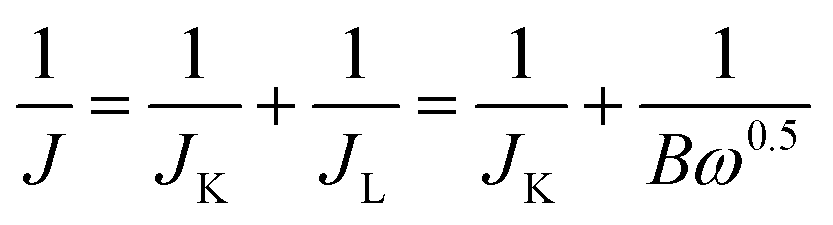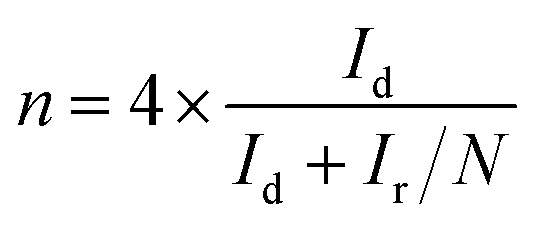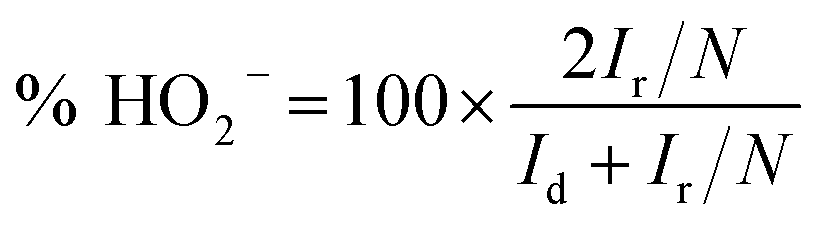Pt/C–LiCoO2 composites with ultralow Pt loadings as synergistic bifunctional electrocatalysts for oxygen reduction and evolution reactions†
Received
21st December 2015
, Accepted 17th February 2016
First published on 22nd February 2016
Abstract
Oxygen reduction and evolution reactions (ORR and OER) are of prime importance for many energy conversion and storage devices, such as regenerative fuel cells and rechargeable metal–air batteries. However, the sluggish kinetics of the ORR and OER strongly limit the efficiency and performance of these electrochemical systems and jeopardize the route of commercialization. Therefore, the design and development of bifunctional electrocatalysts with high activity for both the ORR and OER is challenging but urgent and crucial. Here, we took advantage of Pt/C and LiCoO2 with outstanding ORR activity and high intrinsic OER activity, respectively, to develop a composite material with ultralow Pt loading as a bifunctional catalyst for the ORR and OER in alkaline media. This catalyst was fabricated via simple ultrasonic mixing, exhibiting superb electrocatalytic activity and good stability. Its ORR activity is comparable to that of the commercial Pt/C catalyst and its OER activity is better than that of single LiCoO2, owing to the synergetic effect between Pt and LiCoO2, which has been demonstrated through the X-ray photoelectron spectroscopy (XPS) characterisation technique. Remarkably, surprisingly high ORR mass activity (2.04 A mgPt−1 at 0.8 V vs. RHE) and enhanced bifunctionality (ΔE = 0.91 V) were obtained for the Pt–LiCoO2 composite catalyst with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 for Pt/LiCoO2. Our work opens up a new track to exploit highly efficient catalysts with reduced consumption of Pt, meanwhile maintaining the optimal catalytic activity and durability.
:49 for Pt/LiCoO2. Our work opens up a new track to exploit highly efficient catalysts with reduced consumption of Pt, meanwhile maintaining the optimal catalytic activity and durability.
Introduction
Increasing energy demands and critical issues of climate change and environmental protection have prompted considerable research interest in exploring renewable and clean electrochemical energy conversion and storage systems with high efficiency and low cost.1,2 To this end, rechargeable metal–air batteries and regenerative fuel cells have received tremendous attention because of their advantages of extremely high energy density and environmental benignity.3–5 These electrochemical energy systems are highly attractive for electrified transportation, and smart grid and stationary power sources, which may help us to build up a low-carbon and low-emission sustainable future. Both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are involved in such energy technologies, which play significant roles equally. However, the kinetically sluggish ORR and OER have become the current bottleneck of these energy devices, and largely precluded their large-scale commercial application.6–9 Although intensive and extensive research efforts have been made during the past decades,10–13 it is still very challenging to search for more active and stable electrocatalysts, especially for highly efficient catalysts with dual functionality for aforementioned reactions.
So far, noble metal catalysts such as Pt or Pt alloys have been widely used as oxygen catalysts for the ORR with a desirable performance in low-temperature fuel cells because of their ideal electrocatalytic activities.14–17 However, Pt is known to be expensive and rare in the earth, hindering its practical application; therefore, great efforts have been directed towards the development of alternative low-Pt-loading catalysts, enabling competitiveness economically. The electrode activity for the OER is another big challenge for regenerative fuel cells and rechargeable metal–air batteries, besides the rate-determining step of the ORR over the electrode in fuel cells. Noble metal oxides (e.g., IrO2 and RuO2) are generally applied to meet the requirements of fast kinetics and low overpotentials for the OER,18,19 but still they cannot circumvent the issue of high-price and scarcity found with Pt. It is urgent to develop new types of low-cost but active catalysts for electrocatalytic evolution of oxygen. Along with the intensive research in this scope, many transition metal oxides have been studied as OER electrocatalysts in recent years, such as single-phase oxide, perovskite-type and spinel-type catalysts.20–27 LiCoO2, one of the most well-known cathode materials for lithium-ion batteries,28 has recently been proved to be an excellent electrocatalyst for the OER in alkaline media.29–34 Furthermore, when LiCoO2 was chemically delithiated to form Li1−xCoO2 or developed with some particular microstructure (e.g. nanostructure),33,34 it also exhibited a high ORR activity. However, the preparation methods and techniques are usually quite complex and sophisticated. Therefore, the development of bifunctional electrocatalysts with highly effective catalytic activities for both the ORR and OER via a facile synthesis is imperative to realize large-scale commercialization of regenerative fuel cells and rechargeable metal–air batteries.
Lately, metal oxide supported Pt particles have been used to attempt effective composite catalysts. The obtained catalysts displayed enhanced activity and stability towards the ORR/OER. They mainly utilize the synergetic “spillover” effects between platinum and the support to influence the adsorption/desorption behavior of intermediate species at the catalyst surfaces and altered the electronic structure of platinum, hence promoting the rate determining steps in the corresponding reactions.35–38 Remarkably, a great amount of experimental results have shown that the combination of different types of electrocatalysts to form composite electrodes could result in improved catalytic activity for the ORR and OER, much better than each of the individual components.39–45 It is promising to develop bi-functional electrocatalysts with enhanced activity for both the ORR and OER taking advantage of the synergic effects. Although the ORR and OER mechanisms are complicated, attributed to the multiple reaction steps, the formation of composite electrodes could effectively improve the reaction kinetics due to the synergistic effect of each component for the rate limiting step(s). Previously, we have successfully demonstrated a composite of Pt/C (known for its high activity for the ORR) and perovskite-type Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) oxide (known for its high activity for the OER). The catalytic activity for the ORR and OER in alkaline media was obviously improved compared with the condition of the absence of each other, suggesting the existence of synergistic effects amongst Pt/C and BSCF.45 However, the Pt content in this reported composite is still very high, with a value of at least 20 wt%, which has to be reduced in the electrocatalyst. It is of crucial importance to bring these electrochemical technologies to a competitive commercial position.
In this study, we extended this universal and facile strategy to develop a composite material with Pt/C and LiCoO2 as the bifunctional electrocatalyst for the ORR and OER in alkaline media for the first time. It should be noted that the Pt loading in the as-prepared composite has been reduced to an ultralow level of 2 wt%. To the best of our knowledge, until now, the lowest Pt content in low-Pt-loading catalyst systems has been realized through our attempt and efforts. The composite catalysts delivered superb ORR catalytic activity comparable with that of commercial Pt/C alone, as well as more excellent performance for the OER process than that of LiCoO2 alone. Our study demonstrated that the Pt/C–LiCoO2 composite material is a promising candidate as a bifunctional oxygen catalytic electrode material for rechargeable metal–air batteries and regenerative fuel cell devices.
