
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron–polypyrrole electrocatalyst with remarkable activity and stability for ORR in both alkaline and acidic conditions: a comprehensive assessment of catalyst preparation sequence†
Thanh-Nhan
Tran
,
Min Young
Song
,
Kiran Pal
Singh
,
Dae-Soo
Yang
and
Jong-Sung
Yu
*
Department of Energy Systems Engineering, DGIST, Daegu, 42988, Republic of Korea. E-mail: jsyu@dgist.ac.kr; Fax: +82 53 785 4609; Tel: +82 53 785 6443
First published on 6th May 2016
Abstract
A new facile template-free method is presented to synthesize Fe-treated N-doped carbon (Fe/N–C) catalysts for oxygen reduction reaction (ORR) by employing a synthesis protocol of pyrolysis–leaching–stabilization (PLS) sequence of polypyrrole in the presence of ferric source, which serves dual purposes of an oxidant for pyrrole polymerization and an iron source. Each step in the PLS sequence is assessed in detail in terms of the related structural properties of the resulting carbon catalysts, and their effects on ORR activities are elaborated to confirm the validity of the current synthesis protocol. It is found that the as-prepared carbon catalyst exhibits outstanding high catalytic activity in both alkaline and acidic conditions. The carbon catalyst prepared at a pyrolysis temperature of 900 °C (FePPyC-900) shows remarkably high ORR activity with onset potential of 0.96 V (vs. RHE), which is similar to that of Pt/C, whereas the half-wave potential (E1/2) of FePPyC-900 is 0.877 V, more positive than that of Pt/C at the same catalyst loading amount under alkaline conditions. Furthermore, the FePPyC-900 catalyst also illustrates exceptionally high activity under acidic conditions with onset and half-wave potentials of 0.814 and 0.740 V, respectively, which are almost comparable to those (0.817 and 0.709 V) of the state-of-the-art Pt/C catalyst, which is rarely observed for non-Pt-based carbon catalysts. In addition, the FePPyC-900 catalyst displays much better stability and methanol tolerance than the Pt/C and exhibits a four electron transfer pathway under both alkaline and acidic conditions. Such extraordinary high ORR activity and stability of the FePPyC-samples can be attributed to the implementation of extra stabilization step in addition to conventional sample preparation steps of pyrolysis and subsequent leaching in current PLS synthesis protocol as well as to the use of highly conducting PPy as a single precursor of carbon and nitrogen in the presence of Fe.
1. Introduction
With the increasing concern over environmental safety and depletion of fossil fuels, fuel cell (FC) is considered as a promising source of alternative green power because of its high energy density, high efficiency, and negligible emission of harmful gases.1,2 The proton exchange membrane fuel cell (PEMFC) and alkaline polymer electrolyte fuel cell (APEFC) have attracted global research interest because of their low operation temperature, high efficiency and long life span.3 Platinum (Pt) and Pt-based alloy materials are widely used as electrocatalysts for both hydrogen oxidation reaction and oxygen reduction reaction (ORR) in PEMFC,4,5 although Pt is expensive and naturally less abundant. Furthermore, the sluggish ORR kinetics at the cathode necessitates large usage of noble Pt to accelerate the ORR at the cathode, which greatly increases the cost of fuel cells and thus makes widespread commercialization of the fuel cell technology unlikely.6,7 Therefore, the development of precious metal-free catalyst with both high activity and stability to substitute the Pt-based catalysts has been regarded as a more probable alternative to significantly reduce the cost and realize commercialization of fuel cells.In the past few decades, a range of alternative non-noble metal catalysts (NNMCs), such as metal-free or transition metal-treated heteroatom-doped carbon,8–14 transition metal oxide/carbides/nitrides,15–17 perovskite oxides,18,19 transition metal-coordinated macrocyclic compounds,20–22 and MOF-derived catalyst, have been investigated as alternatives to costly Pt catalyst.23 In particular, transition metal-treated N-doped carbon catalysts (M/N–Cs) have attracted attention as electrocatalysts for the ORR due to high catalytic activity, durability and wide availability along with economical merits. To date, iron (Fe) and/or cobalt (Co)-treated composites with N or P doping have demonstrated good catalytic activities and durability towards ORR in an acidic medium.24–26 There are many types of nitrogen precursors for N-doped carbon, including ammonia (NH3),27 polyacrylonitrile ((C3H3N)n),28 polyaniline ((C6H5NH)n),24 ethylenediamine (C2H4(NH2)2),29 cyanamide (CH2N2),30 urea, and phthalocyanine.20 In addition, polypyrrole ((C4H5N)n) (PPy) is an excellent carbon source with N moiety since it is a conducting polymer and can also be employed as a matrix for incorporating metallic catalysts.31 Furthermore, the aromaticity of the pyrrole ring is favourable to form graphitic carbon structures after carbonization, which can improve ORR activity significantly. Yuasa et al.32 in 2005 first reported on Co-PPy as an electrochemical cathode catalyst, which kindled worldwide focus on transition metal and N-doped carbon research.33 There have also been several studies reporting Fe-treated N-doped carbon derived from Fe-incorporated PPy for ORR applications.15,34–39 However, almost all these papers have shown the ORR activity only under alkaline conditions, except for one study by M. Sevilla et al., which presented the ORR activity of Fe-PPy under both alkaline and acidic conditions.39 The ORR activity of their Fe-PPy catalyst under acidic conditions was found to be quite lower than that of the Pt-based catalyst. In general, the activity of transition metal-treated N-doped carbon is reported to be still much lower than that of the Pt-based catalyst under acidic conditions.
Furthermore, the synthesis process is very important for fabricating good electrocatalysts. Recently, the synthesis sequence of 1st heat treatment-acid etching-2nd heat treatment has been used for the synthesis of transitional metal-incorporated N-doped carbon.30,40–43 Typically, this process includes first heating the mixture of N, C and transitional metal sources, and then etching the unstable species with an acid, and afterward the second heating process is carried out. M. Ferrandon et al. reported that acid etching can remove the iron species adjacent to the active sites to expose them for the ORR reaction, and the second heat treatment increases the stability of the final Fe/N–C catalyst material.43 However, each of the synthesis steps has not been fully investigated for better understanding the effectiveness of the synthesis procedure. On the other hand, most studies have shown the changes in the surface area, electronic conductivity and active site content in different synthesis procedures, mostly without the inclusion of the final stabilization step, which can greatly influence the electrocatalytic activity of Fe/N–C catalysts.44
In this study, Fe-treated N-doped carbon (Fe/N–C) electrocatalysts were prepared from a simple template-free synthesis protocol of a pyrolysis–leaching–stabilization (PLS) sequence for polypyrrole (PPy) and ferric chloride (FeCl3) as C, N, and Fe precursors. Each step in the PLS sequence was assessed in detail in terms of the related structural properties of the resulting carbon catalysts, and their effects on the ORR activities were elaborated to confirm the validity of the current synthesis protocol. PPy was chosen as a single precursor for C and N since it is an excellent conducting polymer and thus can yield an extended graphitic carbon framework with high electrical conductivity upon carbonization. We find that FePPyC-900 prepared by the PLS sequence of the Fe-PPy precursor at 900 °C exhibits excellent ORR performance with near 4-electron transfer and low H2O2 yield, which is comparable to that of commercial E-TEK 20 wt% Pt/C catalyst under both alkaline and acid conditions along with much better long term stability and high methanol tolerance. In particular, the exceptionally high ORR activity comparable to that of the state-of-the-art Pt/C was observed even in an acidic medium, which has been rarely reported before. The results clearly indicate that the PLS sequence is very effective for catalyst preparation and the as-prepared FePPyC catalyst represents a viable alternative to precious Pt catalyst in fuel cells.
2. Experimental section
2.1 Material preparation
FePPyC catalysts were prepared by a new synthesis protocol consisting of a series of pyrolysis–leaching–stabilization (PLS) sequence for Fe-PPy precursors. First, 3.00 ml of monomer pyrrole was dissolved in 60.0 ml of 0.5 M HCl solution and allowed to stir for 15 min at room temperature, and 10 ml FeCl3 (0.74 mol l−1) solution was then added slowly into the abovementioned mixture as an oxidant for pyrrole polymerization and further stirred for 12 h. The mixture containing the polymer and transition metal was filtered by a vacuum filter. The product was dried at 80 °C for 12 h, and the dried precursor was heated to 800, 900, and 1000 °C for 1 h at a heating rate of 10 °C min−1 in an argon atmosphere for carbonization and graphitization, and the sample was then cooled naturally to room temperature at the same atmosphere. To remove redundant phases, mainly unstable metallic iron and iron compounds, the pyrolyzed product was leached in 0.5 M H2SO4 at 80 °C for 8 h. After thorough washing several times with deionized water and absolute ethanol, the product was dried at 80 °C for 12 h. The leached product was pyrolyzed again at the same temperature for 1 h for stabilization (structural integration), as mentioned above, to obtain the FePPyC-T (T = 800, 900, and 1000 as pyrolysis temperature) samples. For comparison, a Fe-free N-doped carbon catalyst was also synthesized from pure pyrrole monomer using hydrogen peroxide as an oxidant for polymerization (denoted as PPyC) instead of FeCl3. The same PLS method of pyrolysis, acid leaching and further stabilization was applied to obtain Fe-free PPyC-T.2.2 Material characterization
The X-ray diffraction (XRD) patterns were measured using a Rigaku Smartlab diffractometer with CuKα radiation using a Ni β-filter at a scan rate of 4°/min. The X-ray source was obtained at 40 kV and 30 mA. Raman spectroscopy measurements (Renishaw) were recorded using an Ar ion laser (λ = 514.5 nm). The scanning electron microscopy (SEM) images were obtained using a Hitachi S-4700 microscope operated at an acceleration voltage of 10 kV. The transmission electron microscope (TEM) was measured on EM 912 Omega at 120 kV. The nitrogen adsorption–desorption isotherms were operated at −196 °C using a Micromeritics ASAP 2020 system. The specific surface areas of the samples were determined from nitrogen adsorption data in the relative pressure range from 0.05 to 0.2 using the Brunauer–Emmett–Teller (BET) equation. The total pore volume was obtained from the amount of gas adsorbed at the relative pressure of 0.99. Pore-size distribution was calculated using Micromeritics software based on the Barrett–Joyner–Halenda (BJH) Method. X-ray photoelectron spectroscopy (XPS) analyses were operated with an AXIS-NOVA (Kratos) X-ray photoelectron spectrometer using a monochromated Al Kα X-ray source (hν = 1486.6 eV) operated at 150 W under a base pressure of 2.6 × 10−9 Torr. The XPS spectra were deconvoluted using the curve fitting program, XPSpeak 4.1. This software was used for all XPS data processing. A special cell with a four-probe configuration was designed and constructed by our group for measurements of the electrical conductivity of materials under controlled pressure. The electrical conductivity is first measured using four probes under a fixed pressure using a Keithley model 6220 and 2182A as the DC current source and voltmeter and then converted to the conductivity as described earlier.45 The sample volume was calculated from measured sample thickness and cross section area of the pressure chamber.2.3 Electrochemical characterization
Electrochemical experiments were carried out at room temperature in a three-electrode cell using a rotating disk electrode (RDE) or rotating ring-disk electrode (RRDE) connected to an electrochemical analyzer (Biologic VMP3). The RRDE measurements were conducted using an electrode with a fitted glassy carbon disk and a platinum ring in an oxygen-saturated 0.1 M KOH or 0.5 M H2SO4. The prepared catalysts or commercial E-TEK 20 wt% Pt/C catalyst (5.0 mg) were dispersed in 1.0 ml of a mixture solution of water and 5 wt% Nafion (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nafion = 9:1). The as-prepared 10 μL catalyst ink was then dropped onto the glassy carbon disk (4 mm) of the RRDE or RDE (0.4 mg cm−2) and dried at 50 °C to prepare the working electrode. An Ag/AgCl with saturated KCl and a Pt wire were used as a reference and a counter electrode, respectively. The potentials of the Ag/AgCl reference electrode were calibrated to the reversible hydrogen electrode (RHE). The potential presented in this study refers to the RHE potential. The potential difference between Ag/AgCl and RHE electrode was calculated using eqn (1).
