
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlginate/graphene double-network nanocomposite hydrogel beads with low-swelling, enhanced mechanical properties, and enhanced adsorption capacity†
Yuan
Zhuang
a,
Fei
Yu
c,
Hong
Chen
d,
Jie
Zheng
d,
Jie
Ma
*ad and
Junhong
Chen
*ab
aState Key Laboratory of Pollution Control and Resource Reuse, School of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, P. R. China. E-mail: jma@tongji.edu.cn; Tel: +86-21-6598-1831
bDepartment of Mechanical Engineering, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. E-mail: jhchen@uwm.edu
cCollege of Chemistry and Environmental Engineering, Shanghai Institute of Technology, Shanghai 2001418, China
dDepartment of Chemical and Biomolecular Engineering, The University of Akron, Akron, Ohio 44325, USA
First published on 10th June 2016
Abstract
As a novel material, double network hydrogels have attracted great attention in recent years for their excellent mechanical properties; however, several other characteristics are yet to be improved. Here we report the synthesis of a novel alginate/reduced graphene oxide (RGO) double-network (GAD) hydrogel through a facile method, and investigate the GAD's mechanical properties, stability, and adsorption capacity in comparison with those of an alginate/RGO single network hydrogel (GAS). To produce the GAD, the first network of alginate is formed with randomly distributed graphene oxide (GO), resulting in the GAS; then the GAS is treated by a hydrothermal reduction, through which the GO is reduced and self-assembles into a second RGO network interpenetrating with the first, alginate network, forming the double-network GAD. The mechanism of the GAD formation is investigated and the property differences between the GAS and the GAD are examined. The resulting GAD exhibits a higher Young's modulus than the GAS, and the modulus increases with GO concentrations. The GAD also has a lower swelling ratio than the GAS, which leads to improved gel stability in highly concentrated alkali/salt solutions. The GAD beads exhibit an excellent adsorption capacity (Cu2+, 169.5 mg g−1 and Cr2O72−, 72.5 mg g−1) for heavy metal ions, far better than that of the GAS. Even after 10 regeneration cycles, both GAS and GAD can still retain their considerable adsorption capacity for metals. The results of this work are of great significance to double network gel research, especially for environmental applications. With good stability, adsorption capacity, and regeneration ability, the double network gel could be a promising adsorbent nanomaterial for pollutant removal from aqueous solutions.
Introduction
Alginate, a natural polysaccharide extracted from seaweeds, has a linear chain structure of (1–4)-linked β-D-mannuronic acid (M) and α-L-guluronic acid (G) residues arranged in a block-wise fashion, giving it hydrophilicity, biocompatibility, nontoxicity and exceptional formability.1 Alginate can chelate with divalent cations to form hydrogels. The gel formation is driven by interactions between G-blocks, which form tightly held junctions in the presence of divalent cations.2 Alginate gels are widely used in a variety of applications such as in biomedical and other functional materials;3,4 however, ionic alginate gels have limited stability as the ion exchange between divalent ions in the alginate and monovalent ions in solutions could destabilize and rupture the gel.5 Moreover, the use of hydrogels has been severely limited because conventional hydrogels inevitably “swell” under specific conditions, which leads to drastic degradation of their mechanical properties.6 Conventional methods to stabilize alginate hydrogels include covalent crosslinking and introduction of high concentrations of crosslinking cations to ensure tight connections between polymer chains.7 Recently, to improve the mechanical properties of alginate hydrogels, the formation of novel network structures in gels, such as nanocomposites8 and double networks,9 has been proposed. Gong et al.10 first reported double-network hydrogels.11 Typically, double-network gels consist of two interpenetrating polymer networks with contrasting structural and mechanical properties. The first network is highly stretched and densely crosslinked, making it stiff and brittle while the second network is flexible and sparsely crosslinked, making it soft and stretchable.Graphene oxide (GO), a popular carbon nanomaterial, can be turned into reduced GO (RGO) hydrogels through the self-assembly of GO sheets by hydrothermal treatments. There are many chemical functionalities on the GO sheets such as hydroxyl, epoxy, and carboxyl groups, allowing GO to be further functionalized for hybridization with other materials to form composite materials.12–16 GO and alginate both contain negative ions, which could cause electrostatic repulsion between them and allow GO to be well dispersed.17 Moreover, GO nanosheets can crosslink the polymer chains through surface grafting and hydrogen bonding interactions.18,19 Fan et al.18 prepared a double-network nanocomposite consisting of alginate, GO and polyacrylamide, but the double network in this material was formed by alginate and polyacrylamide, while GO was only used as an additive. Sun et al.20 synthesized extremely stretchable and tough hydrogels using ionically crosslinked alginate and covalently crosslinked polyacrylamide. Huang et al.21 prepared a double-network hydrogel of graphene and poly(acrylic acid) with excellent mechanical properties. However, in previous studies, synthetic high-molecular weight polymers were the main choices for the formation of double networks, because the goal in the traditional double-network studies was to obtain gels with better mechanical properties. GO could form 3D networks crosslinked by polymers, and studies have investigated the nanocomposites of graphene and natural polymers;22 however, until now, there have been few reports on GO forming double networks with natural polymers. Moreover, it is worth noting that GO nanosheets could form a loose 3D network originating from the coordinating interactions between oxygen-containing groups of GO and calcium ions, which has largely been ignored in previous studies of GO and alginate composites. Thus the formation of a stable double network consisting of alginate and graphene is feasible and attractive.
In this study, we demonstrate a facile approach to continuously prepare a graphene/alginate double-network nanocomposite gel (GAD) by combining the unique characteristics of the conventional double-network hydrogel and the nanocomposite hydrogel. In the composite, alginate was crosslinked first to form a single network, and then GO formed another network across the alginate network through reduction. The formation of the second GO network improves the material properties, which makes the hydrogel stronger and more stable. The resulting GAD was used as an adsorbent material for the removal of heavy metals from aqueous solutions. The experimental results revealed that the GAD has excellent adsorption capacity for heavy metals. The GAD features minimal swelling upon water immersion, and the resulting GAD shows excellent alkali/salt resistance performance attractive for practical applications.