Experimental
Synthesis and physicochemical characterization
LiCoO2 was synthesized by a traditional solid-state reaction method reported previously.33 Stoichiometric amounts of Li2CO3 (Sigma-Aldrich) and Co3O4 (Sigma-Aldrich) were ground and mixed thoroughly using high energy ball milling (Fritsch Pulverisette 6) in absolute ethanol at 400 rpm for 1 h, followed by drying and sintering at 800 °C for 5 h in air. Platinum on carbon, Pt/C, with a Pt loading of 20 wt% was used in this study, which was purchased from Sigma-Aldrich. The samples were characterized by X-ray diffraction (XRD, Rigaku Smartlab 3 kW) using filtered Cu Kα radiation (λ = 1.5406 Å) in a step-scan mode within the 2θ range of 10–90° at 0.02° per step. X-ray photoelectron spectroscopy (XPS) analysis was conducted on a Kratos AXIS Ultra DLD system under UHV conditions with an Al Kα X-ray, and the data were analyzed using the Kratos Vision and CasaXPS software, with the cross-calibration to C 1s = 284.8 eV. The morphology of the catalysts was investigated by transmission electron microscopy (TEM, JEOL 2100).
Electrode preparation
Three kinds of Pt–LiCoO2 composite electrocatalysts with different mass ratios of Pt to LiCoO2 (1:9, 1:19 and 1:49) were prepared and studied. In order to compare with the commercial Pt/C electrocatalyst (20 wt% loading), the same mass ratios of catalyst to carbon (2:8) were chosen for all the Pt–LiCoO2 composite and single LiCoO2 electrocatalysts. Taking Pt–LiCoO2 (1:9) as an example, 1 mg of Pt/C (20 wt% loading), 1.8 mg of LiCoO2 and 7.2 mg of carbon black (Super P, Alfa Aesar) were ultrasonically mixed in a solution containing 1 ml of absolute ethanol and 0.1 ml of Nafion solution (5 wt% in isopropanol and water solution) for 2 h, forming a catalyst ink with a concentration of ∼1.818 mgcatalyst mL−1. Next, 7 μL of the catalyst ink was pipetted onto the surface of a glassy carbon rotating disk electrode (GC-RDE, 0.196 cm2, Pine Research Instrumentation, USA) or a rotating ring-disk electrode (RRDE, GC disk with a 320 μm gap Pt ring, Pine Research Instrumentation, USA) and dried in air, yielding a catalyst loading of ∼0.0649 mg cm−2disk. Before coating the catalyst, the RDE or RRDE was polished with 0.05 μm and/or 0.3 μm of Al2O3 slurry, and rinsed with ethanol and ultrapure water.
Electrochemical measurements
All the electrochemical measurements were conducted in a three-electrode glass cell (Pine Research Instrumentation, USA) using a CHI 760E bipotentiostat (CH Instruments, Inc., USA). The electrolyte was 0.1 M KOH solution. The catalyst-coated RDE or RRDE was employed as the working electrode, a platinum wire as the courter electrode, and an Ag/AgCl (4 M KCl filled) electrode as the reference electrode. All the potential values mentioned in this study were referenced to that of the reversible hydrogen electrode (RHE) with the relation of E(RHE) = E(Ag/AgCl) + 0.948 V, according to the calibration measurement in H2-saturated 0.1 M KOH electrolyte with two platinum wires as working and counter electrodes by measuring hydrogen oxidation/evolution currents on the working electrode. Cyclic voltammograms (CVs) were carried out at a scan rate of 50 mV s−1 in an O2-saturated electrolyte. Linear sweep voltammograms (LSVs) were recorded at a scan rate of 5 mV s−1 in the O2-saturated electrolyte from 1.148 to 0.348 V (ORR) with varying rotating speeds (2400, 2000, 1600, 1200, 800 and 400 rpm) or from 1.148 to 1.948 V (OER) at 1600 rpm. The kinetic parameters for the ORR were calculated by the Koutecky–Levich equation:| | 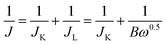 | (1) |
| | | B = 0.62nFCoD2/3oυ−1/6 | (2) |
where J is the measured current density, JK and JL correspond to the kinetic and diffusion-limiting current densities, ω is the electrode rotating rate (rad s−1), n is the electron transfer number, F is the Faraday constant, Co is the bulk concentration of O2 in the electrolyte, Do is the diffusion coefficient of O2, and υ is the kinetic viscosity of the electrolyte.
RRDE voltammograms for the ORR were obtained at a scan rate of 5 mV s−1 from 1.148 to 0.348 V (disk electrode) at different rotating rates in the O2-saturated electrolyte. The ring potent was a constant value of 1.448 V, which is considered to be sufficiently high to oxidize any HO2− intermediate.46,47 The electron transfer number (n) and the production percentage of the HO2− intermediate were determined using the following equations:
| | 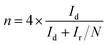 | (3) |
| | 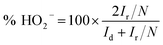 | (4) |
where
Id and
Ir are the disk and ring current, respectively, and
N is the current collection efficiency of the Pt ring (here,
N = 0.37).
Results and discussion
Bulk and surface structures
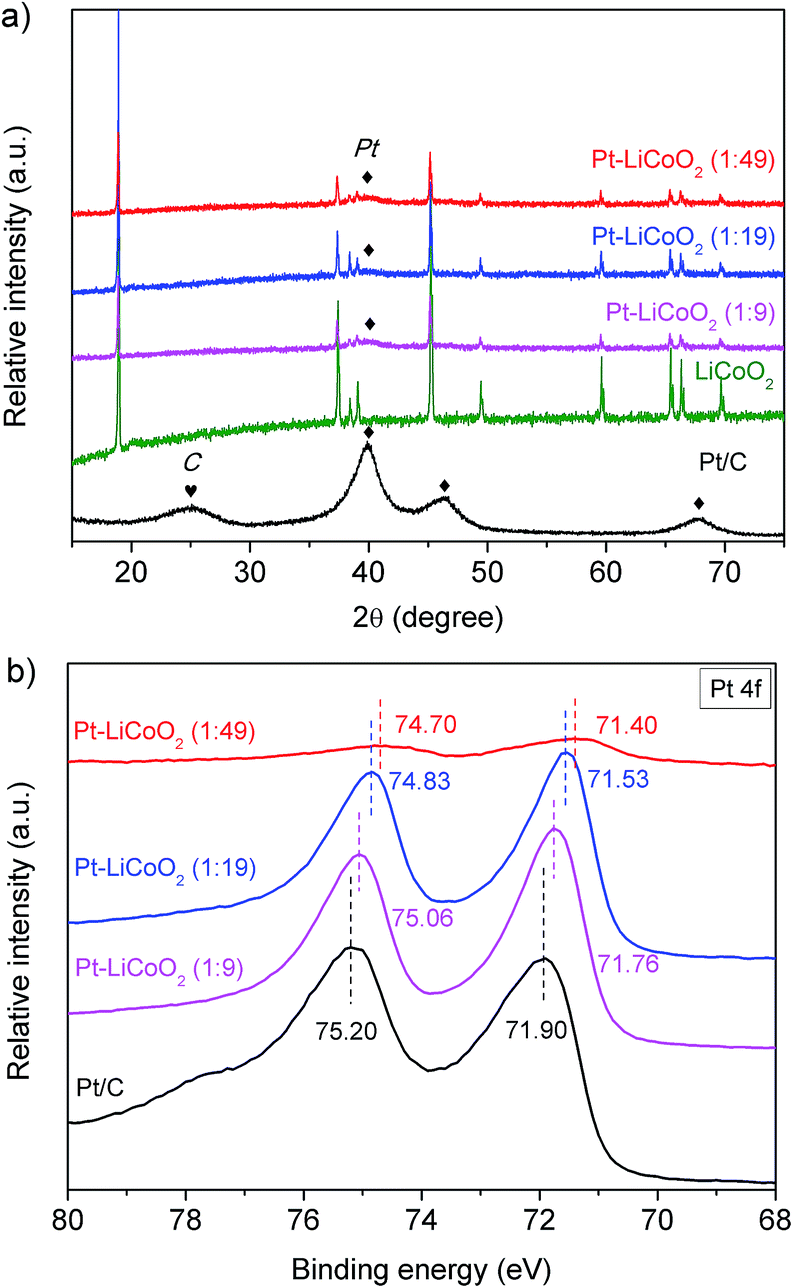
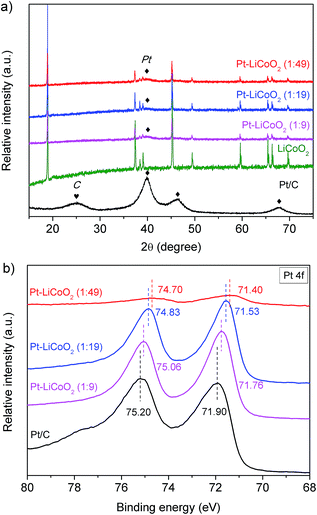
Fig. 1a shows the room-temperature XRD patterns of the Pt–LiCoO2 composites with different mass ratios of Pt to LiCoO2 fabricated by simple ultrasonic mixing, and the pristine LiCoO2 synthesized through high-temperature calcination at 800 °C and commercial Pt/C were also provided as the reference. Impurity phases in the obtained LiCoO2 sample were not detected, indicating the formation of pure single-phase LiCoO2. All the diffraction peaks can be well indexed to a layered α-NaFeO2 structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. This well-known structure consists of alternating planes of coordinated Li and Co ions, which are separated by close-packed oxygen layers.48,49 According to Rietveld refinement of the XRD data, the lattice parameters were found to be a = b = 2.817(6) Å and c = 14.05(8) Å (c/a = 4.988), which were in alignment with the literature results,48–50 and further suggested a well-formed layered structure of LiCoO2. The XRD of commercial Pt/C presents broad diffraction peaks, indicating the nanoscale crystalline characteristic of the Pt particles. It is in good accordance with the ∼3 nm Pt particles in the commercial Pt/C (TEM image in Fig. S1†). After forming the composites with LiCoO2, the intensity of Pt diffraction peaks dropped to a very low level that only the main peak at the 2θ of 39.8° was observed, which could be attributed to the small amount of Pt in the composite catalysts. For the LiCoO2 in the three composites, very similar lattice parameters as well as the values of c/a to the pristine LiCoO2 were obtained, which were a = b = 2.816(2) Å, c = 14.05(0) Å, c/a = 4.989 for Pt–LiCoO2 (1:9), a = b = 2.817(2) Å, c = 14.05(3) Å, c/a = 4.988 for Pt–LiCoO2 (1:19), and a = b = 2.816(7) Å, c = 14.05(2) Å, c/a = 4.989 for Pt–LiCoO2 (1:49). The results suggest that the phase structure of the LiCoO2 cannot be affected after the introduction of Pt/C.
