
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perfluoroalkyl-substituted conjugated polymers as electron acceptors for all-polymer solar cells: the effect of diiodoperfluoroalkane additives†
Andong
Zhang
a,
Qiang
Wang
b,
Ralf A. A.
Bovee
b,
Cheng
Li
*a,
Jianqi
Zhang
*c,
Yi
Zhou
d,
Zhixiang
Wei
c,
Yongfang
Li
ad,
René A. J.
Janssen
b,
Zhaohui
Wang
*a and
Weiwei
Li
*a
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 10090, China. E-mail: licheng1987@iccas.ac.cn; wangzhaohui@iccas.ac.cn; liweiwei@iccas.ac.cn
bMolecular Materials and Nanosystems & Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cNational Center for Nanoscience and Technology, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: zhangjq@nanoctr.cn
dLaboratory of Advanced Optoelectronic Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, China
First published on 14th April 2016
Abstract
A series of six diketopyrrolopyrrole (DPP) based conjugated polymers with a varying content of solubilizing perfluoroalkyl chains were synthesized. Based on a systematic investigation of the influence of the solvent on the photovoltaic performance, it is found that 1,6-diiodoperfluorohexane (IC6F12I) is an effective solvent additive to enhance the power conversion efficiency (PCE) of DPP polymers with perfluoroalkyl side chains. The polymers consist of thiazole-flanked DPP units that alternate along the main chain with varying ratios of thiophene (T) and perfluoroalkyl benzodithiophene (FBDT) units. The polymers possess high molecular weights, narrow band gaps and good crystalline properties. The DPP polymers were used as electron acceptors in bulk heterojunction solar cells with another DPP polymer as the electron donor. A solvent mixture of CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-chloronaphthalene (1-CN) is found to provide the best PCE of 2.9% in non-fluorine based DPP polymer solar cells, but yields a low PCE of 0.52% for perfluoroalkyl-containing polymer solar cells. Perfluoroalkyl-containing polymer solar cells fabricated from CHCl3 with IC6F12I as the processing additive show a significantly improved PCE of 2.1%. The morphology analysis of the blend films reveals that IC6F12I as an additive improves the micro-phase separation between the polymer donor and acceptor, which results in enhanced charge generation.
:1-chloronaphthalene (1-CN) is found to provide the best PCE of 2.9% in non-fluorine based DPP polymer solar cells, but yields a low PCE of 0.52% for perfluoroalkyl-containing polymer solar cells. Perfluoroalkyl-containing polymer solar cells fabricated from CHCl3 with IC6F12I as the processing additive show a significantly improved PCE of 2.1%. The morphology analysis of the blend films reveals that IC6F12I as an additive improves the micro-phase separation between the polymer donor and acceptor, which results in enhanced charge generation.
Introduction
There has been increasing interest in non-fullerene conjugated materials to replace fullerene derivatives as electron acceptors in organic solar cells in recent years.1–3 A significant number of conjugated small molecules4–20 and polymers21–35 with excellent electron transport properties and aligned energy levels have been designed and synthesized, which are well-suited as electron acceptors in organic photovoltaic devices. Currently, perylenediimide and naphthalenediimide based materials are considered as the most promising non-fullerene acceptors,36,37 since these materials have shown high electron mobilities of about 1 cm2 V−1 s−1 (ref. 38) and deep lowest unoccupied molecular orbital (LUMO) levels which are similar to those of fullerene derivatives. As a consequence, perylenediimide- and naphthalenediimide-based small molecules and conjugated polymers as electron acceptors in organic solar cells show impressively high power conversion efficiencies (PCEs) above 7%,4–6,39 approaching the performance of fullerene-based solar cells with PCEs over 10%.40,41In terms of the large possible variation in materials, conjugated polymers possess an advantage compared to fullerene derivatives, such as [6,6]-phenyl-C61-butyric acid methyl ester (PCBM), for which variations in the optical band gap and LUMO energies are more restricted. In recent years, conjugated polymers have been widely exploited as electron donors in polymer solar cells (PSCs),42,43 and design principles from these studies can also be utilized for the development of new acceptor polymers. In particular, when introducing electron-rich and electron-deficient moieties into a conjugated backbone, the so-called donor–acceptor polymers exhibit distinct variations in frontier orbital energy levels, absorption spectra, and charge-carrier mobilities, which allow the conjugated polymers to be used as electron donors or acceptors in solar cell devices. For instance, the diketopyrrolopyrrole (DPP) unit is widely used as an electron-deficient building block to construct narrow band gap polymers with near-infrared absorption.44,45 DPP polymers also exhibit high hole mobilities, exceeding 10 cm2 V−1 s−1 in favourable cases,46 and impressive crystallinity,47,48 such that PSCs based on DPP polymers reach PCEs above 8%.49–51 Meanwhile, organic field-effect transistors (FETs) based on DPP polymers also present high electron mobilities above 5 cm2 V−1 s−1,46,52 which are higher than those of fullerene derivatives,53 indicating their potential for use as electron acceptors in PSCs. One recent example is the design of a polymer in which the DPP is flanked by two thiazole rings to effectively lower the LUMO level.34 The resulting DPP polymer can act as an electron acceptor in combination with another DPP polymer as the electron donor in PSCs to reach a PCE of 2.9%. At present this represents the highest PCE when using DPP polymers as electron acceptors. It will be of importance to further explore new DPP polymer acceptors for efficient photovoltaic devices.
Recently, we designed and synthesized a DPP polymer acceptor PDPP2TzFBDT (Scheme 1) that has similar frontier energy levels to those of PCBM and can be potentially used as a universal acceptor for PSCs.54 However, our initial attempt to use PDPP2TzFBDT as an electron acceptor with PDPP5T34 as an electron donor was not successful and resulted in very poor performance with PCEs of 0.19%. PDPP2TzFBDT has long perfluoroalkyl chains that are partially responsible for the deep LUMO levels, but the lipophobic perfluoroalkyl chains also cause poor miscibility with PDPP5T, resulting in large phase separation in blends of these two polymers, poor charge generation, and consequently low PCEs. Similar behaviour was also reported in donor polymer:fullerene systems, in which the donor polymers that bear long perfluoroalkyl chains showed large phase-separated domains and hence provide poor PCEs in PSCs.55,56 Therefore, it is important to find a way to improve the micro-phase separation in these blend films in order to apply perfluoroalkyl-based conjugated polymers in organic photovoltaic devices.
 | ||
| Scheme 1 Chemical structures of the DPP polymers and their synthetic routes. (i) Stille polymerization by using Pd2(dba)3/PPh3 in toluene/DMF (10:1, v/v) at 115 °C. Note: m:n was determined from the feed ratio of the monomers 2 and 3. | ||
In this work, we explore the use of diiodoperfluoroalkanes as additives in solution-processed PSCs based on perfluoroalkyl DPP acceptor polymers. Based on the similarity principle, we assume that polymers with perfluoroalkyl units have better solubility in perfluoroalkanes as additives, which would prevent the fast precipitation and therefore reduce the domain size of the polymers. A similar function of o-dichlorobenzene (o-DCB) as an additive in DPP polymer solar cells was also observed.57 We synthesized several DPP acceptor polymers, in which the thiazole-flanked DPP segment was linked with thiophene (T) and perfluoroalkyl benzodithiophene (FBDT) units in different ratios (Scheme 1). The polymers were found to have small optical band gaps (Eg) and good crystalline properties, depending on the ratio of T and FBDT units. The resulting polymers were used as electron acceptors in polymer–polymer solar cells with PDPP5T as the electron donor, which were solution-processed from chloroform (CHCl3) with 1-chloronaphthalene (1-CN) or -diiodoperfluoroalkanes as processing additives. The solar cells based on PDPP5T:PDPP2TzFBDT fabricated from CHCl3:IC6F12I exhibited a much improved PCE of 2.1% compared to cells from CHCl3 or CHCl3:1-CN due to their better micro-phase separation. The results reveal that diiodoperfluoroalkanes can be used to effectively tune the morphology of perfluoroalkyl-based polymer solar cells. This encourages the design of perfluoroalkyl-based conjugated polymers for efficient solar devices.
