
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dandelion-shaped TiO2/multi-layer graphene composed of TiO2(B) fibrils and anatase TiO2 pappi utilizing triphase boundaries for lithium storage†
Weixin
Song
a,
Jun
Chen
b,
Xiaobo
Ji
b,
Xuemei
Zhang
a,
Fang
Xie
a and
D. Jason
Riley
*a
aDepartment of Materials, Imperial College London, London SW72AZ, UK. E-mail: jason.riley@imperial.ac.uk
bCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
First published on 4th May 2016
Abstract
A three-dimensional dandelion-shaped TiO2/multi-layer graphene compound (TiO2/MLG) composed of TiO2(B) fibrils and anatase pappus structures has been synthesized as a potential anode material for Li storage. Electron microscopy indicates that the composite contains triphase boundaries between anatase, TiO2(B) and graphene, which are responsible for the enhancement of energy storage and the decrease of electrode polarization. Cyclic voltammetric investigations indicate that both Li+ insertion and pseudocapacitance contribute to charge storage. Ultrahigh specific capacities of 243 and 182 mA h g−1 have been obtained at 0.1 and 1 A g−1, respectively. Moreover, the excellent capacity retention can reach 99.6% after 100 cycles with almost 100% coulombic efficiency at 0.1 A g−1. The importance of the triphase boundary in enhancing the storage of charge and transport of Li+ is demonstrated.
Introduction
As a consequence of an increasing demand for energy, improved energy storage is required to enable future power systems. Lithium-ion batteries offer high energy and power density and as a result are attracting great interest with a focus on designing electrodes that can enhance energy-storage performance.1,2 Titanium dioxide (TiO2), one of the most promising anode materials, has been intensively studied, not only due to its low cost, abundance and non-toxicity but also on account of the negligible volume deformation during the Li-ion insertion/de-insertion processes (intercalation is preferred to describe lithium storage in a laminar host structure)3 and excellent gravimetric capacity.3–5 The operational potential of TiO2 is high (>1 V vs. Li+/Li), which can enhance battery safety by reducing the probability of the formation of lithium dendrites at the anode surface and avoiding the formation of a solid electrolyte interface (SEI).6–8 To-date various polymorphs of TiO2 including anatase (tetragonal),9 rutile (tetragonal),5 brookite (orthorhombic)10 and bronze (TiO2-B, monoclinic)11 have been extensively studied as anode materials in lithium-ion batteries. The other allotropes, which either are easily transformed to other types like ramsdellite (TiO2-R)12 and hollandite (TiO2-H)13 or need high-pressure and high-temperature synthesis conditions, such as α-PbO2-structured TiO2(II),14 baddeleyite,15 columbite,16 cubic,17 TiO2(OI)18 and TiO2(OII),19 have been rarely researched. Despite the numerous studies, TiO2 has not been used extensively as a high rate anode material owing to its limited ability to store charge and poor conductivity.An intrinsic drawback of TiO2 is the material's poor electronic conductivity (10−13 S cm−1)5,20,21 and limited ionic conductivity.22 Several solutions have been proposed to overcome this issue including: (i) addition of a conducting phase, e.g., carbon coating23 or compositing with transition metal oxides,24 (ii) reduction of the ionic diffusion length by fabricating nanoscale TiO2 structures such as wires,25 tubes,26 cubes,27 rods28 and flowers,8 and (iii) doping through either the introduction of Ti3+ or oxygen vacancies5,29 or with heteroatoms B,30 C,6 N,31 S,32 Ni,33 Zn34 or Sn.35 Charge stored in a Li+/heterophase anode system may be enhanced36–39 through interfacial charge storage at both the solid–liquid interface and internal solid–solid interfaces. Jamnik and Maier have demonstrated increased charge storage at TiO2–metal interfaces as a result of the metal acting as an electron sink and the TiO2 storing excess Li+. In TiO2 photochemical studies, it has been demonstrated that owing to the offset in the conduction band levels, photogenerated charge carriers can be stored at the junction between anatase and TiO2(B).40–42 This band offset has been employed in TiO2 engineering to yield a material in which Li+ and electrons are separated across TiO2(B)/anatase TiO2 interfaces within the anode leading to increased charge storage.
In the present work we have fabricated a new TiO2–graphene composite designed to enhance the prospects of this low cost, abundant, non-toxic material as an anode in a Li-battery. The TiO2 is dandelion shaped with nanosized TiO2(B) fibrils capped with anatase TiO2 pappi. The as-prepared structure has extensive solid–electrolyte and solid–solid junctions to maximize interfacial charge storage. Multilayered graphene (MLG) is added to the TiO2 phases to yield a composite with enhanced conductivity and an increased number of solid–solid junctions.
Experimental
Materials preparation: dandelion-shaped TiO2/multi-layer graphene (TiO2/MLG) and the compared TiO2 anode materials were synthesized by a solvent-thermal method. For TiO2/MLG preparation, 0.24 g polyvinylpyrrolidone (PVP, Sigma, average mole weight: 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was dissolved in 40 mL acetic acid solution (Sinopharm Chemical Reagent Beijing Co. Ltd.) by strong stirring, followed by the addition of 0.05 g multi-layer graphene (∼10 nm thickness, Hefei Weijing Material Technology Co. Ltd.) and continuous stirring for 20 min. Then, 1 mL tetrabutyl titanate (TBT, Sinopharm Chemical Reagent Beijing Co. Ltd.) was added dropwise to the above dark solution in 5 min, and the mixture was sealed to sonicate at a power of 99 W for 20 min at 50 °C. The obtained suspension was transferred to a 60 mL Teflon-lined stainless-steel autoclave, and maintained at 150 °C for 24 h. After being cooled to room temperature, the precipitate was collected by centrifuge, washed with ethanol and water several times and dried at 60 °C in an oven. The dried sample was then calcined in the tube furnace at 400 °C for 4 h flowing with an argon atmosphere, and the temperature rose from room temperature with an increase rate of 3° min−1. After grinding, the TiO2/MLG powder was obtained. The compared TiO2 sample was prepared using the same method but without the use of multi-layer graphene.