:Nafion = 9:1). The as-prepared 10 μL catalyst ink was then dropped onto the glassy carbon disk (4 mm) of the RRDE or RDE (0.4 mg cm−2) and dried at 50 °C to prepare the working electrode. An Ag/AgCl with saturated KCl and a Pt wire were used as a reference and a counter electrode, respectively. The potentials of the Ag/AgCl reference electrode were calibrated to the reversible hydrogen electrode (RHE). The potential presented in this study refers to the RHE potential. The potential difference between Ag/AgCl and RHE electrode was calculated using eqn (1).| E(RHE) = EAg/AgCl + 0.198 + 0.059 × pH | (1) |
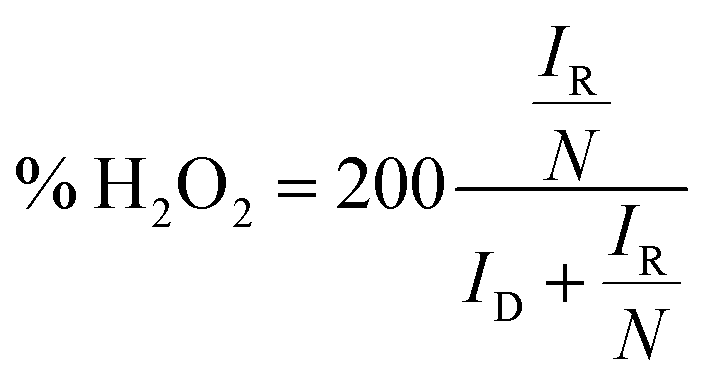 | (2) |
The electron transfer number (n) was determined from the RRDE measurements using the following eqn (3).
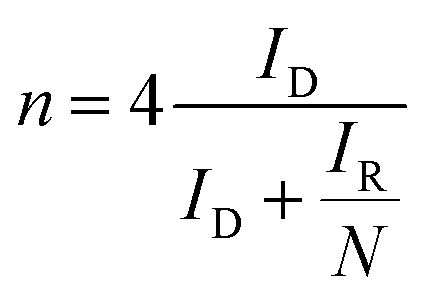 | (3) |
Chronoamperometric (CA) analysis for FePPyC-900 and Pt/C catalysts was evaluated at a constant potential of 0.85 V for O2-saturated 0.1 M KOH or 0.62 V for O2-saturated 0.5 M H2SO4. The methanol crossover effect was checked by introducing 3.0 M methanol to the O2-saturated 0.1 M KOH or 0.5 M H2SO4 electrolyte at a constant potential of 0.85 or 0.62 V under alkaline and acid conditions, respectively.
3. Results and discussion
Fig. 1 illustrates the PLS synthesis process of the FePPyC-T catalyst. In brief, pyrrole monomer was dispersed in an HCl solution, to which an iron(III) chloride solution was added for the polymerization of pyrrole. The mixture was then filtered to obtain the solid phase, followed by drying in an oven until the sample weight did not change, usually for ∼12 h at 80 °C. The first heat treatment was carried out at 800, 900, and 1000 °C, respectively, under the argon atmosphere for 1 h for carbonization and graphitization. The heat-treated product was than leached in 0.5 M H2SO4 at 80 °C for 8 h to remove any unstable phases. The second heat treatment was carried out to make a loose carbon matrix, formed during acid leaching, further graphitized and stabilized under the same condition, as mentioned above in the first heat treatment step of the synthesis, to obtain the final FePPyC-T. For comparison, a Fe-free PPyC-T was also prepared using the identical PLS process with hydrogen peroxide as an oxidant for polymerization instead of FeCl3, as mentioned in the Experimental section.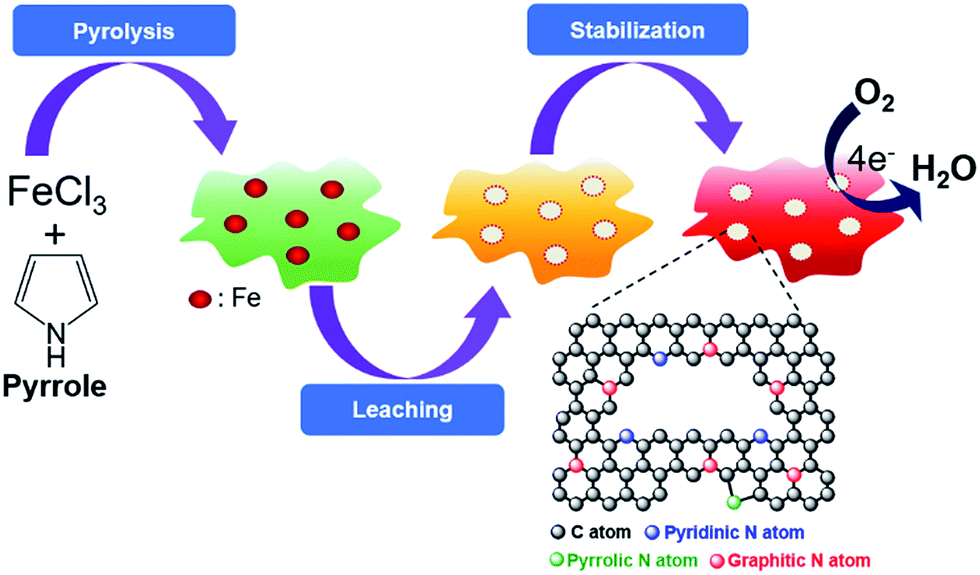 | ||
| Fig. 1 Schematic for the synthesis of FePPyC-T catalyst. Step-1: oxidative polymerization of pyrrole by the addition of FeCl3 solution, and first pyrolysis of the Fe–polypyrrole under an argon atmosphere for 1 h for carbonization and graphitization. Step-2: acid leaching for removing unstable Fe species. Step-3: second pyrolysis for structural integration and stability. | ||
The ORR activity of the Fe-treated N-doped carbon catalysts is influenced by pyrolysis temperature.26 Pyrolysis at relatively a low temperature from 500 to 700 °C may lead to Fe–N4 configuration as ORR active sites, whereas heat treatment at higher temperatures can increase the electrical conductivity of the carbon material and form Fe–N4−x centers with a lower N content, which are reported to form new active sites.43 Thus, there may be a trade-off between the N content and electrical conductivity, which is decided mainly by the reaction temperature.11,26 In general, pyrolysis at temperatures higher than 700 °C is considered to be more desirable for better ORR performance despite some loss of the N content in the carbon framework. To find an optimal pyrolysis temperature, the Fe–PPy mixture precursor was pyrolyzed at various temperatures, ranging from 800 to 1000 °C in this study.
First, the effect of carbonization on the morphology of the prepared catalyst was evaluated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Fig. 2a and e show the SEM and TEM images of the Fe-free PPyC-900 carbon material. It can be seen that PPyC-900 shows a dense bulky structure, and no porous structure is observed. Fig. 2b–d are SEM images of FePPyC-800, FePPyC-900, and FePPyC-1000, respectively, which show the formation of aggregated small carbon particles interconnected together, which are likely to form a porous structure between the carbon particles. Fig. 2f–h show TEM images of FePPyC-800, FePPyC-900, and FePPyC-1000, respectively, which also show the formation of aggregated small carbon particles interconnected together. We surmise that the formation of this type of interconnected structure can lead to interconnected open pores for efficient mass transfer during ORR and eventually help increase the catalytic activity of the catalyst.17
 | ||
| Fig. 2 SEM (a–d) and TEM (e–h) images of PPyC-900, FePPyC-800, FePPyC-900, and FePPyC-1000, respectively. | ||
In this study, FePPyC-T catalysts were prepared by the PLS synthesis sequence. The effect of each step in the PLS sequence on structural properties of the resulting carbon catalysts is elaborated to confirm the validity of the current synthesis protocol. Fig. S1 of (ESI)† shows the TEM and HR-TEM images of FePPyC-900bf obtained before acid leaching. A particle with a size of around 35 nm (Fig. S1a of ESI†) is clearly seen. A high resolution (HR)-TEM image of the selected region of the particle in Fig. S1b of ESI† presents a lattice fringe of d = 2.05 Å, which corresponds to plane spacing of Fe(110) plane. In addition, Fig. S2 of ESI† illustrates a typical TEM image and elemental mapping of FePPyC-900bf, which indicate the uniform distribution of Fe nanoparticles (NPs) and N in carbon. Furthermore, TEM image and element mapping images for FePPyC-900af obtained after acid leaching are shown in Fig. S3 of ESI.† After acid leaching, all the large Fe particles are leached out from FePPy-900af, but Fe is still found to be well dispersed in the N-doped FePPy-900af carbon matrix.
