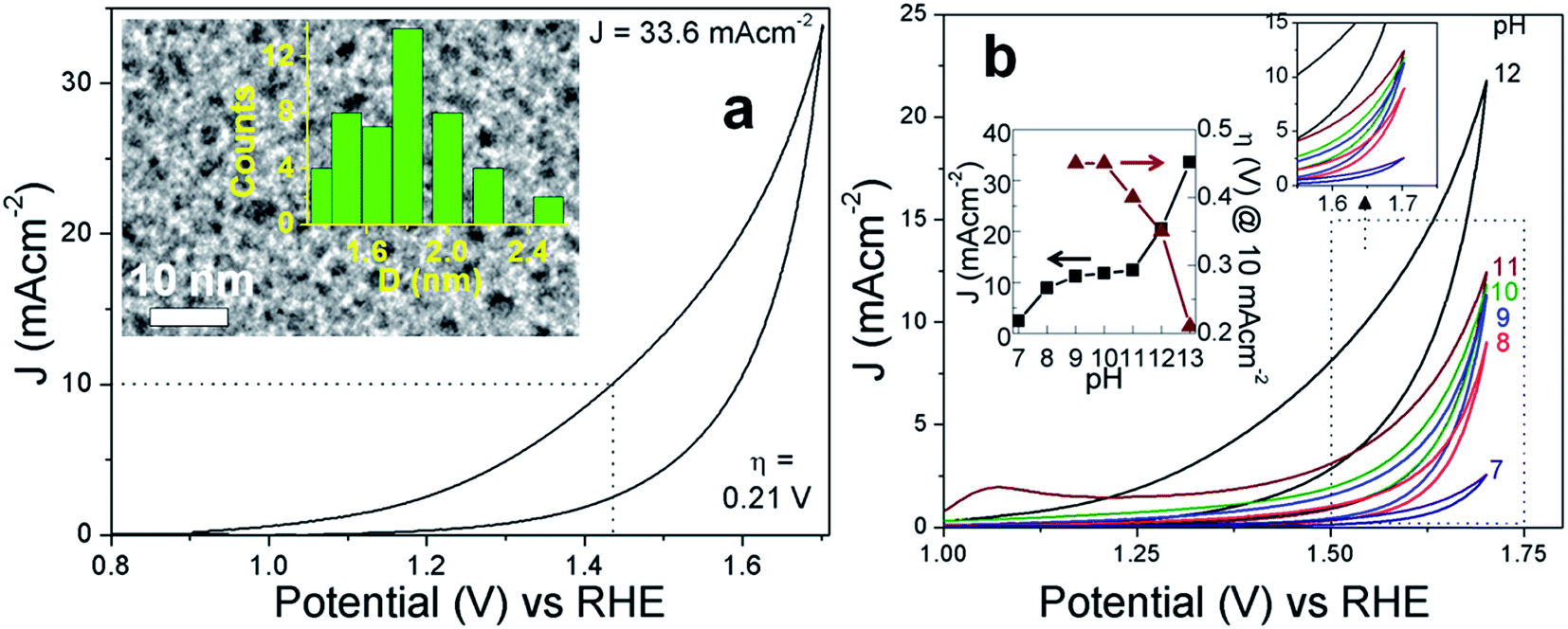 | ||
| Fig. 2 (a) CV of C5 at pH 14. The inset shows the TEM image of C5 with the diameter histogram. (b) pH dependent CVs of C5. The inset shows the variation of J and η with pH. | ||
 | ||
| Fig. 3 TEM images of (a) C10 and (b) C30. The insets show the diameter histogram. (c) CVs at pH 14. The inset shows the variation of J and η. (d) Nyquist plots. The inset shows the analogous equivalent circuit. (e) Tafel slopes. The inset shows the variation of the Tafel slope and η. (f) ECSA in cm2 mg−1 and (g) chronopotentiometric curves. | ||
The investigation of faradaic resistance was performed by electrochemical impedance spectroscopy which gives an insight into the OER kinetics at an overpotential of 0.3 V. The electrochemical interface is provided by the equivalent circuit shown in Fig. 3d (inset).66Fig. 3d shows the Nyquist plots where C5 exhibits the least resistance; the total Ohmic resistance of the cell (RT) and the charge-transfer resistance (RCT) being 10 and 130 ohm, respectively, indicate the highest charge transfer between electrodes and oxidized/reduced species in accordance with the CV and LSV results. C10 has an RCT of 155 ohm, 1.2 times more than that of C5. RCT is 280, 340, 465 and 538 ohm for C15, C20, C25 and C30, respectively. Likewise, RT for C10, C15, C20, C25 and C30 is 13, 27, 29, 29 and 53 ohm, respectively. The high resistance in the Nyquist plots reflects the poor electrical conductivity of the C-dots which is largely due to the presence of a significant fraction of amorphous elemental carbon, 5-HMF and furan derivatives. However, this disadvantage is overcome by the ability of the trapped anti-oxidant to readily scavenge the intermediate radicals and the presence of keto-groups acting as the catalytic sites.