m space group. This well-known structure consists of alternating planes of coordinated Li and Co ions, which are separated by close-packed oxygen layers.48,49 According to Rietveld refinement of the XRD data, the lattice parameters were found to be a = b = 2.817(6) Å and c = 14.05(8) Å (c/a = 4.988), which were in alignment with the literature results,48–50 and further suggested a well-formed layered structure of LiCoO2. The XRD of commercial Pt/C presents broad diffraction peaks, indicating the nanoscale crystalline characteristic of the Pt particles. It is in good accordance with the ∼3 nm Pt particles in the commercial Pt/C (TEM image in Fig. S1†). After forming the composites with LiCoO2, the intensity of Pt diffraction peaks dropped to a very low level that only the main peak at the 2θ of 39.8° was observed, which could be attributed to the small amount of Pt in the composite catalysts. For the LiCoO2 in the three composites, very similar lattice parameters as well as the values of c/a to the pristine LiCoO2 were obtained, which were a = b = 2.816(2) Å, c = 14.05(0) Å, c/a = 4.989 for Pt–LiCoO2 (1:9), a = b = 2.817(2) Å, c = 14.05(3) Å, c/a = 4.988 for Pt–LiCoO2 (1:19), and a = b = 2.816(7) Å, c = 14.05(2) Å, c/a = 4.989 for Pt–LiCoO2 (1:49). The results suggest that the phase structure of the LiCoO2 cannot be affected after the introduction of Pt/C.
 |
| | Fig. 1 (a) Powder XRD patterns and (b) Pt 4f XPS spectra of Pt/C, LiCoO2 and the as-prepared Pt–LiCoO2 composites with different mass ratios. | |
The XRD patterns are very similar to one another, which only reveal the bulk composition information. Therefore, more information on the surface of the as-prepared catalysts was collected from the XPS characterization. As shown in Fig. 1b, typical characteristic peaks of Pt 4f7/2 and Pt 4f5/2 were observed at binding energies (BEs) of 71.90 and 75.20 eV, respectively for the commercial Pt/C. In the case of Pt–LiCoO2 composites, the spectra of Pt 4f shifted to a lower BE with the reduction of the Pt content in the composites, a similar phenomenon was also observed by Zhu et al.45 It suggests that the electronic structure of Pt was probably modified through the interaction between Pt and LiCoO2. In addition, Co 2p and O 1s XPS spectra of the electrocatalysts are also given in Fig. S2 of the ESI.† All the catalysts show a Co 2p3/2 main peak at around 780 eV with a satellite peak at 790 eV and a Co 2p1/2 main peak at around 795 eV with a satellite peak at 805 eV, which demonstrates that the oxidation state is Co3+ in the LiCoO2 and Pt–LiCoO2 composites.49,51 According to the investigation of Dahéron et al., there was no Co2+ in these samples because the strong broadening of the main peak and very intense satellite peaks at 786 eV (Co 2p3/2) and 803 eV (Co 2p1/2) related to the characterization of Co2+ coordinated by oxygen are not detected here.51 For the Pt–LiCoO2 (1–9) sample, the core peaks of Co 2p slightly shifted to higher BEs compared to the pristine LiCoO2, indicating possible partial oxidation of surface Co3+ to Co4+, and it has been reported that the presence of Co4+ could facilitate the OER.52,53 The XPS spectra of O 1s displayed two peaks at BEs of around 529.5 and 531.4 eV for all the samples, which correspond to the lattice oxygen and adsorbed species at the surface. Compared with the pristine LiCoO2, the ratio of peak intensities of surface to lattice oxygen increased with the increase of Pt content in the composites. It means that a stronger interaction existed between the composite catalysts and adsorbed oxygen-containing species due to the synergistic effect, which is beneficial for the process of the ORR.
Electrochemical behaviour for the ORR
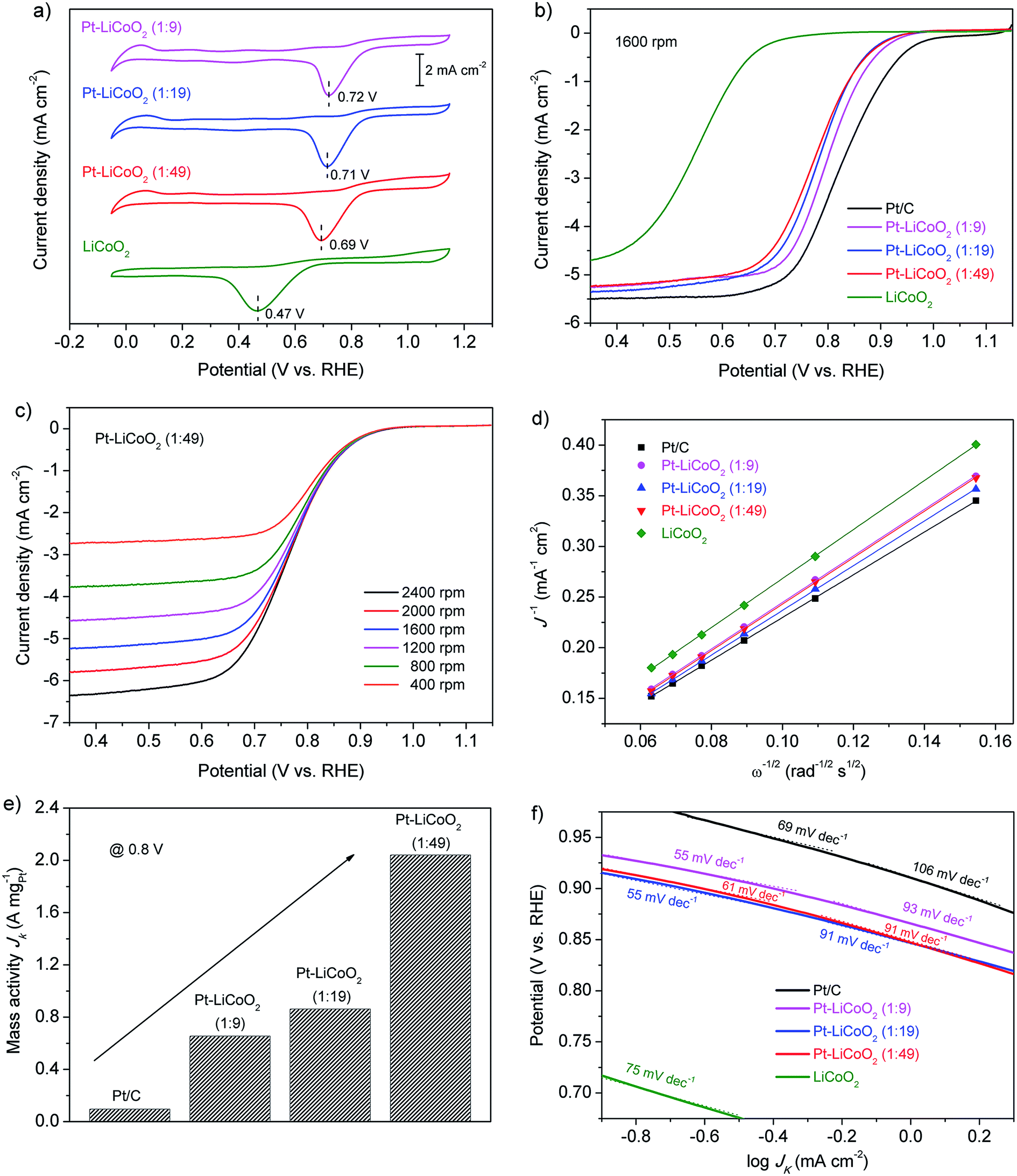