000) was dissolved in 40 mL acetic acid solution (Sinopharm Chemical Reagent Beijing Co. Ltd.) by strong stirring, followed by the addition of 0.05 g multi-layer graphene (∼10 nm thickness, Hefei Weijing Material Technology Co. Ltd.) and continuous stirring for 20 min. Then, 1 mL tetrabutyl titanate (TBT, Sinopharm Chemical Reagent Beijing Co. Ltd.) was added dropwise to the above dark solution in 5 min, and the mixture was sealed to sonicate at a power of 99 W for 20 min at 50 °C. The obtained suspension was transferred to a 60 mL Teflon-lined stainless-steel autoclave, and maintained at 150 °C for 24 h. After being cooled to room temperature, the precipitate was collected by centrifuge, washed with ethanol and water several times and dried at 60 °C in an oven. The dried sample was then calcined in the tube furnace at 400 °C for 4 h flowing with an argon atmosphere, and the temperature rose from room temperature with an increase rate of 3° min−1. After grinding, the TiO2/MLG powder was obtained. The compared TiO2 sample was prepared using the same method but without the use of multi-layer graphene.
Characterization: the crystallographic structure of the as-prepared materials was studied by X-ray powder diffraction (XRD) using a Bruker D8 diffractometer with monochromatic Cu Kα radiation (λ = 1.5406 Å), and the diffraction data were recorded in the 2θ range of 10–80°. The morphological structures of the anode materials were investigated by a FEI Quanta 200 scanning electron microscopy (SEM) and JEOL 2010F transmission electron microscopy (TEM). The thermogravimetric analysis/differential scanning calorimetry (TGA/DSC) of the samples was carried out on a Diamond TG thermo-analyzer.
Electrochemical tests: the anode was fabricated with the active material, acetylene black, and binder (polyvinylidene fluoride, PVDF) in a weight ratio of 8:1:1 by using NMP as the solvent and copper foil as the current collector (∼2.5 mg cm−2), followed by drying in vacuum at 110 °C for 24 h. The R2016 coin cell was assembled in an argon-filled glove box using metallic lithium as the cathode and Celgard 2500 membrane as the separator. The electrolyte was 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), diethyl carbonate (DEC) (v/v/v, 1/1/1). Cyclic voltammetry (CV) and galvanostatic charge/discharge cycling tests were carried out in a setting voltage range by using an electrochemical workstation (CHI660C) and a CT2001A LAND battery tester, respectively. Electrochemical impedance spectroscopy (EIS) was studied using a Modulab (Solartron Analytical) with the amplitude of 5 mV in the frequency range from 1 MHz to 10 mHz. All electrochemical tests were carried out at room temperature.
Results and discussion
The X-ray diffraction (XRD) pattern of the as-prepared composite is shown in Fig. 1. The diffraction pattern can be indexed to the three constituent phases of the composite, namely graphite (P63/mmc, JPCD no. 65-6212), anatase TiO2 (I41/amd, JPCD no. 65-5714) and TiO2(B) (C2/m, JPCD no. 46-1238). The observed graphite peak is evidence of the incorporation of the multi-layer graphene (MLG) which was used as a reagent during the preparation. The strong (002) diffraction peak at 26.5° illustrates that the MLG structure was maintained throughout the hydrothermal and calcination processes employed in fabrication.43,44 TiO2(B) is formed from the thermal dehydration of layered hydrogen titanate,45,46 resulting from the low-rate of hydrolysis of TBT in acetic acid. The slow hydrolysis of the TBT precursor with the help of the PVP dispersant is beneficial for the self-assembly of anatase and TiO2(B). Rietveld refinement of the diffraction pattern using a two-phase analysis model, see Fig. S1,† indicates that the phase content of anatase TiO2 and TiO2(B) are 70.7% and 29.3%, respectively.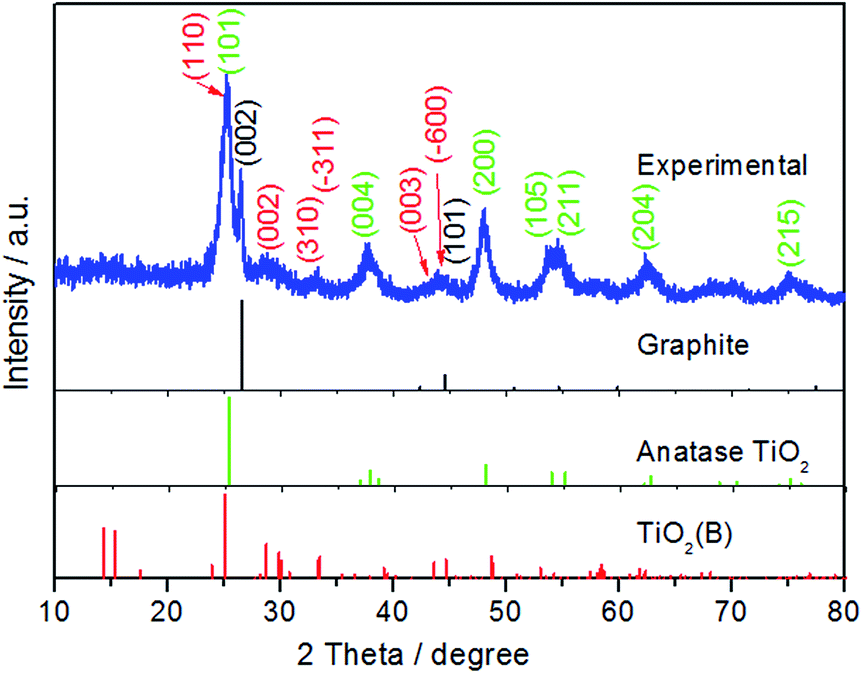 | ||
| Fig. 1 XRD patterns of the as-prepared sample. | ||
In order to estimate the amount of MLG in the composite sample, thermal analysis/differential scanning calorimetry (TGA/DSC) was carried out in air with a heating speed of 5 °C min−1 (Fig. S2†). The slight weight loss before 120 °C is ascribed to the evaporation of adsorbed water molecules. As the complete decomposition temperature of graphene has been generally considered to be around 700 °C,47–50 the weight loss at this temperature has been assumed to correspond to the content of MLG in the TiO2/MLG composite, which is estimated to be 5.8%. In addition, the wide exothermic peaks of TiO2/MLG reflects the oxidization of the carbon skeleton of MLG, which can be affected by the formed C–Ti bond and the distribution of TiO2 creating localized spots to facilitate the oxidization reaction.51–53 Besides MLG, the phase transformation of TiO2 from TiO2(B) to anatase to rutile in the range of 350–600 °C causes fluctuations in the exothermic peaks.54,55