Fig. 3e represents the Tafel plots of C5–C30 and the Tafel slopes obtained from linear fitting of the plots are tabulated in Table 1. The linearity of the Tafel plots is maintained in the overpotential range of 0.2–0.4 V, above and below which the plots depart from linearity. C5 shows linearity over the longest overpotential range, showing its relatively higher electrokinetic activity. The Tafel slope obtained for C5 is 68(±4) mV dec−1, which is less than that of surface-oxidized MWCNTs.3 The Tafel slope is 1.3 and 1.8 times higher in C10 and C30, respectively. The electrochemically active surface area (ECSA) was measured as the slope of the curve obtained by plotting the charging current density differences of anodic and cathodic sweeps at different scan rates versus the corresponding scan rates. The equation used for this calculation is: double layer capacitance (CDL) = Vs/Ic where Vs is the scan rate and Ic is charging current density differences of anodic and cathodic sweeps. For every C-dot, Ic was recorded at four different scan rates, namely 50, 100, 150 and 200 mV s−1. The detailed CDL obtained with corresponding scan rates is given in Table S4.† As shown in Fig. 3f and Table 1, C5 and C10 have the highest ECSA indicating the presence of a large number of catalytically active sites. The ECSA accounts for both the surface and the core volume in the C-dots available for charge transfer between the electrode surface and the reactants. It is observed that the ECSA of C5 (17.5 cm2) is slightly less than that of one of the best performing conventional IrOx,10 and comparable to that of the N doped carbon catalyst.7 The C-dot catalyzed bulk electrolysis was tested by chronopotentiometry maintaining the current density constant at 10 mA cm−2 for 4 h (Fig. 3g). For all the C-dots, the overpotential increases with time since the long term stability of metal oxide catalysts is unexpected for the C-dots.10 C5 however demonstrated the best performance. In C5, an initial hike of overpotential to 0.35 V within 10 min is followed by a slow rise to 0.5 V after 4 h. Similarly, C10 shows an initial rise from 0.36 to 0.43 V within 10 min and a slow increase to 0.64 V after 4 h. The sudden drop in activity within 10 min suggests the conversion of trapped 5-HMF into the furan derivatives (vide infra Section 3.3). Lesser the concentration of trapped 5-HMF greater is its rate of consumption, decreasing the OER activity over time. Since the initial overpotential value could not be stabilized over time, the faradaic efficiency is expectedly low for the C-dots. The above trends are reflected from the chronoamperometric studies performed with controlled electrolysis at 1.53 V in 1 M KOH for 4 h (Fig. S14†). For C5, after an initial drop in current density by 3.8 mA cm−2 within 10 min, it stabilizes over 4 h. Rest of the C-dots also show a constant drop in current density with increased time. Up to 4 h, C10, C15, C20, C25 and C30 demonstrate a steady current density of 3.1, 2.3, 2, 1 and 0.2 mA cm−2, respectively. The faster attenuation in the chronoamperometric plots of Fig. S14† as compared to Fig. 3g is due to faster consumption of 5-HMF at a constant higher potential.