XRD patterns of all the samples prepared at difference pyrolysis temperatures reveal a broad peak at around 2θ = 25° indexed to the (002) plane and a small peak at 44° corresponding to (100) plane of graphitic carbon, which are typical of a turbostratic carbon structure, as shown in Fig. 3a.49 In particular, the intensity of the (002) peak is becoming stronger and narrower with increasing temperature. This clearly indicates that on increasing the carbonization temperature, the graphitic nature of the catalysts is also increasing.44,45 A comparison of the XRD patterns of PPyC-900 and FePPyC-900 suggests that FePPyC-900 possesses more graphitic structures, which can be ascribed to the catalytic property of Fe. The XRD pattern of FePPyC-900bf shows the existence of iron phase (Fig. S4a of ESI†), which is consistent with the TEM images seen in Fig. S1 and S2 of ESI.† However, no XRD signals for iron species are found in the sample after acid leaching (FePPyC-900af) and the 2nd pyrolysis (FePPyC-900), which indicates that the acid leaching process removes almost all the iron metal or oxide species from the sample. The remaining iron species are dispersed in the catalyst mainly as mononuclear Fe species with co-existing Fe2+ and Fe3+, which can be further confirmed by XPS.50
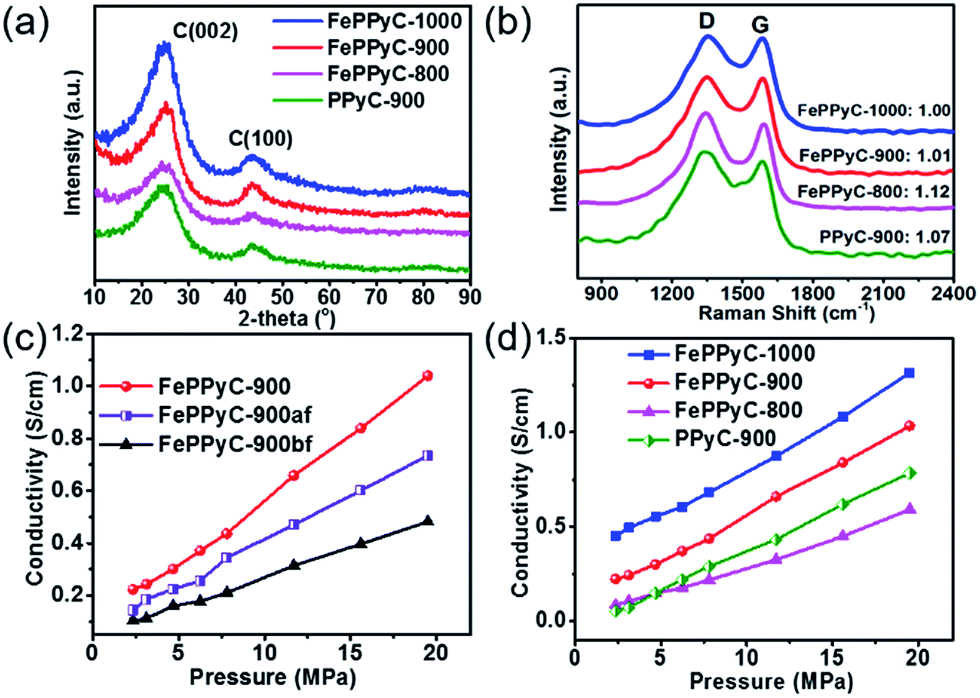 | ||
| Fig. 3 (a) XRD patterns and (b) Raman spectra of the FePPyC-T and PPyC catalysts. Electrical conductivity versus pressure of (c) FePPyC samples prepared in different synthesis steps of the PLS sequence at pyrolysis temperature of 900 °C and (d) FePPyC-T and PPyC-900 catalysts. | ||
Raman spectra of all the samples are shown in Fig. 3b. All the samples displayed two broad bands at 1340 and 1575 cm−1, which can be assigned to the D band and G band, respectively. D band is related to the breaking of symmetry caused by structural disorders and defects, whereas the G band represents the in-plane tangential stretching vibration mode (E2g) of the graphite sheet.51 The positions of both peaks are similar for all samples, showing that the structures of all the carbon samples are similar. The ratio of the D and G band intensities (ID/IG) often indicates the disordered nature of the prepared carbon frameworks and was observed to be 1.12, 1.07, 1.01, and 1.00 for FePPyC-800, PPyC-900, FePPyC-900, and FePPyC-1000, respectively. The decrease in the ID/IG ratios suggests that the carbon structure becomes more ordered and graphitic upon increasing the pyrolysis temperature, which is in agreement with the XRD results. In addition, similar to our XRD results, from a comparison of the ID/IG values between FePPyC-900 and PPyC-900, it can be noted that the treatment of Fe increases structural order and crystallinity in the carbon framework. The increase in crystallinity or graphitic nature can definitely help in improving the electrical conductivity of the samples. As shown in Fig. S4b of ESI,† for the different synthesis steps, the ID/IG ratios of FePPyC-900bf, FePPyC-900af, and FePPyC-900 are found to be 0.88, 1.02 and 1.01, respectively. Higher ID/IG ratio of FePPyC-900af and FePPyC-900 than that of FePPyC-900bf suggests that disorder and defective sites increase in the FePPyC-900af and FePPyC-900 framework, from which Fe species were leached out.52
Electrical conductivity is an essential property required in electroactive materials for efficient ORR. Hence, the effect of Fe and carbonization temperature on the conductivity of the prepared catalysts was investigated using a cell with a four-probe configuration (see ESI† for details).45Fig. 3c shows an assessment of each step of the PLS sequence in terms of the electrical conductivity. As the pressure increases, the conductivity increases for all the samples. FePPyC-900af was found to show higher conductivity than FePPyC-900bf due to acid leaching of the unstable or nonconductive iron oxide species (Fig. 3c). After the second pyrolysis treatment, FePPyC-900 reveals higher conductivity than FePPyC-900af. Fig. 3d illustrates that the conductivity of FePPyC-T samples also increases with increasing pyrolysis temperature. This can be understood by the fact that the increasing pyrolysis temperature certainly improves the graphitization.8,45 In addition, FePPyC-900 exhibits higher conductivity than Fe-free PPyC-900, suggesting that iron treatment plays an important role of increasing the graphiticity of the carbon framework, as proven by XRD and Raman spectra in Fig. 3,26,40 eventually improving the overall conductivity.26,41,44,47
The surface properties of the prepared samples were determined by nitrogen isotherms, as shown in Fig. S5a of ESI.† N2 isotherms for PPyC-900 exhibit type I isotherms typical of microporous materials, whereas the FePPyC-T samples show type IV isotherms characteristics for microporous and mesoporous materials.53 The Brunauer–Emmet–Teller (BET) surface areas of PPyC-900, FePPyC-800, FePPyC-900bf, FePPyC-900af, FePPyC-900, and FePPyC-1000 were determined to be 565.8, 355.6, 425.6, 504.8, 592.2, and 726.1 m2 g−1, respectively. Table S1 of ESI† summarizes the BET surface area, pore volume, and pore diameter of all the prepared samples. FePPyC-900af (504.8 m2 g−1) shows a higher surface area than FePPyC-900bf (425.6 m2 g−1), and the surface area increases further after the second pyrolysis process (592.2 m2 g−1). As observed in Table S1 of ESI,† a similar trend was observed for the total pore volume, suggesting that removing the iron species by acid leaching increases the total surface area and pore volume. In addition, the surface area was found to increase for the Fe-treated samples with increasing temperature. Interestingly, Fe-free PPyC-900 sample reveals a high micropore volume percentage near 95% of the total pore volume, whereas FePPyC-800, FePPyC-900, and FePPyC-1000 present decreased micropore volume percentage of 68.4%, 62.2% and 75.8%, respectively, as shown in Table S1,† which indicate an increased mesoporous volume in the FePPyC-T samples. The pore size distributions of these samples are shown in Fig. S5b,† and it can be seen that the pore sizes of the FePPyC-T samples are larger than that of the Fe-free PPyC-900. The average pore size of PPyC-900, FePPyC-800, FePPyC-900, and FePPyC-1000 is in the range of 3.0–6.6 nm. Such increased mesopore volume and average pore size in the FePPyC-T samples can be attributed to Fe removal from the carbon framework during acid washing, which results in large pores in the carbon framework. These mesopore structures are expected to be helpful for improving the ORR activity.