The dandelion-shaped TiO2/MLG with TiO2(B) fibrils and anatase TiO2 pappi was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Fig. 2(a) and (b) show that TiO2/MLG is composed of connected ultrathin nanowires and nanosheets, with a thickness of several nanometers. The SEM image of TiO2/MLG is shown in Fig. S3† with the spherical dandelion morphology. The selected area electron diffraction (SAED) pattern shown in Fig. 2c displays broad diffraction rings that can be assigned to a composite of the two TiO2 polymorphs, namely anatase TiO2 and TiO2(B), and the broad rings assigned to (110), (202), (020), (113), (−621) and (423) planes for TiO2(B) can also be identified to the (101), (103), (200), (105), (204) and (224) planes of anatase TiO2.4 The high resolution transmission electron microscopy (HRTEM) image of TiO2/MLG displayed in Fig. 2d clearly demonstrates the crystal domains of the three phases in a nano-region, and the boundary structures between anatase TiO2 and TiO2(B). Within the triple phase region indicated by an ellipse in Fig. 2d, it is found that the lattice fringes of the (003) plane of TiO2(B) and the (004) plane of anatase TiO2 are adjacent with a 125° angular mismatch, and there is a 5° angular mismatch between the (100) plane of graphene and the (003) plane of TiO2(B) and 120° between the (100) plane of graphene and the (004) plane of anatase TiO2. As depicted in Fig. 2e, the interface mismatch rate, δ, between two mismatched phases can be calculated using eqn (1) where δ reflects the interface matching intensity or the possibility of interface match.4
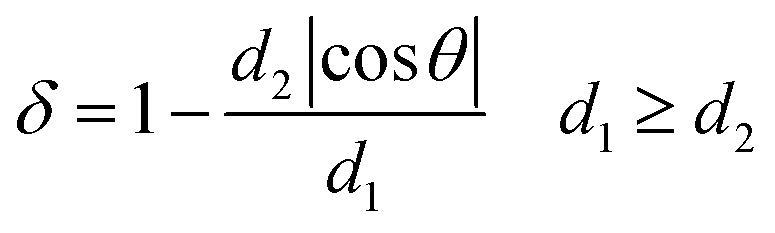 | (1) |
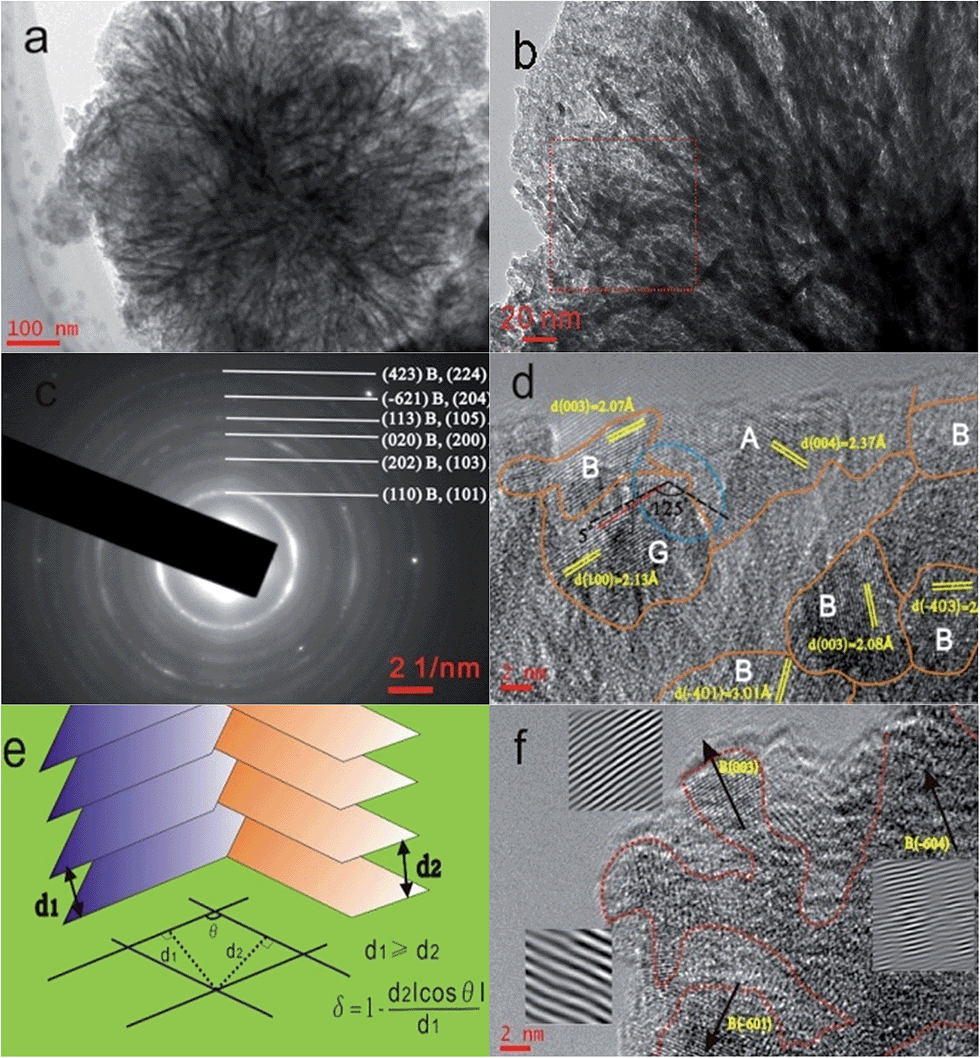 | ||
| Fig. 2 (a) TEM image of TiO2/MLG morphology. (b) Magnified TEM image around the outer region of TiO2/MLG. (c) Corresponding SAED pattern of the rectangular area in (b). A and B represent anatase TiO2 and TiO2(B), respectively. (d) HRTEM image with interface lines between phase regions. A, B and G in the image indicate anatase TiO2, TiO2(B) and graphene phase, respectively. (e) Representation of the interface mismatch rate, δ, between two adjacent phases. (f) HRTEM image of the focused limb part with the magnified lattice plane by FFT and inverse FFT. | ||
Cyclic Voltammetry (CV) of TiO2/MLG was performed in a voltage range of 1–3 V vs. Li/Li+, and the CV curves are shown in Fig. 3a and b. The first CV cycle, beginning from the open circuit voltage, shows different electrochemical behavior to the subsequent cycles, consistent with conditioning of the anode material during the first round of Li insertion/de-insertion. The second and fourth cycles are consistent in shape illustrating excellent reversibility of the reactions for the as-prepared material.4,7 The cathodic peak located at 1.37 V vs. Li/Li+ in the first cycle is found to disappear in the following, which has been explained as the irreversible reactions of some impurities.4 Compared with the CV peaks of TiO2(B),7 the cathodic peak at around 1.4 V in the first cycle results from the open channel saturation of TiO2(B) by the inserted Li+ ions. In the anodic scan of the second cycle there are three distinct peaks labeled A, B1 and B2 in Fig. 3a. At higher scan rates, Fig. 3b, peaks B1 and B2 are less well defined and eventually merge into a single entity labeled B. Analysis of peaks A and B as the scan rate is varied from 0.2 mV s−1 to 1.2 mV s−1 indicates that the peak current of the former is a linear function of the square root of the scan rate, Fig. 3(c), whilst the height of the latter is directly proportional to the scan rate, Fig. 3d. The as-prepared TiO2/MLG includes a mixture of anatase and TiO2(B) presenting diffusion- and surface-controlled electrochemical reactions at different potentials.3,4,7 The position of peak A, 1.68 V vs. Li/Li+, and the variation in peak height with scan rate are consistent with lithium insertion into anatase TiO2.5 Peak B can be assigned to a pseudocapacitive process on the surface of TiO2(B).56 A higher current associated with anatase TiO2 is in agreement with the composition of the composite.