To verify the oxygen evolution, O2 formation in the solution phase was measured over time during electrocatalysis with the aid of a Clark type oxygen electrode (YSI, Inc.) (Fig. S15†). The electrolyte solution was degassed with nitrogen and the experiment was performed in a gas tight cell with a stirred 15 mL 1 M KOH solution at pH 14. Vigorous evolution of the gases was observed during the entire course of the electrochemical reaction. The difference between the dissolved oxygen measured before and after electrocatalysis at a constant voltage of 1.6 V versus RHE is represented as the dissolved oxygen produced over a span of 50 min (Table S5 and Fig. S16†). With C5, over a period of 42 min, the dissolved oxygen concentration increased from 100 to 270 μm i.e. a rise by 170%. For C10–C30, the corresponding rise is 140–160%. The control experiment without the C-dot catalyst shows that only 37 μM O2 is dissolved after 50 min, when water was slowly oxidized by the bare glassy carbon electrode at the same potential and pH. The estimation of dissolved O2 undermines the actual evolved oxygen and the faradaic efficiency based on the dissolved O2 for C5 at an overpotential of 0.35 V for 3000 s is estimated to be 20%. Carbon based materials are usually prone to oxidation and in water medium the dissolved CO2 can form carbonic acid which can lower the solution pH. The probability of such an event was tested by measuring the pH of the reaction mixture at an interval of 500 s up to 3000 s at a stretch (Fig. S17†). The constant pH of every reaction mixture over 3000 s excludes the possibility of the formation of CO2.
O groups alter the electronic environment of the neighbouring carbon atoms, thus facilitating the adsorption of the radical intermediates favourably leading to oxygen evolution.
 | ||
| Fig. 4 TEM images of (a) C-B, (b) C-E, (c) G5 and (d) G10. The inset in (c) shows a single G-dot. (e) CVs of C-A–C-E, G5 and G10 at pH 14. The inset shows the variation of J and η. (f) Tafel slopes. The inset shows the variation of the Tafel slope and η. (g) CVs at pH 14 after 300 cycles. | ||
From the Tafel plots in Fig. 4f and the slopes in Table 1, it can be observed that C-A to C-D, G5 and G10 show similar kinetics like C10–C30, C-E being relatively better and close to C5. On average, the ECSA of C-A and C-E is close to that of C10–C20 and that of C-B to C-D is comparable to those of C25 and C30. The graphitized G5 and G10 with a near-perfect arrangement of the sp2 hybridized carbon matrix and relatively lesser topological defects have the least number of catalytic sites and hence the ECSA is the lowest, corroborating the high Tafel slopes. The stability of the three best performing C-dots, C5, C10 and C-E, was also tested by the recyclability tests up to the 300th cycle (Fig. 4g). It was observed that the catalytic performance decreases after each batch of 100 cycles, although C5 shows relatively better performance. Post 100 cycles, C5, C10 and C-E demonstrate current densities of 30, 25 and 16 mA cm−2 and an overpotential of 0.25, 0.38 and 0.35 V, respectively at 10 mA cm−2. Post 200 cycles, C5 and C10 yield current densities of 26 and 20 mA cm−2 and an overpotential of 0.28 and 0.39 V, respectively at 10 mA cm−2. Even after 300 irreversible CV cycles, C5 has an overpotential of 0.31 V, whereas that of C10 is 0.42 V at 10 mA cm−2. Although it is a non-metal catalyst, the OER activity of C5 is exceptional, whereas that of the other C-dots is pretty decent, as compared to the reports available to date.