The elemental compositions of all the samples were monitored by X-ray photoelectron spectroscopy (XPS). Fig. 4a shows XPS survey scans of PPyC-900 and FePPyC-T samples, which indicate the presence of carbon C 1s, oxygen O 1s, nitrogen N 1s, and iron Fe 2p. In particular, heteroatoms, such as N, S, P, and B, doped into the carbon framework are believed to be responsible for the improved activity towards various electrochemical reactions.9,11,45,54,55 Among them, electronegative N is most extensively investigated as a dopant and found to influence the physicochemical properties of carbon. Moreover, it is also commonly accepted that the presence of Fe can convert the carbon framework into carbon with a more graphitized structure.24,44,47
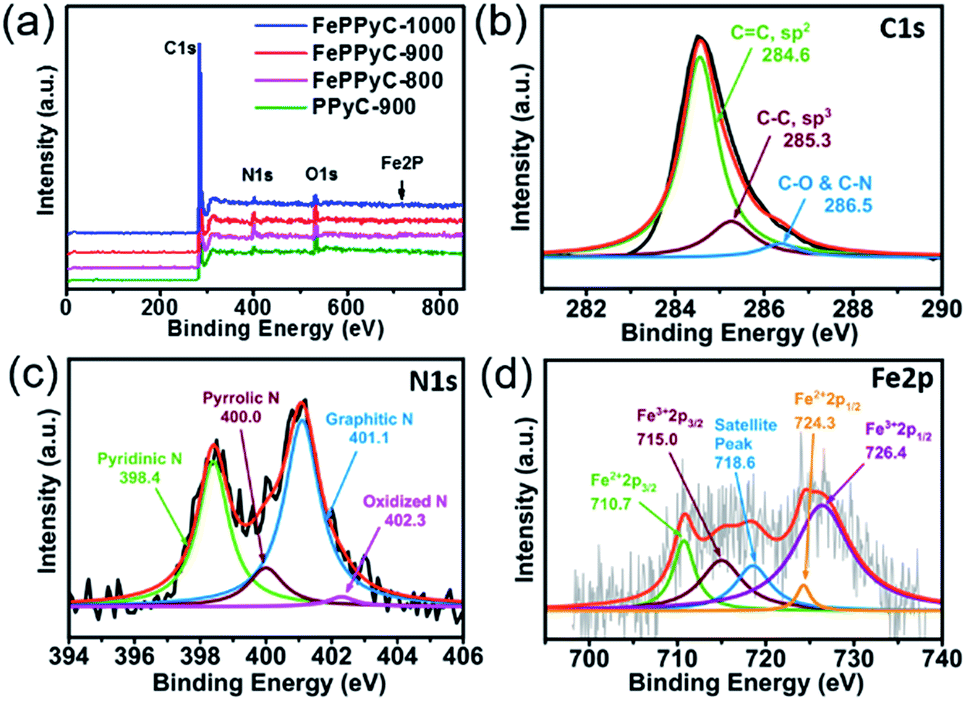 | ||
| Fig. 4 (a) XPS survey scans of PPyC-900 and FePPyC-T samples prepared at various carbonization temperatures. Deconvoluted XPS spectra of (b) C 1s, (c) N 1s, and (d) Fe 2p of FePPyC-900. | ||
In Table 1, FePPyC-900af shows a lower iron content than FePPyC-900bf, indicating that acid washing removes the unstable iron metal.43 Furthermore, the carbon content increases from 89.4% to 92.8% with increasing pyrolysis temperature, whereas the nitrogen content decreases from 6.1% to 2.6%. The iron content seems to be decreasing with increasing reaction temperature, which may be due to the shielding effect of the carbon layer, towards the XPS incident beam, formed on the Fe particle at higher temperatures.
| Sample | C 1s (%) | O 1s (%) | N 1s (%) | Fe 2p (%) |
|---|---|---|---|---|
| PPyC-900 | 89.42 | 6.64 | 3.94 | 0 |
| FePPyC-800 | 89.48 | 4.09 | 6.11 | 0.32 |
| FePPyC-900 | 91.45 | 4.04 | 4.25 | 0.26 |
| FePPyC-1000 | 92.88 | 4.30 | 2.62 | 0.20 |
| FePPyC-900bf | 90.70 | 4.88 | 3.93 | 0.49 |
| FePPyC-900af | 90.90 | 3.96 | 4.89 | 0.25 |
As can be seen in Fig. 4b and S6 of ESI,† the major carbon (C 1 s) species detected by XPS are sp2 C at 284.6 eV and sp3 C at 285.3 eV, and a minor signal at 286.5 eV ascribing to different bonding of carbon with nitrogen or oxygen was also observed in all the samples.56,57 The relative percentage of the different carbon species for C 1 s in all the samples are summarized in Table S2 of ESI.† As expected, sp2 C increases with the presence of Fe and increasing carbonization temperature in the prepared samples. This can be understood by the fact that the increasing pyrolysis temperature and presence of Fe certainly improves the graphitization along with increase in sp2 hybridization and thus increases the electrical conductivity, as proven in Fig. 3d.44,47
Fig. 4c, S7 and S8 of ESI† show that the N1s is deconvoluted into four different peaks such as pyridinic nitrogen N1 (∼398.6 eV), pyrrolic nitrogen N2 (∼400 eV), graphitic nitrogen N3 (∼401.1 eV), and oxidized nitrogen N4 (∼402.3 eV).48,58,59 The relative amount of N bonding configurations significantly changes with increasing pyrolysis temperature. As shown in Fig. S9a of ESI,† FePPyC-900af and FePPyC-900 show similar relative intensity of pyridinic-N, but reveal higher intensity than FePPyC-900bf, whereas FePPyC-900af shows higher pyrrolic-N intensity than FePPyC-900 and FePPyC-900bf. Moreover, FePPyC-900bf and FePPyC-900 show quite similar graphitic-N intensity, but much higher intensity than FePPyC-900af. This can be attributed to the acid washing after first pyrolysis, which exposes edge-sited N species such as pyridinic-N and pyrrolic-N. During the second pyrolysis process, some of the pyrrolic-N converts to graphitic-N without much change in pyridinic-N. The increase in the graphitic-N amount after the second pyrolysis may contribute to enhanced electrical conductivity of FePPyC-900 compared to FePPyC-900af, as evidenced in Fig. 3c.
In particular, the pyridinic-N and pyrrolic-N decrease with increasing temperature, as shown in Fig. S9b of ESI,† implying that these N species are less stable at higher temperatures. On the contrary, amount of graphitic-N increases significantly with increasing pyrolysis temperature. The graphitic-N species is found to be relatively more stable and becomes predominant at temperatures higher than 900 °C. In general, pyridinic-N and graphitic-N are reported to be active for the ORR.55 Oxidized-N, known to be catalytically inactive for the ORR, also increases steadily from 800 °C to 1000 °C. Interestingly, upon a comparison of PPyC-900 and FePPyC-900, it can be seen that the pyrrolic-N species decrease, whereas pyridinic-N increases in FePPyC-900, and graphitic-N is not changed much, implying that Fe may help generate more edge pyridinic N-species, which can eventually help to enhance the ORR performance of FePPyC-900.47,60
Fig. 4d shows the Fe 2p spectrum of FePPyC-900 catalyst, and the signal intensity is very weak. According to previous reports,25,26,61 the Fe 2p peak is deconvoluted into five peaks at 710.7, 715.0, 718.6, 724.3, and 726.4 eV. The photoelectron peaks at 710.7 and 715.0 eV correspond to binding energy of 2p3/2 orbitals of Fe(II) and Fe(III) ion, respectively. The peaks at 724.3 and 726.4 eV can be assigned to 2p1/2 binding energy of Fe(II) and Fe(III), respectively. The peak at 718.6 eV corresponds to a satellite peak. This indicates that both Fe(II) and Fe(III) co-exist in the catalyst. It can be seen that the total area of the deconvoluted peaks corresponding to Fe3+ is higher than that of Fe2+, indicating a higher atomic percentage of Fe(III), which may be due to the use of Fe(III) chloride as an iron precursor.
The electrocatalytic ORR activity is determined for the samples prepared in different synthesis steps of the PLS sequence. Herein, each step of the PLS sequence was assessed in terms of the ORR activity. For FePPyC-900af (after acid leaching but before second pyrolysis), the onset potential and limiting current density are improved from those of FePPyC-900bf (after the first pyrolysis, but before acid leaching) as shown in Fig. S10a and b of ESI.† Best of all, FePPyC-900 shows the most positive onset potential along with a much higher current density under both alkaline and acidic conditions after second pyrolysis, indicating the importance of the second pyrolysis after acid leaching.62 The ORR activity of the catalyst was significantly enhanced after acid leaching and additional pyrolysis, which can help to make the loose carbon matrix, formed during acid leaching, stabilized and further graphitized. N species doped into the carbon framework can also contribute to the enhancement of the ORR activity. According to the report by Lai et al.,63 both pyridinic-N and graphitic-N species, in particular, pyridinic-N plays a more important role as an active center for the ORR. Fig. 5 shows the interdependence of the ORR activity (onset potential) on the specific surface area, electrical conductivity, and amount of both pyridinic- and graphitic-N species of FePPyC-900bf, FePPyC-900af, and FePPyC-900 samples. Interestingly, each step of a series of pyrolysis–leaching–stabilization (PLS) sequences brings out the different structural properties of the resulting carbon samples, and their effects on the ORR activity are illustrated in Fig. 5. FePPyC-900 has much higher pyridinic- and graphitic-N species than FePPyC-900bf and FePPyC-900af. Moreover, FePPyC-900 shows much higher BET surface area and electrical conductivity than FePPyC-900bf and FePPyC-900af. Therefore, the synergistic contribution of all these properties improved the ORR performance of FePPyC-900.