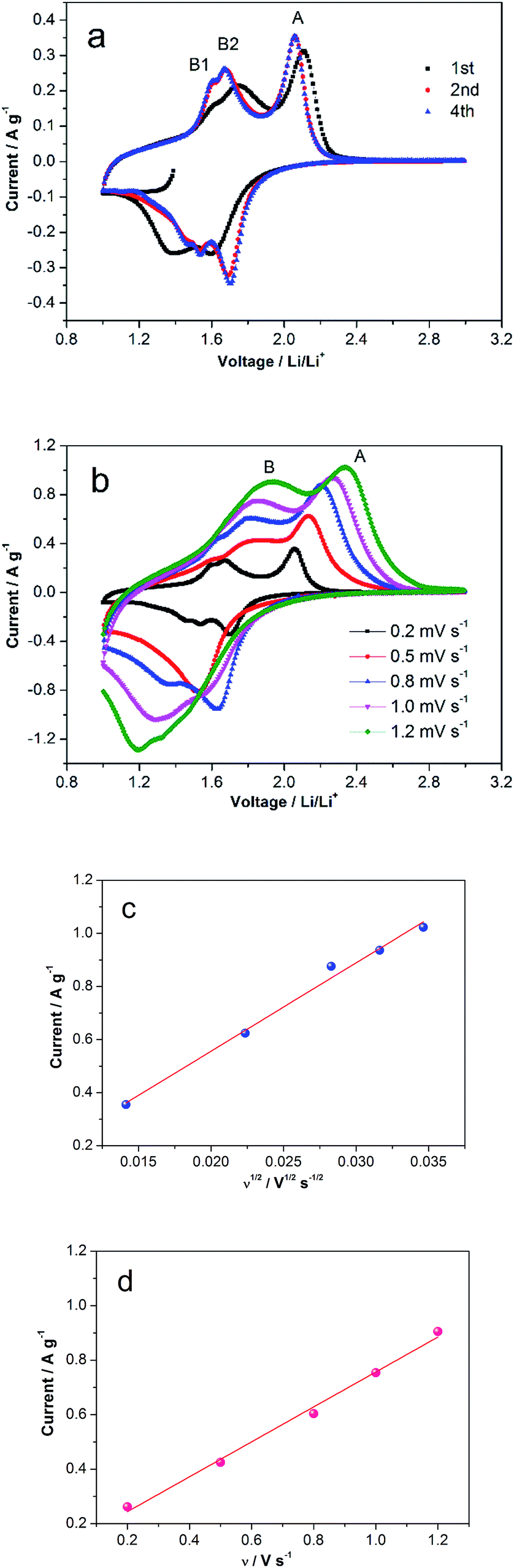 | ||
| Fig. 3 CV curves of TiO2/MLG. (a) The first four cycles at the scan rate of 0.2 mV s−1. (b) CV curves at different scan rates from 0.2 to 1.2 mV s−1. Linear relationships of (c) the A peak currents vs. square root of scan rates, and (d) the S peak currents vs. scan rates from (b). | ||
The galvanostatic charge/discharge profiles in the voltage range of 1 to 3 V vs. Li/Li+ of TiO2/MLG and TiO2 prepared using the same synthetic method but without MLG are shown in Fig. 4a. The discharge curves of TiO2/MLG and TiO2 can be divided into three regions.36,57 The first region (Region 1) with a steep decrease in potential is ascribed to the homogeneous Li+ insertion into bulk TiO2 until the solid-solution limit of Li+ ions in TiO2, such behavior is indicative of a nano-sized titania material.58 Region 2 is characteristic of the lithiation of anatase. It can be seen that the average voltage plateau of TiO2/MLG (1.75 V) is 20 mV higher than that of TiO2 (1.73 V), from which it can be deduced that the boundary structures between TiO2 and graphene have the potential to lower the electrode polarization resulting from the good electronic and ionic conductivity44 in the biphase TiO2 (Li-rich and Li-poor) co-existence plateau.36 The contributed capacities of 126 and 106 mA h g−1 for TiO2/MLG and TiO2 in the two regions result from the random insertion of Li+ into over half the available interstitial octahedral sites of anatase.4,57 Region 3, at low potentials, corresponds to the loss of pseudocapacitive charges from the interfaces in the system. The TiO2/MLG composite anode and the TiO2 anode have very similar behavior in regions 1 and 2 of the discharge curve indicating that the degree of lithiation of the nanoparticles is independent of the amount of MLG. In region 3 the TiO2/MLG composite displays a higher specific capacity at all potentials. In the second and third cycles, region 3 of the TiO2/MLG composite can be divided into two slopes of different gradients whilst for the TiO2 anode a single gradient is observed, as shown in Fig. S5.† The columbic efficiency of TiO2/MLG in the first cycle is 80.4%, slightly higher than the 78.8% observed for the TiO2 anode.