3.3 OER mechanism
Since trapping of the highest concentrations of 5-HMF shows quite unprecedented OER activities, the actual contribution of 5-HMF was tested by employing the molecule alone as a catalyst for water oxidation. 0.001 g of commercial 5-HMF was mixed with 250 μL distilled water, 250 μL of ethanol and 25 μL of 5% aqueous Nafion solution. 5 μL of the resulting suspension was carefully drop cast onto the surface of the GC disk followed by vacuum drying for 24 h. Under similar conditions to those of the C-dots, 5-HMF shows decent performance with an overpotential of 0.45 V at 10 mA cm−2 and the maximum achieved current density is 14 mA cm−2 (Fig. S19†). This again highlights the utility of accommodating 5-HMF in the C-dots. This electrochemical activity of 5-HMF is unlikely due to self-electro-oxidation since the electro-oxidation peak for 5-HMF and related compounds appears at lower voltage (0.8–1.2 V versus RHE) after several voltammetric cycles and no such peak was observed in the present case.67,68 Also, the OER onset potential for all the C-dots is found to be in the range of 0.96–1.0 V versus RHE, close to the thermodynamic potential of 1.23 V (Fig. S18†). Therefore in the present potential range of study up to 1.7 V versus RHE, the organic molecules are not electro-oxidized and electrochemical side reactions rarely occur. In the C-dots and G-dots without any anti-oxidant, the carbonyl bands and the topological defects are sufficient to catalyze the forward reactions towards O2 evolution. Our results are in agreement with the previous reports,3,57 since irrespective of the presence of any anti-oxidant, all the C-dots and G-dots demonstrate superior OER activity as compared to the well known homogeneous and heterogeneous catalysts. Also, the extremely low QY indicates that the C-dots and G-dots are probably not N-doped since high QY is generally associated with N/S doped C-dots.69 The nitrogen content measured by elemental analyses (Table S6 and Fig. S20†) does not match with the trend of OER activity of different C-dots and G-dots. The slightest N-doping is due to the use of precursors such as PVP, ethylenediamine, CTAB and ethanolamine.In alkaline medium, the sequential oxidation reactions are: 2H2O → H2O + ˙OH (ads) + H+ + e− (step I) → H2O + ˙O (ads) + 2H+ + 2e− (step II) → ˙OOH (ads) + 3H+ + 3e− (step III) → O2 + 4H+ + 4e− (step IV).3,4 The OER activity is dependent on the adsorption and desorption kinetics of the intermediate radicals at the electropositive sites on the catalyst surface. The CO groups with an electron-withdrawing inductive effect alter the electronic environment of the surrounding carbon atoms for adsorption of the radicals. The advantage of nurturing 5-HMF molecules in the C-dots is two-fold. Firstly, the 5-HMF molecules in close vicinity to these CO groups on the C-dot surface have an exceptional ability to scavenge the radical intermediates leading to faster adsorption kinetics. Secondly, 5-HMF is randomly distributed in the entire “small” volume of the C-dots and due to its strong affinity towards the ˙OH/˙O/˙OOH intermediates, enhances their adsorption at the catalytically active sites. The three possible pathways by which 5-HMF can scavenge the radicals are shown in Fig. 5a. The most feasible pathway (A) involves extraction of H˙ from the C6 position of 5-HMF, where the allyl radical (I) can be stabilized through conjugation which finally gets oxidized to 2,5-diformylfuran.70 The latter with CO and –CHO groups in turn acts as the active sites for radical adsorption.