To evaluate the electrocatalytic activity of the obtained composite catalysts with different compositions for the ORR, CVs tests were first conducted in O2-saturated 0.1 M KOH solution. It was also compared with that of pristine LiCoO2 and commercial Pt/C (Fig. S3†). As shown in Fig. 2a & S3 of the ESI,† all the detected samples presented distinct single oxygen reduction peaks, suggesting the conspicuous catalytic activity for the ORR. The shape of CV curves for all the composite catalysts was more similar to that of LiCoO2 than that of Pt/C, because the proportion of LiCoO2 in the composites was larger. However, the ORR peaks of the composite catalysts appeared at around 0.71 V, which shifted greatly to positive potentials compared with that of LiCoO2 with a value of 0.47 V, and were very close to the cathodic peak of Pt/C (0.81 V), indicating the excellent electrocatalytic activity towards the ORR. Similar trends were further verified by the RDE measurements. Fig. 2b shows a comparison of LSV curves for each of the catalysts performed at 1600 rpm. The ORR on the pristine LiCoO2 electrode initiated at around 0.70 V, and then a continuous increase in the current density was witnessed without an obvious current plateau. All the prepared Pt–LiCoO2 composite catalysts displayed similar potentials for the commencement of the ORR (∼0.96 V), which were more positive than that of the LiCoO2 electrode, and much closer to that of commercial Pt/C (∼1.0 V), indicating a more facile ORR process. The wide current plateau on all the Pt–LiCoO2 composites is considered as the strong limiting diffusion current and suggests a diffusion-dominated process that is related to a favourable four-electron ORR pathway, as is also observed on the Pt/C catalyst. Additionally, as compared with a half-wave potential of 0.554 V obtained from the LiCoO2 electrode, the half-wave potentials significantly shifted to 0.799, 0.781 and 0.775 V, positively, for Pt–LiCoO2 (1:9, 1:19 and 1:49) composites, respectively, which are only approximately 0.05 V more negative than that of Pt/C (0.825 V). Furthermore, Pt–LiCoO2 composite catalysts presented a comparable limiting current density with that observed on Pt/C at 0.35 V, even for the sample with the Pt content of only 2 wt%, which is 1.14 times that of the pristine LiCoO2 electrode. All of the above results evidence that the ORR catalytic activity of obtained composites was greatly enhanced by the synergistic effect between Pt and LiCoO2.
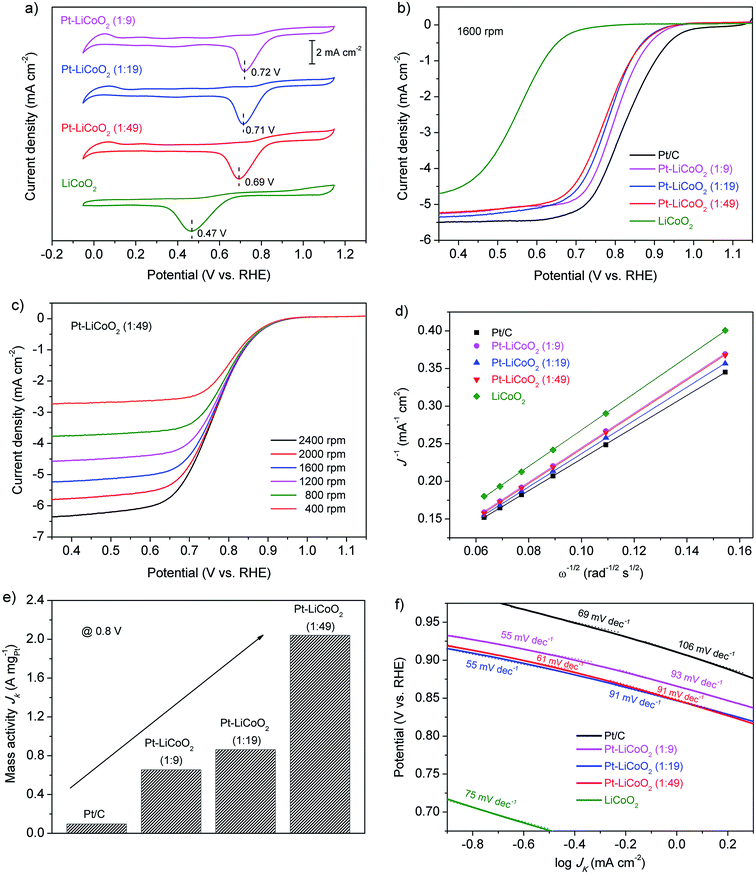 |
| | Fig. 2 (a) CV curves of the ORR on LiCoO2 and the as-prepared Pt–LiCoO2 composites with different mass ratios in an O2-saturated 0.1 M KOH solution at a scan rate of 50 mV s−1. (b) LSV curves of various electrocatalysts on the RDE at 1600 rpm in an O2-saturated 0.1 M KOH solution at a scan rate of 5 mV s−1. (c) LSV curves of Pt–LiCoO2 (1:49) on RDE at different rotating rates. (d) K–L plots at 0.35 V for various electrocatalysts on the basis of the RDE data in (c) and Fig. S4 of the ESI.† (e) ORR mass activity of various catalysts at 0.8 V. (f) Tafel plots based on the LSV curves in (b). | |
A more detailed study of the RDE measurements at rotating rates from 400 to 2400 rpm was conducted, allowing further insight into the ORR kinetics and electrocatalytic processes on the aforementioned catalysts (Fig. 2c and S4 of the ESI†). Fig. 2d shows the K–L plots of various catalysts constructed from rotation rate dependent current density. The linearity of the K–L plots indicates first-order reaction kinetics towards the concentration of dissolved oxygen, and near parallelism of the fitting lines, especially for the composite catalysts and Pt/C, suggests an efficient four-electron-dominated ORR pathway on all of the catalysts. Considering the catalysts' cost and the economic feasibility, the ORR mass activity on the basis of the Pt content of the as-prepared composite catalysts is calculated, which can reflect the ORR intrinsic activity of catalysts.54 The kinetic currents at a potential of 0.8 V were used to determine the activities. As shown in Fig. 2e, a significant improvement in mass activity was observed for the Pt–LiCoO2 (1:49) catalyst (2.04 A mgPt−1) in comparison with the benchmark Pt/C (0.095 A mgPt−1) at 0.8 V, which was almost 21 times higher than the corresponding value of Pt/C. It evinces the superb ORR intrinsic catalytic activity of the as-prepared composite catalysts by the beneficial synergistic effect even though a very small amount of Pt/C was mixed well with LiCoO2. Besides, the Tafel slope was adopted as another figure of merit. As can be seen in Fig. 2f, the excellent catalytic activity for the ORR on the composite electrodes was further proved by the smaller slopes of Tafel plots with the values of 55, 55 and 61 mV dec−1 for Pt–LiCoO2 (1:9, 1:19 and 1:49) composites, respectively, than that of Pt/C (69 mV dec−1), again suggesting faster ORR kinetics of the composite samples.