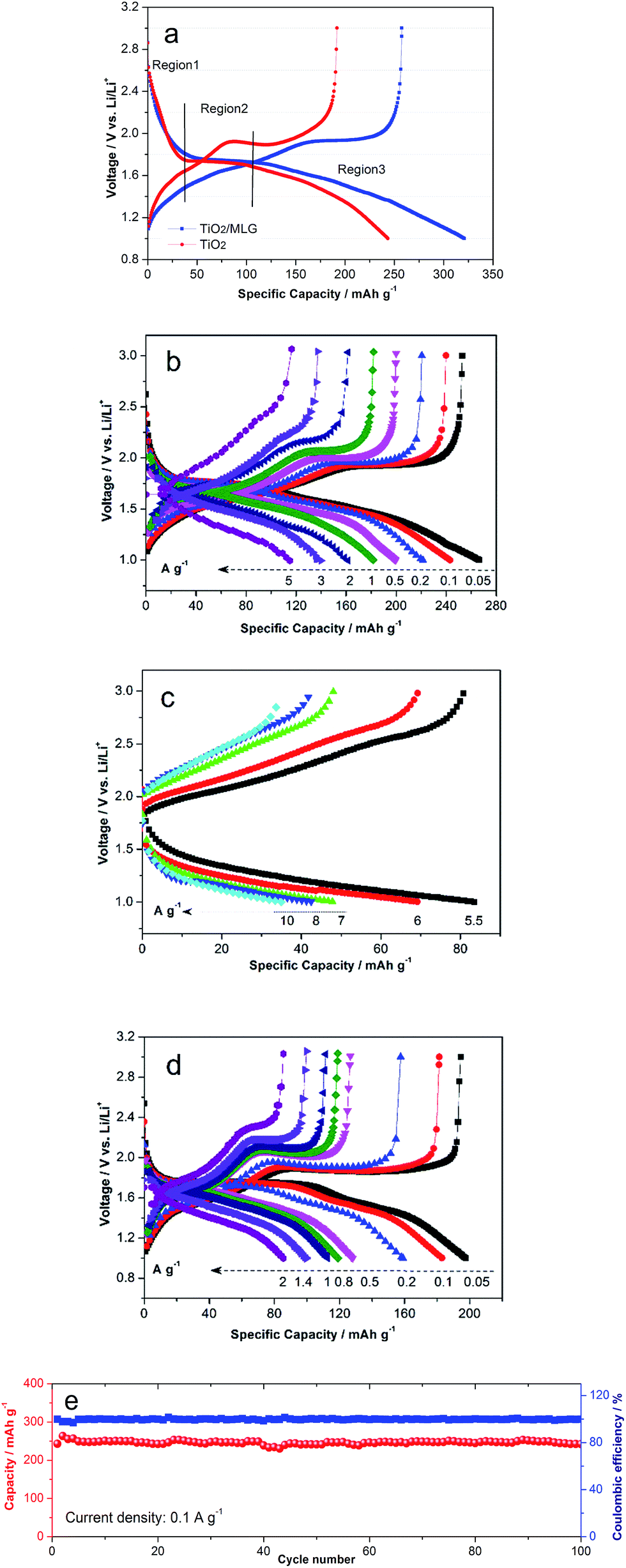 | ||
| Fig. 4 Galvanostatic charge/discharge profiles. (a) The first cycle of TiO2/MLG and TiO2 at a current density of 0.05 A g−1. (b) Rate performances of TiO2/MLG from 0.05 to 2 A g−1 and (c) from 5.5 to 10 A g−1. (d) Rate performances of TiO2 from 0.05 to 2 A g−1. (e) Cycling performances of TiO2/MLG at 0.1 A g−1. | ||
Fig. 4b shows the discharge capacities of TiO2/MLG at current densities of 0.05, 0.1, 0.2, 0.5, 1, 2, 3 and 5 A g−1 to be 266, 243, 221, 200, 182, 161, 140 and 115 mA h g−1. For the higher discharge rates of 5.5, 6, 7, 8, and 10 A g−1 the capacities of TiO2/MLG are 84, 69, 48, 43 and 35 mA h g−1, as indicated in Fig. 4c. For comparison, the specific discharge capacities of TiO2 displayed in Fig. 4d are 198, 183, 159, 128, 119, 113, 100 and 86 mA h g−1 for current densities of 0.05, 0.1, 0.2, 0.5, 0.8, 1, 1.4 and 2 A g−1, respectively. It is found that the specific capacity of TiO2/MLG at 1 A g−1 retains 75% of the value at 0.1 A g−1, while TiO2 keeps 62%. The cyclic performances of TiO2/MLG in Fig. 4e was investigated to evaluate the electrode stability. Only a slight loss of capacity was observed after 100 cycles at a current density of 0.1 A g−1. The corresponding capacity retention of TiO2/MLG is 99.6% with high coulombic efficiency almost approaching 100%, even for high current rates of 5 and 10 A g−1 as illustrated in Fig. S6.† The coulombic efficiencies of the 1200 cycles of TiO2/MLG are extremely coincident when compared with that of the reference TiO2 (Fig. S7†), which confirms the high electrode stability of TiO2/MLG during cycling. With reference to the conclusions from Wu et al.,4 the high electrochemical stability of the electrode is attributed to the three-dimensional dandelion-shaped microsphere structure which provides short diffusion distance for ion transport, high contact area between the electrolyte and electrode and the boundaries of TiO2/MLG which are capable of accommodating strain during cycling, leading to high Li+ diffusion kinetics as well as stable electrochemical reactions.