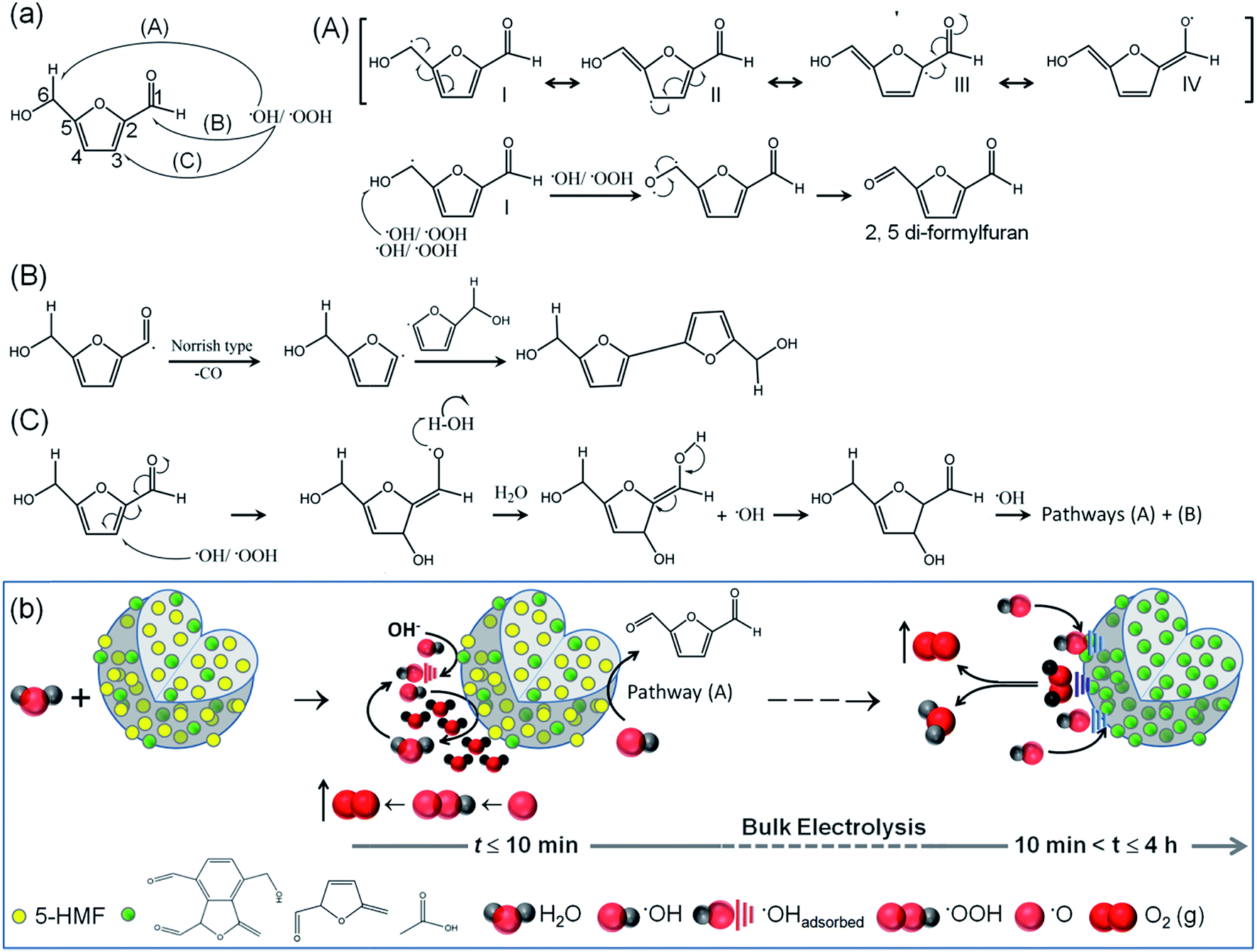 | ||
| Fig. 5 (a) Mechanistic pathways (A–C) of the extraction of ˙OH by 5-HMF. (b) Schematic representation of bulk electrolysis steps with C5–C15 C-dot catalysts. At time, t < 10 min, the discharge of OH− ions occurs at the active sites containing CO groups and continued extraction of ˙OH intermediates by 5-HMF molecules increases the local H2O concentration near the active sites leading to vigorous O2 evolution. Simultaneously, the 5-HMF is continuously consumed to yield 2,5-diformylfuran derivatives. When the electrolysis is continued beyond 10 min, the furan derivatives solely dictate the adsorption and desorption of intermediates and govern the OER kinetics, in the absence of 5-HMF. | ||
As the electrochemical reaction progresses, the 2,5-diformylfuran entities increase in concentration remarkably increasing the active sites of adsorption, at the cost of 5-HMF molecules. The chronopotentiometric curves in Fig. 3g suggest that all the 5-HMF molecules are undoubtedly converted to 2,5-diformylfuran within the first 10 min of bulk electrolysis. Post 10 min of the reaction up to at least 4 h, the OER activity is driven by the standard adsorption and desorption of the radical intermediates to form molecular O2 (Fig. 5b). This clearly explains the steady performance of C5 containing the highest concentration of 5-HMF, even if this C-dot is electrically non-conducting.