Results and discussion
The preparation process of the gels is shown in Scheme 1. The GO could form RGO hydrogels through reduction, and the sodium alginate forms alginate beads as soon as the sodium alginate liquid is dropped into the CaCl2 solution through crosslinking. For the alginate/graphene composite, the sodium alginate/GO mixture forms the single network gel, which consists of 3D alginate networks and 2D GO. Then, after being treated by reduction, the GO forms another 3D network, which interpenetrates but remains independent of the alginate network, and thus the double network gel is obtained. Fig. S1† shows an optical image of hydrogels with different GO concentrations. It can be seen that the diameter of alginate/graphene beads increased with the increase of GO content both for the single network and the double network, indicating strong interactions between GO and alginate. Under an optical microscope, as shown in Fig. S2,† there are parallel circular waves on the surface of the alginate bead. However, in the GAS, many waves appear in other directions besides the parallel ones, indicating that the addition of 2D GO has influence on the surface structure of 3D alginate beads. Such a rough structure may be beneficial for adsorption because the increased surface area makes more and stronger interactions with pollutants. Moreover, in the GAD, the waves are in disorder. The different surfaces between the GAS and the GAD may be due to the different combination types of alginate and GO in the composite. It is the hydrophilic GO that is located on the hydrophilic alginate in the GAS, and the hydrophilic GO becomes hydrophobic 3D RGO in the GAD.23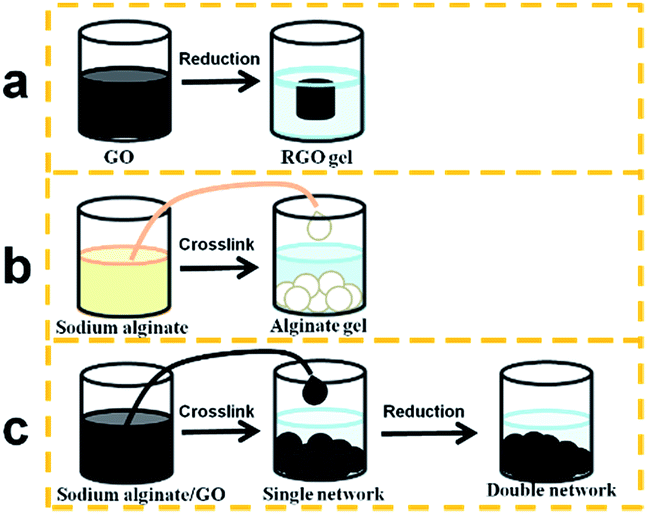 | ||
| Scheme 1 Preparation process of the RGO hydrogel (a), alginate hydrogel bead (b), and alginate/graphene composite hydrogel bead (c). | ||
To further investigate the structure of alginate/graphene beads, Scanning Electron Microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) images of graphene–alginate single networks (GAS) and the GAD are shown in Fig. 1. There are many particle-like structures in the GAS (Fig. 1a), while there are lots of petal-like structures in the GAD (Fig. 1b). As it is known that graphene has a sheet structure, the petal-like structures are likely attributed to the self-assembled graphene sheets. It is plausible that the second network consisting of graphene was formed on the first alginate network, and then the two networks formed a double network together. As a result, the structure turned from particle-like into petal-like. The petal-like structure of GO could expose more edges and more active sites for pollutant adsorption.
 | ||
| Fig. 1 SEM images of (a) GAS and (b) GAD; elemental mapping of (c) GAS and (d) GAD, with insets showing O elemental mapping. | ||
To further investigate the self-assembly process of GO, EDS mapping of the GAS and GAD was performed, as shown in Fig. 1c and d. The petal-like structure of the GAD coincides with the O element distribution. However, the particle-like structure of the GAS has no coincidence with O distribution, indicating that hydrogen bonds play an important role in the formation of 3D RGO. On one hand, hydrogen bonds between alginate and RGO allow them to form a uniform composite as seen in the GAS. On the other hand, hydrogen bonds between the graphene sheets in RGO promote the formation of 3D RGO in the GAD.
The transmission electron microscopy (TEM) images of the GAS and GAD in Fig. S3† show that the GAD has more edges and smaller sheets than the GAS. The increase of graphene edges has also been observed in the translation of pure GO to RGO, further indicating the self-assembly of GO sheets.24
In the Raman spectra (Fig. 2a), two characteristic bands located at around 1350 cm−1 (D band) and 1570 cm−1 (G band) are observed in both GAS and GAD. The D band could reflect the presence of structural defects connected with vacancies, grain boundaries, and amorphous carbon species; the G band is related to the sp2 graphitic domains (non-oxide regions).24 The intensity ratio of the D band to the G band ID/IG of the GAD (0.78) is higher than that of the GAS (0.52), indicating that there are more structural defects and more edges formed in the double network. This change after reduction is consistent with other investigations on the transformation from GO to RGO gels.25,26
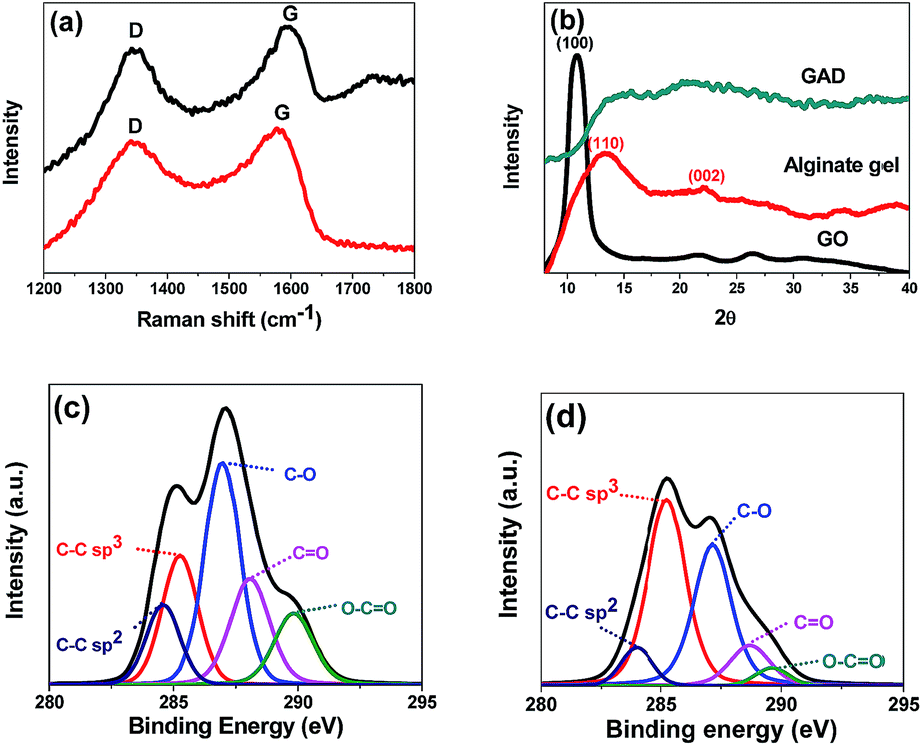 | ||
| Fig. 2 Raman spectra (a) of the GAS and GAD; XRD patterns (b) of GO, alginate, and GAD; The deconvolution of C1s XPS spectra of the GAS (c) and GAD (d). | ||
The XRD patterns of GO, alginate gel and GAD are shown in Fig. 2b. In GO, the peak at 2θ = 10.6° is a characteristic peak of GO with an interlayer spacing of 8.34 Å.27 In the alginate gel, the two typical peaks around 15° (110) and 22.5° (002) correspond to the lateral packing among molecular chains and the layer spacing along the molecular chain direction. In the GAD, the disappearance of GO characteristic peaks after 3D graphene formation is also found in other studies of GO hydrogel preparation.22 Such changes indicate the absence of direct interaction or ordered stacking between GO sheets to form 3D networks.22
XPS was conducted to further study the chemical structures of the GAS and GAD (Fig. 2c). In the deconvolution of C1s XPS spectra, the peaks at binding energies of 284.5, 286.5, 287.8 and 289.0 eV are attributed to the non-oxide C (C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), C–O bonds, carbonyl C (CO), and carboxylate C (O–CO).28,29 The GAD has stronger C–C than that of the GAS, due to the π–π interaction during the 3D graphene network formation. Moreover, the GAD has a stronger sp3 C–C bond than the GAS, indicating that there are more structural defects, consistent with the Raman results.