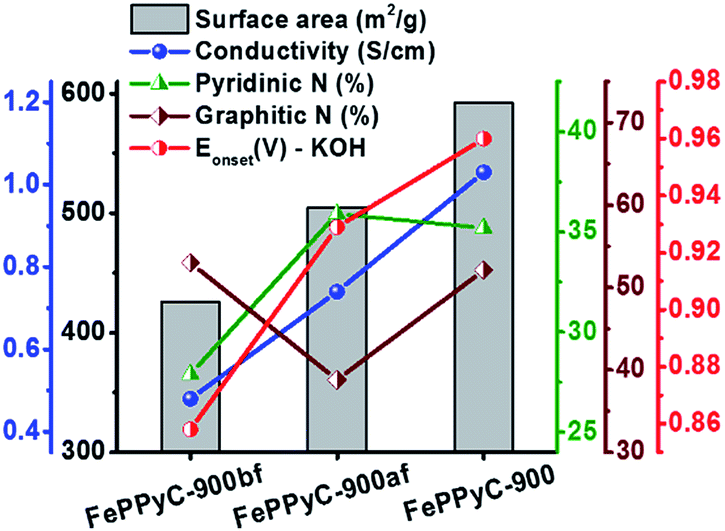 | ||
| Fig. 5 Assessment of each step of the PLS sequence in terms of the electrical conductivity (at 20 Mpa), nitrogen content, and specific surface area along with the onset potential vs. RHE for the FePPyC-900bf, FePPyC-900af and FePPyC-900 samples under alkaline conditions, indicating the effect of each synthesis step on the material properties and ORR activity. | ||
Electrocatalytic ORR activity is measured using cyclic voltammetry (CV) and rotating ring disk electrode (RRDE) techniques. The CV curve of FePPyC-900 catalyst in the N2-saturated 0.1 M KOH solution displays a featureless curve. When oxygen gas is purged in the solution, the CV curve exhibits a noticeable ORR peak at 0.85 V, as shown in Fig. S11a of ESI.† In Fig. S11b of ESI,† the CV curve of FePPyC-900 in N2-saturated 0.5 M H2SO4 shows a pair of symmetric peaks at around 0.54 V and 0.66 V, which may be due to the redox reaction of quinone/hydroquinone or Fe(II)/Fe(III).64,65 With an O2-saturated acidic solution, a CV curve with different peak shape, marked by a strong reduction peak at 0.62 V occurs. These results suggest that FePPyC-900 shows significantly high ORR activity in both acid and alkaline electrolytes.
The CV curves of the PPyC-900 and FePPyC-T samples are presented in Fig. S11c and d of ESI.† It can be seen that the FePPyC-T samples show nearly rectangular CV curves, which indicates less electrode polarization in FePPyC-T than in Fe-free PPyC-900 under both alkaline and acidic conditions. This result clearly indicates that Fe plays a crucial role in enhancement of the electrochemical activity in the FePPyC-T samples by increasing the electrical conductivity (see Fig. 3d). Moreover, from the areas in the CV curves, FePPyC-T samples were found to show a higher electrochemical surface area than Fe-free PPyC-900, suggesting that the FePPyC-T samples prepared in the presence of Fe have much more active sites than the PPyC-900 (Fig. S11c and d of ESI†). In particular, the FePPyC-900 (0.847 V vs. RHE) has a more positively-shifted ORR peak potential than FePPyC-800 (0.623 V) and FePPy-1000 (0.801 V) under alkaline conditions, indicating the highest ORR activity in FePPyC-900 among the FePPyC-T samples prepared at different carbonization temperatures. Interestingly, the ORR peak potential of 0.847 V for FePPyC-900 is one of the most positively shifted values reported so far to the best of our knowledge. Table S3 of ESI† compares the ORR peak potentials for different electrocatalysts prepared in this work under alkaline conditions.
The ORR activity of FePPyC-900 in both alkaline and acidic electrolytes was further confirmed by the CV curves at a slow scan rate of 10 mV s−1, as shown in Fig. S12 of ESI.† The voltammogram of FePPyC-900 in N2-saturated 0.1 M KOH and 0.5 M H2SO4 shows quasi-rectangular patterns without obvious redox peaks, whereas well-defined ORR peaks are seen at 0.87 V and 0.69 V (vs. RHE) for CV in 0.1 M KOH and 0.5 M H2SO4 electrolyte, respectively, when O2 was introduced, suggesting efficient electrocatalytic activity of FePPyC-900 for ORR in both alkaline and acidic media.
Fig. 6 shows the ORR activities of the PPyC-900 and FePPyC-T samples prepared at different pyrolysis temperatures in alkaline and acidic conditions. The ORR activity performance data of all samples are summarized in Table 2. The electrochemical measurements of all the samples show that FePPyC-900 sample has the best ORR activity under both alkaline and acidic conditions. FePPyC-900 shows an onset potential of ∼0.960 V vs. RHE, almost identical to that of 20 wt% Pt/C, and a half wave potential (E1/2) of 0.877 V vs. RHE, which is more positively shifted than 0.844 V of the Pt/C under alkaline conditions. Under acidic conditions, it can be seen that FePPyC-900 shows an onset potential of 0.814 V, only 3 mV less positive compared with that of 20 wt% Pt/C (0.817 V), and a half wave potential of 0.740 V, which is more positive than 0.709 V of 20 wt% Pt/C. FePPyC-900 reveals current density of 6.40 mA cm−2, which is slightly higher than 6.33 mA cm−2 for the 20 wt% Pt/C in KOH solution. In H2SO4 solution, FePPyC-900 shows a current density of 5.70 mA cm−2, which is higher than that of Pt/C (5.40 mA cm−2). This represents extraordinary activity, which is among the best ever reported for metal-treated doped carbon catalysts, and is comparable to the state-of-the-art commercial Pt/C catalyst, even in an acidic medium. In general, the activity of transition metal-treated N-doped carbon is reported to be lower than or comparable to that of Pt/C catalyst under alkaline conditions, but still much lower than that of the Pt-based catalyst under acidic conditions.41,66,67 Therefore, such remarkable ORR activity of the FePPyC-900 sample, particularly under acidic conditions, has not been reported before to the best of our knowledge.
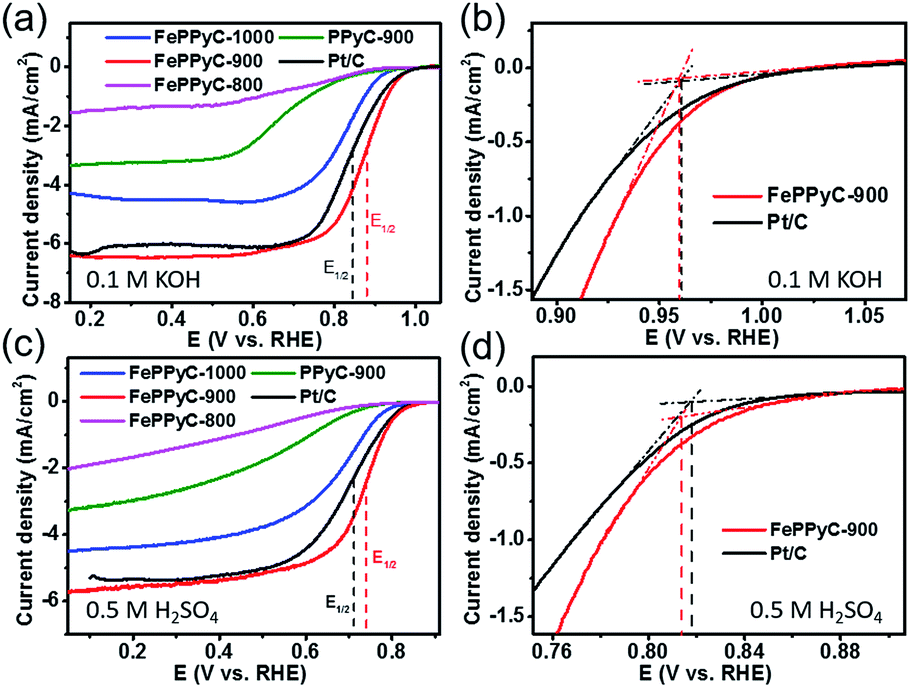 | ||
| Fig. 6 LSV curves of the different samples prepared at different pyrolysis temperatures in O2-saturated (a) 0.1 M KOH and (c) 0.5 M H2SO4. Onset potential determination in expanded LSV curves of FePPyC-900 and 20 wt% Pt/C (E-TEK) in O2-saturated (b) 0.1 M KOH and (d) 0.5 M H2SO4. The LSV experiments were conducted at a rotating speed of 1600 rpm and 10 mV s−1 potential scan rate. | ||
| Sample | Alkaline | Acidic | ||||
|---|---|---|---|---|---|---|
| E onset (V) | E 1/2 (V) | LCD (mA cm−2) | E onset (V) | E 1/2 (V) | LCD (mA cm−2) | |
| PPyC-900 | 0.873 | 0.671 | 3.42 | 0.738 | 0.541 | 3.10 |
| FePPyC-800 | 0.893 | 0.697 | 1.79 | 0.720 | 0.435 | 1.94 |
| FePPyC-900 | 0.960 | 0.877 | 6.40 | 0.814 | 0.740 | 5.70 |
| FePPyC-1000 | 0.921 | 0.816 | 4.46 | 0.789 | 0.673 | 4.34 |
| Pt/C | 0.961 | 0.844 | 6.33 | 0.817 | 0.709 | 5.40 |
Fig. 7 illustrates the variation of the N content, specific surface area and electrical conductivity along with their effects on the ORR activity (onset potential) in the resulting carbon samples as a function of the carbonization temperature in the presence and absence of a Fe precursor. There exists an interesting trade-off between the N content, surface area, and electrical conductivity. As the carbonization temperature increases, the N content decreases, which is unfavourable for the ORR, but at the same time, the surface area and conductivity increases, which are beneficial for the ORR. This trade-off should be optimized for the best performance. FePPy-900 possesses high electrical conductivity and an appropriate N content as well as a high surface area with a mesopore structure, and thus outperforms the other samples.