Although the standard OER mechanism can be easily understood once 5-HMF converts to the furan species, the OER activity of the C-dots containing 5-HMF within the first 10 min of bulk electrolysis is intriguing. According to a plausible mechanism, the initial electrolytic discharge of the OH− ions (step I) at the active sites on the surface of the C-dots will allow 5-HMF near the CO groups to extract ˙OH intermediates and convert them to H2O according to the mechanistic pathway (A) in Fig. 5a. When H2O concentration increases near the active sites it will shift the equilibrium towards the evolution of O2 in steps I and II according to the Le Châtelier's principle. The kinetics of continued extraction of ˙OH, ˙O, ˙OOH radicals in steps I–III and regeneration of H2O are faster with the presence of 5-HMF in C5–C15 as observed from the lower Tafel slopes (Table 1). With a decrease in the 5-HMF concentration from C5 to C30, there is a drop in the local H2O around the catalytic sites which affects the OER activity. Moreover, the H2O molecules formed in situ due to scavenging of ˙OH/˙OOH radicals are assumed to be highly reactive for the processes of ˙OH migration and rearrangement of the local oxygen environment for the OER at lower overpotentials (Fig. 5b). The pathway (B) of scavenging the radicals (Fig. 5a) is less feasible and involves a Norrish type reaction where ˙OH/˙OOH can extract H˙ from C1 and gives a carbonyl radical which consecutively produces the furanyl radical at C2 followed by their fusion to produce a new C–C bond. The kinetics of pathway (C) are rather slow and are less likely to occur since the bond dissociation energy of CC is very high ∼146 kcal per mole.
4. Conclusions
In summary, we have demonstrated C-dots with a trapped anti-oxidant, 5-HMF, as a metal-free OER catalyst that can oxidise water at very low overpotential and with high current density. A limited duration of the microwave reaction of sucrose, oxalic acid and PVP precursors could retain the anti-oxidant and increased microwave time converts the anti-oxidant to its furan derivatives. The trapping of 5-HMF inside the C-dots was confirmed by 13C NMR spectroscopy and LC-MS studies. The C-dot with the highest concentration of 5-HMF oxidizes water at a low overpotential of 0.21(±0.03) V generating current density up to 33.6(±2.3) mA cm−2. With C10–C30 electrocatalysts prepared at an increased duration of microwave irradiation, the current densities progressively decrease and the overpotential at 10 mA cm−2 increases to 0.34–0.43 V. The C-dots and G-dots prepared from other precursors produce maximum current densities of 15–25 mA cm−2 and the overpotentials to reach 10 mA cm−2 are 0.39–0.41 V. Within the first 10 min of bulk electrolysis, 5-HMF in C5, C10 and C15 could extract the ˙OH/˙OOH radicals and can increase the H2O concentration around the catalytically active sites such as the topological defects and carbon atoms attached to CO for vigorous O2 evolution. Post 10 min, 5-HMF is likely consumed and the resulting furan derivatives act as catalytic sites to drive the OER activity. Therefore, one of the limitations of this approach for designing OER catalysts is the unstable nature of 5-HMF which decomposes to the furan derivatives over the course of continuous electrolysis. Secondly, the electro-kinetics are challenged by the poor electrical conductivity of the C-dots which is due to the presence of amorphous elemental carbon, 5-HMF and furan derivatives. However, this disadvantage is overcome by the ability of the trapped anti-oxidant to readily scavenge the intermediate radicals and the presence of keto-groups acting as the catalytic sites. The 5-HMF molecules could not be trapped in the differently synthesized C-dots from precursors other than sucrose, oxalic acid and PVP. Here the keto groups and topological defects facilitate the OER activity albeit at higher overpotentials compared to the anti-oxidant containing C-dots. Although the stability of 5-HMF is a matter of concern and needs to be addressed, we believe that our approach will open up new avenues in the OER and has the potential to be extended to other anti-oxidants produced from carbohydrates (Table S7†). The antioxidants can be impregnated or intercalated within functionalized carbon nanomaterials such as carbon nanotubes and graphene or other well known OER catalysts.