Experimental
Materials and measurements
All synthetic procedures were performed under an argon atmosphere. Commercial chemicals were used as received.THF and toluene were distilled from sodium under a nitrogen atmosphere. The monomers 3,6-bis(5-bromothiazol-2-yl)-2,5-bis(2-octyldodecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1)54 and (4,8-bis(5-perfluorohexylthiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) (3),54 and the polymers PDPP2TzT,35 PDPP2TzFBDT54 and PDPP5T58 were synthesized according to the literature procedures. 2,5-Bis(trimethylstannyl)thiophene (2) was purchased from Aldrich Chemical Co. and recrystallized from methanol before use in polymerization reactions.
1H-NMR spectra of the polymers were recorded on a Bruker AVIII 500WB NMR Spectrometer at 100 °C with 1,1,2,2-tetrachloroethane-d2 as the solvent and tetramethylsilane (TMS) as the internal standard. The molecular weight was determined by gel permeation chromatography (GPC) at 140 °C on a PL-GPC 220 system (Agilent Technologies with a Knauer PDA detector) using a PLgel 10 μm MIXED-B LS column and o-DCB as the eluent against polystyrene standards. A low concentration of 0.1 mg mL−1 polymer in o-DCB was applied to reduce aggregation. Optical absorption spectra were recorded on a JASCO V-570 spectrometer with a slit width of 2.0 nm and a scan speed of 1000 nm min−1. Cyclic voltammetry (CV) was performed under an inert atmosphere at a scan rate of 0.1 V s−1 and 1 M tetrabutylammonium hexafluorophosphate in acetonitrile as the electrolyte. An ITO glass slide covered with a thin layer polymer (approx. 20 nm) was used as the working electrode. The counter and reference electrodes were a Pt wire and Ag/AgCl, respectively. Atomic force microscopy (AFM) images were recorded using a Digital Instruments Nanoscope IIIa multimode atomic force microscope in the tapping mode under ambient conditions. The tips were purchased from Bruker (Model: SCANASYST-AIR with one cantilever, T = 650 nm, L = 115 μm, W = 25 μm and spring constant of 0.4 N m−1). 2D grazing-incidence wide angle X-ray scattering (2D-GIWAXS) measurements were performed by using a Xenocs WAXS/SAXS system, with an X-ray wavelength of 1.5418 Å. The incident angle was 0.2°. The sample-to-detector distance was 127.5 mm. The scattered X-rays were detected by using a Dectris Pilatus 100k counting detector. The counting time was 4 h. All film samples were prepared by spin-coating solutions on Si/SiO2 substrates. Steady state fluorescence spectra were recorded at room temperature using an Edinburgh Instruments FLS980 double-monochromator luminescence spectrometer equipped with a nitrogen-cooled near-IR sensitive photomultiplier (Hamamatsu).
Photovoltaic devices with an inverted configuration were made by spin coating a ZnO sol–gel at 4000 rpm for 60 s onto pre-cleaned, patterned ITO substrates. The photoactive layer was deposited by spin coating a chloroform solution containing PDPP5T and thiazole-based DPP polymers and an appropriate amount of processing additives such as 1-CN, 1,4-diiodoperfluorobutane (IC4F8I), 1,6-diiodoperfluorohexane (IC6F12I) or 1,8-diiodoperfluorooctane (IC8F16I) in air. MoO3 (10 nm) and Ag (100 nm) were deposited by vacuum evaporation at ca. 4 × 10−5 Pa as the back electrode.
The active area of the cells was 0.04 cm2. The J–V characteristics were measured by using a Keithley 2400 source meter unit under AM1.5G spectrum from a solar simulator (Enlitech model SS-F5-3A). The illumination intensity was determined at 100 mW cm−2 using a monocrystalline silicon reference cell with a KG5 filter. The short-circuit current density (Jsc) under AM1.5G conditions was estimated from the spectral response and convolution with the solar spectrum. The external quantum efficiency was measured by using a Solar Cell Spectral Response Measurement System QE-R3011 (Enli Technology Co., Ltd.). The thickness of the active layers in the photovoltaic devices was measured on a Veeco Dektak XT profilometer.
PDPP2Tz10FBDT
To a degassed solution of monomer 1 (83.66 mg, 0.082 mmol), monomer 2 (30.21 mg, 0.074 mmol) and monomer 3 (10.78 mg, 0.008 mmol) in toluene (2 mL) and DMF (0.2 mL), tris(dibenzylidene-acetone)dipalladium(0) (2.25 mg, 2.5 μmol) and triphenylphosphine (2.58 mg, 9.8 μmol) were added. The mixture was stirred at 115 °C for 24 h, after which the reaction mixture was precipitated in methanol and filtered through a Soxhlet thimble. The polymer was extracted with acetone, hexane and chloroform. The chloroform fraction was reduced and the polymer was precipitated in acetone. The polymer was collected by filtering over a 0.45 μm PTFE membrane filter and dried in a vacuum oven to yield PDPP2Tz10FBDT (80 mg, 94%) as a dark solid. GPC (o-DCB, 140 °C): Mn = 77.2 kg mol−1, PDI = 2.05.PDPP2Tz30FBDT
The same procedure as for PDPP2Tz10FBDT was used, but now 1 (49.68 mg, 0.049 mmol), 2 (13.95 mg, 0.034 mmol) and 3 (19.21 mg, 0.015 mmol) were used as the monomers. Yield: 55 mg (93%). GPC (o-DCB, 140 °C): Mn = 65.1 kg mol−1, PDI = 2.05.PDPP2Tz50FBDT
The same procedure as for PDPP2Tz10FBDT was used, but now 1 (42.12 mg, 0.041 mmol), 2 (8.45 mg, 0.021 mmol) and 3 (27.14 mg, 0.021 mmol) were used as the monomers. Yield: 53 mg (93%). GPC (o-DCB, 140 °C): Mn = 66.0 kg mol−1, PDI = 2.72.PDPP2Tz70FBDT
The same procedure as for PDPP2Tz10FBDT was used, but now 1 (53.60 mg, 0.052 mmol), 2 (6.46 mg, 0.016 mmol) and 3 (48.36 mg, 0.037 mmol) were used as the monomers. Yield: 71 mg (90%). GPC (o-DCB, 140 °C): Mn = 61.9 kg mol−1, PDI = 2.38.Results and discussion
Synthesis and characterization
Perfluoroalkyl-DPP based polymers were prepared by Stille polymerization (Scheme 1). The synthesis of two derivatives, PDPP2TzT35 and PDPP2TzFBDT,54 has been described previously. The copolymers PDPP2Tz10FBDT, PDPP2Tz30FBDT, PDPP2Tz50FBDT, and PDPP2Tz70FBDT were synthesized by copolymerizing the dibromo-DPP monomer 1 with the bisstannyl monomers of T (2) and FBDT (3), in which the ratio of the co-monomers 2 and 3 was adjusted to prepare polymers with different contents of perfluoroalkyl units. Stille polymerizations were performed under identical conditions using Pd2(dba)3/PPh3 as the catalyst system and toluene/DMF as the solvent at 115 °C.The polymers show different 1H-NMR spectra (ESI, Fig. S1†), but it is difficult to determine the ratio of the polymer segments. Therefore, the m:n ratio is denoted according to the feed ratio of the co-monomers 2 and 3 (Scheme 1) and does not necessarily represent the actual ratio of T and FBDT units in the polymer backbone. The molecular weight of these polymers has been determined by GPC using o-DCB as the eluent. As shown in Table 1 and Fig. S2 (ESI†), most polymers possess a similarly high molecular weight between 60 and 80 kg mol−1. The solubility of PDPP2TzFBDT is very poor in o-DCB such that only a soluble small molecular weight fraction can be measured by GPC. The similar molecular weight of these DPP copolymers is also beneficial for investigation of solvent influence on photovoltaic devices.