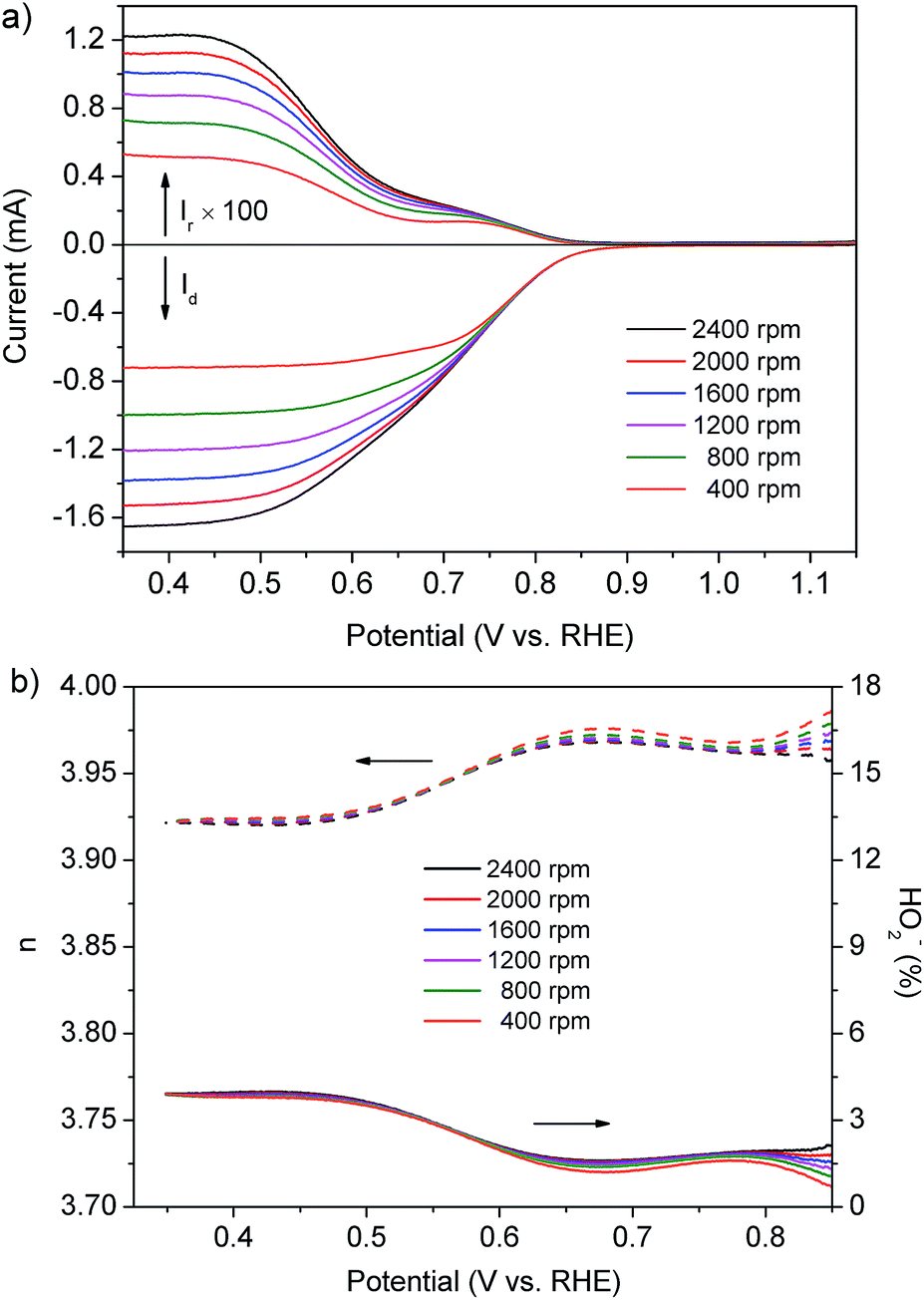
The RRDE technique is extremely effective in evaluating the catalytic activity of electrocatalysts for the ORR by directly monitoring the generation rate of HO2− during the ORR. We also adopted this technique to further verify the ORR pathway on the prepared composite catalyst. Fig. 3a shows the current on the disk electrode (Id) and the ring electrode (Ir, multiplied by 100) using the Pt–LiCoO2 (1:49) catalyst at various rotation speeds. Obviously, the Id was far higher than Ir, suggesting that a small amount of HO2− was produced during the ORR process. As illustrated in Fig. 3b, the amount of formed HO2− was less than 4% and the value of n was above 3.92 for the Pt–LiCoO2 (1:49) catalyst over a wide potential range from 0.35 to 0.85 V at all the rotating rates. It is consistent with the results obtained from the fitted K–L plot, which denotes a 4e− ORR process. Compared with Pt/C (n > 3.94 and the amount of formed HO2− < 2.91%) and pristine LiCoO2 (n > 3.80 and the amount of formed HO2− < 9.96%) as shown in Fig. S5 of the ESI,† we found that the value of n and the generation rate of HO2− on the Pt–LiCoO2 (1:49) catalyst was quite close to the results obtained on commercial Pt/C and superior to LiCoO2, again indicating the much enhanced electrocatalytic efficiency due to the synergistic effect between Pt and LiCoO2.
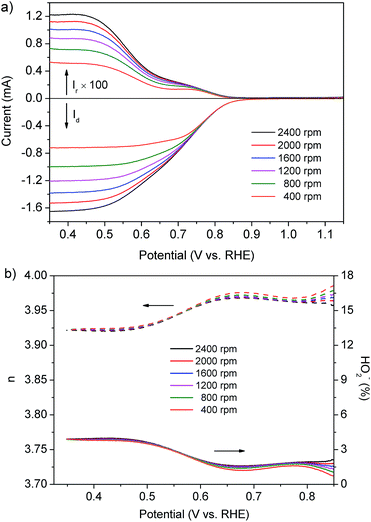 |
| | Fig. 3 (a) Disk (bottom) and ring currents (top, multiplied by 100) of the Pt–LiCoO2 (1:49) catalyst collected on the RRDE at different rotating rates in O2-saturated 0.1 M KOH electrolyte. (b) The electron transfer number (n, dash line) and percentage of HO2− (solid line) of the Pt–LiCoO2 (1:49) catalyst at various potentials based on the RRDE data in (a). | |
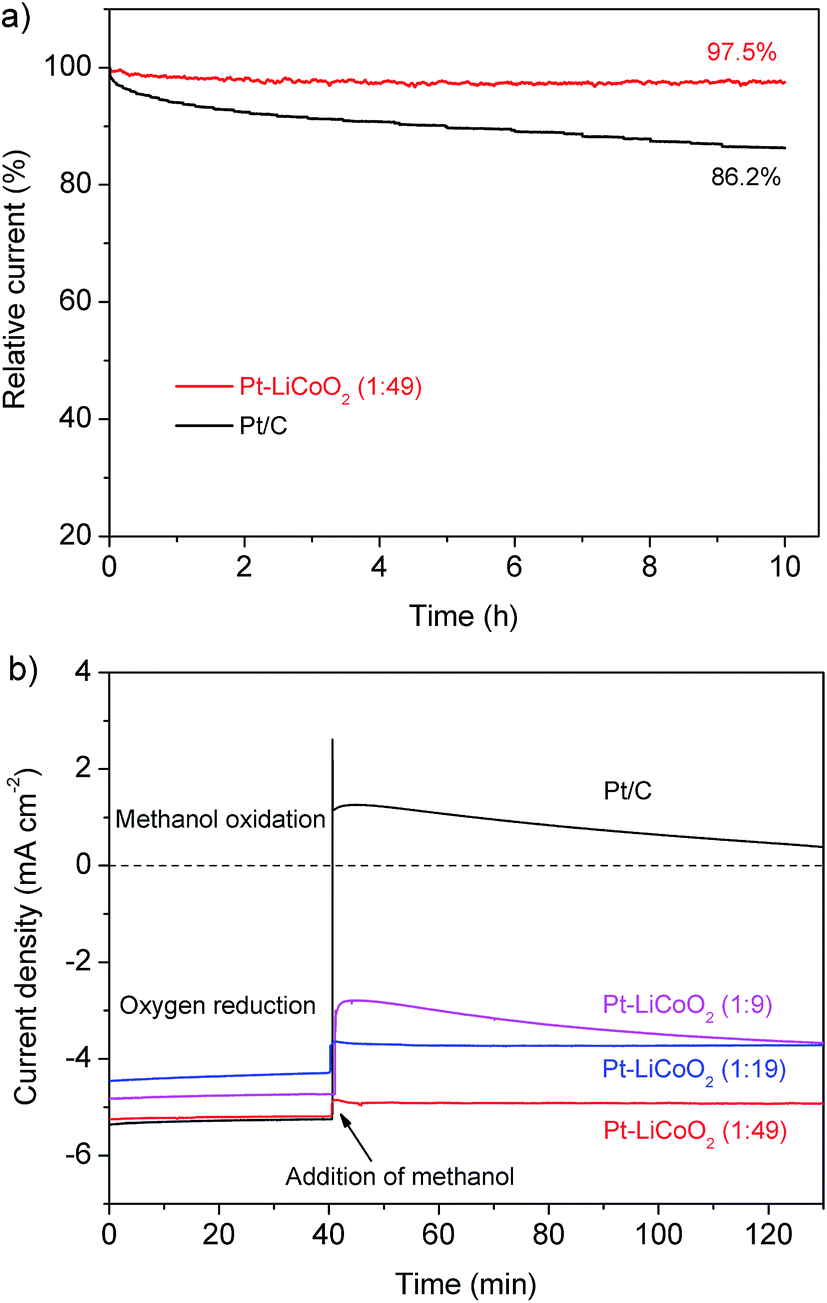
Besides the high ORR catalytic activity that is comparable to that of Pt/C, the Pt–LiCoO2 (1:49) also exhibited a strong durability superior to Pt/C in the potential range of the ORR. For instance, the retention of the current was still up to 97.5% for the newly developed composite catalyst of Pt–LiCoO2 (1:49) after the continuous polarization period of 10 h (Fig. 4a). In contrast, a significant attenuation with the loss of 13.8% of the initial current was observed for the Pt/C catalyst under the same operating conditions. Additionally, the methanol tolerance is also a significant index for the cathode materials of direct-methanol fuel cells (DMFCs). As shown in Fig. 4b, the initial cathodic current for the ORR on the commercial Pt/C electrode suddenly shifted to a reverse anodic current upon the addition of methanol, suggesting the conversion of the dominated process from the ORR to the methanol oxidation reaction. It demonstrates the poor ability of the Pt/C catalyst to resist the poisoning cross-over effects. With the reduction of the platinum content in the composite electrocatalysts, the cross-over effect was gradually suppressed. Especially for the Pt–LiCoO2 (1:49) catalyst, only a slight change was observed after the introduction of methanol into the electrolyte, indicating its high selectivity for the ORR in a methanol-containing environment. LSV curves before and after adding methanol were very similar (Fig. S6†), further proving that the Pt–LiCoO2 (1:49) catalyst possesses excellent tolerance to methanol, thus being a promising electrode material in DMFCs.