Acknowledgements
The Department of Science and Technology (DST), Government of India is duly acknowledged for the financial support under grant no. SR/S1/PC-28/2011. AD and SK thank University Grants Commission (UGC) and Council of Scientific and Industrial Research (CSIR), New Delhi, India for their fellowships, respectively. S. B. acknowledges the academic and research funding of IISER Kolkata.Notes and references
- J. K. Hurst, Science, 2010, 328, 315 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- X. Lu, W. Yim, B. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. D. Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826 CrossRef CAS PubMed.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1756 Search PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- J. Pfrommer, M. Lublow, A. Azarpira, C. Göbel, M. Lücke, A. Steigert, M. Pogrzeba, P. W. Menezes, A. Fischer, T. Schedel-Niedrig and M. Driess, Angew. Chem., Int. Ed., 2014, 53, 5183 CAS.
- O. Diaz-Morales, T. J. P. Hersbach, D. G. H. Hetterscheid, J. N. H. Reek and M. T. M. Koper, J. Am. Chem. Soc., 2014, 136, 10432 CrossRef CAS PubMed.
- M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042 CrossRef CAS PubMed.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. Galán-Mascarós, J. Am. Chem. Soc., 2013, 135, 13270 CrossRef CAS PubMed.
- J. D. Baran, H. Grönbeck and A. Hellman, J. Am. Chem. Soc., 2014, 136, 1320 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454 CrossRef CAS PubMed.
- M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042 CrossRef CAS PubMed.
- A. Annamalai, A. G. Kannan, S. Y. Lee, D.-W. Kim, S. H. Choi and J. S. Jang, J. Phys. Chem. C, 2015, 119, 19996 CAS.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820 CrossRef CAS PubMed.
- N. Zhang, Y. Zhang and Y.-J. Xu, Nanoscale, 2012, 4, 5792 RSC.
- K. Cheng, D. He, T. Peng, H. Lv, M. Pan and S. Mu, Electrochim. Acta, 2014, 132, 356 CrossRef CAS.
- A. Datta, S. Kapri and S. Bhattacharyya, Green Chem., 2015, 17, 1572 RSC.
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109 CrossRef CAS PubMed.
- S. Bhattacharyya, I. Perelshtein, O. Moshe, D. H. Rich and A. Gedanken, Adv. Funct. Mater., 2008, 18, 1641 CrossRef CAS.
- Z. Bullard, E. C. Girão, J. R. Owens, W. A. Shelton and V. Meunier, Sci. Rep., 2015, 5, 7634 CrossRef CAS PubMed.
- S. Kapri, S. Maiti and S. Bhattacharyya, Carbon, 2016, 100, 223 CrossRef CAS.
- R. Singhal, Z. Orynbayeva, R. V. K. Sundaram, J. J. Niu, S. Bhattacharyya, E. Vitol, M. Schrlau, E. Papazoglou, G. Friedman and Y. Gogotsi, Nat. Nanotechnol., 2011, 6, 57 CrossRef CAS PubMed.
- R. Singhal, S. Bhattacharyya, Z. Orynbayeva, E. Vitol, G. Friedman and Y. Gogotsi, Nanotechnology, 2010, 21, 015304 CrossRef PubMed.
- V. Pelletier, S. Bhattacharyya, I. Knoke, F. Forohar, M. Bichay and Y. Gogotsi, Adv. Funct. Mater., 2010, 20, 3168 CrossRef CAS.
- X. Duan, H. Sun, Y. Wang, J. Kang and S. Wang, ACS Catal., 2015, 5, 553 CrossRef CAS.
- H. Fei, R. Ye, G. Ye, Y. Gong, Z. Peng, X. Fan, E. L. G. Samuel, P. M. Ajayan and J. M. Tour, ACS Nano, 2014, 8, 10387 Search PubMed.
- Y. Gong, H. Fei, X. Zou, W. Zhou, S. Yang, G. Ye, Z. Liu, Z. Peng, J. Lou, R. Vajtai, B. I. Yakobson, J. M. Tour and P. M. Ajayan, Chem. Mater., 2015, 27, 1181 CrossRef CAS.