| Polymer | M n (kg mol−1) | PDIb | Solution | Film | E LUMO (eV) | E HOMO (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ peak (nm) | λ onset (nm) | E g (eV) | λ peak (nm) | λ onset (nm) | E g (eV) | |||||
| a Determined with GPC at 140 °C using o-DCB as the eluent. b PDI is the polydispersity index. c E LUMO = −5.23 − Ered. d E HOMO = ELUMO − Eg. e The molecular weight was determined by using a low molecular weight fraction that dissolved in o-DCB at 140 °C. | ||||||||||
| PDPP2TzT | 74.0 | 2.80 | 691, 757 | 805 | 1.54 | 710, 774 | 861 | 1.44 | −4.03 | −5.47 |
| PDPP2Tz10FBDT | 77.2 | 2.05 | 691, 753 | 800 | 1.55 | 701, 760 | 848 | 1.46 | −4.39 | −5.85 |
| PDPP2Tz30FBDT | 65.1 | 2.05 | 689, 745 | 799 | 1.55 | 688, 744 | 823 | 1.51 | −4.27 | −5.78 |
| PDPP2Tz50FBDT | 66.0 | 2.72 | 689, 734 | 799 | 1.55 | 683, 728 | 831 | 1.49 | −4.30 | −5.79 |
| PDPP2Tz70FBDT | 61.9 | 2.38 | 680, 729 | 806 | 1.54 | 679, 731 | 822 | 1.51 | −4.24 | −5.75 |
| PDPP2TzFBDTe | 13.1 | 1.73 | 672, 712 | 772 | 1.61 | 672, 719 | 787 | 1.58 | −4.56 | −6.14 |
Optical and electrochemical properties
The optical absorption spectra of the DPP polymers in CHCl3 solution and in solid state thin films are shown in Fig. 1 and the parameters are summarized in Table 1. All polymers exhibit near-infrared absorption with absorption onsets ranging from 772 to 805 nm in CHCl3 solution and red-shifted absorptions in thin films with onsets between 787 and 861 nm. PDPP2TzT in which the thiazole-flanked DPP unit is alternating with T as the aromatic units has the lowest Eg of 1.44 eV, while PDPP2TzFBDT where the thiazole-flanked DPP unit is alternating with FBDT units shows the largest Eg of 1.58 eV among these polymers. When using a ratio of T and FBDT as 9:1 in PDPP2Tz10FBDT, the Eg slightly increases to 1.46 eV. Interestingly, as the ratio of T and FBDT is further changed to 7:3, 5:5, and 3:7, the polymers PDPP2Tz30FBDT, PDPP2Tz50FBDT, and PDPP2Tz70FBDT have very similar Eg of 1.49 to 1.51 eV in thin films, which are red-shifted compared to that of the copolymer PDPP2TzFBDT. The absorption spectra of the polymers in CHCl3 solution with a concentration of 0.001 g L−1 are shown in Fig. S3 (ESI†). Interestingly, when increasing the content of FBDT units, the intensity of the absorption at 700 nm decreases, indicating a lower absorption coefficient for FBDT-based polymers.
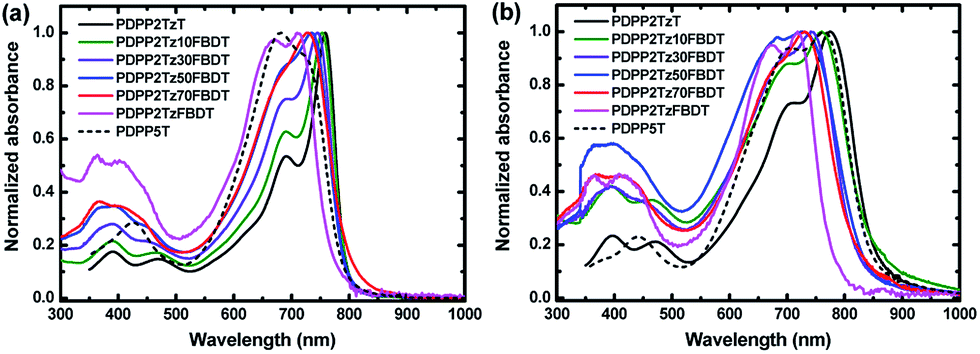 | ||
| Fig. 1 Optical absorption spectra of the DPP polymers (a) in CHCl3 solution and (b) in solid state thin films. | ||
The electrochemical properties of the DPP polymers were determined by CV measurements (ESI†, Fig. S4 and Table 1). The highest occupied molecular orbital (HOMO) level of PDPP2TzT is at −5.47 eV and the LUMO level is at −4.03 eV. When the T unit is replaced by the FBDT unit, the resulting PDPP2TzFBDT polymer exhibits deeper HOMO and LUMO levels at −6.14 and −4.56 eV, respectively. The copolymers with T and FBDT show HOMO levels around −5.75 and −5.85 eV and LUMO levels around −4.24 and −4.39 eV. The results illustrate that FBDT units can effectively lower the energy levels of electron acceptors. It is also noted that the LUMO levels were not linearly reduced with increasing FBDT content, which is possibly from the different aggregation of the polymers in o-DCB and the deviation when exacting reduction potential from CV curves in Fig. S4, ESI.†
Crystalline properties
In order to investigate the crystalline properties and molecular packing in solid state films, 2D-GIWAXS59 was applied on thin films spin coated from CHCl3 on Si/SiO2 substrates and the results are shown in Fig. 2 and Table 2. The q values are directly extracted from the peaks of GIWAXS line cuts. PDPP2TzT shows a distinct (100) diffraction peak in the in-plane direction, which correlates with a lamellar packing distance of 22.4 Å of the 2-octyldodecyl side chains. The (010) diffraction peak in the out-of-plane direction for PDPP2TzT is related to the π–π stacking distance of 3.67 Å of the conjugated backbone. These results reveal that PDPP2TzT is a semi-crystalline polymer with a distinct “face on” orientation of the polymer chains, which is beneficial for charge transport in the vertical direction in bulk heterojunction solar cells.40 The polymers incorporating both T and FBDT into the main chain also exhibit the (100) and (010) diffraction peaks indicating a “face on” orientation for the 9:1 and 7:3 ratios, but the intensity of the diffractions is strongly reduced for the 5:5 and 3:7 co-monomer ratios. This is not unexpected because the introduction of the differently sized T and FBDT units in a random fashion will destroy the translational symmetry along the chain, which precludes obtaining highly ordered polymer domains. For PDPP2TzFBDT the diffraction peaks in the 2D-GIWAXS are restored, demonstrating that it is not the FBDT unit itself that causes the reduced crystallinity of the DPP polymers that were made with both co-monomers. PDPP2TzFBDT exhibits both (100) and (010) diffractions peaks in the out-of-plane direction, while in the in-plane direction the diffraction peaks are much less pronounced. We tentatively attribute this behaviour to the absence of a clear preference for the “face-on” or “edge-on” orientation of the PDPP2TzFBDT polymer chains on the surface. Apparently both types of domains are present, but the orientation of the crystallites is random.