C), C–O bonds, carbonyl C (CO), and carboxylate C (O–CO).28,29 The GAD has stronger C–C than that of the GAS, due to the π–π interaction during the 3D graphene network formation. Moreover, the GAD has a stronger sp3 C–C bond than the GAS, indicating that there are more structural defects, consistent with the Raman results.
Swelling and mechanical properties
Conventional hydrogels often swell in water, which drastically weakens the mechanical toughness of hydrogels. For hydrogels that exhibit hysteresis during deformation, the equilibrium between osmotic and elastic energies is inevitably lost when one network is partly broken.6Fig. 3a shows the swelling ratios of the GAS and GAD with different GO contents at different pH values, all of which increased with increasing pH and decreased with increasing GO contents. Under all conditions, the GAD had a lower swelling ratio than the GAS. Kamata et al.6 proved that the mechanical properties of hydrogels are strongly affected by their degree of swelling, and that the swelling suppression of hydrogels may help maintain their initial shape and retain their mechanical properties. As the GAD had a lower swelling ratio than the GAS, the GAD may have better mechanical properties than the GAS. Thus the GAD was more stable than the GAS, and was not easily destroyable by the swelling force.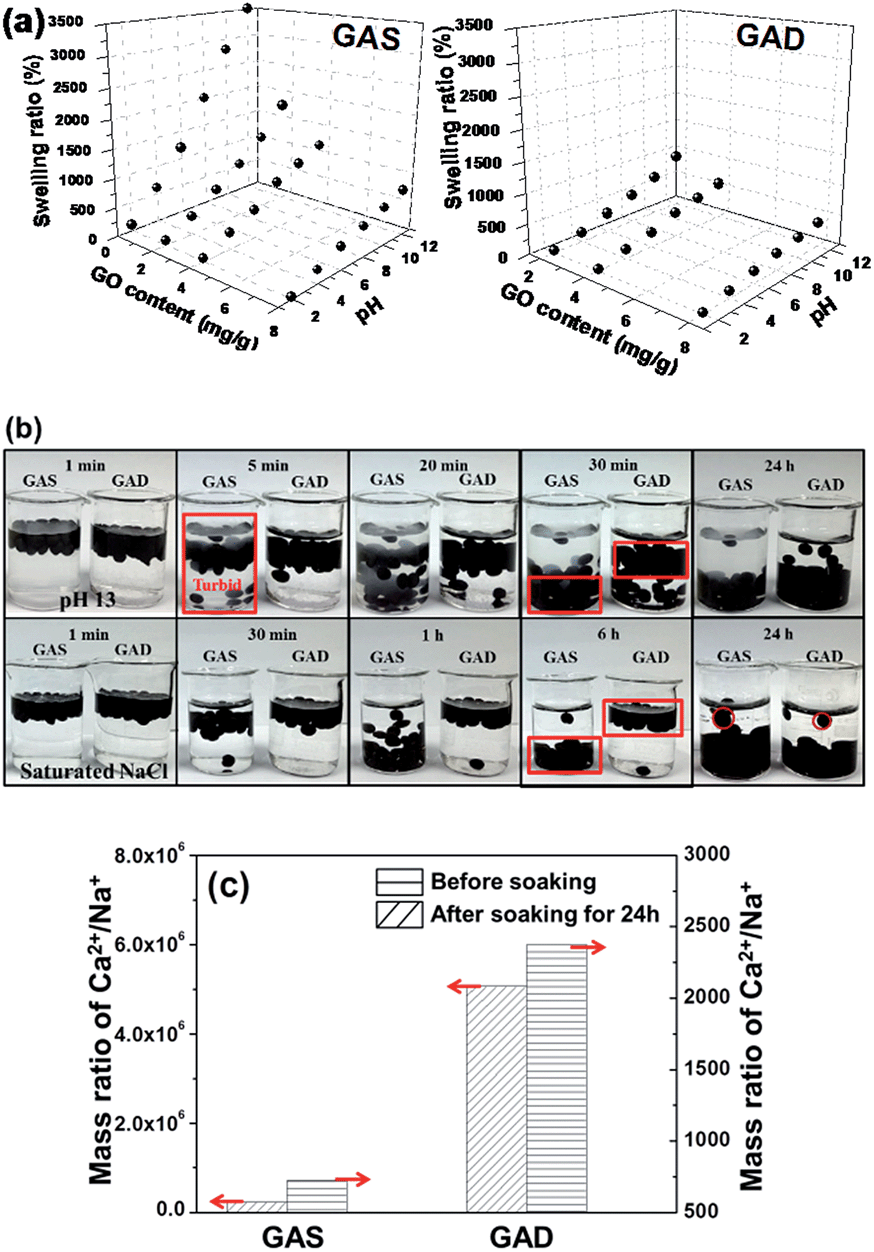 | ||
| Fig. 3 Swelling ratios (a) of the GAS and GAD with different GO content; optical images (b) of the GAS and GAD soaked in NaOH (pH = 13) and saturated NaCl solutions at different times; and the ICP results (c) of the GAS and GAD before and after being soaked in NaCl for 24 h. | ||
Since the alginate-based materials have limited stability and could swell in monovalent ion solutions,30 it is necessary to investigate the swelling ratio (as shown in eqn (1)) of the GO/alginate gel in salt and alkaline solutions. To further study the swelling behaviors between single networks and double networks, we placed the GAS and GAD into a NaOH solution of pH 13 and a saturated solution of NaCl (Fig. 3b). At pH 13, within 5 min, suspensions appeared in the solution containing the GAS due to the over-swelling of alginate in a strong base. Both GAS and GAD began to sink in 5 min, but the GAD sank much slower than the GAS. After 30 min, almost all the GAS beads reached the bottom of the beaker while most of the GAD beads remained in the upper portion of the solution. After 24 h, though most of the beads sank to the bottom for both GAS and GAD, the GAD solution remained clear while the GAS solution was turbid. Since the beads had a higher swelling ratio at a higher pH, the GAD had better pH tolerance than the GAS.