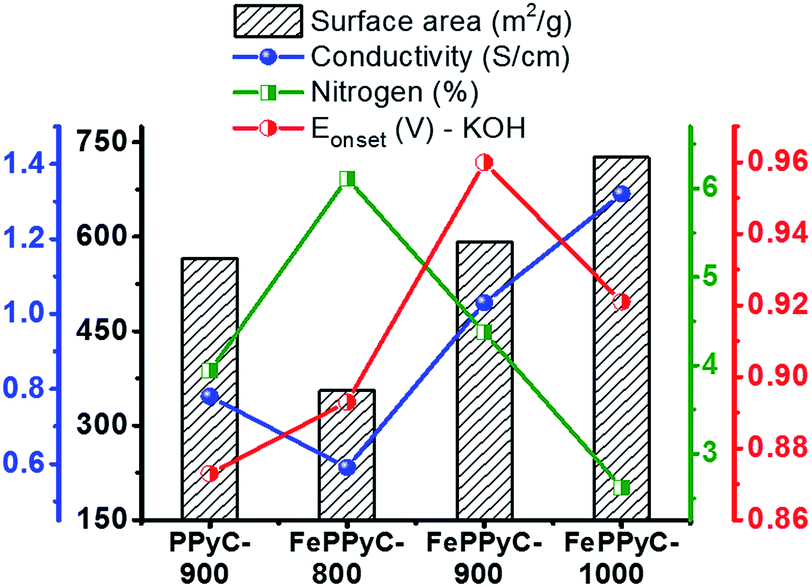 | ||
| Fig. 7 Comparative relation of the electrical conductivity (at 20 Mpa), nitrogen content, and specific surface area along with the onset potential vs. RHE of the PPyC-900 and FePPyC-T samples under alkaline conditions. | ||
Interestingly, despite the higher surface area and electrical conductivity, FePPyC-1000 shows a slightly lower ORR activity than FePPyC-900. This is probably because FePPyC-1000 has a lower N content, which clearly indicates that N is a part of the catalytically active center. Interestingly, FePPyC-800 shows poor ORR activity despite the higher N content most probably because of the low electrical conductivity and surface area, strongly indicating that carbonization temperature should be higher than 800 °C for the efficient development of graphiticity as well as surface area in the resulting carbon. Therefore, the carbonization temperature for the optimal ORR performance must be near 900 °C in the current system. Individual N species may also be an important factor affecting the enhanced ORR activity. As shown in Fig. 4c and S9b of ESI,† FePPyC-900 exhibits a relatively higher percentage of pyridinic- and graphitic-N compared with FePPyC-800 and FePPyC-1000. The high content of pyridinic- and graphitic-N can also contribute to the improved ORR activity of FePPyC-900 catalyst.
The LSV curves of the FePPyC-900 at different rotation rates with a scan rate of 10 mV s−1 are shown in Fig. S13a and c of ESI† for O2-saturated 0.1 M KOH and 0.5 M H2SO4 conditions, respectively. The ORR behaviour of FePPyC-900 at low overpotential (above 0.9 V for alkaline and 0.75 V for acidic conditions) is independent of the rotation rate, suggesting that this potential range corresponds to an electrochemical kinetics process.68 As observed in Fig. S13b and d of ESI,† the Kotechky–Levich plots of FePPyC-900 at different potential are linear and nearly parallel, and the insets of Fig. S13b and d† show the calculated electron transfer number varying (n) from 3.5 to 4.0, which indicates a four electron ORR pathway under both alkaline and acidic conditions.
Furthermore, electron transfer number and hydroxide yield of FePPyC-900 catalyst were also measured using the RRDE measurement at 10 mV s−1 with a rotation speed of 1600 rpm. The H2O2 yield and the electron transfer number n were calculated from eqn (2) and (3) based on the RRDE data, as described in the Experimental section. Fig. S14a and c of ESI† show the ring and disk current density curves of the FePPyC-900 sample in 0.1 M KOH and 0.5 M H2SO4, respectively. The onset potentials of the ring current curves are lower than the counterparts of disk current curves under both alkaline and acidic conditions, which is similar to the behaviour of Pt/C catalysts in previous reports.69,70 The peroxide yield for FePPyC-900 under alkaline conditions increases from 1.1% to 7.2% over the potential range from 0.76 to 0.0 V, and the average n is around 3.89, as shown in Fig. S14b of ESI.† For an acidic electrolyte, the peroxide yield decreases gradually from 1.4% to 0.6% from 0.67 to 0.02 V vs. RHE (Fig. S14d of ESI†), and the average n is ∼3.96, indicating a favourable 4 electron reduction process. The electron transfer number calculated from RRDE method is in good agreement with that based on the Koutecky–Levich plots calculation, indicating the four electron pathway of the ORR process for the FePPyC-900 catalyst in both alkaline and acidic media.
FePPyC-900 in an acidic electrolyte exhibits lower ORR activity (0.814 V onset potential) than that (0.960 V onset potential) in an alkaline electrolyte. Furthermore, the onset potential of hydrogen peroxide oxidation, which is determined by the onset potential of ring current curve, is much more negative than that of ORR under alkaline conditions compared to observation under acidic conditions, as seen in Fig. S14a and c of ESI,† suggesting a somewhat different ORR mechanism in alkaline and acidic media. This also indicates that the formation of hydrogen peroxide is more favourable at a more negative potential under alkaline conditions as shown in high yield of hydrogen peroxide in alkaline electrolyte compared to that in an acidic electrolyte, as observed in Fig. S14 of ESI.† The gap of the electrochemical performance between alkaline and acidic media may be explained by the inner-sphere and outer-sphere electron transfer mechanism between the oxygen molecule and active site on the electrode surface.71 It is acknowledged that N doping into the carbon framework generates charge delocalization and a slightly positive charge at the adjacent carbon atoms, and these charged carbon become active sites for the adsorption of oxygen molecules during the ORR.72 Typically, the oxygen molecule is first adsorbed at the active site (*) in an acidic electrolyte, and then the adsorbed O2 receives an electron from the electrode and a proton to form *–(OOH)ads. This process is called the inner-sphere electron transfer (ISET). Other two protons interact with two oxygen atoms to break the O–OH bond to generate water and –OH species, and the additional proton then reacts with the remaining –OHads to form water, as shown in eqn (4)–(6).
| * + O2 + H+ + e− → *–(OOH)ads | (4) |
| *–(OOH)ads + 2H+ + 2e− → *–(OH)ads + H2O | (5) |
| *–(OH)ads + H+ + e− → * + H2O | (6) |
Furthermore, hydrogen peroxide can be formed when a proton react with *–(OOH)ads in eqn (4) to generate H2O2, which can also re-adsorb to the active site to receive 2e− and 2H+ to generate water.73 In addition, protonation at the nitrogen atom (as an Lewis base site) in N-doped carbon can have a strong influence on the lone-pair electrons of N atoms, decreasing the charge delocalization of adjacent carbon atoms, which leads to decreased catalytic activity in an acidic electrolyte.74
In an alkaline electrolyte, there are two possible pathways. The first is the chemisorption of an oxygen molecule onto the active site followed by receiving electrons and capturing protons from water until the OH− anion is generated without the desorption of a peroxide species (HO2−) intermediate, as shown in eqn (7)–(9), which is analogous to the ISET in an acidic electrolyte.71
| * + O2 → *–(OO)ads | (7) |
| *–(OO)ads + H2O + 2e− → *–(OOH)ads + OH− | (8) |
| *–(OOH)ads + H2O + 2e− → 3OH− | (9) |
The second is the outer-sphere electron transfer mechanism (OSET), in which the solvated oxygen molecule adsorbs on the active site through hydrogen bonding of *–(OH)ads species and received the first electron from the electrode by the tunnelling effect to form oxygenated radical species [O2˙−·(H2O)n]aq, as shown in eqn (10).71,75 At a more negative potential, the solvated [O2˙−·(H2O)n]aq species can be fully stripped off water shell and adsorbed to the active site followed by protonation to generate hydroperoxyl radical (HO2˙), as shown in eqn (11) and (12). An additional electron was then captured by (HO2˙) to form a hydrogen peroxide ion (HO2−), as shown in eqn (13).71 This illustrates the more negative potential for hydrogen peroxide generation compared to the ORR potential in the RRDE measurement, as shown in Fig. S14a of ESI.† In addition, the high hydrogen peroxide yield in the alkaline electrolyte compared to that in the acidic electrolyte at low potential could be due to the stable existence of a superoxide radical in alkaline media, which may stay in the outer-Helmholtz plane.
| *–OHads + [O2·(H2O)n]aq + e− → *–OHads + [O2˙−·(H2O)n]aq | (10) |
| [O2˙−·(H2O)n]aq → (O2˙−)ads + nH2O | (11) |
| (O2˙−)ads + H2O → (HO2˙)ads + OH− | (12) |
| (HO2˙)ads + e− → (HO2−)ads → (HO2−)aq | (13) |
The ORR kinetics on the FePPyC −900 catalyst can be evaluated by Tafel analysis. In Fig. S15a of ESI,† the Tafel slopes of FePPyC-900 and Pt/C at low over potential were found to be 95 mV dec−1 and 106 mV dec−1 under alkaline conditions, respectively. In Fig. S15b of ESI,† the Tafel slope values of FePPyC-900 and Pt/C under acidic conditions were determined to be 78 mV dec−1 and 79 mV dec−1, respectively. Interestingly, under both alkaline and acidic conditions, the FePPyC-900 catalyst shows a lower Tafel slope than those of the state-of-the-art Pt/C. The lower Tafel slope value is desirable for the ORR activity, indicating that FePPyC-900 is more effective than Pt/C for the ORR under both alkaline and acid conditions. The exchange current density can be obtained by extrapolation of the horizontal axis. The lower Tafel slope would generate a higher exchange current density, whereas a higher Tafel slope would give a lower exchange current density.76 Hence, the FePPyC-900 sample has a higher exchange current density than the Pt/C catalyst, indicating higher ORR activity.
In addition, the ORR performance of our catalyst was evaluated by the current-time response. The typical dynamic current response of all samples was measured at various potentials from 0.96 to 0.06 V (vs. RHE) at a rotation rate of 1600 rpm, as shown in Fig. S16.† The FePPyC-900 catalyst shows a much higher current compared to the other samples in different synthesis steps (Fig. S16a†). In addition, with different pyrolysis temperatures, FePPyC-900 also exhibits the best ORR performance with a higher current compared to the other FePPyC-T samples when increasing the negative applied potential, as observed in Fig. S16b.† These results are in good agreement with the LSV curves in Fig. S10a of ESI† and 6a.