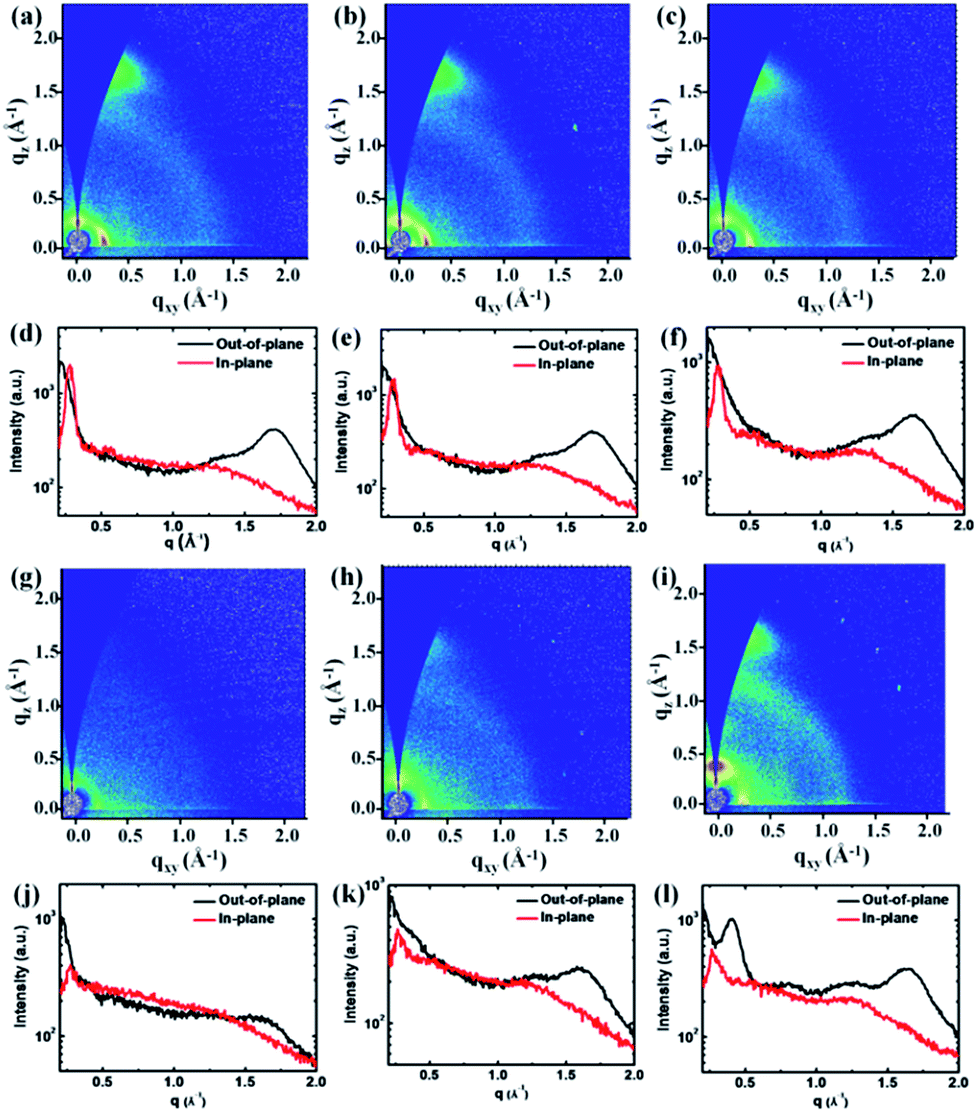 | ||
| Fig. 2 (a)–(c) and (g)–(i) 2D-GIWAXS patterns of the polymer thin films spin coated from CHCl3. (d)–(f) and (j)–(l) The out-of-plane and in-plane cuts of the corresponding 2D-GIWAXS patterns. (a) and (d) PDPP2TzT; (b) and (e) PDPP2Tz10FBDT; (c) and (f) PDPP2Tz30FBDT; (g) and (j) PDPP2Tz50FBDT; (h) and (k) PDPP2Tz70FBDT; (i) and (l) PDPP2TzFBDT. | ||
| Polymer | Lamellar spacing | π–π spacing | ||
|---|---|---|---|---|
| q (Å−1) | d (Å) | q (Å−1) | d (Å) | |
| a Additional diffraction peak was present: qz = 0.41 Å−1 and d = 15.3 Å. | ||||
| PDPP2TzT | 0.28 | 22.4 | 1.71 | 3.67 |
| PDPP2Tz10FBDT | 0.28 | 22.4 | 1.70 | 3.70 |
| PDPP2Tz30FBDT | 0.28 | 22.4 | 1.64 | 3.83 |
| PDPP2Tz50FBDT | 0.27 | 23.3 | 1.63 | 3.85 |
| PDPP2Tz70FBDT | 0.27 | 23.3 | 1.61 | 3.90 |
| PDPP2TzFBDTa | 0.27 | 23.3 | 1.64 | 3.83 |
| PDPP5T | 0.33 | 19.0 | 1.64 | 3.83 |
All thiazole-based DPP polymers presented here exhibit a similar d-spacing of about 23.0 Å for the (100) diffraction peak, originating from the lamellar packing distance induced by the 2-octyldodecyl side chains. The π–π stacking distances gradually increased from 3.67 Å to 3.90 Å for the polymers PDPP2TzT to PDPP2Tz70FBDT due to the increasing number of sterically demanding 5-perfluorohexylthiophene-2-yl substituents, but then decreased to 3.83 Å for PDPP2TzFBDT possibly because of a more regular polymer structure (Table 2). A closer π–π stacking distance provides an improved wave function overlap between neighbouring chains which is beneficial for charge transport. It is also interesting to note that PDPP2TzFBDT shows a new diffraction peak at qz = 0.41 Å−1 with a distance of d = 15.3 Å, which is possibly induced by the lamellar stacking of perfluoroalkyl units (ESI, Fig. S5†).
Finally, as a comparison and because it is used as an electron donor, PDPP5T with five flanking thiophene units shows a (100) diffraction peak in the in-plane direction with d = 19.04 Å and an (010) peak in the out-of-plane direction with d = 3.83 Å, indicating a “face on” orientation (ESI, Fig. S6†).
In summary, the 2D-GIWAXS data reveal that PDPP2TzT and PDPP2TzFBDT are more crystalline than their T/FBDT mixed co-polymers and that the absence of the 5-perfluorohexylthiophene-2-yl substituents in PDPP2TzT enables a closer π–π stacking (3.67 Å) than in PDPP2TzFBDT (3.83 Å), which can enhance charge transport for the former.
Polymer–polymer solar cell performance
The thiazole-flanked DPP polymers were applied as electron acceptors using PDPP5T as the electron donor in polymer–polymer photovoltaic devices with an inverted polarity configuration, in which ITO/ZnO and MoO3/Ag were used as electron and hole extracting contacts, respectively. PDPP5T has a similar absorption spectrum to that of the DPP acceptor polymers (Fig. 1).When the photoactive layers were spin coated from CHCl3 without processing additives low PCEs of 0.11–0.39% (ESI, Table S1†) were obtained. The low performance is attributed to the large micro-phase separation between the donor and acceptor polymers as inferred from the corresponding AFM images (ESI, Fig. S7†). This behaviour is similar to that of PDPP5T:PCBM solar cells when solution processed from CHCl3 solution.57
To improve the blend morphology, the photoactive layers of the PDPP5T:DPP-polymer (1:1 w/w) blends were spin coated from CHCl3 solutions using 1-CN, IC4F8I or IC6F12I as high boiling point solvent additives.