In a saturated solution of NaCl, the difference of the sinking speed between the GAS and the GAD becomes more obvious: after 6 h, almost all of the GAS beads reached the bottom of the beaker while almost all of the GAD beads did not have visible movement. After 24 h, the GAD beads arrived at the bottom without an obvious size change, while the GAS beads clearly became larger. Therefore, the GAD had better stability in high-concentration salt solutions than the GAS. To further investigate the change of the beads in NaCl solutions, an inductively coupled plasma emission spectrometer (ICP) was used to measure the ionic concentrations of the GAS and GAD before and after they were soaked in NaCl for 24 h, and the results are shown in Table S1.† Moreover, as the Ca2+ in the beads could have ion exchange with Na+ in the solution, which would make the bead unstable, we calculated the mass ratio of Ca2+/Na+ as shown in Fig. 3c. In the original beads, the Ca2+/Na+ in the GAD is higher than that in the GAS, indicating the enhanced interaction between alginate and Ca2+ content in the crosslinking process of alginate gel formation. After being soaked in NaCl for 24 h, the GAS and GAD had nearly equivalent Na+ content; however, the GAD had 3 times higher Ca2+ content than the GAS. In addition, the Ca2+/Na+ in the GAD is higher than that in the GAS, indicating a better crosslinking between alginate and Ca2+ in the GAD, which suggests that the 3D graphene could help the GAD retain more Ca2+ during the ionic exchange process in the NaCl solution than the 2D graphene in the GAS.
Moreover, the addition of GO and the formation of double networks obviously suppressed the swelling of the gel beads, because (1) the GO hydrogel shows little swelling behavior; (2) the increased crosslinking density of the GAD over the GAS after hydrothermal treatment by covalent bonding; and (3) hydrogen bonds could restrict the mobility of alginate, thereby improving the swelling resistance.24
For the mechanical test, all the hydrogels were made into cylindrical shapes as shown in Fig. 4a; the diameter of the hydrogels was 1 mm (±0.1 mm). It can be seen that the hydrogels are flexible. Fig. 4b shows the stress–strain profile and Fig. 4c shows the Young's modulus of the single network and double network hydrogels at different GO concentrations. For both single network and double network hydrogels, samples with higher GO concentrations exhibited a higher Young's modulus. Upon GO addition into the alginate-based gel, the mechanical property improvement has also been found in other studies.17,18 For the hydrogel at the same GO concentration, the double network gels have better compressive strength than the single network gels. The increase of strength with increasing GO content and the increase of strength after the double network formation are consistent with the change in the swelling ratio and further prove that swelling behaviour could reduce the mechanical strength. In the stress–strain curves of the single network at GO concentrations of 2 mg mL−1, 4 mg mL−1 and 8 mg mL−1, all the curves show yielding-like behaviour, which indicates the damage of the first network before the macroscopic fracture of the whole double network;31 moreover, for the pure alginate gel and double network, the hydrogels do not have such yielding behaviour, indicating that the GAD had no fracture strain and fracture strength in the compression test.32 Kitiri et al.31 compared the properties of the double network of a different first network, and found that the lack of charge in the first network may result in a random coil configuration, which would lead to a greater deformability under compression. The Young's modulus of the pure alginate hydrogel is 0.29 MPa, and those of the GAS and GAD are 0.93 MPa and 2.14 MPa respectively at a GO concentration of 8 mg mL−1. Thus the GAD achieved a higher strength than the GAS even without adding any crosslinker of the second network. It is because the second RGO network in this study is formed through a self-assembly process rather than a crosslinking process.33 Commonly, the traditional double network consists of a rigid first network and a soft second network;33 however, this study shows that the gel could also get better mechanical properties while the first network is soft and the second network is rigid.