In earlier reports,24,77,78 carbon black (Vulcan XC-72) was used as a supporting carbon for N-doped carbon, and the catalyst consisting of N-doped carbon along with Vulcan carbon showed high ORR activity performance. Herein, we also added a certain amount of Vulcan carbon (XC-72) into our catalyst in the initial step of the synthesis process while keeping the remaining synthesis protocol identical and tested the ORR activity of the resulting FePPyC-900/VC under alkaline and acidic conditions (Fig. S17 of ESI†). Unlike the earlier study, the Vulcan carbon did not increase the ORR activity of the FePPyC-900 catalyst in the current experimental conditions under both alkaline and acidic conditions.
It has been also demonstrated that the Fe and Co dual-containing N–C compositions are considered to be the most promising systems for the development of high-performance ORR electrocatalysts.3,41,79 Thus, according to the current synthesis method of FePPyC-900, FeCoPPyC-900 was synthesized using the identical PLS protocol with both Fe and Co metal precursors. A certain amount of cobalt nitrate (Co(NO3)2) was mixed with a FeCl3 solution to have a mole ratio of Fe:Co = 1:1, and the mixture was then added to the pyrrole solution. The full synthesis procedure followed the same processes as those of FePPyC-900. The LSV curves in Fig. S18 of ESI† illustrate the electrocatalyst performance of FeCoPPyC-900 under both alkaline and acidic conditions. Under alkaline conditions, the onset potential of FeCoPPyC-900 is similar to those of the commercial Pt/C and FePPyC-900. The half-wave potential and limiting current of FeCoPPy-900 are also similar to those of FePPyC-900 and slightly higher than those of Pt/C, suggesting high electrocatalyst activity of the FeCoPPyC-900 catalyst under alkaline conditions, as reported earlier.3,41,79 Under acidic conditions, however, the onset potential and half-wave potential of FeCoPPyC-900 are much lower than those of FePPyC-900 and Pt/C, even though the limiting current of FeCoPPy-900 is better than those of FePPyC-900 and Pt/C. These extra experimental results indicate that the activity of FePPyC-900/VC and FeCoPPyC-900 is not good in an acidic electrolyte compared to that of the benchmark Pt/C, whereas the FePPyC-900 catalyst exhibits excellent ORR performance under both the alkaline and acidic conditions.
Both the tolerance against the methanol crossover and stability of the catalyst material are also important parameters for an evaluation of practical fuel cell applications.80 The methanol tolerance was evaluated by the current–time response at 0.85 V vs. RHE under alkaline conditions. It is shown in Fig. 8a that the ORR current of Pt/C drastically decreases after the addition of 3.0 M methanol, which can be attributed to methanol oxidation instead of oxygen reduction by Pt/C under the current experimental conditions. On the other hand, except for a slight initial decrease, the ORR current of FePPyC-900 is almost unaffected, demonstrating that FePPyC-900 catalyst illustrates significant methanol tolerance unparalleled to Pt/C.56 In addition, the FePPyC-900 catalyst also exhibits excellent long-term stability compared to the commercial Pt/C catalyst for oxygen reduction under 0.1 M KOH conditions (Fig. 8b) and 0.5 M H2SO4 condition (Fig. S19 of ESI†).
 | ||
| Fig. 8 (a) Chronoamperometric responses at 0.85 V in O2-saturated 0.1 M KOH solution (1600 rpm) before and after the addition of 3.0 M methanol, and (b) relative J–t responses at 0.85 V in a O2-saturated 0.1 M KOH solution for FePPyC-900 and 20 wt% Pt/C (E-TEK) electrodes. | ||
The stability of FePPyC-900 was evaluated further using the accelerated durability test (ADT) by potential cycling within a range of 0.6–1.1 V for O2-saturated 0.1 M KOH and 0.5–1.0 V (vs. RHE) for O2-saturated 0.5 M H2SO4. As shown in Fig. S20a and b of ESI,† the E1/2 of FePPyC-900 decreases to 23 and 35 mV after 2000 and 5000 potential cycles, respectively, whereas the E1/2 of the Pt/C decreases to 37 mV after 2000 cycles and further decreases to 50 mV after 5000 cycles under alkaline conditions. In Fig. S20c and d of ESI,† the E1/2 of FePPyC-900 in O2-saturated 0.5 M H2SO4 negatively shifted with 34 and 44 mV after 2000 and 5000 potential cycles, respectively, which are much lower than the 53 mV and 74 mV negative shift observed for the Pt/C catalyst. This also supports the excellent durability of FePPyC-900 under both alkaline and acidic conditions.
As shown in Fig. S19 and S20 of ESI,† it was observed that even after the long term cycling and accelerated durability test (ADT), the prepared catalyst shows only a minute decrease in the activity. This drop in activity can be attributed to the carbon corrosion and/or deactivation of N species as N-doped carbon lattice is a main active center for the ORR. TEM and SEM analyses of FePPyC-900 were performed before and after ADT, as shown in Fig. S21.† It was found that the carbon surface, which was smooth and well interconnected before ADT still maintained a similar morphology and interconnectivity even after ADT, even though the carbon surface may be coated with a nafion binder. This may be the reason for only a minute decrease in the activity after ADT. Although it is very difficult to estimate the extent of carbon corrosion only by the TEM image, it is expected that some carbon corrosion takes place and thus decreases the ORR activity after ADT. As reported previously, carbon corrosion can be attributed to the decay of activity during the ORR performance.81–83 In addition, the slow deactivation of N species may be also a reason because the protonation at the N atom in N-doped carbon can have a strong influence on the lone-pair electrons of the N atoms, decreasing the charge delocalization of the adjacent carbon atoms, which leads to decreased catalytic activity.
After all, the FePPyC-900 demonstrates much better methanol tolerance and long-term stability than state-of-the-art Pt/C under both alkaline and acidic conditions. Electrocatalytic activities of as-prepared FePPyC-900 electrocatalyst and other related Fe-treated N-doped carbon catalysts in earlier works are summarized in Tables S4 and S5 (ESI†) under alkaline and acid conditions, respectively. FePPyC-900 represents remarkable activity, which is among the best activity ever reported for Fe-treated N-doped carbon catalysts, and thus illustrates a viable alternative to a costly Pt/C catalyst in terms of synthesis procedure and economical point as well as ORR activity and stability under both alkaline and acid conditions.
4. Conclusions
A highly efficient noble metal-free electrocatalyst for ORR was developed by novel synthesis protocol of pyrolysis–leaching–stabilization (PLS) sequence at different pyrolysis temperatures using polypyrrole as a single precursor for N and carbon, and ferric chloride as an oxidant and iron source. PPy was chosen as a single precursor for C and N because it is an excellent conducting polymer and can yield an extended graphitic carbon framework with high electrical conductivity upon carbonization. Each step of the PLS sequence was assessed in detail in terms of various physical properties of the resulting catalysts to validate the efficacy of the synthesis protocol. The full PLS sequence was found to be very effective for the preparation of an efficient Fe-treated N-doped carbon electrocatalyst. In particular, the additional stabilizing pyrolysis step after acid leaching exhibits remarkable effect for better ORR activity as elaborated when a PLS sample (FePPyC-900) is compared with the corresponding first pyrolysis sample (FePPyC-900bf) and subsequent acid-leached sample (FePPyC-900af). The catalyst prepared at 900 °C (FePPyC-900) displayed highest catalytic activity towards oxygen reduction, which is comparable to that of state-of-the-art commercial 20 wt% Pt/C under both alkaline and acidic conditions. In particular, it was found that the ORR activity of the FePPyC-900 catalyst is almost comparable to that of Pt/C even in an acidic medium, which has rarely been reported as a non-Pt electrocatalyst to the best of our knowledge. This high ORR performance of FePPyC-900 can be attributed to the high degree of graphiticity, high electrical conductivity, high surface area, and high density of the active pyridinic- and graphitic-N species facilitated by the new synthesis protocol. Furthermore, FePPyC-900 exhibits better stability and methanol tolerance than the commercial Pt/C for ORR under both alkaline and acidic conditions with a four electron transfer pathway, suggesting the complete reduction of oxygen to water. Further modification such as additional carbon support and addition of Co as another metal to help in the current system was not found to be beneficial. As a result, our as-prepared FePPyC electrocatalyst can become a viable alternative catalyst to precious Pt/C catalyst in PEMFCs with high ORR performance and long term stability.Acknowledgements
We gratefully acknowledge the financial support by the NRF Grant (NRF-2010-0029245) and the Global Frontier R&D Program on Center for Multiscale Energy System (NRF 2011-0031571) funded by the Ministry of Education, Science and Technology of Korea. Special thanks are given to the KBSI at Jeonju, Chuncheon and Busan for SEM, TEM and XPS measurements. We thank Hyean-Yeol Park, Tong-Hyun Kang, Jong-Deok Park, and Sung-Soo Kim for helping in BET and Raman measurements.Notes and references
- S. Giddey, S. P. S. Badwal, A. Kulkarni and C. Munnings, Prog. Energy Combust. Sci., 2012, 38, 360–399 CrossRef CAS
.