When the active layer was spin coated from CHCl3 with 3% 1-CN as the additive, PDPP5T:PDPP2TzT cells resulted in a PCE of 2.8% with a Jsc of 7.1 mA cm−2, an open-circuit voltage (Voc) of 0.81 V, and a fill factor (FF) of 0.49. Using the same solvent mixture, the solar cells based on PDPP5T with PDPP2Tz10FBDT, PDPP2Tz30FBDT, PDPP2Tz50FBDT, and PDPP2Tz70FBDT as electron acceptors showed gradually decreasing PCEs from 2.3% down to 1.5%, 0.52%, and 0.26%. The lower PCEs were mainly caused by a reduction of Jsc from 5.7 to 0.82 mA cm−2. PDPP5T:PDPP2TzFBDT cells had a slightly increased PCE of 0.43% when spin coated from CHCl3:1-CN solution, but is still much lower than that of PDPP5T:PDPP2TzT cells (Table 3).
:DPP-polymer. The hole and electron mobilities from SCLC measurements are also included
| Acceptor | Solvent | J sc (mA cm−2) | V oc (V) | FF | PCEa (%) | PCEbest (%) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) | μ h/μe |
|---|---|---|---|---|---|---|---|---|---|
|
a
J
sc was calculated by integrating the EQE spectrum with the AM1.5G spectrum.
b The poor wettability of the mixed solution on the surface of ITO/ZnO prevents forming continuous thin films for solar devices. The weight ratio of PDPP5T to the acceptor DPP-polymers is 1:1. The thickness of the active layers is around 75 nm.
|
|||||||||
| PDPP2TzT | CHCl3:1-CN 3% |
7.1 ± 0.3 | 0.81 ± 0.00 | 0.49 ± 0.03 | 2.8 ± 0.04 | 2.9 | 2.0 × 10−4 | 1.3 × 10−4 | 2 |
| CHCl3:IC6F12I 10%b |
— | — | — | — | — | — | — | — | |
| PDPP2Tz10FBDT | CHCl3:1-CN 3% |
5.7 ± 0.5 | 0.79 ± 0.08 | 0.49 ± 0.02 | 2.3 ± 0.4 | 2.4 | 1.7 × 10−4 | 6.7 × 10−5 | 3 |
| CHCl3:IC6F12I 10%b |
— | — | — | — | — | — | — | — | |
| PDPP2Tz30FBDT | CHCl3:1-CN 3% |
4.1 ± 0.4 | 0.75 ± 0.01 | 0.46 ± 0.02 | 1.5 ± 0.2 | 1.7 | 9.7 × 10−5 | 1.6 × 10−5 | 6 |
| CHCl3:IC6F12I 10% |
1.3 ± 0.1 | 0.78 ± 0.01 | 0.37 ± 0.01 | 0.38 ± 0.04 | 0.42 | 7.0 × 10−5 | 4.0 × 10−5 | 2 | |
| PDPP2Tz50FBDT | CHCl3:1-CN 3% |
1.6 ± 0.2 | 0.75 ± 0.01 | 0.43 ± 0.03 | 0.52 ± 0.08 | 0.68 | 1.9 × 10−4 | 5.6 × 10−6 | 33 |
| CHCl3:IC6F12I 10% |
1.9 ± 0.3 | 0.75 ± 0.01 | 0.36 ± 0.09 | 0.50 ± 0.08 | 0.56 | 4.4 × 10−5 | 1.1 × 10−5 | 4 | |
| PDPP2Tz70FBDT | CHCl3:1-CN 3% |
0.8 ± 0.1 | 0.78 ± 0.02 | 0.40 ± 0.04 | 0.26 ± 0.04 | 0.32 | 2.0 × 10−4 | 1.6 × 10−6 | 125 |
| CHCl3:IC6F12I 10% |
1.8 ± 0.1 | 0.76 ± 0.05 | 0.42 ± 0.03 | 0.48 ± 0.04 | 0.54 | 1.5 × 10−4 | 2.4 × 10−6 | 65 | |
| PDPP2TzFBDT | CHCl3:1-CN 3% |
1.3 ± 0.2 | 0.68 ± 0.03 | 0.43 ± 0.03 | 0.43 ± 0.09 | 0.52 | 4.9 × 10−4 | 1.9 × 10−6 | 265 |
| CHCl3:IC6F12I 10% |
7.4 ± 0.3 | 0.67 ± 0.06 | 0.41 ± 0.02 | 2.03 ± 0.02 | 2.1 | 9.2 × 10−5 | 3.7 × 10−6 | 25 | |
We then tested diiodoperfluoroalkanes as processing additives. For PDPP5T:PDPP2TzFBDT cells we used IC4F8I, IC6F12I and IC8F12I and found that both IC4F8I and IC6F12I significantly increase the PCE. With IC8F16I as the additive, the solar cells showed very poor J–V characteristics with large leakage current. This may due to the high melting point (∼75 °C) of IC8F16I, producing holes in the active layers when it was removed via a high vacuum process. Above 5%, the volume ratio of IC6F12I in CHCl3 has little influence on the device performance of PDPP5T:PDPP2TzFBDT cells (ESI, Table S3†). After optimization, the PCE of PDPP5T:PDPP2TzFBDT cells spin coated from CHCl3:IC6F12I was found to be 2.0%, with Jsc = 7.4 mA cm−2, Voc = 0.67 and FF = 0.41 at a film thickness of 75 nm (Fig. 3a). The external quantum efficiency (EQE) of the optimized PDPP5T:PDPP2TzFBDT cells shows a broad photoresponse from 300 to 850 nm with a maximum EQE over 0.3 in the near-infrared spectral region where the polymer absorbs light (Fig. 3b). PDPP5T:PDPP2TzFBDT cells fabricated from CHCl3 with 20% IC4F8I also provided PCEs up to 1.8% (ESI, Table S3†).
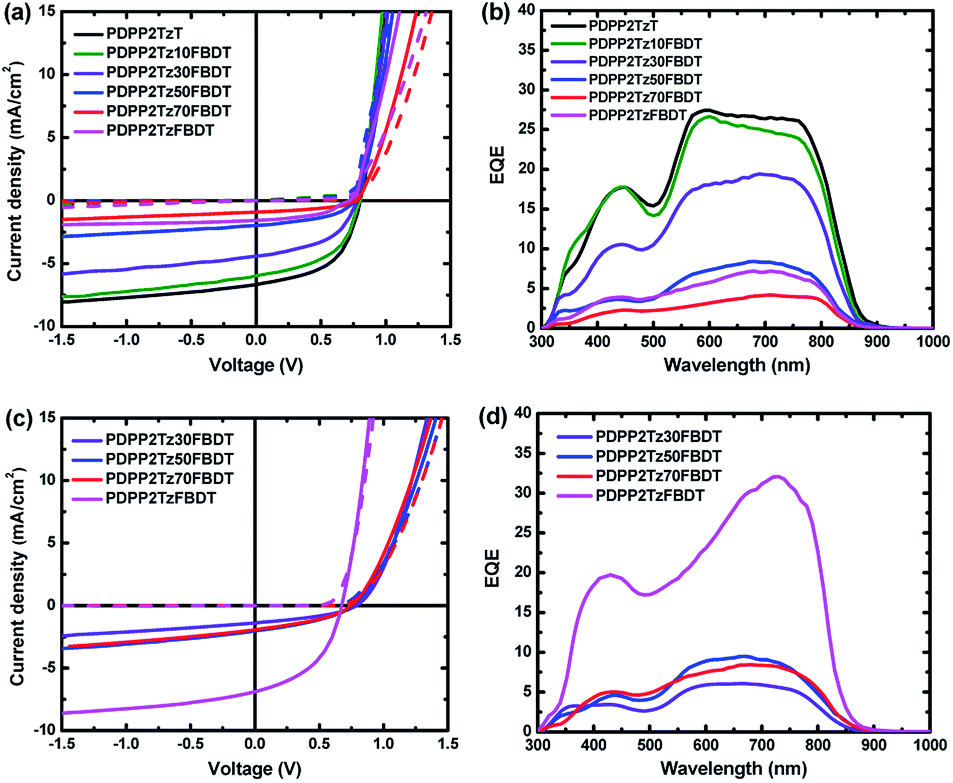 | ||
| Fig. 3 (a) and (c) J–V characteristics in the dark (dashed lines) and under white light illumination (solid lines). (b) and (d) EQE of the optimized PDPP5T:DPP-polymer solar cells. (a) and (b) The active layers fabricated from CHCl3 solution with 3% 1-CN. (c) and (d) The active layers fabricated from CHCl3 solution with 10% IC6F12I. | ||
We then optimized the other PDPP5T:DPP-polymer solar cells using CHCl3:IC6F12I (9:1) as the solvent mixture (Fig. 3 and Table 3). We failed to fabricate solar cells for PDPP2TzT and PDPP2Tz10FBDT with PDPP5T using CHCl3:IC6F12I as solvent due to the poor wettability of the solutions on the ITO/ZnO surface, but for the polymers with an increased number of FBDT units, working devices were obtained. The current density–voltage (J–V) characteristics and EQE of the optimized solar cells are shown in Fig. 3 and Table 3. PDPP5T:PDPP2Tz30FBDT cells fabricated from CHCl3:IC6F12I provided a low PCE of 0.38% with Jsc = 1.3 mA cm−2 compared to the PCE of 1.5% for the same blend processed from CHCl3:1-CN. For the PDPP2Tz50FBDT and PDPP2Tz70FBDT based cells with a higher content of FBDT units, the PCEs obtained with IC6F12I as the co-solvent were similar to the PCEs obtained with 1-CN.