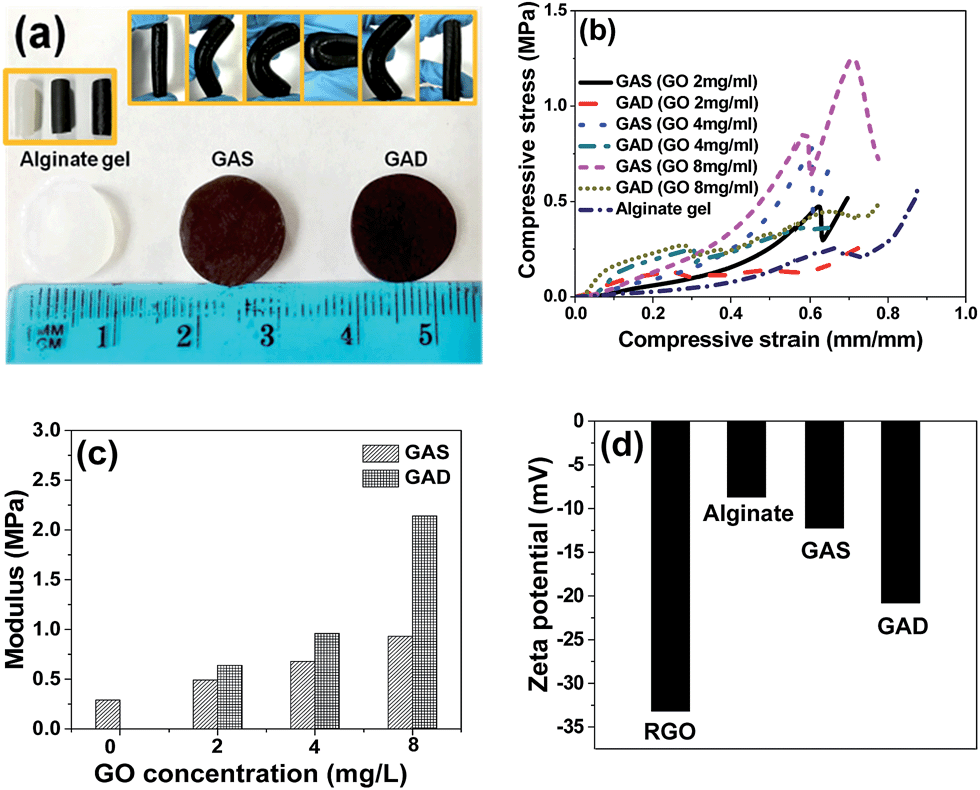 | ||
| Fig. 4 (a) Photograph of alginate, GAS and GAD hydrogels for the mechanical property test, (b) compressive stress–strain profile and (c) Young's modulus of hydrogels at different GO concentrations, (d) zeta potential of RGO, alginate gel, GAS and GAD (GO concentration 8 mg mL−1) at pH 8. | ||
To study the influence of surface charges on the hydrogel properties, zeta potential values of RGO, alginate gel, GAS and GAD are shown in Fig. 4d. The zeta potential of the first network alginate gel is −8.68 mV and the second network RGO is −33.19 mV, and thus the second network contains more charges than the first network. As a result, the random coil configuration would occur in the compression process of the GAD. Moreover, the GAD has a higher zeta potential value than the GAS, which indicates the higher ionization degree of the double network than that of the single network. The charge change may be related to the graphene self-assembly process in the second network formation. And the electrostatic repulsion between the alginate network and the RGO network would be strengthened in the double network. Therefore, the two networks had more power to support each other to form a stronger hydrogel.
Adsorption properties
To investigate the adsorption properties of the GAS and GAD, toxic anionic and cationic heavy metal ions Cr2O7 (ref. 2–34) and Cu2+ (ref. 35) were used as target pollutants. The influence of GO concentration on metal adsorption on the GAS and GAD is shown in Fig. 5a, and it can be seen that the adsorbents have higher adsorption capacities at higher GO concentrations. As shown in Fig. 5b, the removal rates for all the pollutants on the adsorbents increase with increasing solid-to-liquid ratio, while the adsorption capacities have an opposite trend. It is because there are more active sites offered by the adsorbent at a higher adsorbent dosage; moreover, the rate of increase is faster at a lower solid-to-liquid ratio, which is because more active sites would overlap at a higher solid-to-liquid ratio. Considering the increase of removal rate and the decrease of adsorption capacity, 1 mg mL−1 is chosen as the optimum solid-to-liquid ratio. The Langmuir isotherm, which is the most commonly used isotherm model in adsorption studies, assumes that there is a monolayer formed by the adsorbate on the homogenous adsorbent surface, and there is no interaction between the adsorbent and adsorbed adsorbate molecules.36 Moreover, the Langmuir model could also be used to estimate the maximum adsorption capacity.37 Based on Langmuir adsorption, the isotherms of Cr2O72− and Cu2+ on the GAS and GAD are shown in Fig. 5c. The heavy metal adsorption results of the GAS and GAD also fit well with the Langmuir model which indicates the energetic homogeneity of the bead surface.38 Langmuir and Freundlich models were used to fit the metal adsorption process as shown in Fig. S4a and b,† and the parameters are shown in Table S2.† The GAD had a great adsorption capacity for Cu2+ (169.5 mg g−1) and for Cr2O72− (72.46 mg g−1). The Langmuir model assumes an adsorption homogeneity,39,40 and the results fit the Langmuir model better than the Freundlich model, which indicates the good uniformity of the samples. The adsorption of Cu2+ on the GAD is relatively higher than that of most of the other adsorbents such as alginate clay,41 porous alginate beads,42 superparamagnetic alginate,43 magnetic alginate beads,38 collagen/cellulose beads,44 activated carbon, and alginate/activated carbon.45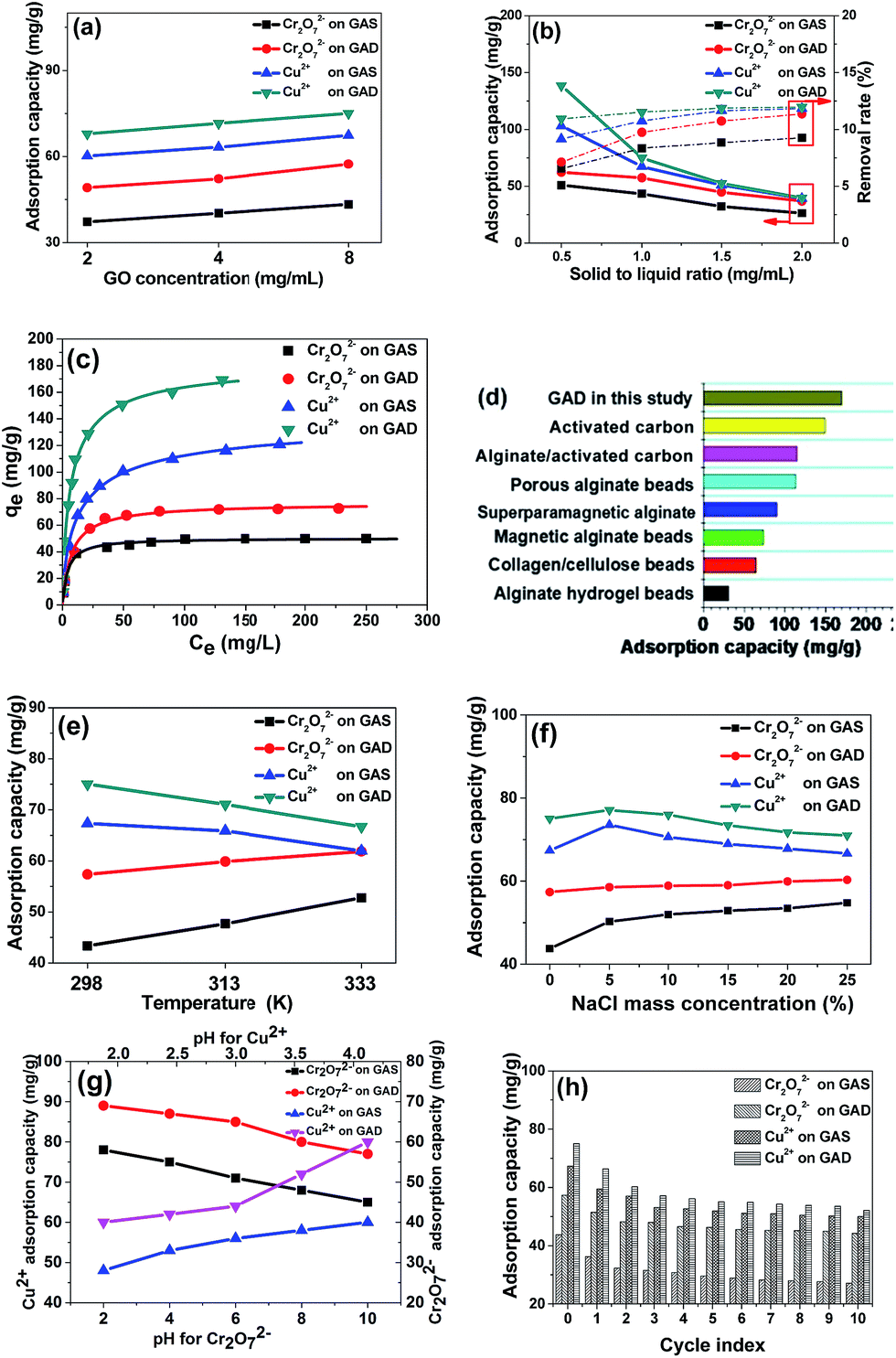 | ||
