
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanofiller-tuned microporous polymer molecular sieves for energy and environmental processes†
Qilei
Song
*ab,
Shuai
Cao
c,
Robyn H.
Pritchard
a,
Hazim
Qiblawey
d,
Eugene M.
Terentjev
a,
Anthony K.
Cheetham
c and
Easan
Sivaniah
*ae
aCavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK. E-mail: q.song@imperial.ac.uk; esivaniah@icems.kyoto-u.ac.jp
bDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
cDepartment of Materials Science & Metallurgy, University of Cambridge, Cambridge CB3 0FS, UK
dDepartment of Chemical Engineering, College of Engineering, Qatar University, Qatar
eInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Kyoto 606-8501, Japan
First published on 25th November 2015
Abstract
Microporous polymers with molecular sieving properties are promising for a wide range of applications in gas storage, molecular separations, catalysis, and energy storage. In this study, we report highly permeable and selective molecular sieves fabricated from crosslinked polymers of intrinsic microporosity (PIMs) incorporated with highly dispersed nanoscale fillers, including nonporous inorganic nanoparticles and microporous metal–organic framework (MOF) nanocrystals. We demonstrate that the combination of covalent crosslinking of microporous polymers via controlled thermal oxidation and tunable incorporation of nanofillers results in high-performance membranes with substantially enhanced permeability and molecular sieving selectivity, as demonstrated in separation of gas molecules, for example, air separation (O2/N2), CO2 separation from natural gas (CH4) or flue gas (CO2/N2), and H2 separation from N2 and CH4. After ageing over two years, these nanofiller-tuned molecular sieves became more selective and less permeable but maintained permeability levels that are still two orders of magnitude higher than conventional gas separation membranes.
Introduction
Molecular separations are important for energy, environmental and chemical process industries, such as air separation, natural gas purification, H2 recovery and purification, carbon dioxide capture, desalination of water, and chemical separations in oil refinery processes. These processes have large benefits for society but also present environmental challenges that require cost and energy efficient technological solutions. Conventional separation processes, such as distillation or solvent scrubbing, require a lot of energy input to liquefy gases (cryogenic distillation for air separation), or evaporate liquids (thermal desalination of water, amine scrubbing, or distillation in oil refining processes) since these separations involve phase changes. Membrane separation is a promising alternative technology to these energy-intensive molecular separations. Both gases and liquids can be pressurized and fed to membranes that allow the selective separation of molecules in a single phase, avoiding the energy input for distillation or evaporation. Thus membrane separation processes, with high permeability and selectivity are more attractive for these molecular-scale separations owing to their high energy efficiency, low environmental impact, and compact units with small footprint.1Commercial molecular separation membranes are dominated by polymeric materials, the molecular transport in which follows the solution-diffusion mechanism, and where the permeability coefficient (P) is a product of solubility (S) and diffusion coefficient (D), P = S × D. The selectivity of different species (A over B), is defined by the ratio of their permeability, αA/B = PA/PB = (SA/SB) × (DA/DB), where SA/SB is the solubility selectivity and DA/DB is the diffusivity selectivity. However, conventional selective polymeric membranes present considerably low gas permeability and an inherent trade-off between permeability and selectivity exists, known as upper bound.2,3 This upper bound was explained theoretically by Freeman,4 who also predicted two strategies to enhance both the permeability and selectivity of polymeric membrane materials: (i) improving the solubility selectivity (SA/SB)5 or (ii) increasing the stiffness of polymer chains while maintaining large interchain spacing, such as thermally rearranged (TR) polymers,6,7 and polymers of intrinsic microporosity (PIMs).8–16
PIMs polymers are a class of polymers with rigid and contorted backbone structure.8–16 The chemical structure of a representative PIM-1 is shown in Fig. 1a. In the solid state, rigid polymer chains (as visualized in Fig. 1b) cannot pack efficiently, forming interconnected cavities behaving like micropores. The pore structure in PIMs molecular sieves can be visualized as the hour-glass shaped interconnected cavities, as shown in Fig. 1c. The molecule-sized pores and intermolecular interactions allow high solubility of gas molecules, particularly condensable gases, while the bottlenecks or gates interconnecting pores behave as sieves for separating gas molecules with different sizes. Owing to the unique pore structure, these polymeric molecular sieves achieve both higher permeability and higher selectivity.1,17 Further structural modifications of PIMs include substituting side functional groups that may introduce stronger intermolecular forces (i.e. hydrogen bonding),13,18,19 or design of more rigid polymer backbone structure,14,15,20–23 producing more selective or permeable molecular sieves.
 | ||
| Fig. 1 Thermal-oxidative crosslinking of PIM-1 polymer nanocomposites incorporated with nanofillers. (a) Chemical structure of PIM-1 polymer. (b) 3D model of PIM-1 polymer chain segment. (c) Schematic diagram of molecular sieve membranes fabricated from PIMs polymer showing hour-glass-shaped interconnected cavities for rapid and selective transport of gas molecules (e.g. CO2 and CH4). (d) Molecular structure of ZIF-8. Yellow regions indicate Connolly surface probed by H2 molecules. (e) Schematic diagram showing rigid polymer chains incorporated with nanofillers are covalently crosslinked to three-dimensional networks upon thermal-oxidative processing at suitable temperature (350–450 °C) in the presence of trace amount of oxygen. (f) SEM image of ZIF-8 nanocrystals. Cross-sectional SEM images of (g) PIM-1/ZIF-8 after annealing at 120 °C under vacuum (1 mbar), (h) TOX-PIM-1/ZIF-8 crosslinked at 385 °C for 24 h under vacuum (1 mbar), (i) PIM-1/ZIF-8 after annealing at 300 °C for 48 h under vacuum (1 mbar). (j) PIM-1/SiO2 annealed at 120 °C, (k) TOX-PIM-1/SiO2 annealed at 385 °C for 24 h under vacuum (1 mbar). Samples were fractured in liquid nitrogen and coated with a thin layer of gold. | ||
Mixed matrix membranes (MMMs), formed by incorporating molecular sieves or nanoscale fillers into polymer matrices, have also attracted significant interest.24–27 When the size of fillers in the polymer matrix is reduced to nanoscale, i.e. approximate to that of polymer chains, the properties of nanocomposite materials are strongly dependent on the interface between the polymer and fillers, where the local polymer chain mobility and conformation and their interactions vary significantly.26 Novel microporous materials, such as metal–organic frameworks (MOFs),28 with well-defined pores as shown in Fig. 1d, have been demonstrated as building blocks for molecular-sieving membranes,29–39 or as fillers in MMMs.40–47 Whilst often beneficial to membrane performance, the structure–property relationships of MMMs is complicated to predict, with diffusion and adsorption parameters varying from the bulk polymeric phase, to the interface with the fillers and potentially the internal surfaces of fillers.