The effect of perfluoroalkyl-based additives on the device performance was further investigated by using different donor polymers, PTB7-Th60 and PDPP2T-DTP61 (ESI, Fig. S8†). Solar cells based on PTB7-Th or PDPP2T-DTP as the donor and PDPP2TzFBDT as the acceptor show high performance with PCEs of 2.5% and 1.0% when fabricated from CHCl3 with IC6F12I as additives. In comparison, the PCEs of the cells processed from CHCl3 with 1-CN are 2.1% and 0.6% (ESI, Table S4 and Fig. S9†). The results confirm that IC6F12I as the additive can enhance the photovoltaic performance based on acceptor polymers bearing perfluoroalkyl side units such as PDPP2TzFBDT. However, all cells show relatively low absolute PCE compared to other high efficiency non-fullerene solar cells, indicating a suboptimal morphology.
Charge transport in the blends
To study the influence of different additives on the charge transport in the blend films, we determined hole and electron mobilities from space charge limited current (SCLC) measurements using a device configuration consisting of ITO/MoO3/active layer/Au for hole-only devices and ITO/ZnO/active layer/LiF/Al for electron-only devices. PDPP5T:PDPP2TzT layers fabricated from CHCl3:1-CN have well balanced hole and electron mobilities around 10−4 cm2 V−1 s−1 (Table 3). With the same solvent mixture, the hole mobilities of other layers were similar to those of the PDPP2TzT cell, but the electron mobilities gradually decreased to 10−6 cm2 V−1 s−1 with increasing content of FBDT in acceptors. The unbalanced hole and electron mobilities could explain the low PCEs in FBDT-based polymer solar cells from CHCl3:1-CN. When using IC6F12I as the additive, the electron mobilities were slightly improved compared to those from CHCl3:1-CN, but the hole mobilities were reduced (Table 3). As a consequence, more balanced hole and electron mobilities can be achieved. As an example the μh/μe ratio of the PDPP5T:PDPP2TzFBDT blend decreases from 265 to 25 when replacing 1-CN by IC6F12I. The more balanced charge transport is accompanied by an enhancement of the PCE from 0.52% to 2.1%.
Morphology investigation
Because the efficiency of solar cells is intimately related to the morphology, the photoactive layers were further investigated by AFM, 2D-GIWAXS and photoluminescence (PL). When the films were fabricated from CHCl3 without additives, large domains were observed in AFM images (ESI, Fig. S7†). These disappeared in thin films processed from CHCl3 with additives (Fig. 4). Fig. 4 shows that for PDPP5T:PDPP2Tz and PDPP5T:PDPP2Tz10FBDT blends processed from CHCl3:1-CN the lateral dimensions and the height of the surface corrugation are smaller (Fig. 4a and b) than for the blends with a higher FBDT content (Fig. 4c–f), suggesting that the latter blends have a coarser micro-morphology with larger domains.
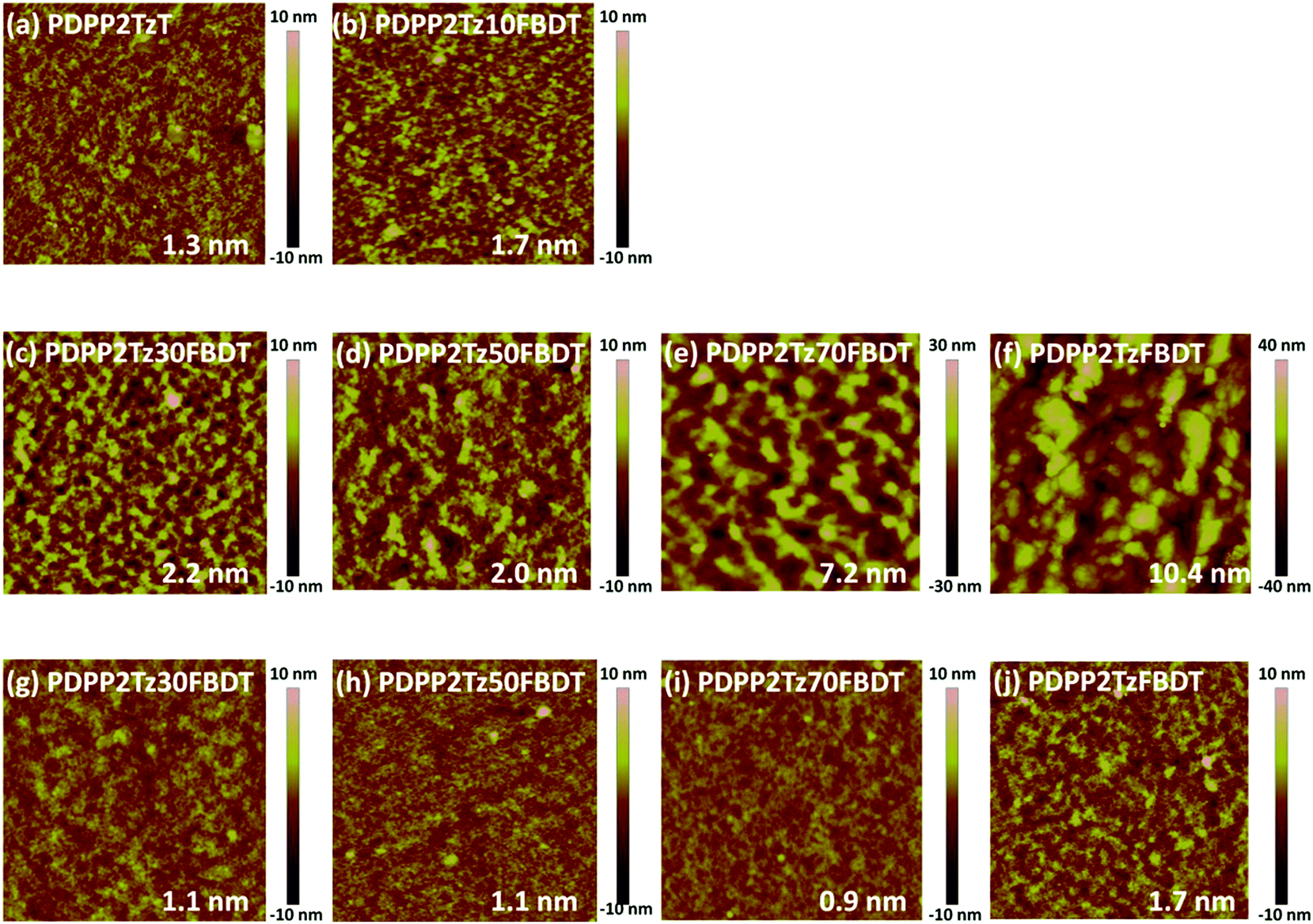 | ||
| Fig. 4 AFM height images (3 × 3 μm2) of PDPP5T:DPP-polymer (1:1) blends spin coated from (a)–(f) CHCl3 containing 3 vol% 1-CN and (g)–(j) CHCl3 containing 10 vol% IC6F12I. The root mean square (RMS) roughness Rq is also included in the panels. | ||
When the same blends were fabricated from CHCl3:IC6F12I the AFM images showed a strongly reduced surface roughness (Fig. 4g–j) compared to those from CHCl3:1-CN (Fig. 4c–f) and smaller lateral dimensions, strongly suggesting an enhanced mixing of the donor and acceptor polymers in the bulk-heterojunction systems. For PDPP5T:PDPP2TzFBDT (Fig. 4j), the surface corrugation is enhanced compared to the other blends.