Electrochemical impedance spectroscopy (EIS) was used to explore the variation of impedance of TiO2/MLG during electrochemical cycling. Fig. 5(a) and (b) show the Nyquist plots for the electrodes cycled 0 and 1000 times with corresponding calculated plots which are obtained from the equivalent circuit model in the inset, respectively. For the components in the equivalent circuit, the values, with errors, that were calculated to yield the best fits to the experimental data are listed in Table S1 and S2.† In the equivalent circuit of both, R1 represents the internal resistance of the cell that arises from both the electrode and electrolyte. CPE2 (constant phase element) in Fig. 5a represents the pseudocapacitance that is generated on the complicated surface of internal triphase boundaries4 plus electrodes. R2 represents the charge transfer and possible ion diffusion, and CPE1 is the capacitor in the bulk associated with Li+ ion insertion. In Fig. 5b, two new components are introduced namely CPE3 and R3. CPE3 is defined as the Warburg resistance with the factor, n = 0.5 (Table S2†) and correspondingly, CPE1 becomes a pure capacitor with n = 1. The Warburg resistance results from the solid-state ion diffusion in the electrode channels. The presence of R3 points to the development of a surface resistance during cycling, which can be attributed to the open interfacial structure in the electrode or SEI film on the electrode surface.4 According to Tables S1 and S2,† the resistances, R1 and R2 have decreased but the capacitance of CPE1 and CPE2 have increased after multiple electrochemical cycles when compared with those of the fresh cell. The impedance data indicate that the structural rearrangement of the TiO2/MLG electrode during cycling promotes the boundary storage to be predominant over bulk storage for Li+ ions. The interfacial structure provides more open channels for ion diffusion with cycling.
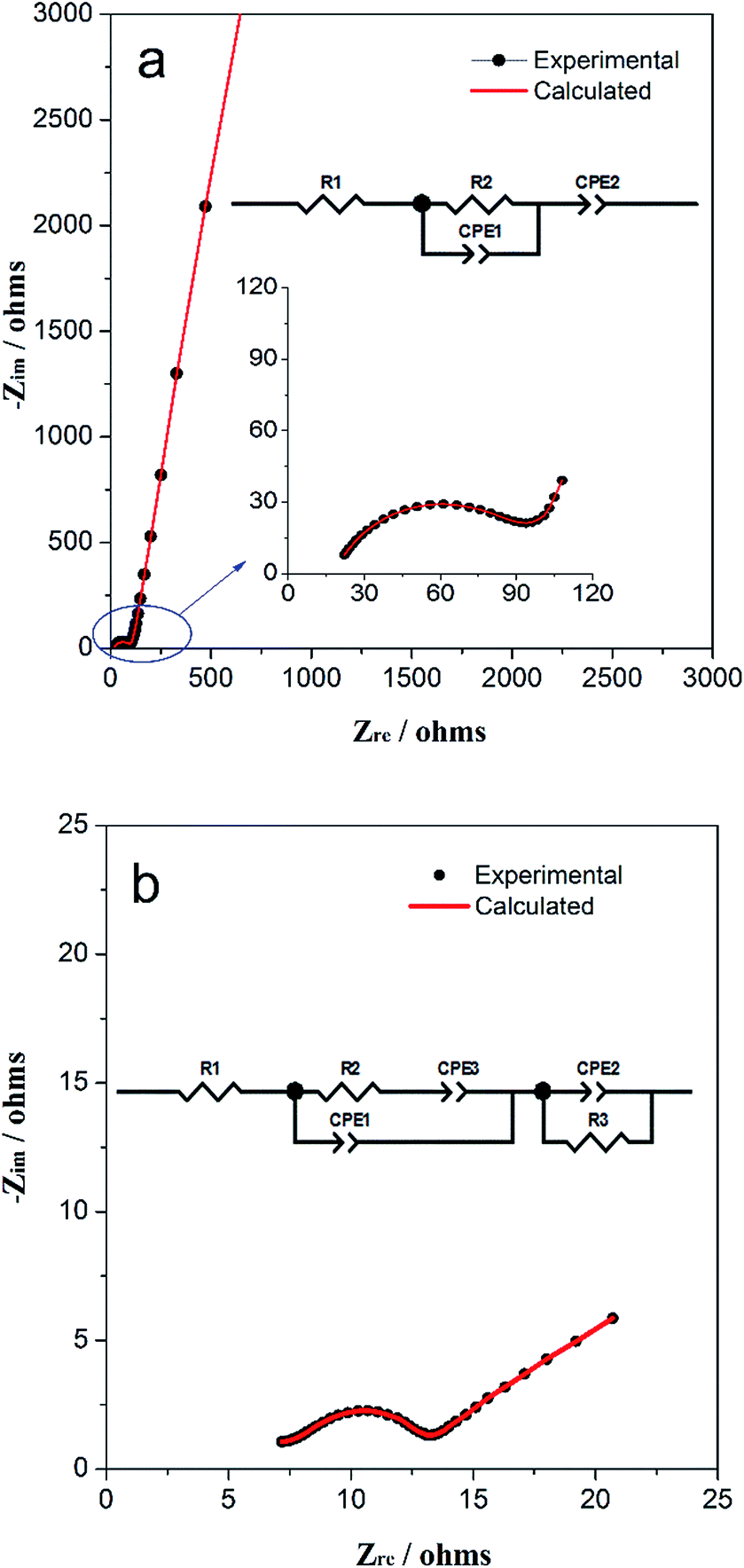 | ||
| Fig. 5 Nyquist plot of the TiO2/MLG cell after (a) 0 and (b) 1000 cycles with corresponding calculated fitting results. The inset is the corresponding equivalent circuit model. | ||
Fig. 6 is a schematic representation of the TiO2/MLG composite. The as-prepared material has an abundance of tri-phase boundaries between anatase TiO2, TiO2(B) and graphene. Wang et al.42 have demonstrated that charge storage at the anatase TiO2–TiO2(B) junction can enhance Li+ storage in anode materials. The high specific capacities observed for the biphasic TiO2 material prepared in the absence of MLG points to a synergistic interfacial effect improving charge storage. The specific capacitance of the dandelion structured TiO2 is further enhanced when MLG is introduced into the composite. The interfaces within nanosized materials can offer more sites for Li+ storage36 and the boundaries between MLG and TiO2 phases also provide sites for Li storage. The MLG itself with nearly 30 layers will only contribute negligible capacity in the voltage range of 1–3 V vs. Li/Li+.59 There are abundant phase boundaries arising from the polycrystal in the TiO2/MLG dandelion structure, contributing to large extent of lattice mismatch, as well as extra sites for Li+ ion and electron storage and some disordered planes probably provide more active sites for Li+ ion storage and transfer.4,11 The TiO2/MLG composite is able to store charge through both Li+ insertion and a pseudocapacitive mechanism, as evidenced by the i–V curves obtained when the potential is cycled. We postulate that the improved rate performance of the TiO2/MLG composite relative to the TiO2 material results from the fact that the edge-abundant interfaces of graphene can promote Li+ ion diffusion and electron transport along the triphase boundaries, as schematically presented in Fig. 6.