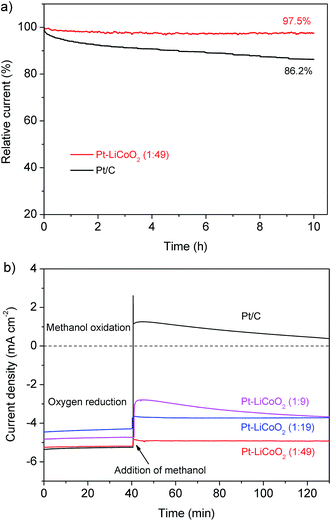 |
| | Fig. 4 (a) Current–time chronoamperometric responses of Pt–LiCoO2 (1:49) and Pt/C catalysts supported on the RDE (1600 rpm) at 0.65 V. (b) Chronoamperometric responses of various electrocatalysts supported on the RDE (1600 rpm) at 0.65 V in O2-saturated 0.1 M KOH electrolyte without methanol (0–40 min) and with 1 M methanol (40–130 min). | |
Electrochemical behaviour for OER
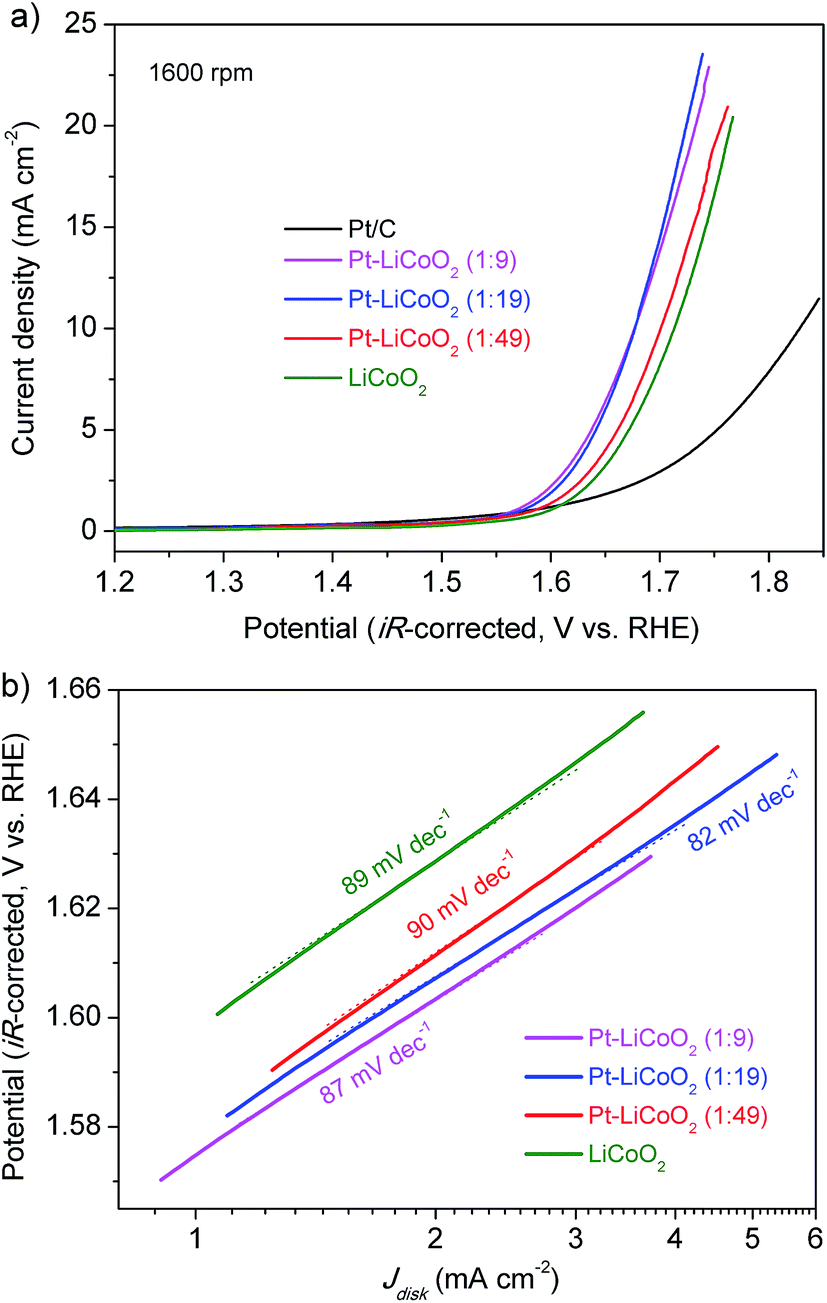
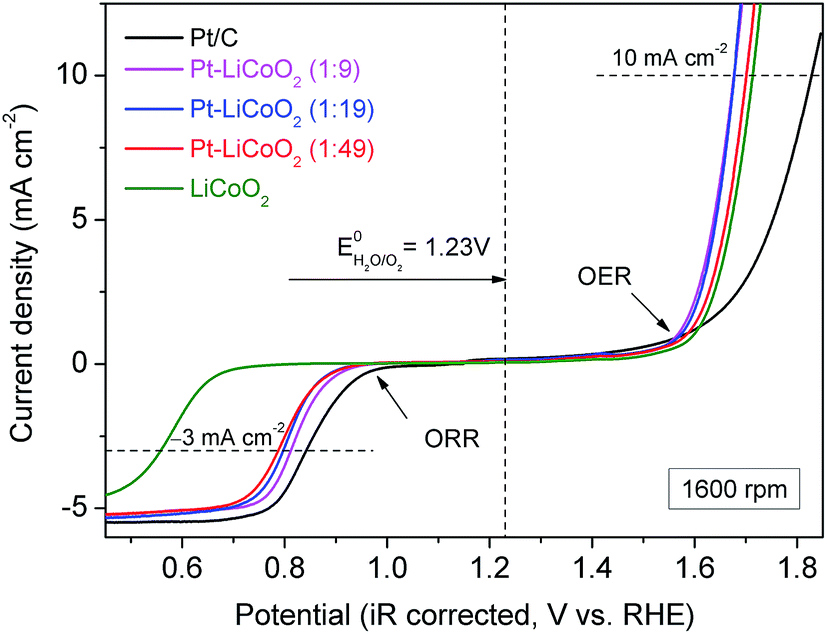
The electrocatalytic activity of the as-prepared composite catalysts for the OER was investigated by extending the potential window to the water oxidation range. Typical LSV curves are shown in Fig. 5a for Pt–LiCoO2 composites, and Pt/C and LiCoO2 catalysts at 1600 rpm. All the composite catalysts afforded higher OER current than either pristine LiCoO2 or Pt/C, suggesting their improved catalytic performances toward the OER. It is meaningful to compare the overpotential (η) for achieving a current density of 10 mA cm−2, which is a metric standard relevant to solar fuel synthesis.55 As shown in Fig. 5a, the values of η were calculated to be 0.60, 0.44, 0.44, 0.47 and 0.48 V for Pt/C, Pt–LiCoO2 (1:9, 1:19 and 1:49) and pristine LiCoO2 respectively based on the standard potential for the electrolysis of water to oxygen being 1.23 V. Our composite catalysts rendered small values of η, especially for Pt–LiCoO2 (1:9) and (1:19), showing the lowest η with the value of 0.44 V among all the tested catalysts. The OER kinetics of electrocatalysts were estimated by comparing the slopes of the corresponding Tafel plots. As can be seen in Fig. 5b, the smallest Tafel slope of 82 mV dec−1 was achieved for the Pt–LiCoO2 (1:19) catalyst, whereas the pristine LiCoO2 showed a higher value of 89 mV dec−1, similar to the reported value in the literature.56 These results clearly demonstrated that the composite catalysts possess high OER catalytic activity. The stability of the catalysts in the alkaline electrolyte was explored by carrying out 50 continuous potential cycles on the RDE at a rotation rate of 1600 rpm (Fig. S7 of the ESI†). The attenuation of the maximum current density within the tested potential range was 25% for the pristine LiCoO2 electrode after 50 cycles, while the current on the Pt–LiCoO2 (1:49) electrode decreased by 14% under the same conditions. It most likely results from the interaction between Pt and LiCoO2, even though only a slight amount of Pt existed in the composite.