- K. N. Chaudhari, M. Y. Song and J.-S. Yu, Small, 2014, 10, 2625–2636 CrossRef CAS PubMed
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed
- L. Zhang, J. Zhang, D. P. Wilkinson and H. Wang, J. Power Sources, 2006, 156, 171–182 CrossRef CAS
- S. M. Alia, S. Pylypenko, K. C. Neyerlin, D. A. Cullen, S. S. Kocha and B. S. Pivovar, ACS Catal., 2014, 4, 2680–2686 CrossRef CAS
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed
- D.-S. Yang, S. Chaudhari, K. P. Rajesh and J.-S. Yu, ChemCatChem, 2014, 6, 1236–1244 CAS
- I. Shaukatali, H.-S. Choi, W. Peng, M. Y. Song and J.-S. Yu, Electrochem. Commun., 2013, 30, 9–12 CrossRef
- F. Razmjooei, K. P. Singh, M. Y. Song and J.-S. Yu, Carbon, 2014, 78, 257–267 CrossRef CAS
- D.-S. Yang, D. Bhattacharjya, M. Y. Song and J.-S. Yu, Carbon, 2014, 67, 736–743 CrossRef CAS
- K. P. Singh, M. Y. Song and J.-S. Yu, J. Mater. Chem. A, 2014, 2, 18115–18124 CAS
- X. Gong, S. S. Liu, C. Y. Ouyang, P. Strasser and R. Z. Yang, ACS Catal., 2015, 5, 920–927 CrossRef CAS
- J. T. Zhang and L. M. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS
- Y. Su, H. Jiang, Y. Zhu, X. Yang, J. Shen, W. Zou, J. Chen and C. Li, J. Mater. Chem. A, 2014, 2, 7281–7287 CAS
- Y. Wang, X. Cui, L. Chen, C. Wei, F. Cui, H. Yao, J. Shi and Y. Li, Dalton Trans., 2014, 43, 4163–4168 RSC
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed. Engl., 2014, 53, 3675–3679 CrossRef CAS PubMed
- T. Takeguchi, T. Yamanaka, H. Takahashi, H. Watanabe, T. Kuroki, H. Nakanishi, Y. Orikasa, Y. Uchimoto, H. Takano, N. Ohguri, M. Matsuda, T. Murota, K. Uosaki and W. Ueda, J. Am. Chem. Soc., 2013, 135, 11125–11130 CrossRef CAS PubMed
- K.-N. Jung, J.-H. Jung, W. B. Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902–9907 CAS
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2004, 108, 11375–11383 CrossRef CAS
- Y. Y. Jiang, Y. Z. Lu, X. Y. Lv, D. X. Han, Q. X. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS
- W. M. Li, J. Wu, D. C. Higgins, J. Y. Choi and Z. W. Chen, ACS Catal., 2012, 2, 2761–2768 CrossRef CAS
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan, G. Xin, J. Zheng and X. Li, J. Mater. Chem. A, 2014, 2, 14064–14070 CAS
- J. Zhang, D. He, H. Su, X. Chen, M. Pan and S. Mu, J. Mater. Chem. A, 2014, 2, 1242–1246 CAS
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed
- X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178–2180 CrossRef CAS
- S. Ye and A. K. Vijh, Electrochem. Commun., 2003, 5, 272–275 CrossRef CAS
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed
- D. Nguyen-Thanh, A. I. Frenkel, J. Wang, S. O'Brien and D. L. Akins, Appl. Catal., B, 2011, 105, 50–60 CrossRef CAS
- M. Yuasa, A. Yamaguchi, H. Itsuki, K. Tanaka, M. Yamamoto and K. Oyaizu, Chem. Mater., 2005, 17, 4278–4281 CrossRef CAS
- X. Yuan, X.-L. Ding, C.-Y. Wang and Z.-F. Ma, Energy Environ. Sci., 2013, 6, 1105–1124 CAS
- H. Liu, Z. Shi, J. Zhang, L. Zhang and J. Zhang, J. Mater. Chem. A, 2009, 19, 468–470 RSC
- G. Ciric-Marjanovic, S. Mentus, I. Pasti, N. Gavrilov, J. Krstic, J. Travas-Sejdic, L. T. Strover, J. Kopecka, Z. Moravkova, M. Trchova and J. Stejskal, J. Phys. Chem. C, 2014, 118, 14770–14784 CAS
- L. Z. Gu, L. H. Jiang, J. T. Jin, J. Liu and G. Q. Sun, Carbon, 2015, 82, 572–578 CrossRef CAS
- S. Z. Ren, S. B. Ma, Y. Yang, Q. Mao and C. Hao, Electrochim. Acta, 2015, 178, 179–189 CrossRef CAS
- G. Wu, K. Artyushkova, M. Ferrandon, A. J. Kropf, D. Myers and P. Zelenay, ECS Trans., 2009, 25, 1299–1311 CAS
- M. Sevilla, L. Yu, T. P. Fellinger, A. B. Fuertes and M.-M. Titirici, RSC Adv., 2013, 3, 9904–9910 RSC
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefevre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J.-P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal., B, 2013, 130–131, 143–151 CrossRef CAS
- J. Herranz, F. Jaouen, M. Lefèvre, U. I. Kramm, E. Proietti, J.-P. Dodelet, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, P. Bertrand, T. M. Arruda and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 16087–16097 CAS
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CAS
- M. Y. Song, D.-S. Yang, K. P. Singh, J. Yuan and J.-S. Yu, Appl. Catal., B, 2016, 191, 202–208 CrossRef CAS
- N. K. Chaudhari, M. Y. Song and J.-S. Yu, Sci. Rep., 2014, 4, 5221 CAS
- Y. Wu, S. Zhao, K. Zhao, T. Tu, J. Zheng, J. Chen, H. Zhou, D. Chen and S. Li, J. Power Sources, 2016, 311, 137–143 CrossRef CAS
- D.-S. Yang, M. Y. Song, K. P. Singh and J.-S. Yu, Chem. Commun., 2015, 51, 2450–2453 RSC
- C. a. Cao, X. Zhuang, Y. Su, Y. Zhang, F. Zhang, D. Wu and X. Feng, Polym. Chem., 2014, 5, 2057–2064 RSC
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Mater. Chem. A, 2016, 4, 3858–3864 CAS
- Z. S. Wu, W. Ren, L. Xu, F. Li and H. M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed
- J. Wu, X. J. Zheng, C. Jin, J. H. Tian and R. Z. Yang, Carbon, 2015, 92, 327–338 CrossRef CAS
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS
- Y. Lijun, J. Shujuan, Z. Yu, Z. Lei, C. Sheng, W. Xizhang, W. Qiang, M. Jing, M. Yanwen and H. Zheng, Angew. Chem., 2011, 123, 7270–7273 CrossRef
- J. Campos-Delgado, I. O. Maciel, D. A. Cullen, D. J. Smith, A. Jorio, M. A. Pimenta, H. Terrones and M. Terrones, ACS Nano, 2010, 4, 1696–1702 CrossRef CAS PubMed
- D.-S. Yang, D. Bhattacharjya, M. Y. Song, F. Razmjooei, J. Ko, Q.-H. Yang and J.-S. Yu, ChemCatChem, 2015, 7, 2882–2890 CrossRef CAS
- T. Hu, X. Sun, H. Sun, G. Xin, D. Shao, C. Liu and J. Lian, Phys. Chem. Chem. Phys., 2014, 16, 1060–1066 RSC
- D. Long, W. Li, L. Ling, J. Miyawaki, I. Mochida and S.-H. Yoon, Langmuir, 2010, 26, 16096–16102 CrossRef CAS PubMed
- Q. Shi, F. Peng, S. Liao, H. Wang, H. Yu, Z. Liu, B. Zhang and D. Su, J. Mater. Chem. A, 2013, 1, 14853–14857 CAS
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS
- H. Kong, J. Song and J. Jang, Chem. Commun., 2010, 46, 6735–6737 RSC
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More, D. J. Myers and P. Zelenay, J. Mater. Chem., 2011, 21, 11392–11405 RSC
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS
- H. Xiao, Z.-G. Shao, G. Zhang, Y. Gao, W. Lu and B. Yi, Carbon, 2013, 57, 443–451 CrossRef CAS
- Z. Y. Mo, S. J. Liao, Y. Y. Zheng and Z. Y. Fu, Carbon, 2012, 50, 2620–2627 CrossRef CAS
- B. Cao, G. M. Veith, R. E. Diaz, J. Liu, E. A. Stach, R. R. Adzic and P. G. Khalifah, Angew. Chem., Int. Ed. Engl., 2013, 52, 10753–10757 CrossRef CAS PubMed
- H. T. Chung, G. Wu, Q. Li and P. Zelenay, Int. J. Hydrogen Energy, 2014, 39, 15887–15893 CrossRef CAS
- J. Sanetuntikul, C. Chuaicham, Y.-W. Choi and S. Shanmugam, J. Mater. Chem. A, 2015, 3, 15473–15481 CAS
- M. Li, X. Bo, Y. Zhang, C. Han, A. Nsabimana and L. Guo, J. Mater. Chem. A, 2014, 2, 11672–11682 CAS
- C. He, J. J. Zhang and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3231–3236 CAS
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CAS
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201–1204 CrossRef CAS PubMed
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed
- K. Wan, Z.-P. Yu, X.-h. Li, M.-y. Liu, G. Yang, J.-h. Piao and Z.-x. Liang, ACS Catal., 2015, 5, 4325–4332 CrossRef CAS
- C. H. Choi, H.-K. Lim, M. W. Chung, J. C. Park, H. Shin, H. Kim and S. I. Woo, J. Am. Chem. Soc., 2014, 136, 9070–9077 CrossRef CAS PubMed
- Z. Chen, D. Higgins and Z. Chen, Carbon, 2010, 48, 3057–3065 CrossRef CAS
- K. Kakaei, Fuel Cells, 2012, 12, 939–945 CrossRef CAS
- C. Jeyabharathi, P. Venkateshkumar, M. S. Rao, J. Mathiyarasu and K. L. N. Phani, Electrochim. Acta, 2012, 74, 171–175 CrossRef CAS
- Q. Lin, X. Bu, A. Kong, C. Mao, F. Bu and P. Feng, Adv. Mater., 2015, 27, 3431–3436 CrossRef CAS PubMed
- M. Y. Song, H. Y. Park, D.-S. Yang and J.-S. Yu, ChemSusChem, 2014, 7, 1755–1763 CrossRef CAS PubMed
- L. M. Roen, C. H. Paik and T. D. Jarvi, Electrochem. Solid-State Lett., 2004, 7, A19–A22 CrossRef CAS
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS
- J.-H. Park, S.-D. Yim, T. Kim, S.-H. Park, Y.-G. Yoon, G.-G. Park, T.-H. Yang and E.-D. Park, Electrochim. Acta, 2012, 83, 294–304 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta01543c |
| This journal is © The Royal Society of Chemistry 2016 |