- I.-Y. Jeon, H-J. Choi, S-M. Jung, J-M. Seo, M-J. Kim, L. Dai and J-B. Baek, J. Am. Chem. Soc., 2012, 135, 1386 CrossRef PubMed.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362 RSC.
- M. Zhang, Q. Yao, W. Guan, C. Lu and J. M. Lin, J. Phys. Chem. C, 2014, 118, 10441 CAS.
- Y. Han, H. Huang, H. Zhang, Y. Liu, X. Han, R. Liu, H. Li and Z. Kang, ACS Catal., 2014, 4, 781 CrossRef CAS.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272 RSC.
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297 CrossRef CAS PubMed.
- H. He, X. Wang, Z. Feng, T. Cheng, X. Sun, Y. Sun, Y. Xia, S. Wang, J. Wang and X. Zhang, J. Mater. Chem. B, 2015, 3, 4786 RSC.
- M. Xu, S. Xu, Z. Yang, M. Shu, G. He, D. Huang, L. Zhang, L. Li, D. Cuia and Y. Zhang, Nanoscale, 2015, 7, 15915 RSC.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118 RSC.
- S. Mitra, S. Chandra, P. Patra, P. Pramanik and A. Goswami, J. Mater. Chem., 2011, 21, 17638 RSC.
- A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Chem. Commun., 2012, 48, 407 RSC.
- X. Zhai, P. Zhang, C. Liu, T. Bai, W. Li, L. Dai and W. Liu, Chem. Commun., 2012, 48, 7955 RSC.
- H. Li, X. He, Y. Liu, H. Huang, S. Lian, S.-T. Lee and Z. Kang, Carbon, 2011, 49, 605 CrossRef CAS.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747 CrossRef CAS PubMed.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921 RSC.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578 CrossRef CAS PubMed.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405 CrossRef CAS PubMed.
- X. Liu, S. Cui, Z. Sun and P. Du, Electrochim. Acta, 2015, 160, 202 CrossRef CAS.
- N. Murai, M. Yonaga and K. Tanaka, Org. Lett., 2012, 14, 1278 CrossRef CAS PubMed.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286 RSC.
- M. M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204 RSC.
- H. Pawara and A. Lali, RSC Adv., 2014, 4, 26714 RSC.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518 CrossRef CAS PubMed.
- Y. X. Li, Y. Li, Z. J. Qian, M. M. Kim and S. K. Kim, J. Microbiol. Biotechnol., 2009, 19, 1319 CAS.
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103 RSC.
- N. L. Weinberg and H. R. Weinberg, Chem. Rev., 1968, 68, 449 CrossRef CAS.
- R. Chen, G. Sun, C. Yang, L. Zhang, J. Miao, H. Tao, H. Yang, J. Chen, P. Chen and B. Liu, Nanoscale Horiz., 2016, 1, 156 RSC.
- H. Fei, Y. Yang, Z. Peng, G. Ruan, Q. Zhong, L. Li, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083 CAS.
- A. A. Baradii, K. B. Kokoh, H. Huser, C. Lamy and J.-M. Léger, Electrochim. Acta, 1999, 44, 2779 CrossRef CAS.
- E. Detsi, J. B. Cook, B. K. Lesel, C. L. Turner, Y.-L. Liang, S. Robbennolt and S. H. Tolbert, Energy Environ. Sci., 2016, 9, 540 CAS.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- L. Zhao, F. Di, D. Wang, L.-H. Guo, Y. Yang, B. Wan and H. Zhang, Nanoscale, 2013, 5, 2655 RSC.
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M. M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644 CAS.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850 RSC.
- S. Jiang, Z. Li, H. Wang, Y. Wang, L. Menga and S. Song, Nanoscale, 2014, 6, 14262 RSC.
- O. Kargbo, Y. Jin and S.-N. Ding, Curr. Anal. Chem., 2015, 11, 4 CrossRef CAS.
- D. Könning, W. Hiller and M. Christmann, Org. Lett., 2013, 14, 624 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta04737h |
| This journal is © The Royal Society of Chemistry 2016 |