The blended thin films were further analysed using 2D-GIWAXS measurements. A PDPP5T:PDPP2TzFBDT film spin coated from CHCl3:IC6F12I showed a lower intensity for the in-plane (100) and out-of-plane (010) diffraction peaks (Fig. 5b and d) as compared to the diffraction of the same blend spin coated from CHCl3:1-CN (Fig. 5a and c). Hence, the higher mixing induced by IC6F12I reduces the crystallization of the polymers.
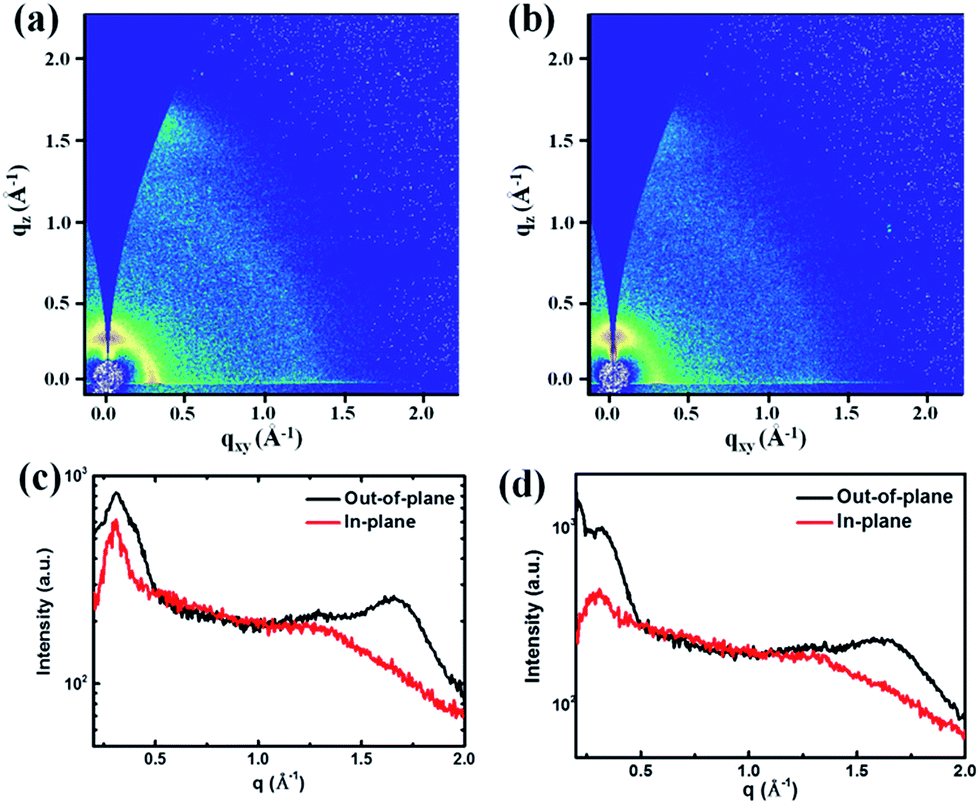 | ||
| Fig. 5 (a) and (b) 2D-GIWAXS patterns of PDPP5T:PDPP2TzBDT (1:1) thin films spin coated from different solutions. (c) and (d) The out-of-plane and in-plane cuts of the corresponding 2D-GIWAXS patterns. (a) and (c) Solution processed from CHCl3 with 3% 1-CN as the additive. (b) and (d) Solution processed from CHCl3 with 10% IC6F12I as the additive. | ||
Steady state PL was applied to further study the morphology difference originating from the spin coating solvent (Fig. 6). The pure polymer and blend thin films were excited at 760 nm and show fluorescence between 800 and 1200 nm. The PL spectra were corrected for the fraction of absorbed photons at the excitation wavelength using the absorption spectra of the same films (ESI, Fig. S10†). Fig. 6 shows that the PL intensity of blend films is significantly reduced compared to that of pure films, indicating the charge transfer from PDPP5T to thiazole-bridged DPP polymers. When processed from CHCl3:IC6F12I, the blend films have generally a lower PL intensity compared to the PL of films from CHCl3:1-CN. In particular, the PL intensity of PDPP5T:PDPP2TzFBDT spin coated from CHCl3:IC6F12I was greatly quenched (Fig. 6f). In this case the luminescence of the PDPP5T:PDPP2TzFBDT blend is almost the same as that of pure PDPP5T when the film is spin coated from CHCl3:1-CN. A higher PL quenching indicates a better charge generation which is expected when the mixing of the two components is enhanced.
 | ||
| Fig. 6 Photoluminescence spectra of the PDPP5T, thiazole based DPP polymers and blends of PDPP5T:DPP polymers (1:1) fabricated from CHCl3 with 3% 1-CN or 10% IC6F12I. The thin films have a similar thickness around 80–95 nm and were excited at 760 nm for measurements. | ||
Role of diiodoperfluoroalkane additives
The use of perfluoroalkyl based processing additives to CHCl3 resulted in more intimately blended films for perfluoroalkyl-substituted DPP acceptor polymers with donor polymers such as PDPP5T than with 1-CN as the additive. The PCE, however, was only significantly enhanced for PDPP2TzFBDT. For the other co-polymers, with varying ratios of T and FBDT units, the blends become better mixed using IC6F12I but the PCE is not really improved. As a result of their irregular structure the mixed T/FBDT co-polymers are less crystalline and possibly the resulting morphology is too well-mixed.Although diiodoperfluoroalkane additives effectively enhance the PCE of PDPP5T:PDPP2TzFBDT solar cells, the PCE is still lower than that of the PDPP5T:PDPP2Tz blends without the perfluoro substituents on the acceptor polymer. The main reason is the lower Voc and lower FF. The latter is likely related to the lower electron mobility of PDPP5T:PDPP2TzFBDT films compared to PDPP5T:PDPP2Tz films (Table 3). Hence, designing perfluoroalkyl-conjugated polymers with high electron mobilities and optimizing the morphology by looking for new solvents will be the routes to further improve the PCEs of these cells.