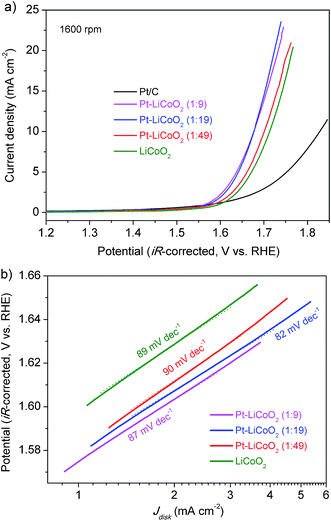 |
| | Fig. 5 (a) LSV curves and (b) corresponding Tafel plots of the various electrocatalysts on the RDE at 1600 rpm in O2-saturated 0.1 M KOH electrolyte at a scan rate of 5 mV s−1. | |
Bifunctional activity assessment
Finally, the overall oxygen electrocatalytic activity of the as-prepared composite catalysts is summarized in Fig. 6. To evaluate the bifunctional activity of the catalysts, the potential difference between the ORR and OER is a common indicator, defined as ΔE = EOER @ 10 mA cm−2 − EORR @ −3 mA cm−2. It is noteworthy that an ORR current density of −3 mA cm−2 is around the half-wave potential of the state-of-the-art Pt/C catalyst, while an OER current density of 10 mA cm−2 is almost equivalent to that of an ideal solar cell device with 10% efficiency.57 The smaller difference in the potential values indicates the better bifunctional activity of the catalyst. Table 1 lists the values of ΔE of our catalysts and other reported state-of-the-art bifunctional catalysts in 0.1 M KOH solution at a rotation rate of 1600 rpm. The ΔE values were 0.86, 0.87 and 0.91 V for Pt–LiCoO2 (1:9, 1:19 and 1:49) catalysts, respectively. Remarkably, these values fall into the lowest ΔE observed for all the excellent electrocatalysts in Table 1, only slightly higher than that of the Pt/C to BSCF/C catalyst = 1:4 (0.83 V), the best catalyst listed in Table 1. It should be noted that both the catalyst loading and the Pt content of our catalysts were lower than those of Pt/C to BSCF/C = 1:4. These are the possible reasons that resulted in the slightly lower bifunctionality of our electrocatalysts. Regardless, the as-prepared Pt–LiCoO2 composite catalysts prepared by the simple ultrasonic mixing rendered superb bifunctional activity for the ORR and OER. It suggests that the as-prepared composites could be the optimal bifunctional catalysts. As mentioned above, the high bifunctionality of the series of Pt–LiCoO2 composites could result from the synergistic effect between Pt and LiCoO2, achieved by the alteration of the electronic structure of Pt on the one hand, and by the modification of the adsorption/desorption behaviours of oxygen-containing species.
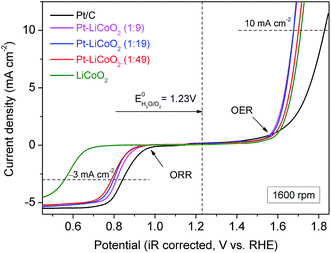 |
| | Fig. 6 Oxygen electrode activities of electrocatalysts within the potential window of the ORR and OER on the RDE in O2-saturated 0.1 M KOH electrolyte. | |
Table 1 Assessment of bifunctionality of Pt/C, Pt–LiCoO2 composites with different mass ratios, LiCoO2 and other catalysts reported in the literature in 0.1 M KOH solution at a rotation rate of 1600 rpm
| Sample |
ORR E @ −3 mA cm−2 (V) |
OER E @ 10 mA cm−2 (V) |
ΔE (V) |
Catalyst loading (mg cm−2disk) |
|
LT = low temperature; N-rGO = nitrogen-doped reduced graphene oxide; PCN = phosphorus-doped g-C3N4; CFP = carbon-fiber paper.
|
| 20 wt% Pt/C |
0.84 vs. RHE |
1.83 vs. RHE |
0.99 |
0.065 |
| Pt–LiCoO2 (1:9) |
0.81 vs. RHE |
1.67 vs. RHE |
0.86 |
0.065 |
| Pt–LiCoO2 (1:19) |
0.80 vs. RHE |
1.67 vs. RHE |
0.87 |
0.065 |
| Pt–LiCoO2 (1:49) |
0.79 vs. RHE |
1.70 vs. RHE |
0.91 |
0.065 |
| LiCoO2 |
0.56 vs. RHE |
1.71 vs. RHE |
1.15 |
0.065 |
| Pt/C to BSCF/C = 1:4 (ref. 45) |
0.76 vs. RHE |
1.59 vs. RHE |
0.83 |
0.26ORR |
| 0.19OER |
| LT-LiCoO2 (ref. 33)a |
∼0.55 vs. RHE |
∼1.70 vs. RHE |
1.15 |
0.25 |
| H–Pt/CaMnO3 (ref. 37) |
— |
— |
1.01 |
0.085 |
| 20 wt% Ir/C (ref. 55) |
0.69 vs. RHE |
1.61 vs. RHE |
0.92 |
0.028 |
| 20 wt% Ru/C (ref. 55) |
0.61 vs. RHE |
1.62 vs. RHE |
1.01 |
0.028 |
| Mn oxide (ref. 55) |
0.73 vs. RHE |
1.77 vs. RHE |
1.04 |
— |
| LaNiO3−δ (ref. 58) |
−0.32 vs. Ag/AgCl |
∼0.71 vs. Ag/AgCl |
1.03 |
0.26 |
| La0.3(Ba0.5Sr0.5)0.7Co0.8Fe0.2O3−δ (ref. 59) |
∼−0.32 vs. Hg/HgO |
∼0.68 vs. Hg/HgO |
1.00 |
0.64 |
| Fe3O4/graphene (ref. 60) |
∼0.6 vs. RHE |
1.78 vs. RHE |
1.18 |
0.20 |
| MnCoFeO4/N-rGO (ref. 61)a |
0.78 vs. RHE |
1.71 vs. RHE |
0.93 |
0.10 |
| La(Co0.55Mn0.45)0.99O3−δ/N-rGO (ref. 62)a |
∼−0.18 vs. Ag/AgCl |
∼0.78 vs. Ag/AgCl |
0.96 |
0.25 |
| α-MnO2 (ref. 63) |
0.76 vs. RHE |
1.72 vs. RHE |
0.96 |
0.20 |
| PCN on CFP (ref. 64)a |
∼0.67 vs. RHE |
1.63 vs. RHE |
0.96 |
0.20 |
Conclusions
In summary, active and stable bifunctional electrocatalysts for the ORR and OER were successfully fabricated via simply ultrasonic mixing Pt/C with LiCoO2. The Pt–LiCoO2 (1:49) catalyst with an ultralow Pt content of 2 wt% exhibited an ORR catalytic activity comparable to that of commercial Pt/C with a Pt content of 20 wt%. Notably, a super high ORR mass activity of 2.04 A mgPt−1 at 0.8 V was achieved for the Pt–LiCoO2 (1:49) catalyst, which was 21-fold higher than that of Pt/C. Meanwhile, the as-prepared composite catalyst also exhibited favourable OER activity with a lower overpotential and Tafel slope. Good stability and methanol tolerance for the ORR and OER were demonstrated from the composite catalysts in this study. The outstanding electrocatalytic activity could arise from the interaction between Pt/C and LiCoO2, which synergistically enhanced the bifunctionality of catalysts. The proposed simple method for preparing electrocatalysts with bifunctionality is a promising route to prepare practical Pt-based electrocatalysts with ultralow noble metal content, furthermore reducing the cost of electrochemical energy systems. The facile synthesis method and the effective performance enable our catalysts to be competitive candidates as the oxygen electrode of new energy devices, such as regenerative fuel cells and rechargeable metal–air batteries. Moreover, the present synthetic strategy can be generalized to produce highly active bifunctional catalysts by combining other ORR and OER catalysts with lower cost and higher activity.