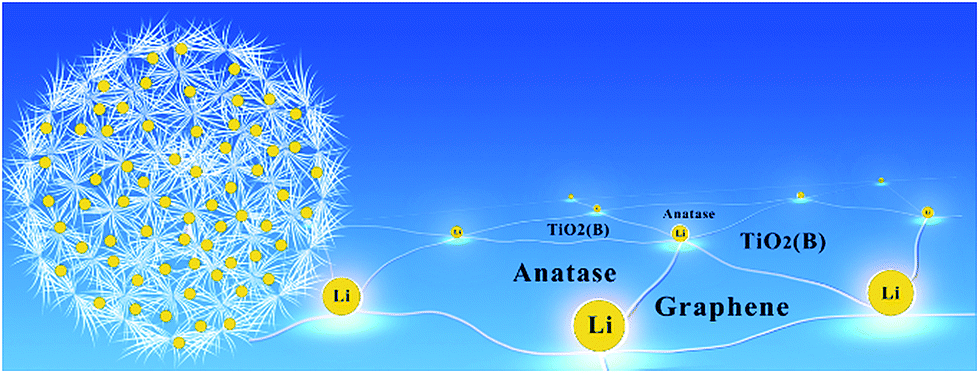 | ||
| Fig. 6 Schematic representation of interfacial storage for Li+ ions of TiO2/MLG. | ||
Conclusions
Dandelion-shaped TiO2/MLG composed of TiO2(B) fibrils and anatase pappi was synthesized as a promising anode material for Li storage. Both the material characterization and cyclic voltammetry presenting diffusion- and surface-controlled electrochemical reaction at different potentials, evidenced the anatase/TiO2(B) heterophase in TiO2/MLG, and the open channel saturation of TiO2(B) by inserted Li+ ions at around 1.4 V was used to explain the irreversible cathodic peak. A mismatch rate, δ, between two adjacent phases with mingled boundaries, was introduced to describe the interplanar mismatch degree. The edge-abundant interfaces of graphene involved in the triphase boundaries were reported to enhance the electrochemical storage properties of TiO2/MLG and lower the electrode polarization by the improvement of Li+ diffusion and electron transport. The discharge capacities of TiO2/MLG at current densities of 0.05, 0.1, 0.2, 0.5, 1, 2, 3 and 5 A g−1 are 266, 243, 221, 200, 182, 161, 140 and 115 mA h g−1, and the capacity retention is 99.6% with high coulombic efficiency almost approaching 100% after 100 cycles at the current density of 0.1 A g−1. The resistances of TiO2/MLG decreased with cycling and more boundaries were opened to enhance ionic and electron conductivity.Acknowledgements
Financial support from the Imperial College PhD Scholarship, Engineering and Physical Sciences Research Council (EPSRC, Grant: EP/L015277/1) is greatly appreciated.Notes and references
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 5926–5940 RSC.
- B. Kang and G. Ceder, Nat. Mater., 2009, 458, 190–193 CrossRef CAS PubMed.
- L. Kavan, J. Solid State Electrochem., 2014, 18, 2297–2306 CrossRef CAS.
- Q. Wu, J. Xu, X. Yang, F. Lu, S. He, J. Yang, H. J. Fan and M. Wu, Adv. Energy Mater., 2015, 5, 1401756–1401765 Search PubMed.
- J. Chen, W. Song, H. Hou, Y. Zhang, M. Jing, X. Jia and X. Ji, Adv. Funct. Mater., 2015, 25, 6793–6801 CrossRef CAS.
- S. Goriparti, E. Miele, M. Prato, A. Scarpellini, S. Marras, S. Monaco, A. Toma, G. C. Messina, A. Alabastri, F. D. Angelis, L. Manna, C. Capiglia and R. P. Zaccaria, ACS Appl. Mater. Interfaces, 2015, 7, 25139–25146 CAS.
- B. Laskova, M. Zukalova, A. Zukal, M. Bousa and L. Kavan, J. Power Sources, 2014, 246, 103–109 CrossRef CAS.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- C. Kim, R. Buonsanti, R. Yaylian, D. J. Milliron and J. Cabana, Adv. Energy Mater., 2013, 3, 1286–1291 CrossRef CAS.
- M. Zhao, L. Li, H. Lin, L. Yang and G. Li, Chem. Commun., 2013, 49, 7046–7048 RSC.
- Y. Ren, Z. Liu, F. Pourpoint, A. R. Armstrong, C. P. Grey and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 2164–2167 CrossRef CAS PubMed.
- J. Akimoto, Y. Gotoh, Y. Oosawa, N. Nonose, T. Kumagai, K. Aoki and H. Takei, J. Solid State Chem., 1994, 113, 27–36 CrossRef CAS.
- M. Latroche, L. Brohan, R. Marchand and M. Tournoux, J. Solid State Chem., 1989, 81, 78–82 CrossRef CAS.
- K. Spektor, D. T. Tran, K. Leinenweber and U. Häussermann, J. Solid State Chem., 2013, 206, 209–216 CrossRef CAS.
- H. Sato, S. Endo, M. Sugiyama, T. kikegawa, O. Shimomura and K. Kusaba, Science, 1991, 251, 786–788 CAS.
- Y. Al-Khatatbeh, K. K. M. Lee and B. Kiefer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 134114 CrossRef.
- M. Mattesini, J. S. de Almeida, L. Dubrovinsky, N. Dubrovinskaia, B. Johansson and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 212101 CrossRef.
- N. A. Dubrovinskaia, L. S. Dubrovinsky, R. Ahuja, V. B. Prokopenko, V. Dmitriev, H. P. Weber, J. M. Osorio-Guillen and B. Johansson, Phys. Rev. Lett., 2001, 87, 275501 CrossRef CAS PubMed.
- L. S. Dubrovinsky, N. A. Dubrovinskaia, V. Swamy, J. Muscat, N. M. Harrison, R. Ahuja, B. Holm and B. Johansson, Nature, 2001, 410, 653–654 CrossRef CAS PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- D. D'Elia, École Nationale Supérieure des Mines de Paris, 2011 Search PubMed.