| Fig. 5 (a) Influence of GO concentration on Cr2O72− and Cu2+ adsorption on the GAS and GAD (initial metal concentration 100 mg mL−1, 298 K, solid-to-liquid ratio 1 mg mL−1, pH 4), (b) influence of solid-to-liquid ratio on metal (Cr2O72− and Cu2+) adsorption on the GAS and GAD (initial metal concentration 80 mg mL−1, 298 K, GO concentration 8 mg mL−1, pH 4), (c) Langmuir adsorption isotherms of metal (Cr2O72− and Cu2+) adsorption on the GAS and GAD (298 K, GO concentration 8 mg mL−1, solid-to-liquid ratio 1 mg mL−1, pH 4), (d) comparison of the adsorption capacity of Cu2+ with other materials, (e) influence of temperature on metal (Cr2O72− and Cu2+) adsorption on the GAS and GAD (initial metal concentration 100 mg mL−1, GO concentration 8 mg mL−1, solid-to-liquid ratio 1 mg mL−1, pH 4), (f) influence of NaCl on metal (Cr2O72− and Cu2+) adsorption on the GAS and GAD (initial metal concentration 100 mg mL−1, 298 K, GO concentration 8 mg mL−1, solid-to-liquid ratio 1 mg mL−1, pH 4), (g) influence of pH on metal (Cr2O72− and Cu2+) adsorption on the GAS and GAD (initial metal concentration 100 mg mL−1, 298 K, GO concentration 8 mg mL−1, solid-to-liquid ratio 1 mg mL−1), (h) regeneration of the GAS and GAD (initial metal concentration 100 mg mL−1, 298 K, GO concentration 8 mg mL−1, solid-to-liquid ratio 1 mg mL−1, pH 4). | ||
The Cu2+ adsorption capacity on different kinds of adsorbents is shown in Fig. 5d, which reflects the excellent adsorption capacity of the GAD. As shown in Fig. 5e, Cu2+ favours a lower temperature while Cr2O72− favours a higher temperature; this is because the high temperature would reduce the electrostatic interaction, and thus the hydrogel has lower electrostatic attraction for Cu2+ and lower electrostatic repulsion for Cr2O72−.
As NaCl widely exists in the environment and has an influence on the hydrogel stability, we studied the influence of NaCl on adsorption. As shown in Fig. 5f, the existence of NaCl also improves the adsorption of Cr2O72− on both the GAS and the GAD; however, a higher NaCl concentration could reduce the adsorption for Cu2+. The phenomenon of Na+ promoting pollutant adsorption on alginate has also been observed in other studies.46 For Cu2+ adsorption, there is an increase in active sites arising from the ion exchange between Na+ and Ca2+ at low NaCl concentrations; however, at higher NaCl concentrations, the Na+ could compete with the Cu2+ during the ion exchange with Ca2+. Thus the adsorption capacity for Cu2+ begins to decrease.
The influence of pH is an important factor during metal adsorption.47 As shown in Fig. 5g, the pH range for Cu2+ is 2 to 4 to avoid precipitation at higher pH. For Cu2+, it could hydrolyze and lead to the precipitation of Cu(OH)2 when the pH is higher than 4. As Cr2O72− is an anion, it would not precipitate in the same pH range. It can be seen that the adsorption capacity of Cr2O72− decreases with increasing pH, while the Cu2+ has an opposite trend. This is because, the –COOH in the hydrogel, when at a higher pH, has a higher degree of dissociation, and thus the hydrogel has higher electrostatic attraction for Cu2+ and lower electrostatic repulsion for Cr2O72−.
Considering that regeneration and reusability are important factors of an adsorbent, repeated adsorption–desorption studies of the two metal ions on the GAS and GAD were conducted as shown in Fig. 5h. It can be seen that for both Cr2O72− and Cu2+, the GAS and GAD had good reusability. After 10 cycles, the adsorption capacities of Cr2O72− and Cu2+ on the GAD were maintained at 48.23 and 92.12 mg g−1, respectively. Therefore, there are several important interactions for the metal ion adsorption on the GAS and GAD. The abundant –COOH in both alginate and graphene shows good affinity towards Cr2O72− and Cu2+ through complexation and coordination; on the other hand, there is ion exchange interaction between Ca2+ in alginate and Cu2+ in the solution.48 Moreover, the formation of the double network turns out to be effective for the improvement of adsorption.
To better investigate the adsorption mechanism, the zeta potential of the GAD at different pH values is shown in Fig. S5† from which it can be seen that the GAD has stronger negative charges at a higher pH. At high pH, the influence is more obvious, which corresponds to the influence of pH on adsorption, further proving that the electrostatic interaction plays an important role in the metal adsorption on the GAD.