Conclusions
A series of conjugated polymers based on thiazole-flanked DPP units were synthesized by incorporating different ratios of T and FBDT units into the main chain to tailor the perfluoroalkyl content of the polymers. The perfluoroalkyl based polymers were applied as electron acceptors in polymer solar cells with PDPP5T as the electron donor. While a CHCl3:1-CN solvent mixture provides a PCE of 2.9% for PDPP5T:PDPP2TzT layers without perfluoroalkyl units, this solvent combination only provides a PCE of 0.52% for PDPP5T:PDPP2TzFBDT with perfluoroalkyl units. We showed that the PCE of PDPP5T:PDPP2TzFBDT can be dramatically enhanced to 2.1% by using a CHCl3:IC6F12I solvent mixture. Detailed analysis using AFM, 2D-GIWAXS and PL measurements reveals that IC6F12I as the additive is helpful to enhance the mixing of the donor and perfluoroalkyl-based acceptor in the blended films that enhance the charge generation. The results demonstrate that diiodoperfluoroalkane solvents can be efficient processing additives to improve the performance of solution processed perfluoroalkyl based polymer solar cells.
Acknowledgements
This work was supported by the Recruitment Program of Global Youth Experts of China. The measurements of NMR were performed at the Center for Physicochemical Analysis and Measurements in ICCAS. The help from Dr Junfeng Xiang is acknowledged. The work was further supported by the National Natural Science Foundation of China (21574138, Y5A1141501) and the Strategic Priority Research Program (XDB12030200) of the Chinese Academy of Sciences. The work of Q. W. forms part of the research programme of the Dutch Polymer Institute (DPI), project #762.Notes and references
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- S. M. McAfee, J. M. Topple, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2015, 3, 16393–16408 CAS.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- Y.-J. Hwang, H. Li, B. A. E. Courtright, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2016, 28, 124–131 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed /1–8.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435–441 CAS.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu and A. K. Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed.
- P. E. Hartnett, A. Timalsina, H. S. S. R. Matte, N. Zhou, X. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed.
- O. K. Kwon, J.-H. Park, D. W. Kim, S. K. Park and S. Y. Park, Adv. Mater., 2015, 27, 1951–1956 CrossRef CAS PubMed.
- L. Y. Liu, J. Y. L. Lai, S. Chen, Y. Li, K. Jiang, J. Zhao, Z. Li, H. Hu, T. Ma, H. Lin, J. Liu, J. Zhang, F. Huang, D. Yu and H. Yan, J. Mater. Chem. A, 2015, 3, 13632–13636 Search PubMed.
- B. Jiang, X. Zhang, C. Zhan, Z. Lu, J. Huang, X. Ding, S. He and J. Yao, Polym. Chem., 2013, 4, 4631–4638 RSC.
- Y. Liu, L. Zhang, H. Lee, H.-W. Wang, A. Santala, F. Liu, Y. Diao, A. L. Briseno and T. P. Russell, Adv. Energy Mater., 2015, 5, 1500195 Search PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, H. Lu, J. Wu, G. Li, C. Li, S. Li and Z. Bo, J. Mater. Chem. C, 2015, 3, 6979–6985 RSC.
- X.-F. Wu, W.-F. Fu, Z. Xu, M. Shi, F. Liu, H.-Z. Chen, J.-H. Wan and T. P. Russell, Adv. Funct. Mater., 2015, 25, 5954–5966 CrossRef CAS.
- J. J. W. Jung and W. H. Jo, Chem. Mater., 2015, 27, 6038–6043 CrossRef CAS.
- O. K. Kwon, J.-H. Park, S. K. Park and S. Y. Park, Adv. Energy Mater., 2015, 5, 1400929 Search PubMed.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS PubMed.
- D. Mori, H. Benten, H. Ohkita, S. Ito and K. Miyake, ACS Appl. Mater. Interfaces, 2012, 4, 3325–3329 CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- Y.-J. Hwang, B. A. E. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenekhe, Adv. Mater., 2015, 27, 4578–4584 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- Y.-J. Hwang, T. Earmme, B. A. E. Courtright, F. N. Eberle and S. A. Jenekhe, J. Am. Chem. Soc., 2015, 137, 4424–4434 CrossRef CAS PubMed.
- C. Lee, H. Kang, W. Lee, T. Kim, K.-H. Kim, H. Y. Woo, C. Wang and B. J. Kim, Adv. Mater., 2015, 27, 2466–2471 CrossRef CAS PubMed.
- H. Li, Y.-J. Hwang, T. Earmme, R. C. Huber, B. A. E. Courtright, C. O'Brien, S. H. Tolbert and S. A. Jenekhe, Macromolecules, 2015, 48, 1759–1766 CrossRef CAS.
- I. H. Jung, W.-Y. Lo, J. Jang, W. Chen, D. Zhao, E. S. Landry, L. Lu, D. V. Talapin and L. Yu, Chem. Mater., 2014, 26, 3450–3459 CrossRef CAS.
- S. Subramaniyan, T. Earmme, N. M. Murari and S. A. Jenekhe, Polym. Chem., 2014, 5, 5707–5715 RSC.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Energy Environ. Sci., 2014, 7, 2939–2943 CAS.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- W. Li, Y. An, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2015, 3, 6756–6760 CAS.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2015, 28, 1884–1890 CrossRef PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed /1–8.
- S. Zhang, L. Ye, W. Zhao, B. Yang, Q. Wang and J. Hou, Sci. China: Chem., 2015, 248–256 CrossRef CAS.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- S. Y. Qu and H. Tian, Chem. Commun., 2012, 48, 3039–3051 RSC.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- Z. Yi, S. Wang and Y. Liu, Adv. Mater., 2015, 27, 3589–3606 CrossRef CAS PubMed.
- C. Xiao, G. Zhao, A. Zhang, W. Jiang, R. A. J. Janssen, W. Li, W. Hu and Z. Wang, Adv. Mater., 2015, 27, 4963–4968 CrossRef CAS PubMed.
- H. A. Um, D. H. Lee, D. U. Heo, D. S. Yang, J. Shin, H. Baik, M. J. Cho and D. H. Choi, ACS Nano, 2015, 9, 5264–5274 CrossRef CAS PubMed.
- H. Choi, S.-J. Ko, T. Kim, P.-O. Morin, B. Walker, B. H. Lee, M. Leclerc, J. Y. Kim and A. J. Heeger, Adv. Mater., 2015, 27, 3318–3324 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2014, 137, 1314–1321 CrossRef PubMed.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341–8344 CrossRef CAS PubMed.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- T. B. Singh, N. Marjanović, P. Stadler, M. Auinger, G. J. Matt, S. Günes, N. S. Sariciftci, R. Schwödiauer and S. Bauer, J. Appl. Phys., 2005, 97, 083714 CrossRef.
- A. Zhang, C. Xiao, D. Meng, Q. Wang, X. Zhang, W. Hu, X. Zhan, Z. Wang, R. A. J. Janssen and W. Li, J. Mater. Chem. C, 2015, 3, 8255–8261 RSC.
- P. Homyak, Y. Liu, S. Ferdous, F. Liu, T. P. Russell and E. B. Coughlin, Chem. Mater., 2015, 27, 443–449 CrossRef CAS.
- G. He, X. Wan, Z. Li, Q. Zhang, G. Long, Y. Liu, Y. Hou, M. Zhang and Y. Chen, J. Mater. Chem. C, 2014, 2, 1337–1345 RSC.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS PubMed.
- W. Li, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2012, 134, 13787–13795 CrossRef CAS PubMed.
- P. M. Buschbaum, Adv. Mater., 2014, 26, 7692–7709 CrossRef PubMed.
- J.-D. Chen, C. Cui, Y.-Q. Li, L. Zhou, Q.-D. Ou, C. Li, Y. Li and J.-X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, A. Zhang, M. M. Wienk and R. A. J. Janssen, Chem. Commun., 2015, 51, 4290–4293 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00962j |
| This journal is © The Royal Society of Chemistry 2016 |