- S. Moitzheim, C. S. Nimisha, S. Deng, D. J. Cott, C. Detavernier and P. M. Vereecken, Nanotechnology, 2014, 25, 504008 CrossRef CAS PubMed.
- T. Zhou, Y. Zheng, H. Gao, S. Min, S. Li, H. K. Liu and Z. Guo, Adv. Sci., 2015, 2, 1500027 Search PubMed.
- S. Li, M. Ling, J. Qiu, J. Han and S. Zhang, J. Mater. Chem. A, 2015, 3, 9700–9706 CAS.
- X. Yan, Y. Li, M. Li, Y. Jin, F. Du, G. Chen and Y. Wei, J. Mater. Chem. A, 2015, 3, 4180–4187 CAS.
- K. Siuzdak, R. Bogdanowicz, M. Sawczak and M. Sobaszek, Nanoscale, 2015, 7, 551–558 RSC.
- X. Yang, Y. Yang, H. Hou, Y. Zhang, L. Fang, J. Chen and X. Ji, J. Phys. Chem. C, 2015, 119, 3923–3930 CAS.
- Z. Hong, M. Wei, T. Lan, L. Jiang and G. Cao, Energy Environ. Sci., 2012, 5, 5408–5413 CAS.
- G. Li, Z. Lian, X. Li, Y. Xu, W. Wang, D. Zhang, F. Tian and H. Li, J. Mater. Chem. A, 2015, 3, 3748–3756 CAS.
- H. Tian, F. Xin, X. Tan and W. Han, J. Mater. Chem. A, 2014, 2, 10599–10606 CAS.
- S. Wei, R. Wu, J. Jian, F. Chen and Y. Sun, Dalton Trans., 2015, 44, 1534–1538 RSC.
- W. Jiao, N. Li, L. Wang, L. Wen, F. Li, G. Liu and H.-M. Cheng, Chem. Commun., 2013, 49, 3461–3463 RSC.
- W. Zhang, Y. Gong, N. P. Mellott, D. Liu and J. Li, J. Power Sources, 2015, 276, 39–45 CrossRef CAS.
- Z. Ali, S. N. Cha, J. I. Sohn, I. Shakir, C. Yan, J. M. Kim and D. J. Kang, J. Mater. Chem., 2012, 22, 17625–17629 RSC.
- M. Lübke, I. Johnson, N. M. Makwana, D. Brett, P. Shearing, Z. Liu and J. A. Darr, J. Power Sources, 2015, 294, 94–102 CrossRef.
- J.-Y. Shin, D. Samuelis and J. Maier, Adv. Funct. Mater., 2011, 21, 3464–3472 CrossRef CAS.
- J. Maier, Nat. Mater., 2005, 4, 805–815 CrossRef CAS PubMed.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215–5220 RSC.
- Y. F. Zhukovskii, P. Balaya, E. A. Kotomin and J. Maier, Phys. Rev. Lett., 2006, 96, 058302 CrossRef PubMed.
- D. Yang, H. Liu, Z. Zheng, Y. Yuan, J.-c. Zhao, E. R. Waclawik, X. Ke and H. Zhu, J. Am. Chem. Soc., 2009, 131, 17885–17893 CrossRef CAS PubMed.
- N. Fu, Y. Wu, Z. Jin and G. Lu, Langmuir, 2010, 26, 447–455 CrossRef CAS PubMed.
- R. Wang, X. Xue, W. Lu, H. Liu, C. Lai, K. Xi, Y. Che, J. Liu, S. Guo and D. Yang, Nanoscale, 2015, 7, 12833–12838 RSC.
- W. Song, X. Cao, Z. Wu, J. Chen, Y. Zhu, H. Hou, Q. Lan and X. Ji, Langmuir, 2014, 30, 12438–12446 CrossRef CAS PubMed.
- W. Song, X. Ji, Z. Wu, Y. Zhu, Y. Yang, J. Chen, M. Jing, F. Li and C. E. Banks, J. Mater. Chem. A, 2014, 2, 5358–5362 CAS.
- G. Armstrong, A. R. Armstrong, J. Canales and P. G. Bruce, Chem. Commun., 2005, 2454–2456 RSC.
- T. P. Feist and P. K. Davies, J. Solid State Chem., 1992, 101, 275–295 CrossRef CAS.
- N. Li, G. Liu, C. Zhen, F. Li, L. Zhang and H.-M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef CAS.
- X. Huang, K. Wang, K. Jia and X. Liu, Polym. Adv. Technol., 2015, 26, 1267–1274 CrossRef CAS.
- H. Sun, S. Liu, S. Liu and S. Wang, Appl. Catal., B, 2014, 146, 162–168 CrossRef CAS.
- J. S. Chen, Z. Wang, X. C. Dong, P. Chen and X. W. Lou, Nanoscale, 2011, 3, 2158–2161 RSC.
- L. Wang, L. Shen, Y. Li, L. Zhu, J. Shen and L. Wang, Int. J. Photoenergy, 2013, 2013, 1–7 Search PubMed.
- Q. Huang, S. Tian, D. Zeng, X. Wang, W. Song, Y. Li, W. Xiao and C. Xie, ACS Catal., 2013, 3, 1477–1485 CrossRef CAS.
- Y. Gao, X. Pu, D. Zhang, G. Ding, X. Shao and J. Ma, Carbon, 2012, 50, 4093–4101 CrossRef CAS.
- M. Hu, M. Fang, C. Tang, T. Yang, Z. Huang, Y. Liu, X. Wu and X. Min, Nanoscale Res. Lett., 2013, 8, 548 CrossRef PubMed.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2010, 46, 855–874 CrossRef.
- M. Zukalová, M. Kalbáč, L. Kavan, I. Exnar and M. Graetzel, Chem. Mater., 2005, 17, 1248–1255 CrossRef.
- Y.-G. Guo, Y.-S. Hu and J. Maier, Chem. Commun., 2006, 2783 RSC.
- Y. S. Hu, L. Kienle, Y. G. Guo and J. Maier, Adv. Mater., 2006, 18, 1421–1426 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta02548j |
| This journal is © The Royal Society of Chemistry 2016 |