Conclusion
A new type of double-network nanocomposite hydrogel GAD consisting of the first alginate network and the second graphene network was synthesized and characterized. The GAD has been shown to have enhanced mechanical properties and alkali/salt resistance, lower swelling ratio and better adsorption performance than the single network GAS. The formation of a double network obviously prevents the GO sheets from agglomeration, reduces alginate swelling, and improves the mechanical properties and stability in high concentration alkali/salt solutions. Although previous studies showed that alginate encapsulated nanomaterials had a great adsorption capacity, this study delves into the formation of the double network and discusses the relation between the structural and adsorption properties. Moreover, the application of GAD hydrogels for heavy metal removal demonstrates that the double network in the GAD not only greatly improves their adsorption capacity for heavy ions, but also maintains good reusability after many cycles of adsorption/desorption of heavy ions. Therefore, the obtained results indicate that the GAD double network nanocomposites have great potential for improving gel stability and application in the removal of pollutants from aqueous solutions.Experimental
Materials
All chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) in analytical purity and were used in the experiments without any further purification. All solutions were prepared using deionized water.Preparation of GO/alginate gels
Graphite oxide was obtained using a modified Hummers' method,29,49–51 dispersed in deionized water, and sonicated in an ultrasonication bath for 12 h to obtain a GO solution.Next, 1 g sodium alginate was dissolved into 100 mL GO solution and stirred for 5 h, and then 100 mL sodium alginate and the GO mixture was slowly dropped into 200 mL 10 mg mL−1 CaCl2 solution using a peristaltic pump accompanied by magnetic stirring to obtain the alginate/GO single network beads, as shown in the ESI Video.† The beads were washed several times with deionized water and then freeze-dried. The single-network beads were produced with a GO concentration of 8 mg mL−1 and were termed as GAS. For comparison, an alginate solution without GO also was dropped into 10 mg mL−1 CaCl2 solution accompanied by magnetic stirring to obtain alginate beads.
To obtain the GAD, the GAS hydrogels were placed into 2 mg mL−1 ascorbic acid solution and heated in a water bath at 85 °C for 12 h to form double-network hydrogels termed as GAD. For comparison, a GO solution without alginate was also heated in a water bath after adding ascorbic acid at 85 °C for 12 h to form reduced graphene oxide (RGO) hydrogels. Moreover, single networks and double networks at GO concentrations 2 mg mL−1 and 4 mg mL−1 were also produced to compare with the GAS and GAD. In this paper, besides the study of GO concentration influence, all the GAS and GAD are at a GO concentration of 8 mg mL−1.
To fit the mechanical test instrument, the GAS and GAD were also made into a cylindrical shape through the mixture gelation in the 10 mL injection syringe with a diameter of 1 cm for 48 h.
A swelling study was done for the beads using a common formula for the swelling ratio as shown in eqn (1):52
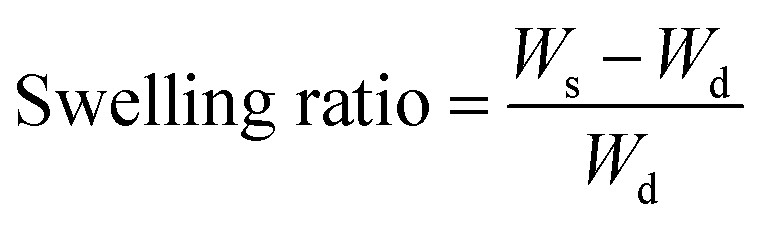 | (1) |
Characterization methods
The surface morphologies of the GAS and GAD were characterized using field-emission scanning electron microscopy (SEM, Hitachi, S-4800) and transmission electron microscopy (TEM, JEOL, JEM-2010). Measurements of micro-Raman spectra were carried out using a Raman spectroscopy system (LabRam, 1B) with a 532 nm wavelength incident laser light and 20 mW power. The X-ray diffraction (XRD) spectra were collected on a Bragg–Brentano diffractometer (Rigaku, D/Max-2200) with monochromatic Cu Kα radiation (λ = 1.5418 Å) of a graphite curve monochromator, and the data were collected from 2θ = 2–40° at a continuous scan rate of 2° min−1 for phase identification. The zeta potential of the material was tested by using a zeta potential analyzer (Brookhaven, PALS ver 3.57) at 23 °C; the concentrations of all the material solutions were 0.3 mg mL−1. The compressive strength was tested using an Instron 3345 single column universal tester with a compression rate of 10 mm min−1.The Na+, Ca2+, Cu2+, and Cr2O72− concentrations in solutions were tested by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES), inductively coupled plasma mass spectrometry (ICP-MS) Agilent 7700 (Agilent, USA) and atomic fluorescence spectrometry (AFS) FP6-A (PERSEE, China).
Adsorption
All the adsorption experiments were studied in a thermostatic shaker at 200 rpm. The metal adsorption was performed at pH = 4. After adsorption, the adsorbent was separated from the solution using a 0.45 μm membrane. The initial concentrations were set in the range of 1–200 mg L−1 for metals. The adsorption capacity (mg g−1) was calculated using eqn (2). | (2) |
Langmuir adsorption isotherms were used to evaluate the adsorption equilibrium as shown in eqn (3). The Langmuir isotherm assumes that the adsorbate forms a monolayer on the homogenous surface of the adsorbent, and there is no interaction between the adsorbed molecules.
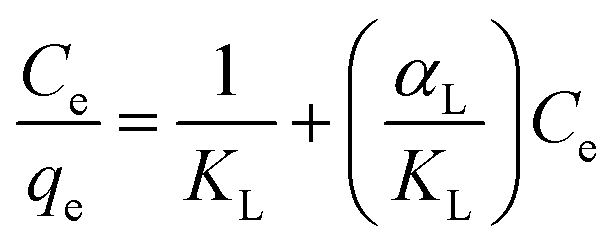 | (3) |
Regeneration of the GAS and GAD was carried out to evaluate their reusability.48 First, a batch sorption test was conducted for 24 h. Next, the used beads were regenerated using a 4% HCl solution for 24 h, followed by washing with distilled water for 3 h to remove hydrogen ions from their surface.
Acknowledgements
This research is supported by The National Natural Science Foundation of China (No. 21207100 and 51072135) and the National Science Foundation (No. 1510099 and 1607475).Notes and references
- Y. Li, F. Liu, B. Xia, Q. Du, P. Zhang, D. Wang, Z. Wang and Y. Xia, J. Hazard. Mater., 2010, 177, 876–880 CrossRef CAS PubMed.
- P. Sikorski, F. Mo, G. Skjak-Braek and B. T. Stokke, Biomacromolecules, 2007, 8, 2098–2103 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- A. Grigoryev, V. Sa, V. Gopishetty, I. Tokarev, K. G. Kornev and S. Minko, Adv. Funct. Mater., 2013, 23, 5903–5909 CrossRef CAS.
- B. Thu, P. Bruheim, T. Espevik, O. Smidsrod, P. SoonShiong and G. SkjakBraek, Biomaterials, 1996, 17, 1069–1079 CrossRef CAS PubMed.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- M. Matyash, F. Despang, C. Ikonomidou and M. Gelinsky, Tissue Eng., Part C, 2014, 20, 401–411 CrossRef CAS PubMed.
- Y. Wang, W. Wang and A. Wang, Chem. Eng. J., 2013, 228, 132–139 CrossRef CAS.
- C. Fan, L. Liao, C. Zhang and L. Liu, J. Mater. Chem. B, 2013, 1, 4251 RSC.
- J. P. Gong, T. Kurokawa, T. Narita, G. Kagata, Y. Osada, G. Nishimura and M. Kinjo, J. Am. Chem. Soc., 2001, 123, 5582–5583 CrossRef CAS PubMed.
- E. Ducrot, Y. L. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- J. J. Shao, W. Lv and Q. H. Yang, Adv. Mater., 2014, 26, 5586–5612 CrossRef CAS PubMed.
- S. Zhang, M. Zeng, W. Xu, J. Li, J. Li, J. Xu and X. Wang, Dalton Trans., 2013, 42, 7854–7858 RSC.
- S. Zhang, M. Zeng, J. Li, J. Li, J. Xu and X. Wang, J. Mater. Chem. A, 2014, 2, 4391–4397 RSC.
- X. Yang, J. Li, T. Wen, X. Ren, Y. Huang and X. Wang, Colloids Surf., A, 2013, 422, 118–125 CrossRef CAS.
- Y. Zhuang, F. Yu, J. Chen and J. Ma, J. Environ. Chem. Eng., 2015, 4, 147–156 CrossRef.
- Y. He, N. Zhang, Q. Gong, H. Qiu, W. Wang, Y. Liu and J. Gao, Carbohydr. Polym., 2012, 88, 1100–1108 CrossRef CAS.
- J. Fan, Z. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433 RSC.
- A. Nieto, R. Dua, C. Zhang, B. Boesl, S. Ramaswamy and A. Agarwal, Adv. Funct. Mater., 2015, 25, 3916–3924 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- P. Huang, W. Chen and L. Yan, Nanoscale, 2013, 5, 6034–6039 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- S. Kim, Y. Yoo, H. Kim, E. Lee and J. Y. Lee, Nanotechnology, 2015, 26, 405602 CrossRef PubMed.
- K. Cao, Z. Jiang, J. Zhao, C. Zhao, C. Gao, F. Pan, B. Wang, X. Cao and J. Yang, J. Membr. Sci., 2014, 469, 272–283 CrossRef CAS.
- A. K. Das, R. K. Layek, N. H. Kim, D. Jung and J. H. Lee, Nanoscale, 2014, 6, 10657–10665 RSC.
- J. Ma, M. X. Yang, F. Yu and J. Zheng, Sci. Rep., 2015, 5, 5749–5760 Search PubMed.
- M. Ionita, M. A. Pandele and H. Iovu, Carbohydr. Polym., 2013, 94, 339–344 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Ma, C. Li, F. Yu and J. Chen, J. Power Sources, 2015, 273, 1048–1055 CrossRef CAS.
- Z. Huang, S. Liu, B. Zhang and Q. Wu, Carbohydr. Polym., 2014, 113, 430–437 CrossRef CAS PubMed.
- E. N. Kitiri, M. Rikkou-Kalourkoti, M. Sophocleous and C. S. Patrickios, Eur. Polym. J., 2015, 69, 573–583 CrossRef CAS.
- W. W. Yao, C. Z. Geng, D. Han, F. Chen and Q. Fu, RSC Adv., 2014, 4, 39588–39595 RSC.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- S. A. Katz and H. Salem, JAT, J. Appl. Toxicol., 1993, 13, 217–224 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- F. Yu, S. Sun, S. Han, J. Zheng and J. Ma, Chem. Eng. J., 2016, 285, 588–595 CrossRef CAS.
- A. S. A. Bakr, Y. M. Moustafa, M. M. H. Khalil, M. M. Yehia and E. A. Motawea, Can. J. Chem., 2015, 93, 289–296 CrossRef CAS.
- F. Yu, J. Ma, J. Wang, M. Zhang and J. Zheng, Chemosphere, 2016, 146, 162–172 CrossRef CAS PubMed.
- Y. Huang, J. Li, X. Chen and X. Wang, RSC Adv., 2014, 4, 62160–62178 RSC.
- W. S. Tan and A. S. Ting, Bioresour. Technol., 2014, 160, 115–118 CrossRef CAS PubMed.
- E. G. Deze, S. K. Papageorgiou, E. P. Favvas and F. K. Katsaros, Chem. Eng. J., 2012, 209, 537–546 CrossRef CAS.
- M. M. Lakouraj, F. Mojerlou and E. N. Zare, Carbohydr. Polym., 2014, 106, 34–41 CrossRef CAS PubMed.
- J. Wang, L. Wei, Y. Ma, K. Li, M. Li, Y. Yu, L. Wang and H. Qiu, Carbohydr. Polym., 2013, 98, 736–743 CrossRef CAS PubMed.
- H. G. Park, T. W. Kim, M. Y. Chae and I.-K. Yoo, Process Biochem., 2007, 42, 1371–1377 CrossRef CAS.
- V. Rocher, J. M. Siaugue, V. Cabuil and A. Bee, Water Res., 2008, 42, 1290–1298 CrossRef CAS PubMed.
- Y. Zhao, J. Li, S. Zhang, H. Chen and D. Shao, RSC Adv., 2013, 3, 18952–18959 RSC.
- B. An, H. Lee, S. Lee, S. H. Lee and J. W. Choi, J. Hazard. Mater., 2015, 298, 11–18 CrossRef CAS PubMed.
- J. Ma, W. Shen, C. Li and F. Yu, J. Mater. Chem. A, 2015, 3, 12307–12313 RSC.
- J. Ma, L. Zhou, C. Li, J. Yang, T. Meng, H. Zhou, M. Yang, F. Yu and J. Chen, J. Power Sources, 2014, 247, 999–1004 CrossRef CAS.
- C. L. J. Ma, F. Yu and J. Chen, ChemSusChem, 2014, 7, 3304–3311 CrossRef PubMed.
- N. Gogoi and D. Chowdhury, J. Mater. Chem. B, 2014, 2, 4089 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta02738e |
| This journal is © The Royal Society of Chemistry 2016 |