PIM-1 polymer-based mixed matrix membranes have been prepared by solution mixing of PIM-1 polymer with various fillers including nonporous fillers and microporous materials, such as PIM-1/silica,48 PIM-1/ZIF-8,49 PIM-1/cages,50 PIM-1/POSS,51 PIM-1/MOFs,52 and PIM-1/porous aromatic frameworks (PAFs).53,54 In all of these studies, addition of fillers enhanced gas permeability of resulting composite membranes to certain extents, however the selectivity remained relatively unchanged (e.g. CO2/CH4 selectivity at 10–15) and far below the desirable level for practical gas separation technology, for example, CO2/CH4 selectivity at 30–40 for natural gas purification. PIM-1 polymer, the dominant phase in these composite membranes, limits the baseline of performance in gas separation. Novel approaches to production of molecular sieves and nanocomposites with both high permeability and high molecular selectivity are desirable for applications of the fast-growing class of microporous polymers including PIMs, for example, in membrane separations.
Recently, we reported that linear PIM-1 polymer can be transformed to a new class of highly crosslinked, insoluble, infusible, and microporous polymer networks (TOX-PIMs) via thermo-oxidative crosslinking,55 demonstrating control of membrane molecular sieving properties through control of the reaction kinetics. Such processing leads to membrane materials with excellent selectivity (e.g. CO2/CH4 up to 70) but with an unsurprising loss in overall gas permeability (e.g. CO2 permeability at 1000 Barrer). Since many microporous polymeric materials are subject to ageing that further reduces permeability, in this work, we sought to improve the control of gas permeability in thermo-oxidatively crosslinked TOX-PIM materials by incorporating nanoscale fillers. The resulting nanofiller-tuned crosslinked polymer molecular sieves show an excellent selectivity and maintain the permeability at a significant level, despite the effects of ageing.
Results
Approach
Our strategy of fabricating crosslinked nanocomposite membranes is visualized in Fig. 1e. Nanofillers are incorporated in rigid PIM polymer matrices and then exposed to controlled thermal oxidative crosslinking in the presence of trace amount of oxygen at suitable temperatures (350–450 °C). The thermal-oxidation and crosslinking reactions transform linear polymer chains to highly crosslinked, infusible, and insoluble networks where the size and shape selectivity (DA/DB) of gas molecules is enhanced in the polymer phase while maintaining high solubility (S) and solubility selectivity (SA/SB). On the other hand, nanofillers, regardless of their internal porosity, disrupt the packing of polymer chains and introduce extra free volume and defects at the interface owing to their high external surface area, consequently enhancing the absolute value of gas diffusivity (D) compared to pure polymer networks. Therefore, the combination of enhanced rigidity in polymer networks and nanofillers in composite membranes offers high permeability and sufficiently high selectivity for gas separation.Fabrication and characterisation of nanocomposite membranes
Polymer nanocomposite membranes were prepared using the solution mixing and casting method (ESI†). PIM-1 polymer was synthesized following the chemistry invented by Budd and McKeown (Fig. S1†).8 Nanoparticles were dispersed as colloids in chloroform and thoroughly mixed with PIM-1 polymer dissolved in chloroform giving a colloidal mixture, following our previously reported approach.56 In this study, two representative types of nanofillers were used: porous ZIF-8 nanocrystals (primary size of 70–100 nm, Fig. 1f) and nonporous inorganic nanoparticles (fumed silica, primary size of 12 nm). ZIF-8 was selected as a prototypical porous MOF owing to its easy synthesis in the form of nanoparticles, well-defined micropore structure, and high thermal stability. Silica was chosen as archetypal nonporous filler for polymer systems. After casting and slowly evaporating the solvent, we obtained self-standing, optically transparent and mechanically flexible polymer films (Fig. S2, ESI†). These PIM nanocomposite films were further exposed to thermal-oxidative crosslinking reactions, following the same protocol reported in our previous work.55 In this study, the thermal-oxidative crosslinking processing was performed by heating the polymer films at 385 °C under continuous vacuum (1 mbar) in a vacuum oven. Afterwards, crosslinked polymer and nanocomposite films became mainly insoluble both in traditional solvents for PIM-1, such as chloroform (see Fig. S2, ESI†), tetrahydrofuran or dichloromethane, and non-solvents (acetone, N-methyl-2-pyrrolidone, dimethylformamide, toluene, or alcohols). These crosslinked polymer films show certain degree of swelling in chloroform, confirming their polymeric nature. The optical transparency confirms the high degree of dispersion of small nanoparticles without formation of large particles or agglomeration, which would result in significant scattering of light and loss in transparency. Our high-magnification scanning electron microscopy (SEM) images of nanocomposite membranes, including PIM-1/ZIF-8 (Fig. 1g), PIM-1/SiO2 nanocomposites (Fig. 1j) clearly show that the nanofillers are well dispersed in the polymer, but we also observed the presence of mesopores in the polymer phase or around the nanoparticles. More SEM images are shown in Fig. S3–5 (ESI†). Furthermore, SEM-EDX analyses (Fig. S6 and S7, ESI†) confirmed the high dispersion of nanofillers in the crosslinked polymer matrices. Similar morphologies can be observed in other composites of PIM-1/nano-silica,48 PIM-1/ZIF-8,49 or PIM-1/cages crystals,50 where heterogeneous interfaces exist. The soluble fractions in most crosslinked samples are below 5 wt% and are of low molecular weight (Fig. S8, ESI†) indicating that a minimal degradation of the polymer occurs during the thermo-oxidative process. SEM images confirmed that the thermal treatment leads to densification of mesopores at the interfaces between polymer and nanofillers, as shown in Fig. 2h and k, though mesopores can still be observed. Thermal-oxidative degradations of PIM-1 polymer nanocomposites were confirmed in thermogravimetric analyzer (Fig. S9, ESI†), where the temperature and heating rate, concentration and flow rate of purging gas, and heat transfer could be well controlled. Therefore, the extent of thermal-oxidative crosslinking of polymer films could be carefully tailored by controlling the reaction kinetics. | ||
| Fig. 2 Gas sorption properties. (a) N2 adsorption–desorption isotherms of PIM-1 thin film, ZIF-8 nanocrystals, and PIM-1/ZIF-8 composite films at 77 K. Adsorption/desorption curves are shown as solid/open symbols, respectively. (b) Pore size distribution derived from the N2 adsorption isotherms based on NLDFT model. (c) N2 adsorption–desorption of PIM-1/ZIF-8 and crosslinked TOX-PIM-1/ZIF-8 composite films at 77 K. (d) Gas sorption isotherms of PIM-1/ZIF-8 (open symbols) and TOX-PIM-1/ZIF-8 (solid symbols) composites at 273 K, and (b) derived pore size distribution. All samples were degassed at 120 °C under high vacuum prior to gas sorption measurements. PIM-1 thin film (300 nm) was dried at 120 °C under vacuum; ZIF-8 nanocrystals were dried at 120 °C under vacuum; thick dense PIM-1/ZIF-8 film (∼50 μm) was dried at 120 °C under vacuum, with ZIF-8 loading at 20 wt%. TOX-PIM-1/ZIF-8 film was crosslinked by heating at 385 °C for 24 h under vacuum (1 mbar). | ||
We used X-ray photoemission spectroscopy (XPS) to probe the chemical bonding states in the surface of polymer films. The high-resolution XPS spectra of O1s, N1s, and C1s of PIM-1 and TOX-PIM-1 films are given in Fig. S10 (ESI†). The major difference is the observation of a higher shoulder peak at binding energies of 531.68 eV in O1s spectra, and a new broad weak peak at 288.78 eV in C1s spectra, both of which are associated with the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds owing to presence of residual oxidized groups in the TOX-PIM-1 film. The FTIR spectra of our TOX-PIM-1 and nanocomposite films (Fig. S11 and S12, ESI†) confirmed the presence of residual oxidised groups such as CO and O–H groups, a clear evidence of oxidation. The nitrile (C
O bonds owing to presence of residual oxidized groups in the TOX-PIM-1 film. The FTIR spectra of our TOX-PIM-1 and nanocomposite films (Fig. S11 and S12, ESI†) confirmed the presence of residual oxidised groups such as CO and O–H groups, a clear evidence of oxidation. The nitrile (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) groups are relatively stable and peaks associated with imine groups (CN) are not observable. These changes of chemical bonding correspond to the thermal oxidation of PIM-1 polymer, with several possible initiation sites for chain reactions and crosslinking (Fig. S13, ESI†).
N) groups are relatively stable and peaks associated with imine groups (CN) are not observable. These changes of chemical bonding correspond to the thermal oxidation of PIM-1 polymer, with several possible initiation sites for chain reactions and crosslinking (Fig. S13, ESI†).
It is well known that nanoparticles dispersed in the polymer matrix serve as spacers for polymer chains, so that the polymer phases in polymer–nanoparticle composites are equivalent to thin films.57 Here, the loose packing in composite membranes is also reflected by lower thermal conductivity (Fig. S14, ESI†). We also studied the mechanical properties of nanocomposite films with stress–strain measurements (Table S1, ESI†) and nanoindentation tests (Table S2, ESI†). These crosslinked polymer nanocomposites show a certain degree of decay in mechanical properties, compared to TOX-PIM-1 film. Nevertheless, these nanocomposite films are still mechanically resilient and can be bended or handled for subsequent high-pressure gas permeation tests.
For nanocomposites incorporated with MOF nanofillers, XRD analysis (Fig. S15, ESI†) confirmed the high crystallinity of ZIF-8 nanoparticles in the polymer matrix after annealing at moderate temperatures (<200 °C). However, thermal processing at relatively higher temperature over an extended period induced partial degradation of ZIF-8, as reflected by the broadening and lower peaks in XRD patterns of nanocomposite membranes or ZIF-8 nanocrystals alone (Fig. S16, ESI†). The partial oxidative degradation of ZIF-8 may be related to the removal of methyl groups.58 Since those partially degraded ZIF-8 nanoparticles still retain their size (Fig. S17, ESI†) and serve as spacers in nanocomposites, the physical effects of such degradation on the polymer network are not significant.
Gas sorption properties
The solubility of various gases in polymer membranes were probed with a series of gas sorption measurements. In our previous work,55 we found that thermal-oxidatively crosslinked PIM-1 polymer films show an interesting gate-opening phenomena in nitrogen adsorption at low temperature of 77 K. Therefore, N2 adsorption at 77 K is not effective to probe the pore structure of microporous polymers, due to the kinetically-limited diffusion of large N2 molecules. In contrast, for PIM-1 thin films or open ZIF-8 framework (Fig. 2a), the sorption of nitrogen was not diffusion-limited. Polymer nanocomposite films annealed at moderate temperatures (120 °C) showed enhanced N2 sorption at a low pressure stage, similar to that of PIM-1 thin films (300 nm).Fig. 2b confirms the microporosity of crystalline ZIF-8, and the broad size distribution of micropores in PIM-1 and composites. These N2 adsorption isotherms confirm that nanofiller-spaced polymer chains are loosely-packed and behave equivalently to thin films (surface area ∼ 800 m2 g−1). In contrast, crosslinked composite membranes show restricted sorption at low pressure (Fig. 2c), while the total quantity of adsorption at 1 bar does not significantly change. From a thermodynamic perspective, the polymer at low temperature of 77 K behaves as a ‘frozen’ state with lower segment mobility. Gas sorption isotherms of CO2, CH4 and N2 at 273 K (Fig. 2d) confirm that the gas solubility in thermal-oxidatively crosslinked nanocomposites is maintained at the same level as those unmodified samples. Pore size distributions derived from CO2 sorption isotherms at 273 K based on non-local density functional theory (NLDFT) method (Fig. S19, ESI†) indicates subtle modifications of ultramicropores (<7 Å). These adsorption isotherms confirm our hypothesis that thermal-oxidative crosslinking does not seriously diminish the fractional free volume (FFV); instead, the crosslinking primarily changes the size of gates and channels connecting free volume elements in the polymer phase, which result in significant changes in kinetics of gas diffusion.
Gas transport properties
Gas transport properties of polymer and nanocomposite films were measured with H2 (2.89 Å), CO2 (3.3 Å), O2 (3.46 Å), N2 (3.64 Å), and CH4 (3.82 Å) at 22 °C using a time-lag apparatus described in detail elsewhere.56Fig. 3a presents the gas permeability versus kinetic diameter of gas molecules for representative polymer films, showing their remarkable molecular sieving functions. As shown in Fig. 3a, the PIM-1 membrane is initially highly permeable, but after thermal oxidative crosslinking modification the resulting TOX-PIM-1 films maintain a relatively good permeability to small gas molecules such as H2 and CO2, while blocking large molecules. Compared to pure TOX-PIM-1 membranes, the gas permeabilities of crosslinked nanocomposite membranes containing silica and ZIF-8 nanoparticles are enhanced while the ideal gas selectivity decreases, both of which correlate with the volume fraction of fillers, as shown in Fig. 3d and e. For example, with the loading of silica nanoparticles increases from 1 wt% to 20 wt%, the CO2 permeability is enhanced from ∼1200 Barrer to ∼2615 Barrer, with an evident loss of the CO2/CH4 selectivity from ∼64 to ∼33. For crosslinked nanocomposites with ZIF-8 fillers increasing from 5 wt% to 20 wt%, the CO2 permeability further increases from 2745 Barrer to 3944 Barrer, with the CO2/N2 selectivity is maintained at ∼27 while the CO2/CH4 selectivity slightly decreases from 38 to 27. Detailed data are given in Table S3, ESI.† | ||
| Fig. 3 Gas transport properties. (a) Gas permeability (at 295 K) versus the kinetic diameter of gas molecules. (b) Solubility (at 295 K) versus the critical temperature of gas molecules. (c) Diffusivity (at 295 K) versus the square of effective diameter of gas molecules.59 In (a–c), the loading of SiO2 in TOX-PIM-1/SiO2 is 10 wt%, loading of ZIF-8 in TOX-PIM-1/ZIF-8 is 20 wt%. (d) Gas permeability, (e) ideal selectivity, (f) solubility (S), (g) solubility selectivity, (h) diffusion coefficient (D), and (i) diffusivity selectivity, as a function of the volume fraction of nanofiller in thermal-oxidatively crosslinked PIM-1 nanocomposite films. TOX-PIM-1 and nanocomposite films were oxidatively crosslinked at 385 °C for 24 h under vacuum (1 mbar). Data of nonporous nanosilica (solid symbols) and porous ZIF-8 (open symbols) at different loadings are plotted together in (d–i). TOX-PIM-1/ZIF-8 nanocomposites with loading of ZIF-8 nanocrystals at 5, 10, and 20 wt%, respectively. TOX-PIM-1/SiO2 nanocomposites with loading of silica nanoparticles at 1, 2, 5, 10, and 20 wt%, respectively. The volume fractions are derived based on weight loadings and densities of the polymer and nanofillers. Lines in (a–c), (d), and (e) are added to guide eyes. | ||
Based on the solution-diffusion model and time-lag measurements, gas solubilities and diffusivities of these nanocomposites are derived. As shown in Fig. 3b and f, the gas solubilities at 295 K are close to those of pure PIM-1 polymers or crosslinked networks. This agrees well with the observation of constant gas solubilities measured from gas sorption isotherms at 273 K, for example, CO2 solubility is maintained at about 0.5 cm3 cm−3 cmHg. The diffusivity of various gas molecules are shown in Fig. 3c and h. Clearly, the incorporation of nanofillers enhances the diffusion of gas molecules, for example, the CO2 diffusivity increases from 2.2 × 10−7 cm2 s−1 to 8.3 × 10−7 cm2 s−1. Therefore, the increase of permeability is attributed to enhanced diffusivity (Fig. 2d). With the increasing loading of nanofillers regardless of their internal porosity, the solubility selectivity (SA/SB) remained relatively constant (i.e. CO2/CH4 about 4.0) while the diffusivity selectivity (DA/DB) decreased, for example, diffusivity selectivity of CO2/CH4 dropped from 19 to 7 (Fig. 3i). It is well known that gas diffusivity in molecular sieves could change over multiple magnitudes depending on the shape and geometry of pore apertures (Fig. S20, ESI†). Previous work has shown that nanofillers disrupt the packing of rigid polymer chains and physically change the microstructure and free volume elements of polymer phase.26 Here, the enhanced diffusion and compromised selectivity at high loading of nanofillers is due to a combination of molecular sieving in polymer phase and enhanced diffusion through interfacial defects.
The effects of nanofillers and thermal processing are more evident when we compare the gas transport properties with the Robeson's upper bound (Fig. 4). For composite membranes containing nonporous nanofillers such as fumed silica nanoparticles, the gas permeability is enhanced while the ideal selectivity is compromised as a function of silica loadings. Such observations agree well with previous study on PIM-1/silica composites.48 In the instances of PIM-1/ZIF-8 composites, the gas permeability was also enhanced while the changes in ideal selectivity varied for different gas pairs. The permeability of H2 increases significantly with the loading of ZIF-8, while the corresponding gas selectivity to large gas molecules show slight increase, e.g. H2/N2 from 14 to 18, H2/CH4 from 12 to 15. Similarly, O2 permeability shows a remarkable enhancement from ∼1000 to ∼1500 Barrer while the O2/N2 selectivity increases from 4 to 5. Compared to nanocomposites containing nonporous nanosilica filler, the samples with ZIF-8 show relatively higher selectivity. The gas transport properties of these nanocomposite membranes are similar to previous studies,48–50 and are determined by a number of factors: (i) the increase of free volume in loosely packed polymer due to disruption of nanoparticles and alcohol-swelling treatment, (ii) microcavities and defects at the interface between polymer and nanoparticle, and (iii) rapid diffusion through the porous fillers, all of which could enhance the gas diffusivity and consequently permeability of the composite films.
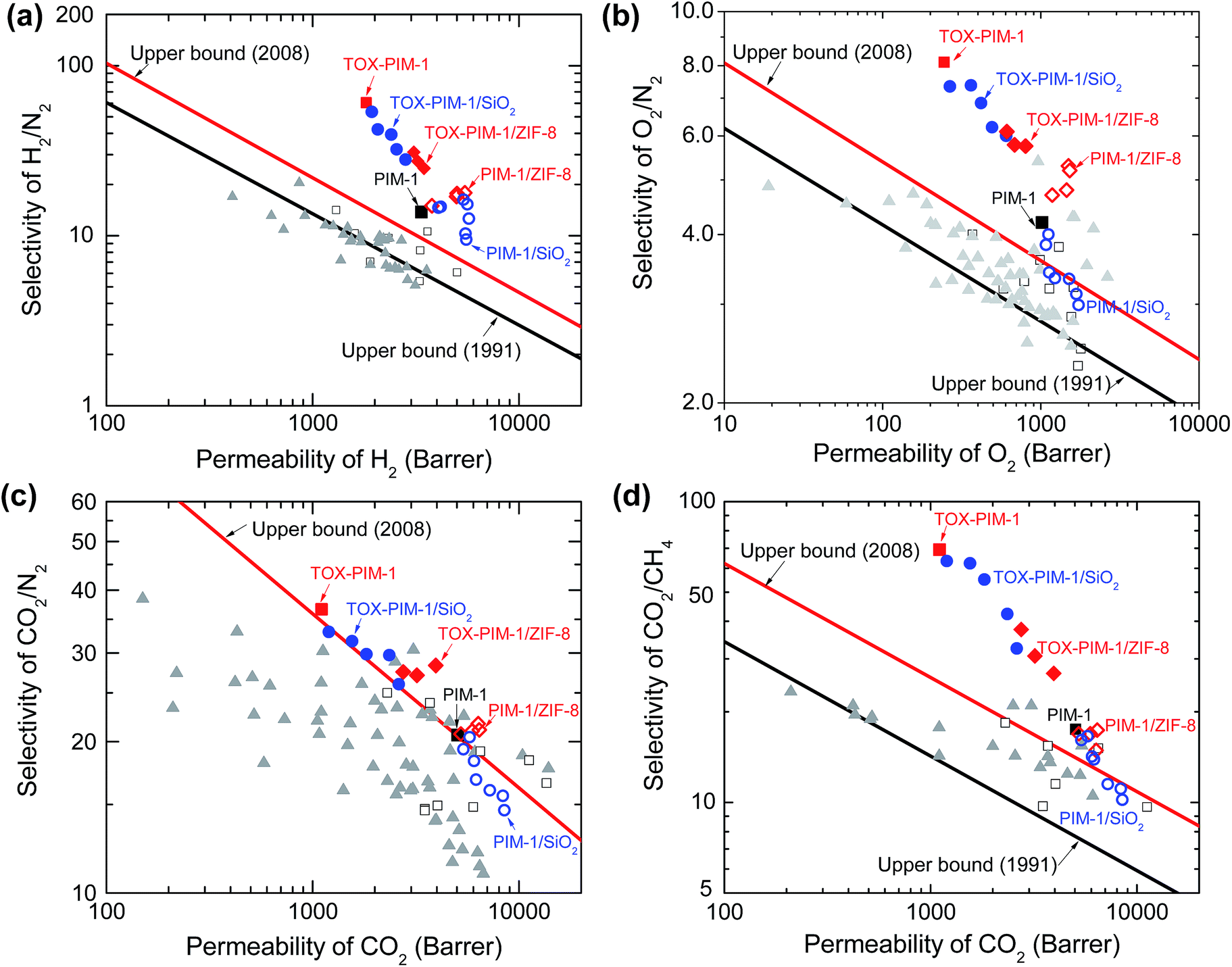 | ||
| Fig. 4 Upper bound plots of selectivity versus permeability for important gas pairs of H2/N2, O2/N2, CO2/N2, and CO2/CH4. Robeson upper bound reported in 1991 and 2008 are plotted.2,3 Open squares: PIM-1 in literature; solid triangles: other PIMs in literature;13–15,20–22 solid squares (black): unmodified PIM-1; solid squares (red): TOX-PIM-1 membranes. Blue open circles: PIM-1/SiO2 composites; blue solid circles: TOX-PIM-1/SiO2; red open diamonds: PIM-1/ZIF-8; red solid diamonds: TOX-PIM-1/ZIF-8. The gas transport properties were measured from pure gas permeation at 4 bar and room temperature of 22 °C. PIM-1 and PIM-1 based nanocomposite films were annealed at 120 °C for 24 h. PIM-1/SiO2 nanocomposites with loading of SiO2 nanoparticles (primary particle size 12 nm) at 1, 2, 5, 10, 20, 30, 40 wt%, respectively. TOX-PIM-1/ZIF-8 and TOX-PIM-1/SiO2 nanocomposite films were oxidatively crosslinked at 385 °C for 24 h under vacuum (1 mbar). Note: individual data points for each nanocomposite loading may be cross-referenced from Fig. 3. | ||
For our TOX-PIM-1 composite films after crosslinking, the selectivities of all gas pairs are enhanced to a very promising level, at a slight trade-off of permeability. For composites exposed to the same processing history as TOX-PIM-1 polymer films, incorporating nanofillers into crosslinked PIM-1 network enhances the permeability while the ideal selectivity of gas pairs decreases as a function of the volume fractions of nanofillers. These nanocomposites are still far more selective than non-crosslinked nanocomposites.48,49 Such significant enhancement of gas selectivity along with the combination of high permeability allows the overall gas separation performance of these nanocomposites to surpass the Robeson upper bound for a range of gas pairs (such as CO2/CH4, O2/N2, H2/N2 and H2/CH4).
Mixed gas transport properties
We also confirmed the enhanced gas permeability and excellent selectivity of thermal-oxidative crosslinked nanocomposite films in mixed gas permeation, with initially equimolar and CO2/CH4 mixture, as shown in Fig. 5a and b. For high free volume glassy polymers, CO2 and other condensable gases adsorbed in the polymer can increase the overall free volume and polymer chain segment mobility, termed as plasticization, which tends to result in lower selectivity at high CO2 partial pressure. At high pressure, the competing sorption of CO2 and CH4 further compromised the gas selectivity. Here, these thermo-oxidatively crosslinked networks show lower gas selectivity at high pressure, which are sufficiently selective for gas separation. The fractions of CH4 in the permeates through these selective films are below 5 vol% (CO2 purity above 95 vol%) (Fig. 5c), whereas it is as high 15 vol% for unmodified PIM-1 polymer membranes. The CO2/CH4 selectivity for commercial membranes (e.g. cellulose acetate) is about 20–30 below 10 bar and decreases to about 10–15 at higher pressure of 30 bar. Here, our membranes give higher selectivity at low pressure, therefore they produce similar gas purity, yet they are far more permeable than commercial membranes.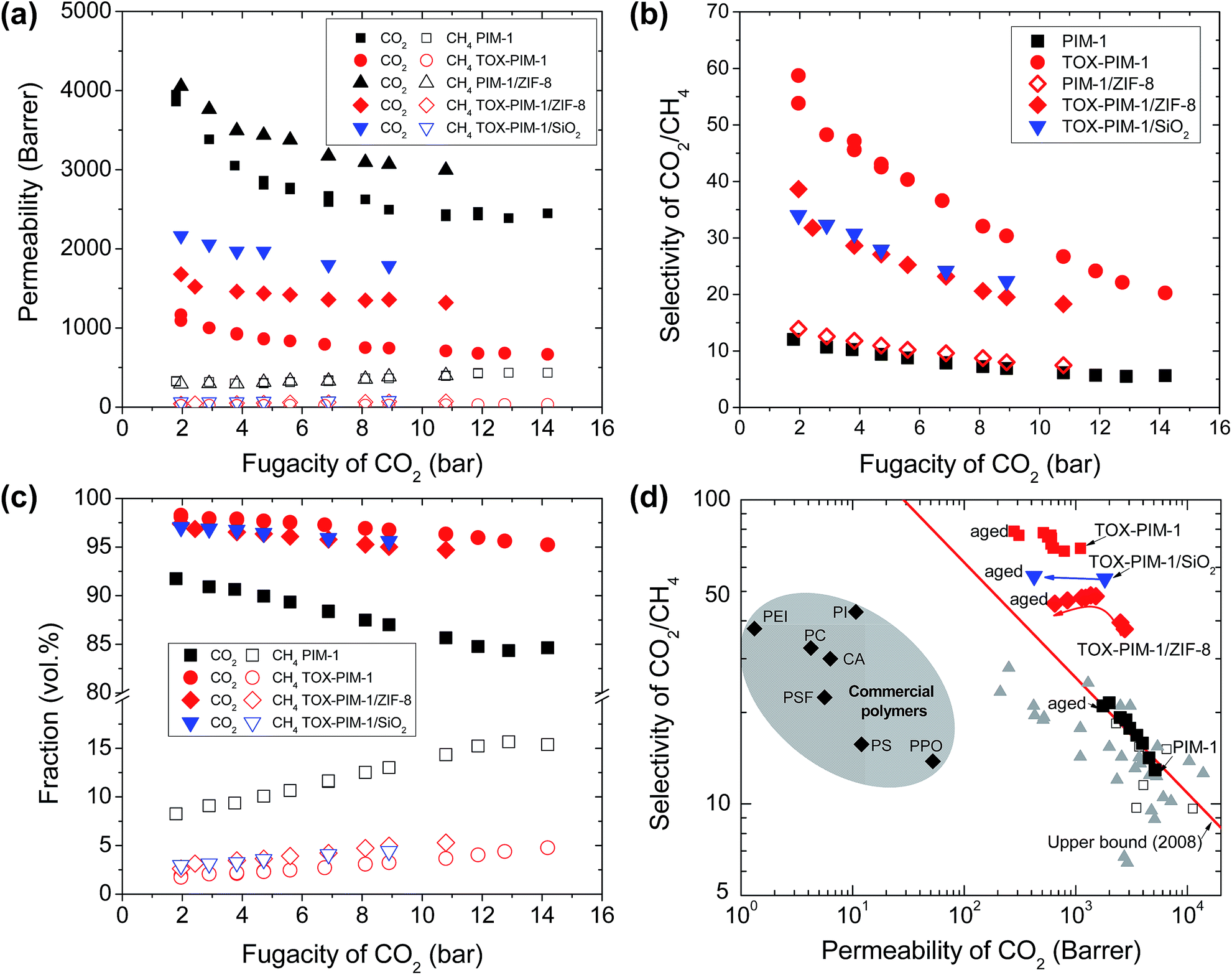 | ||
| Fig. 5 Mixed gas transport properties and aging behaviour. (a) Gas permeability versus the fugacity of CO2 in feeding gas, (b) selectivity CO2/CH4versus fugacity of CO2 in feeding gas mixture, and (c) permeate concentrations for separation of CO2/CH4 (50/50 vol%) gas mixture. (d) Upper bound plot showing the gas transport properties of polymer films during ageing for two years. Commercial polymers are included for comparison: cellulose acetate (CA); polycarbonate (PC); Matrimid® 5218 polyimide (PI); polyetherimide (PEI); polyphenyleneoxide (PPO); polysulfone (PSF), polystyrene (PS), cited from ref. 60. PIM-1 and nanocomposite films were annealed at 120 °C for 24 h under vacuum. Crosslinked PIM-1 and nanocomposites, TOX-PIM-1/ZIF-8 (10 wt%) and TOX-PIM-1/silica (10 wt%), were prepared by heating at 385 °C for 24 h under vacuum (1 mbar). (d) Aging of crosslinked polymer films. Films aged for over two years, PIM-1 for about 750 days, TOX-PIM-1 aged for about 750 days, TOX-PIM-1/SiO2 (5 wt%) aged for about 740 days, TOX-PIM-1/ZIF-8 (5 wt%) aged for about 740 days. For comparison, the data of TOX-PIM-1 membrane reported in a previous work55 are included. | ||
Ageing behaviour
Physical ageing, resulting in gas permeability reduction over time, is a critical issue that limits the industrial applications of membranes fabricated from glassy polymers. Such physical ageing is especially severe for high-free-volume polymers.61 PIMs membranes also suffer from the physical ageing because entangled polymer chains still tend to conform and pack tightly over time. Various approaches have been investigated, such as chemical crosslinking,62,63 and incorporation of additives such as porous aromatic frameworks.53,54 Our recent work showed that TOX-PIM-1 networks still suffer from ageing to some extent, with gas permeability rapidly decreases in 30 days but then slowly stabilized over time.55 In this study, the aged crosslinked nanocomposites and TOX-PIM-1 still show loss in permeability, but the gas permeability and selectivity are maintained at a remarkably high level after ageing over two years (Fig. S21, ESI†), as compared with the upper bound in Fig. 5d. Interestingly, the selectivity of smaller gas molecules to large ones increased to remarkably high level. For example, the H2 permeability decreased to 1250 Barrer, while the H2/CH4 selectivity increased from ∼100 to 350 after ageing. These two-years-aged nanocomposites are still relatively more permeable and less selective compared to aged TOX-PIM-1 films. Furthermore, these aged polymer films maintain high permeability levels that are still two orders of magnitude higher than conventional polymer materials used for gas separation membranes, as presented in Fig. 5d. These data confirm that the thermal-oxidative crosslinking is an effective approach to enhance the rigidity and restrict the pore collapse in microporous polymeric networks.Discussion
Although this study is focused on the crosslinked microporous PIM polymer network incorporated with nanofillers, we also investigated thermo-oxidation at intermediate temperature (i.e. 300 °C), which induced oxidative degradation instead of crosslinking. Oxidative degradation also results in changes in physical and chemical properties, e.g. densification of polymer/filler interface (Fig. 1i), loss of ZIF-8 crystallinity (Fig. S15 and S16, ESI†), lower molecular weight (Fig. S18, ESI†), and diffusion-restricted gas sorption (Fig. S19, ESI†). These changes are related to the heterogeneous structure of nanocomposites where thermal oxidative degradation is prone to happen owing to disrupted polymer thin film and enhanced O2 diffusion. The restricted diffusion as observed in gas sorption may be related to local denser molecular packing, and intermolecular interactions in the oxidized polymer matrix, e.g. hydrogen bonding between oxidized polymer chains which would enhance the local rigidity of polymer chains.55,64 In addition, for those degraded PIM-1/ZIF-8 composites, the polymer–nanofiller interfaces are more complex, e.g. oxidation of polymer, degradation of ZIF-8, and interactions between them, for example, hydrogen bonding may form between oxidized groups in polymer segments (CO) with imidazole (N–H) groups at the boundary of ZIF-8. These complex interactions induced by thermal oxidation would lead to significant changes in the pore structure and gas transport properties. This also indicates that it is possible to combine MOF nanofillers with polymers with well defined surface chemistry and interactions at the interfaces.
A further discussion is on the difference between porous MOFs fillers and inorganic nanoparticles. Owing to the high internal porosity of MOFs and low density, a very small amount of MOF filler (less than 20 wt%) could give sufficient high interfacial areas that drastically change the gas transport properties. Compared to inorganic nanoparticles, MOF fillers are relatively vulnerable to degradation owing to the organic nature, yet their porous structure and functionalities along with tuneable interfacial interactions with polymers would still make them favourable candidates for composites.
One of the important features of polymer nanocomposites is the large surface area of interfaces between nanofillers and polymers. For unmodified PIM-1 and the nanofillers used in this study, their interactions lacks strong forces such as hydrogen bonding or covalent interactions, therefore the interface is mainly dominated by physical dispersion and altered chain mobility and packing. Increasing the volume fraction of nanofillers could significantly increase the interfacial area which controls the properties of nanocomposites including gas diffusion. Still the synergistic combination of covalent crosslinking of polymer phases with enhanced molecular sieving selectivity and enhanced diffusivity at interfaces introduced by dispersed nanoparticles lead to the overall enhanced permeability and sufficiently high selectivity that do not exist for covalently crosslinked polymer alone.
In this study we probed the structure of thick polymer nanocomposite films by gas adsorption and diffusion, whereas thin films would be used practically for membrane separation applications. The gas transport properties in thin film membranes would be different, particularly the ageing behaviour. Previously we have successfully demonstrated that crosslinking is also effective for PIM-1 thin films (∼100 nm) coated on inorganic substrates, research on thermally crosslinked thin film membranes is in progress and will be reported in future.
In a broad context, our work improves the fundamental understanding on molecular transport in the new class of PIMs polymers and nanocomposites. Since PIMs and other microporous polymers have been demonstrated as of great potential for applications, such as gas storage, adsorbents, sensors, catalyst supports, separators for batteries, membranes for gas separation and organic solvent nanofiltration, and hydrocarbons separation for oil refinery processes, we expect that our approach of controlled thermal processing and nanocomposites could be extended to fabrication and processing of the rapid-growing large family of microporous polymers including PIMs and functional nanocomposites for a wide range of applications.
Conclusions
In summary, we demonstrate an approach to the design of polymer molecular sieves fabricated from thermo-oxidatively crosslinked PIM-1 polymer networks incorporated with nonporous inorganic nanoparticles or microporous MOF nanocrystals. Thermal oxidative crosslinking of the PIM-1 polymer phase substantially enhances the gas selectivity of nanocomposites while the gas permeability remains at a remarkably high level. Incorporations of nanoscale fillers, either porous or nonporous, in the crosslinked PIM-1 polymer networks introduce extra channels and heterogeneous interfaces for enhanced diffusion of gas molecules while the gas solubility remains constant, consequently enhancing the overall gas permeability while maintaining sufficiently high selectivity. These highly permeable and selective nanocomposites are promising for a wide range of industrially important molecular separations in energy and environmental processes, such as removal of CO2 from natural gas, shale gas purification, biogas purification, air separation, hydrogen separation and recovery, and other molecular-level separations. Our report, with two-year ageing studies, also indicates the importance of enhancing permeability in this high-free-volume glassy polymer through nanofiller additions. We expect that our approach and findings would have broad scientific implications on understanding the structure and molecular transport phenomena in novel microporous polymeric molecular sieves and polymer nanocomposites, for a broad range of applications beyond gas separations.Acknowledgements
This work was financially supported by the Engineering and Physical Sciences Research Council (EPSRC, UK), European Research Council (ERC) and an NPRP grant from the QNRF (Qatar). Q. S. acknowledges the financial support of China Scholarship Council and an Imperial College Junior Research Fellowship.Notes and references
- D. L. Gin and R. D. Noble, Science, 2011, 332, 674 CrossRef CAS PubMed.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390 CrossRef CAS.
- B. D. Freeman, Macromolecules, 1999, 32, 375 CrossRef CAS.
- H. Lin, E. van Wagner, B. D. Freeman, L. G. Toy and R. P. Gupta, Science, 2006, 311, 639 CrossRef CAS PubMed.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254 CrossRef CAS PubMed.
- H. B. Park, S. H. Han, C. H. Jung, Y. M. Lee and A. J. Hill, J. Membr. Sci., 2010, 359, 11 CrossRef CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610 CrossRef CAS PubMed.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851 CrossRef CAS.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372 CrossRef CAS PubMed.
- C. G. Bezzu, M. Carta, A. Tonkins, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Adv. Mater., 2012, 24, 5930 CrossRef CAS PubMed.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303 CrossRef CAS PubMed.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804 CrossRef CAS PubMed.
- A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau and M. D. Guiver, Macromolecules, 2009, 42, 6038 CrossRef CAS.
- C. R. Mason, L. Maynard-Atem, N. M. Al-Harbi, P. M. Budd, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Macromolecules, 2011, 44, 6471 CrossRef CAS.
- N. Du, H. B. Park, M. M. Dal-Cin and M. D. Guiver, Energy Environ. Sci., 2012, 5, 7306 CAS.
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526 CrossRef CAS PubMed.
- B. S. Ghanem, R. Swaidan, E. Litwiller and I. Pinnau, Adv. Mater., 2014, 26, 3688 CrossRef CAS PubMed.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, J. D. Selbie and D. Fritsch, Adv. Mater., 2008, 20, 2766 CrossRef CAS PubMed.
- W. J. Koros and R. Mahajan, J. Membr. Sci., 2000, 175, 181 CrossRef CAS.
- T. S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483 CrossRef CAS.
- T. C. Merkel, B. D. Freeman, R. J. Spontak, Z. He, I. Pinnau, P. Meakin and A. J. Hill, Science, 2002, 296, 519 CrossRef CAS PubMed.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 211, 311 CrossRef CAS.
- H. Hayashi, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS PubMed.
- Y. Peng, Y. S. Li, Y. J. Ban, H. Jin, W. M. Jiao, X. L. Liu and W. S. Yang, Science, 2014, 346, 1356 CrossRef CAS PubMed.
- A. Huang, W. Dou and J. Caro, J. Am. Chem. Soc., 2010, 132, 15562 CrossRef CAS PubMed.
- H. Bux, F. Liang, Y. Li, J. Cravillon, M. Wiebcke and J. Caro, J. Am. Chem. Soc., 2009, 131, 16000 CrossRef CAS PubMed.
- S. Wang, G. Wang, F. Jiang, M. Luo and H. Li, Energy Environ. Sci., 2010, 3, 1353 CAS.
- H. Bux, C. Chmelik, J. M. van Baten, R. Krishna and J. Caro, Adv. Mater., 2010, 22, 4741 CrossRef CAS PubMed.
- H. Bux, A. Feldhoff, J. Cravillon, M. Wiebcke, Y. S. Li and J. Caro, Chem. Mater., 2011, 23, 2262 CrossRef CAS.
- A. Huang and J. Caro, Angew. Chem., Int. Ed., 2011, 50, 4979 CrossRef CAS PubMed.
- A. Huang, N. Wang, C. Kong and J. Caro, Angew. Chem., Int. Ed., 2012, 51, 10551 CrossRef PubMed.
- A. J. Brown, J. R. Johnson, M. E. Lydon, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem., Int. Ed., 2012, 51, 10615 CrossRef CAS PubMed.
- J. Caro, Curr. Opin. Chem. Eng., 2011, 1, 77 CrossRef CAS.
- A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones and S. Nair, Science, 2014, 345, 72 CrossRef CAS PubMed.
- T.-H. Bae, J. S. Lee, W. Qiu, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem., Int. Ed., 2010, 49, 9863 CrossRef CAS PubMed.
- T. Yang, Y. Xiao and T. S. Chung, Energy Environ. Sci., 2011, 4, 4171 CAS.
- C. Zhang, Y. Dai, J. R. Johnson, O. Karvan and W. J. Koros, J. Membr. Sci., 2012, 389, 34 CrossRef CAS.
- M. J. C. Ordoñez, K. J. Balkus Jr, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28 CrossRef.
- X.-L. Liu, Y.-S. Li, G.-Q. Zhu, Y.-J. Ban, L.-Y. Xu and W.-S. Yang, Angew. Chem., Int. Ed., 2011, 50, 10636 CrossRef CAS PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2014, 14, 48 CrossRef PubMed.
- S. Kanehashi, G. Q. Chen, C. A. Scholes, B. Ozcelik, C. Hua, L. Ciddor, P. D. Southon, D. M. D'Alessandro and S. E. Kentish, J. Membr. Sci., 2015, 482, 49 CrossRef CAS.
- T.-H. Bae and J. R. Long, Energy Environ. Sci., 2013, 6, 3565 CAS.
- J. Ahn, W. J. Chung, I. Pinnau, J. Song, N. Du, G. P. Robertson and M. D. Guiver, J. Membr. Sci., 2009, 346, 280 CrossRef.
- A. F. Bushell, M. P. Attfield, C. R. Mason, P. M. Budd, Y. Yampolskii, L. Starannikova, A. Rebrov, F. Bazzarelli, P. Bernardo, J. Carolus Jansen, M. Lanč, K. Friess, V. Shantarovich, V. Gustov and V. Isaeva, J. Membr. Sci., 2013, 427, 48 CrossRef CAS.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253 CrossRef CAS PubMed.
- W. F. Yong, K. H. A. Kwek, K.-S. Liao and T.-S. Chung, Polymer, 2015, 77, 377 CrossRef CAS.
- S. J. D. Smith, B. P. Ladewig, A. J. Hill, C. H. Lau and M. R. Hill, Sci. Rep., 2015, 5, 7823 CrossRef CAS PubMed.
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669 CrossRef CAS PubMed.
- C. H. Lau, P. T. Nguyen, M. R. Hill, A. W. Thornton, K. Konstas, C. M. Doherty, R. J. Mulder, L. Bourgeois, A. C. Y. Liu, D. J. Sprouster, J. P. Sullivan, T. J. Bastow, A. J. Hill, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2014, 53, 5322 CrossRef CAS PubMed.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef CAS PubMed.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359 CAS.
- A. Bansal, H. Yang, C. Li, K. Cho, B. C. Benicewicz, S. K. Kumar and L. S. Schadler, Nat. Mater., 2005, 4, 693 CrossRef CAS PubMed.
- S. Gadipelli, W. Travis, W. Zhou and Z. Guo, Energy Environ. Sci., 2014, 7, 2232 CAS.
- V. Teplyakov and P. Meares, Gas Sep. Purif., 1990, 4, 66 CrossRef CAS.
- V. Abetz, T. Brinkmann, M. Dijkstra, K. Ebert, D. Fritsch, K. Ohlrogge, D. Paul, K. V. Peinemann, S. P. Nunes, N. Scharnagl and M. Schossig, Adv. Eng. Mater., 2006, 8, 328 CrossRef CAS.
- K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721 CrossRef CAS.
- N. Du, M. M. D. Cin, I. Pinnau, A. Nicalek, G. P. Robertson and M. D. Guiver, Macromol. Rapid Commun., 2011, 32, 631 CrossRef CAS PubMed.
- N. Du, M. M. Dal-Cin, G. P. Robertson and M. D. Guiver, Macromolecules, 2012, 45, 5134 CrossRef CAS.
- Q. Song, S. Cao, P. Zavala-Rivera, L. P. Lu, W. Li, Y. Ji, S. A. Al-Muhtaseb, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2013, 4, 1918 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, synthesis of polymer, molecular weight distribution of polymers, thermal analysis, XPS spectra, FTIR spectra, SEM images of membranes, XRD patterns of ZIF-8 and composites, gas sorption isotherms, and detailed data of gas transport properties. See DOI: 10.1039/c5ta09060a |
| This journal is © The Royal Society of Chemistry 2016 |