Acknowledgements
The work was supported by the Australian Research Council Discovery Project grant DP160104835. Dr Chao Su acknowledges Curtin University for a postdoctoral fellowship. Dr Tao Yang acknowledges the FCT for financial support via the grant SFRH/BPD/86336/2012. The authors also acknowledge the use of equipment, scientific and technical assistance of the WA X-Ray Surface Analysis Facility, funded by the Australian Research Council LIEF grant LE120100026.
Notes and references
- L. Li, Z. Wu, S. Yuan and X. B. Zhang, Energy Environ. Sci., 2014, 7, 2101 CAS.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- J. S. Lee, S. T. Kim, R. Cao, N. S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34 CrossRef CAS.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390 CrossRef CAS PubMed.
- G. Chen, S. R. Bare and T. E. Mallouk, J. Electrochem. Soc., 2002, 149, A1092 CrossRef CAS.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043 CrossRef CAS PubMed.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557 CrossRef CAS PubMed.
- E. Mirzakulova, R. Khatmullin, J. Walpita, T. Corrigan, N. M. Vargas-Barbosa, S. Vyas, S. Oottikkal, S. F. Manzer, C. M. Hadad and K. D. Glusac, Nat. Chem., 2012, 4, 794 CrossRef CAS PubMed.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331 CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530 CrossRef CAS PubMed.
- Y. J. Sa, K. Kwon, J. Y. Cheon, F. Kleitzc and S. H. Joo, J. Mater. Chem. A, 2013, 1, 9992 CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81 CrossRef CAS PubMed.
- J. W. Hong, S. W. Kang, B. S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410 CrossRef CAS PubMed.
- S. Hu, G. Goenaga, C. Melton, T. A. Zawodzinski and D. Mukherjee, Appl. Catal., B, 2016, 182, 286 CrossRef CAS.
- Z. Li, Y. Li, S. P. Jiang, G. He and P. K. Shen, J. Mater. Chem. A, 2014, 2, 16898 CAS.
- S. Siracusano, N. V. Dijk, E. Payne-Johnson, V. Baglio and A. S. Aricò, Appl. Catal., B, 2015, 164, 488 CrossRef CAS.
- R. Cao, W. Lai and P. Du, Energy Environ. Sci., 2012, 5, 8134 CAS.
- K. L. Nardi, N. Yang, C. F. Dickens, A. L. Strickler and S. F. Bent, Adv. Energy Mater., 2015, 5, 1500412 Search PubMed.
- M. Orlandi, S. Caramori, F. Ronconi, C. A. Bignozzi, Z. E. Koura, N. Bazzanella, L. Meda and A. Miotello, ACS Appl. Mater. Interfaces, 2014, 6, 6186 CAS.
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897 CrossRef CAS PubMed.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Adv. Sci., 2016, 3, 1500187, DOI:10.1002/advs.201500187.
- C. Su, W. Wang, Y. Chen, G. Yang, X. Xu, M. O. Tadé and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 17663 CAS.
- R. Liu, F. Liang, W. Zhou, Y. Yang and Z. Zhu, Nano Energy, 2015, 12, 115 CrossRef CAS.
- J. Y. C. Chen, J. T. Miller, J. B. Gerken and S. S. Stahl, Energy Environ. Sci., 2014, 7, 1382 CAS.
- X. Yu, Z. Sun, Z. Yan, B. Xiang, X. Liu and P. Du, J. Mater. Chem. A, 2014, 2, 20823 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- G. Gardner, J. Al-Sharab, N. Danilovic, Y. B. Go, K. Ayers, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2016, 9, 184 CAS.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P. C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345, DOI:10.1038/ncomms5345.
- N. Colligan, V. Augustyn and A. Manthiram, J. Phys. Chem. C, 2015, 119, 2335 CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949, DOI:10.1038/ncomms4949.
- B. Han, D. Qian, M. Risch, H. Chen, M. Chi, Y. S. Meng and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 1357 CrossRef CAS PubMed.
- J. M. Jaksic, D. Labou, G. D. Papakonstantinou, A. Siokou and M. M. Jaksic, J. Phys. Chem. C, 2010, 114, 18298 CAS.
- V. T. T. Ho, C. J. Pan, J. Rick, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2011, 133, 11716 CrossRef CAS PubMed.
- X. Han, F. Cheng, T. Zhang, J. Yang, Y. Hu and J. Chen, Adv. Mater., 2014, 26, 2047 CrossRef CAS PubMed.
- Z. Awaludin, M. Suzuki, J. Masud, T. Okajima and T. Ohsaka, J. Phys. Chem. C, 2011, 115, 25557 CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- S. Mao, Z. Wen, T. Huang, Y. Hou and J. Chen, Energy Environ. Sci., 2014, 7, 609 CAS.
- S. Liu, W. Bian, Z. Yang, J. Tian, C. Jin, M. Shen, Z. Zhou and R. Yang, J. Mater. Chem. A, 2014, 2, 18012 CAS.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902 CAS.
- H. Wang, Y. Yang, Y. Liang, G. Zheng, Y. Li, Y. Cui and H. Dai, Energy Environ. Sci., 2012, 5, 7931 CAS.
- Y. Gao, H. Zhao, D. Chen, C. Chen and F. Ciucci, Carbon, 2015, 94, 1028 CrossRef CAS.
- Y. Zhu, C. Su, X. Xu, W. Zhou, R. Ran and Z. Shao, Chem.–Eur. J., 2014, 20, 15533 CrossRef CAS PubMed.
- J. Sunarso, A. A. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2012, 116, 5827 CAS.
- C. A. Hancock, A. L. Ong, P. R. Slater and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 3047 CAS.
- S. Jeong, S. Park and J. Cho, Adv. Energy Mater., 2011, 1, 368 CrossRef CAS.
- D. Qian, Y. Hinuma, H. Chen, L. S. Du, K. J. Carroll, G. Ceder, C. P. Grey and Y. S. Meng, J. Am. Chem. Soc., 2012, 134, 6096 CrossRef CAS PubMed.
- Y. Zhao, Y. Sha, Q. Lin, Y. Zhong, M. O. Tadé and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 1787 CAS.
- L. Dahéron, R. Dedryvère, H. Martinez, M. Ménétrier, C. Denage, C. Delmas and D. Gonbeau, Chem. Mater., 2008, 20, 583 CrossRef.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed.
- H. Liao and Y. Hou, Chem. Mater., 2013, 25, 457 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150 CrossRef CAS PubMed.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869 CrossRef CAS PubMed.
- W. Zhou and J. Sunarso, J. Phys. Chem. Lett., 2013, 4, 2982 CrossRef CAS.
- J. I. Jung, H. Y. Jeong, J. S. Lee, M. G. Kim and J. Cho, Angew. Chem., Int. Ed., 2014, 53, 4582 CrossRef CAS PubMed.
- B. Zhao, Y. Zheng, F. Ye, X. Deng, X. Xu, M. Liu and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 14446 CAS.
- Y. Zhan, C. Xu, M. Lu, Z. Liu and J. Y. Lee, J. Mater. Chem. A, 2014, 2, 16217 CAS.
- X. Ge, F. W. T. Goh, B. Li, T. S. A. Hor, J. Zhang, P. Xiao, X. Wang, Y. Zong and Z. Liu, Nanoscale, 2015, 7, 9046 RSC.
- Y. Meng, W. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452 CrossRef CAS PubMed.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta10492k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence