
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution processed graphene structures for perovskite solar cells†
Munkhbayar
Batmunkh
ab,
Cameron J.
Shearer
 b,
Mark J.
Biggs
ac and
Joseph G.
Shapter
*b
b,
Mark J.
Biggs
ac and
Joseph G.
Shapter
*b
aSchool of Chemical Engineering, The University of Adelaide, Adelaide, South Australia 5005, Australia
bCentre for Nanoscale Science and Technology, School of Chemical and Physical Sciences, Flinders University, Bedford Park, Adelaide, South Australia 5042, Australia. E-mail: joe.shapter@flinders.edu.au; Tel: +61 8 82012005
cSchool of Science, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
First published on 25th January 2016
Abstract
Organometallic trihalide perovskite light absorber based solar cells have drawn increasing attention because of their recent rapid increase in power conversion efficiency (PCE). These photovoltaic cells have relied significantly on transparent conducting oxide (TCO) electrodes which are costly and brittle. Herein, solution processed transparent conductive graphene films (TCGFs) are utilized, for the first time, as an alternative to traditional TCO electrodes at the electron collecting layer in perovskite solar cells (PSCs). By investigating and optimizing the trade-off between transparency and sheet resistance (Rs) of the graphene films, a PCE of 0.62% is achieved. This PCE is further improved to 0.81% by incorporating graphene structures into both compact and mesoporous TiO2 layers of the solar cell. We anticipate that the present study will lead to further work to develop graphene-based transparent conductive electrodes for future solar cell devices.
Introduction
Photovoltaic (PV) cells are devices that convert sunlight directly into electrical power and have great potential to meet society's continuously increasing energy demands with negligible environmental impact.1 The current PV market is mainly dominated by crystalline silicon (1st generation) and compound semiconductor (2nd generation) based solar cells, which can produce energy with a power conversion efficiency (PCE) that is the highest of all solar cell technologies.2,3 These commercially available solar devices are, however, produced using complex, high-cost manufacturing processes. Recently reported solar cells based on hybrid organometallic halide perovskites are considered the most promising alternatives to the more established solar cell technologies because of their relatively high PCE, and simpler, cheaper fabrication processes.4–7Organic–inorganic halide structures (such as CH3NH3PbX3 (X = Cl, I or Br)), called perovskite materials, have been known for several decades and have recently attracted much attention from the PV community owing to some key exceptional properties.8 These properties include the ability to absorb significant levels of incident light across a wide part of the solar spectrum, and the ability to effectively carry the photoelectrons created from the incident light away into a circuit.9 The PCE of perovskite solar cells (PSCs) has rapidly increased from less than 4% to more than 20% in only 6 years,10–13 making the efficiency comparable with current commercial technologies.12,13
A typical PSC is composed of a transparent conducting oxide (TCO) (indium-doped and/or fluorine-doped tin oxide (ITO or FTO)) electrode, a thin compact hole blocking (TiO2) layer, a perovskite layer with or without a porous metal oxide scaffold layer, a hole transporting layer (HTL) and a metal contact (Au or Ag).11,14,15 In such a device structure, the TCO electrode plays a vital role in collecting electrons from the semiconducting TiO2 and transferring them to the external circuit. However, limited resources of the materials used in typical TCO electrodes and consequent high cost are major issues.16 Additionally, their brittle nature and high structural defects are a major concern for PSC technologies where ease of transportation, handling and installation are important.17 Therefore, the replacement of TCO electrodes with cheaper and robust alternatives is desirable.
Graphene has attracted considerable interest for potential applications in various optoelectronic devices due to its properties including excellent conductivity, low cost and high flexibility.18,19 Moreover, compared to ITO and FTO, graphene has several advantages such as abundance, high transparency in the near-infrared region and high stability in the presence of acid or base.16,20 These unique properties suggest graphene films could be a possible replacement for TCO electrodes. To date, two main processes have been developed for the fabrication of graphene films.21 The first is based on chemical vapor deposition (CVD) of graphene using a metal sheet catalyst (Cu or Ni), followed by transfer printing to target substrates. However, CVD is expensive and its operation is complicated while it also requires high temperatures (>750 °C). Alternatively, solution processed graphene has been considered a promising future electrode material because it can be deposited on large-area flexible substrates and is compatible with roll-to-roll manufacturing techniques.22 Based on these advantages, solution processed graphene films have been used as transparent electrodes for inorganic–organic hybrid solar cells,23,24 organic photovoltaic cells25,26 and dye-sensitized solar cells (DSSCs).27 In addition, CVD processed-graphene based transparent conductive films have very recently been employed as hole collecting electrodes in PSCs even though they are costly and difficult to produce.28,29 However, until now, there has been no effort in the application of graphene based transparent and conductive films to replace traditional TCO electrodes in PSCs despite recent reviews30,31 and a computational study32 suggesting some promise.
In the work reported here, transparent conductive graphene films (TCGFs) prepared from low-temperature processed and chemically derived graphene (or solution processed graphene, Scheme 1) have been employed as a substitute for the electron collecting TCO electrode to test their feasibility in PSCs. Furthermore, the incorporation of graphene structures into semiconducting oxide scaffolds has been shown to be a promising strategy to enhance the efficiency in DSSCs.33 After optimizing sheet resistance (Rs) and light transmittance for PSC performance, we further improved the PCE by employing graphene into both compact and mesoporous TiO2 layers of the devices.
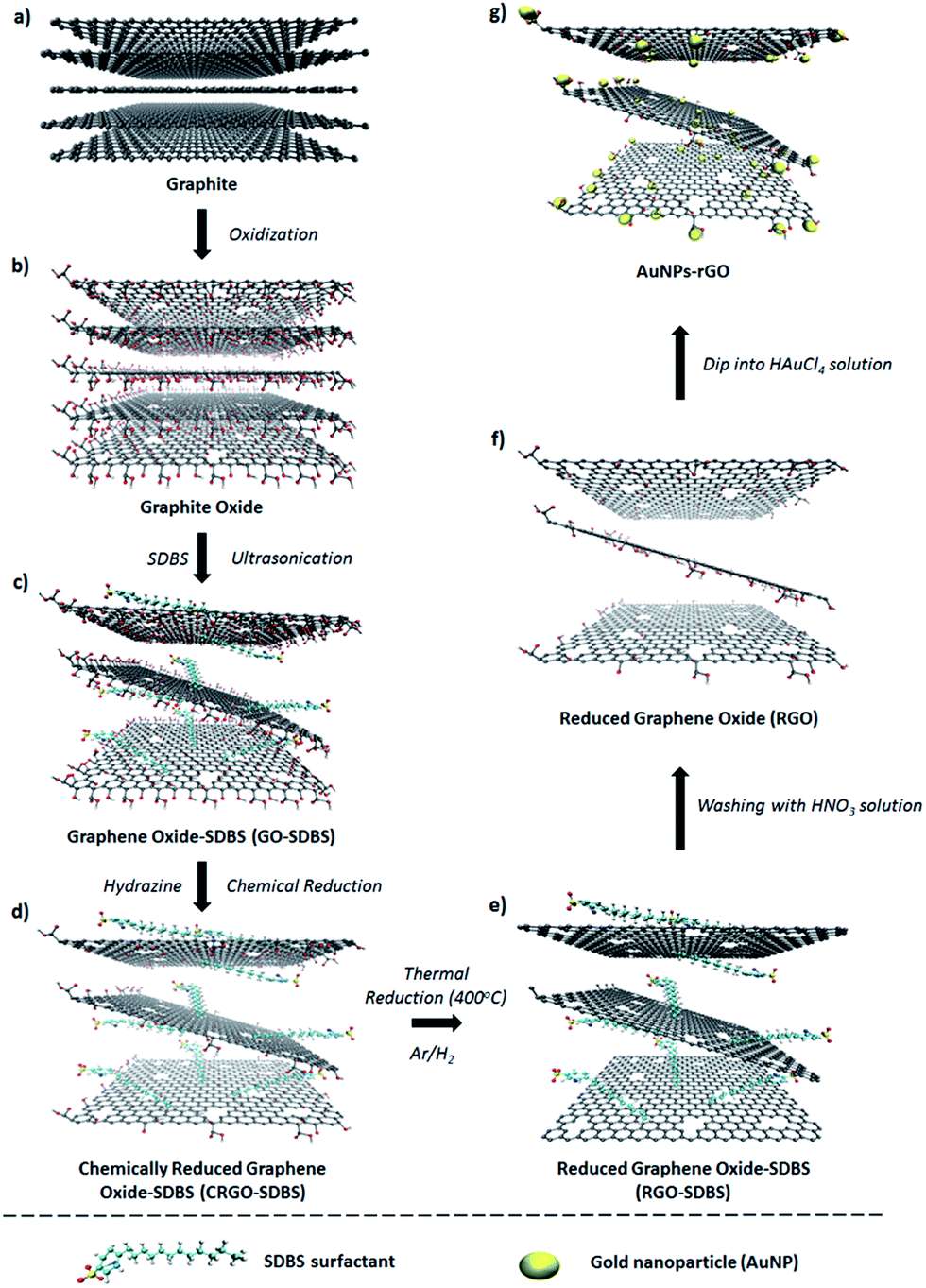 | ||
| Scheme 1 Schematic of the preparation procedure of graphene films. | ||
Results and discussion
Preparation and characterization of graphene films
Graphite oxide was synthesized from natural graphite by an improved Hummers method34 followed by exfoliation to produce graphene oxide (GO) sheets (Scheme 1a–c). A detailed description of the process is given in the experimental section. The prepared GO is known to be electrically non-conductive and the removal of its functional groups is necessary to obtain conductive graphene-based materials.17 In general, GO can be reduced by using chemical agents such as hydrazine or sodium borohydride.35 However, the insolubility of the GO after such chemical reduction limits its further application. In order to tackle this limitation; we added sodium dodecylbenzene sulfonate (SDBS) surfactant into the graphite oxide solution before the exfoliation step (Scheme 1c).36,37In a typical experiment, large-area GO with or without SDBS surfactant was produced by the exfoliation of the previously prepared graphite oxide solution (Scheme 1c). It should be noted that the prepared GO aqueous dispersion was very stable without any precipitation for several months, which is known to be due to the presence of hydrophilic groups (e.g., hydroxyl, epoxy, or carboxyl) on the surface of graphene.21 Subsequently, the chemical reduction of GO aqueous solution was carried out with hydrazine solution in the presence of SDBS. For comparison, the same procedure was also performed in the absence of SDBS. Chemically reduced graphene oxide (CRGO-only) without surfactant disperses poorly in aqueous conditions because of its hydrophobic surface after the removal of oxygen containing functional groups during the reduction process.23 Subsequently, strong π–π interaction between CRGO flakes leads to agglomeration and poor dispersion (inset of Fig. 1a). The atomic force microscopy (AFM) image (Fig. 1a) shows that the CRGO-only flakes without SDBS are aggregated or stacked on each other and their lateral size was measured to be smaller than 1 μm, which is consistent with the results reported in the literature.38,39 In contrast, the SDBS supported CRGO (Scheme 1d, termed “CRGO–SDBS”) showed dramatically improved dispersion in aqueous solution. As illustrated in the inset of Fig. 1b, no precipitate was observed and the solution was stable for several months. More importantly, the flake size of the CRGO–SDBS (Fig. 1b) was significantly larger than that of CRGO-only (Fig. 1a).40 It is known that sonication and conventional chemical reduction steps of GO create many structural defects, decrease the flake size and increase the degree of sp3 hybridization.21 Interestingly, in this study, the SDBS acts to prevent CRGO from fracturing during ultrasonication resulting in large-size graphene sheets. The large-sized graphene structures should, in principle, exhibit lower Rs when used in transparent conductive films because the larger flakes will have less charge scattering related to charge hopping through sheet–sheet contacts in the film.41,42
 | ||
| Fig. 1 AFM images (5 × 5 μm2) of chemically reduced graphene oxide (CRGO) (a) without and (b) with SDBS. Insets show digital photographs of the corresponding samples in an aqueous 1 mg mL−1 solution. | ||
Structural information for these samples was obtained using Raman spectroscopy. It is well known that the intensity ratio (ID/IG) is usually used to determine the level of defects.43 Raman spectra (see Fig. S1†) shows that the ID/IG value of the CRGO–SDBS is lower than that of the CRGO without surfactant, confirming that the chemical (hydrazine) reduction of GO in the presence of SDBS creates less defects on the CRGO compared to the number produced without any surfactant present. Although the use of SDBS during the chemical reduction process has the additional advantage of preventing defect production in the CRGO and providing large graphene sheets, the presence of residual SDBS surfactant may degrade the electrical properties of the graphene films because of its highly insulating nature.36 Therefore, removing SDBS surfactant from the prepared films is of great importance for maximizing the electrical conductivity of the films. In addition, it is well known that the chemical reduction with hydrazine alone is not sufficient to fully reduce the oxygen containing functional groups from the graphene layers.44
In order to improve the quality of graphene structures, the films were prepared from the CRGO–SDBS solution using a vacuum-filtration and transfer technique44 and have been thermally annealed at a temperature of 400 °C under the protection of an Ar and H2 gas flow. Interestingly, we observed that the filtration time for the CRGO–SDBS solution was relatively longer than that for the CRGO-only samples. We attribute this phenomenon to the size of the graphene sheets with the larger CRGO–SDBS sheets blocking the filter paper pores faster. After the thermal annealing of CRGO–SDBS film, the resultant product (Scheme 1e) is denoted “RGO–SDBS”.
The extent of reduction of the prepared samples was studied by attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) and X-ray photoelectron spectroscopy (XPS). ATR-FTIR spectra of GO, CRGO-only, CRGO–SDBS and RGO–SDBS are presented in Fig. 2a. All the observed peaks can be ascribed to O–H stretching mode, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carboxyl or carbonyl stretching vibration, CC stretching, O–H deformations in the C–OH groups, C–OH stretching and C–O stretching vibrations in C–O–C in epoxide from GO.34 After chemical reduction, the peak intensities of the oxygen containing functional groups in both CRGO-only and CRGO–SDBS become very weak compared to that of GO, but not completely gone, indicating that only partial reduction of GO was obtained using hydrazine monohydrate solution (Scheme 1d). However, the CRGO–SDBS exhibits new prominent characteristic peaks at 2960 cm−1, 2928 cm−1 and 2870 cm−1 which correspond to C–H vibrations in SDBS. These absorption peaks in the CRGO–SDBS sample indicates that the SDBS is adsorbed on the CRGO.40 After thermal annealing, the majority of oxygen peaks associated with the functional groups in CRGO–SDBS became very weak, confirming the successful reduction of the GO by the combination of chemical and thermal processes (Scheme 1e, confirmed by curve fitting of C 1s peaks in XPS spectra shown in Fig. S2†). However, the absorption peaks due to the presence of SDBS remain unchanged after annealing at 400 °C for 1 h. This result suggests that the insulating SDBS was not removed by the low-temperature thermal treatment.
O carboxyl or carbonyl stretching vibration, CC stretching, O–H deformations in the C–OH groups, C–OH stretching and C–O stretching vibrations in C–O–C in epoxide from GO.34 After chemical reduction, the peak intensities of the oxygen containing functional groups in both CRGO-only and CRGO–SDBS become very weak compared to that of GO, but not completely gone, indicating that only partial reduction of GO was obtained using hydrazine monohydrate solution (Scheme 1d). However, the CRGO–SDBS exhibits new prominent characteristic peaks at 2960 cm−1, 2928 cm−1 and 2870 cm−1 which correspond to C–H vibrations in SDBS. These absorption peaks in the CRGO–SDBS sample indicates that the SDBS is adsorbed on the CRGO.40 After thermal annealing, the majority of oxygen peaks associated with the functional groups in CRGO–SDBS became very weak, confirming the successful reduction of the GO by the combination of chemical and thermal processes (Scheme 1e, confirmed by curve fitting of C 1s peaks in XPS spectra shown in Fig. S2†). However, the absorption peaks due to the presence of SDBS remain unchanged after annealing at 400 °C for 1 h. This result suggests that the insulating SDBS was not removed by the low-temperature thermal treatment.
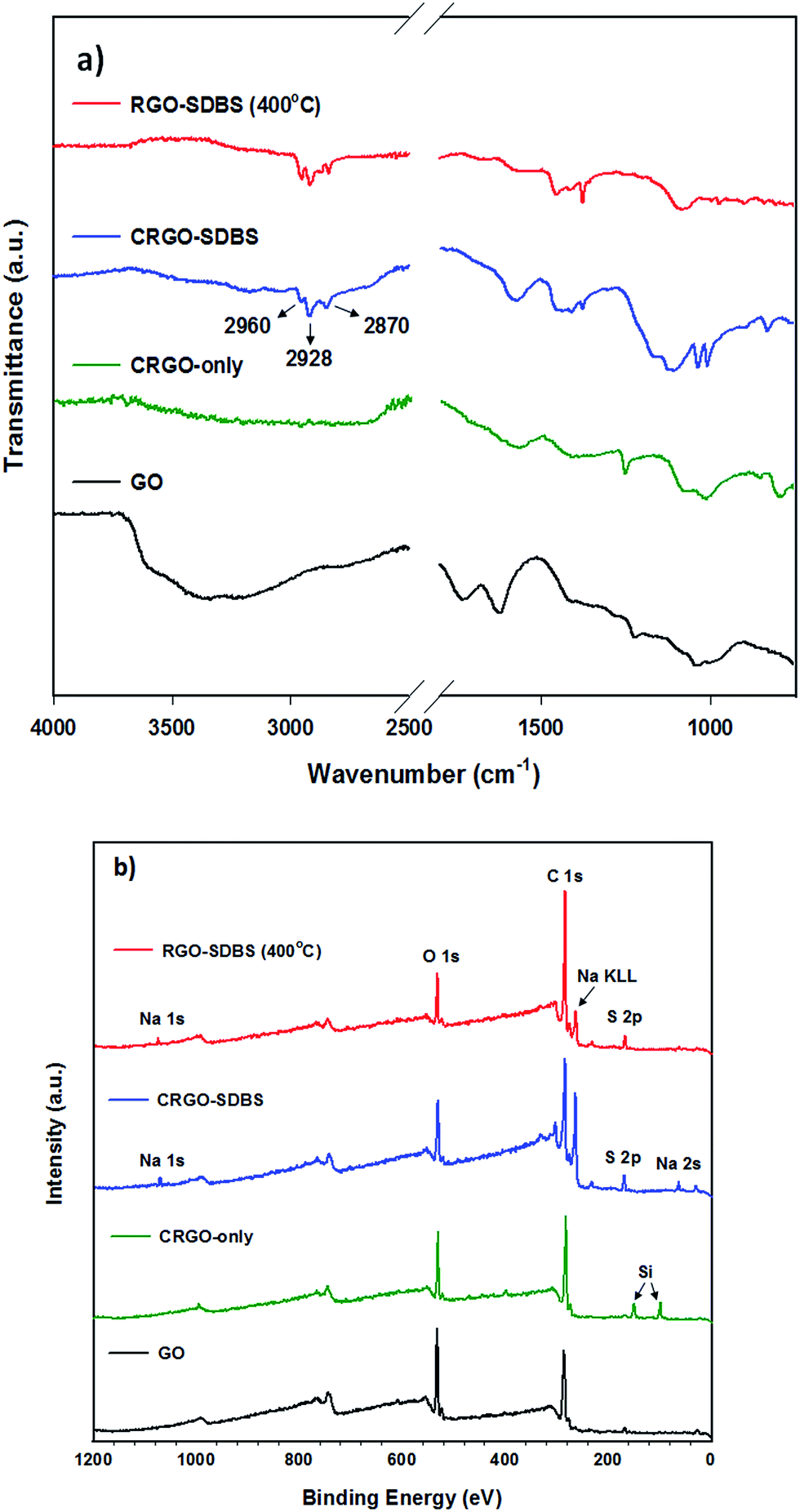 | ||
| Fig. 2 (a) ATR-FTIR and (b) XPS survey spectra of GO, CRGO-only, with SDBS and thermally reduced CRGO–SDBS (RGO–SDBS). | ||
XPS survey spectra of CRGO–SDBS and RGO–SDBS (Fig. 2b) show peaks (in addition to 283.5 eV (C 1s) and 530.5 eV (O 1s)) at binding energies of around 166 eV (S 2p), 262 eV (Na KLL) and 1059.5 eV (Na 1s), further illustrating that the SDBS remains on the CRGO structure after annealing at 400 °C. Nevertheless, it should be noted that the peak intensities of both ATR-FTIR and XPS for CRGO–SDBS structure decreased slightly after annealing at 400 °C. Another noticeable feature from the XPS survey spectra in Fig. 2b is that the appearance of Si 2s and Si 2p peaks at around 99.0 eV and 149.5 eV, respectively for the CRGO without SDBS. These Si peaks can be explained by the poor solubility of the CRGO solution. Due to the large aggregation of CRGO in the solvent, the CRGO sample did not completely cover the silicon substrate. Additionally, the thermal stability of SDBS was investigated using thermo-gravimetric analysis (TGA) (see Fig. S3†). Our finding from TGA analysis was in good agreement with the ATR-FTIR and XPS and suggests that the thermal annealing at 400 °C cannot remove the residual surfactants from the graphene. Therefore, further treatment is required to completely remove the SDBS.
According to previous studies,45–48 the application of concentrated acid solution can be an effective way to completely remove the residual SDBS surfactant and other organic contaminants from the graphene films. Therefore, we used concentrated nitric acid (HNO3, 68%) solution (Scheme 1f). It is widely accepted that the use of HNO3 has the advantage of not only eliminating the insulating surfactant, it also enhances the electrical properties of carbon films by an oxidative doping effect.49,50 In addition to these effects, chemical HNO3 treatment can also cause some weak edge defects with oxygen containing functional groups (see Scheme 1f, termed as “RGO”),45,51,52 which could be very useful for further treatment to maximize the film performance. In order to produce high-performance graphene films, we also introduced metallic gold nanoparticles (AuNPs) onto our RGO by dipping HNO3-functionalized RGO films into HAuCl4 solution (Scheme 1g, called “AuNPs–RGO”). The removal of SDBS and the deposition of AuNPs of the RGO films were characterized by using XPS, scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX).
Fig. 3a shows that the peaks of RGO–SDBS sample at binding energy of 166 eV (S 2p), 262 eV (Na KLL) and 1059.5 eV (Na 1s) have disappeared after treatment with HNO3 and HAuCl4 solutions, indicating of successful removal of the surfactant from the RGO. Additionally, in Fig. 3a, the appearance of two new prominent peaks at around 83.5 eV (Au 4f) and 200 eV (Cl 2p) indicates the successful AuNPs deposition and some residual HAuClx. Moreover, the SEM image (inset of Fig. 3a) clearly shows that the AuNPs were formed on the RGO after dipping the partially functionalized RGO film (Scheme 1f) into HAuCl4 solution. It is worth noting that the deposition of AuNPs on the RGO was achieved without the assistance of any reducing agents due to the HNO3 post-treatment. Therefore the edge defects (OH−, COOH−etc.) in RGO introduced by HNO3 treatment play an important role in reducing Au3+ to Au0.52,53 Moreover, the EDX elemental analysis (Fig. 3b) was carried out on the selected area of SEM image of the prepared sample and further confirms the removal of residual SDBS from the RGO and the formation of AuNPs on the RGO films.
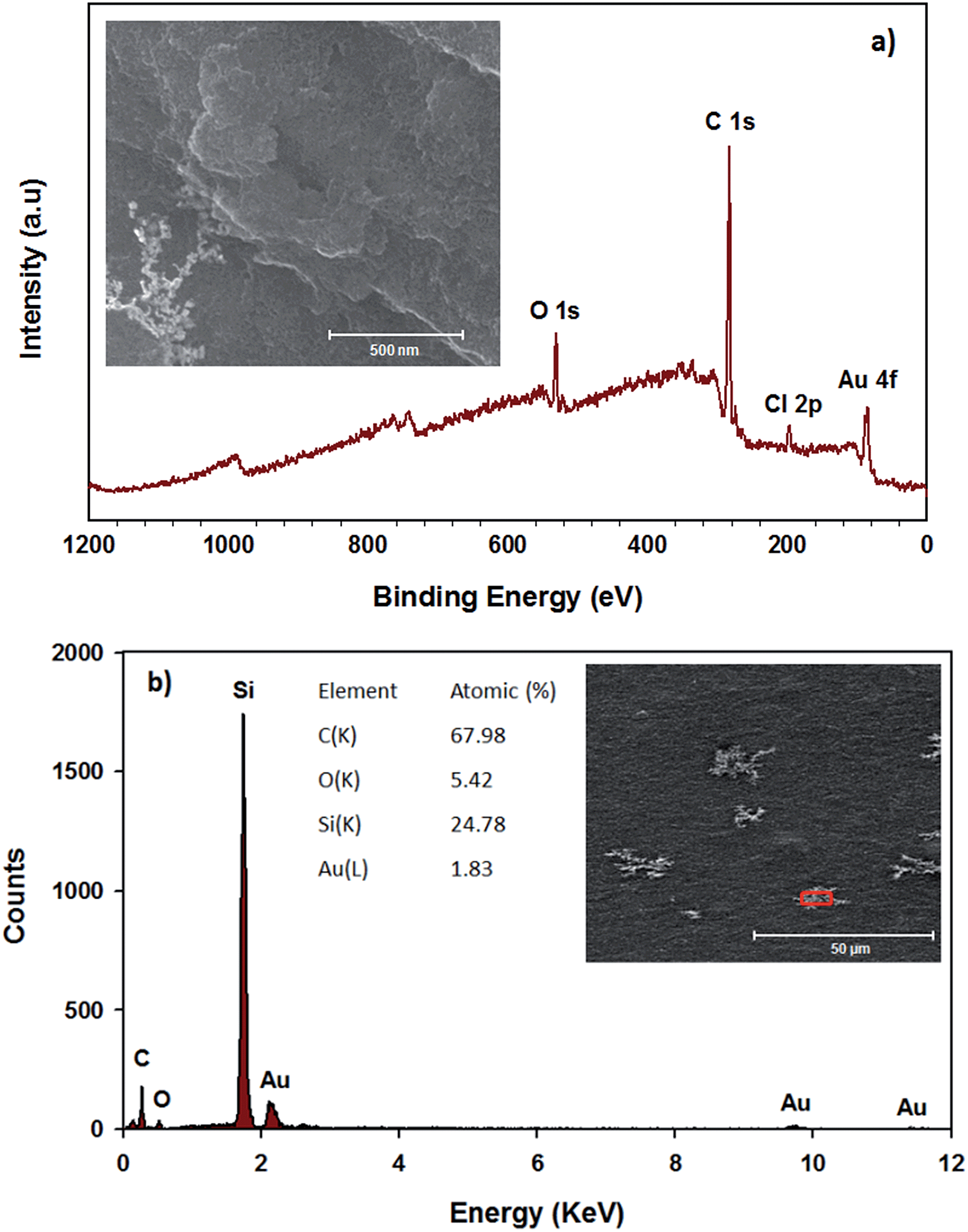 | ||
| Fig. 3 (a) XPS survey spectra (inset: SEM image of AuNPs–RGO) and (b) EDX analysis (red box in the inset is the selected area for analysis) of RGO–SDBS film after HNO3 and HAuCl4 treatments. | ||
Optical and electrical properties of the graphene films
An ideal PV device – one with the highest PCE – is achieved by having the lowest sheet resistance of the TCF, Rs, while achieving the highest transparency. Thin graphene films can exhibit high optical transparency, but they suffer from relatively high Rs. The Rs can be reduced by making the graphene films thicker, but this leads to an increase in the film opacity. There is clearly an optimum film thickness. We sought this thickness by changing the volume of filtered CRGO–SDBS solution. Fig. 4a illustrates the Rs of graphene films prepared from four different structures plotted as a function of filtration volume. These graphene structures are (a) CRGO–SDBS films (Scheme 1d), (b) RGO–SDBS films (Scheme 1e), (c) RGO films (Scheme 1f, HNO3-treated), and (d) AuNPs–RGO films (Scheme 1g). Additionally, the wavelength-dependent optical transparencies of each film with different thicknesses and their corresponding Rs values are shown in Fig. S4.† It can be seen from Fig. S4a† that the transparency of the films decreased with increasing filtered volume of the RGO solution. As shown in Fig. 4a, the Rs of our CRGO–SDBS films were in the range from 2 MΩ sq−1 to 12 MΩ sq−1 depending on the thickness. Interestingly, these Rs values are found to be slightly lower than that of previously reported chemically reduced GO films,44,54,55 despite our films containing insulating SDBS. We attribute this better performance of our CRGO–SDBS films to the production of large-size graphene sheets.40 Although our CRGO–SDBS films showed lower Rs compared to other studies, such Rs values are still too high for satisfactory solar devices.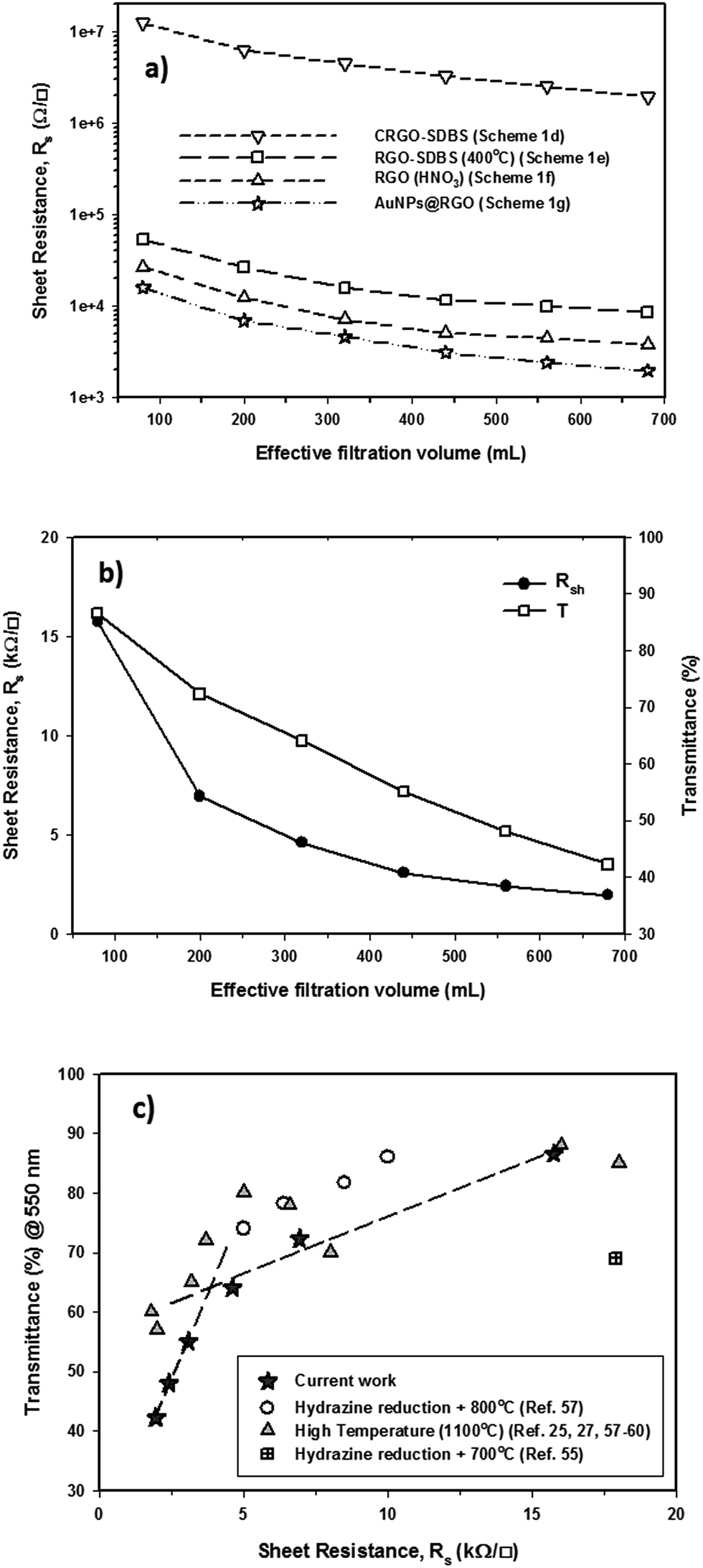 | ||
| Fig. 4 (a) Rsvs. thickness of the graphene films prepared from four different structures; (b) Rs and transmittance (at λ = 550 nm) of selected TCGFs with different thicknesses; (c) comparison of Rs as a function of transmittance (at λ = 550 nm) between our AuNPs–RGO films and other studies. Dash lines show the two regions of differing resistance for the graphene films and the threshold transmittance and corresponding Rs. | ||
As also demonstrated in Fig. 4a, the Rs of CRGO–SDBS film decreased by more than 2 orders of magnitude for a given thickness after the thermal treatment. This improvement in the electrical properties is known to be due to the better graphitization, deoxygenation and cross-linking of the graphene sheets.27,39 However, the thermal reduction of the CRGO–SDBS film reduced the transparency by 4–5% (Fig. S4b†). The darkening of the films after thermal annealing is due to the restoration of the π-electron system in the graphene structure and some impurities from the re-deposition of carbonaceous material which desorbs during thermal treatment and then adsorbs on both sides of the substrate.55 Although residual insulating SDBS is still present in the film after thermal treatment, we were able to achieve a Rs of as low as 8.5 kΩ sq−1 using this film such as that shown in Scheme 1e. Therefore, the removal of the SDBS surfactants with HNO3 was expected to improve the performance of our films.
As expected, the Rs of the RGO–SDBS films were significantly reduced (by more than 2-fold) after treating with concentrated HNO3 (see Fig. 4a). This dramatic improvement in the electrical properties is most likely due to the removal of any remaining SDBS from the film. Another possible reason behind the enhanced conductivity is the chemical doping effect of HNO3 on graphene films.17,49,56 In particular, the Rs value of the RGO–SDBS films was reduced from 8.5 kΩ sq−1 to 3.74 kΩ sq−1 at the same thickness after treating with HNO3 solution. More importantly, the HNO3 treatment not only enhanced the electrical conductivity of the films, it also increased the transparency by around 5% for any given thickness (Fig. S4c†). The increase in the transparency of the films after washing with HNO3 could be ascribed to the removal of remaining impurities of the films, particularly on the underside of the glass.
After depositing the AuNPs on RGO films, the Rs and transmittance of the dried films were measured. Fig. 4a shows that the Rs of RGO films decreased by about 1.8 times after introducing AuNPs onto the films, while no degradation in transmittance was observed (Fig. S4d†) compared to the HNO3-treated RGO films. The improved conductivity could be due to the fact that the AuNPs deposited on RGO created bridges between adjacent sheets, both in-plane and out-of-plane. A low electrical conductivity of graphene film mainly arises from the high inter-sheet contact resistance (deriving from charge hopping) between the edges of graphene sheets.41 In our AuNPs deposited RGO films, the AuNPs play a vital role in conjugating adjacent graphene sheets and subsequently reducing the overall Rs of the film.
The correlation of Rs and transmittance at λ = 550 nm of our AuNPs–RGO films to their volume of filtered solution is depicted in Fig. 4b. Through the systematic treatments, we obtained an Rs of as low as 1.96 kΩ sq−1 for the thick graphene film with transmittance of 42.3%. In contrast, a high optical transparency of 86.6% was achieved for the thin film, but its Rs is 15.7 kΩ sq−1. It should be noted that our Rs values are comparable to previous reports of solution processed graphene films produced by using hydrazine reduction and high-temperature annealing process (800–1100 °C) (Fig. 4c).25,27,55,57–61 Therefore, these TCGFs exhibit great potential for use as transparent electrodes in PV devices. The films based on AuNPs–RGO structures such as that illustrated in Scheme 1g have been chosen for the fabrication of PSC devices. Moreover, we calculated a figure of merit (σDC/σOP) for these TCGFs (Table 1) and the film with Rs = 3.08 kΩ sq−1 at T = 55% showed a high figure of merit (0.176). This σDC/σOP value was higher than that of thinner films, which is expected to correlate with high performance of solar cells.
| Device | R s@T | σ DC/σOP | J sc (mA cm−2) | V oc (V) | FF | η (%) |
|---|---|---|---|---|---|---|
| (1) (80 mL) | 15.7 kΩ sq−1@86.6% | 0.161 | 0.56 | 0.692 | 0.25 | 0.1 |
| (2) (200 mL) | 6.93 kΩ sq−1@72.3% | 0.154 | 1.25 | 0.695 | 0.26 | 0.23 |
| (3) (320 mL) | 4.61 kΩ sq−1@64.1% | 0.164 | 2.02 | 0.700 | 0.29 | 0.41 |
| (4) (440 mL) | 3.08 kΩ sq−1@55.0% | 0.176 | 2.55 | 0.690 | 0.35 | 0.62 |
| (5) (560 mL) | 2.41 kΩ sq−1@48.0% | 0.176 | 2.43 | 0.690 | 0.36 | 0.60 |
| (6) (680 mL) | 1.96 kΩ sq−1@42.3% | 0.177 | 2.21 | 0.694 | 0.37 | 0.57 |
Fabrication and characterization of PV devices
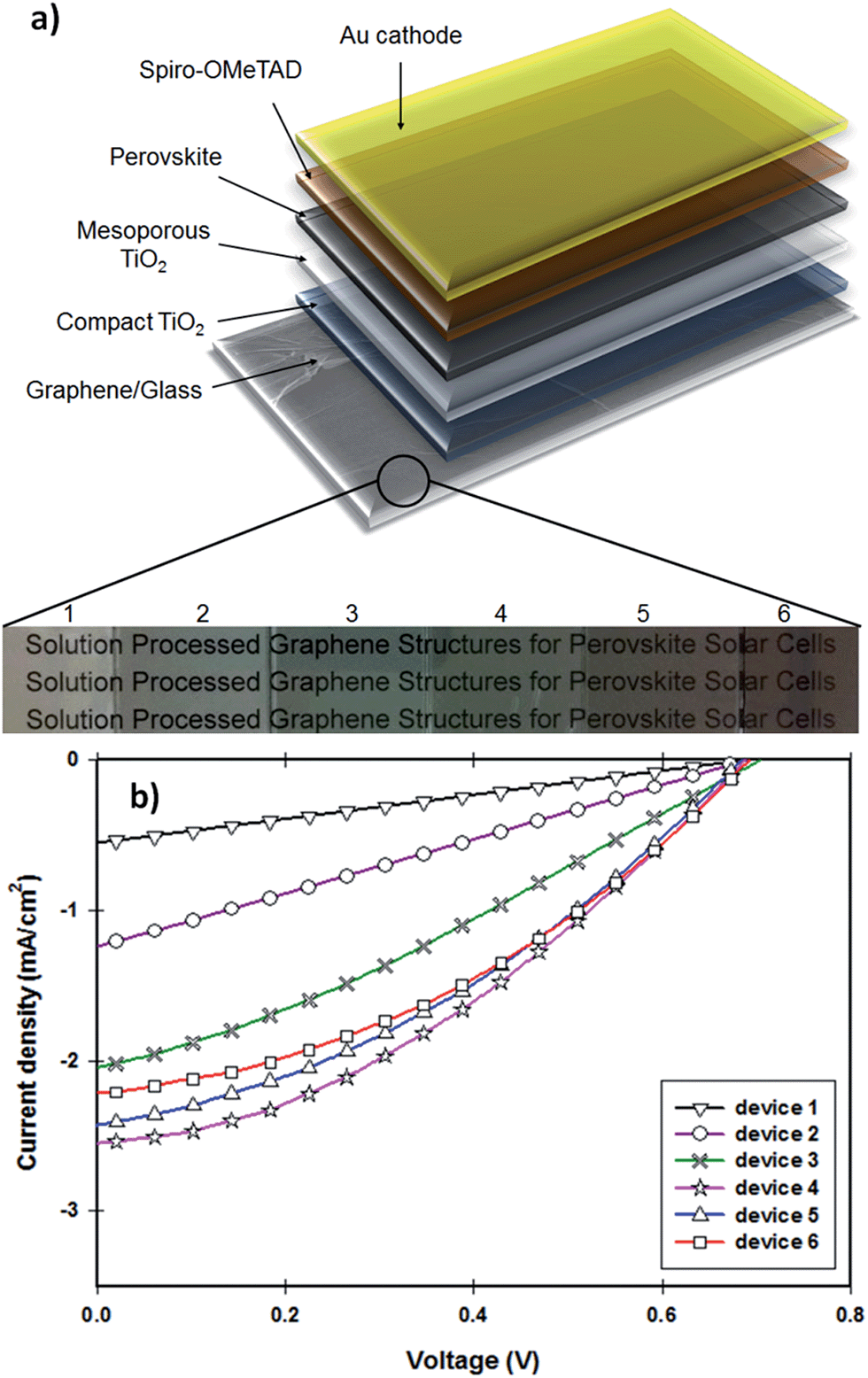 | ||
| Fig. 5 (a) Device structure, (b) photocurrent density–voltage (J–V) curve of the fabricated solar cells with transparent graphene electrodes. PSC devices with 0.075 cm2 active area were illuminated under AM 1.5G simulated sunlight (100 mW cm−2). | ||
The photocurrent density–voltage (J–V) characteristics of the PSCs fabricated with different TCGFs are shown in Fig. 5b and the corresponding PV parameters such as open-circuit voltage (Voc), short-circuit current density (Jsc), fill factor (FF) and PCE have been summarized in Table 1. The measured Voc values of all devices are essentially constant at 0.695 ± 0.05 V, indicating that the thickness of graphene films does not influence this parameter. This is reasonable since the Voc parameter is mainly determined by the energy level difference between the conduction band of electron transporting material and the potential energy of the HTM. In contrast, significant changes in the Jsc and FF were observed. Because of its comparatively high Rs, device 1 showed the lowest Jsc (0.56 mA cm−2) and FF (0.25) values, despite the transparency of graphene film being quite high. Interestingly, the FF value of our PSCs continuously increased from device 1 to device 6, likely to be due to the improvement in the Rs of the graphene films. Therefore the maximum FF value (0.37) was achieved for the device 6 which is made of our most-conductive graphene film with lowest transparency. However, the measured Jsc value (2.21 mA cm−2) of device 6 was not the highest observed. Unlike the FF parameter, no continuous increase was observed for the Jsc value of our devices when the thickness of graphene films increases. In particular, from device 1 to device 4 (an increase in the thickness of graphene films), the Jsc increases from 0.56 to 2.55 mA cm−2 owing to the reduction of Rs. However, when the transmittance of the film drops below 55%, Jsc of the cells decreases (device 5 & 6) despite the films having reduced Rs. This decrease in Jsc is due to the absorption of incident light by the TCGF before it reaches the active perovskite layer. Indeed, the optimum PV parameters for the TCGFs-based PSC were achieved for the graphene film with 3.08 kΩ sq−1@55.0%T. The observed Jsc, Voc and FF values for this PSC (device 4) were 2.55 mA cm−2, 0.69 V and 0.35, respectively, yielding a power conversion efficiency of 0.62%.
For comparison, an FTO electrode based PSC device was also fabricated under the same conditions as devices 1–6 and its J–V curve is plotted in Fig. S5.† The FTO based device exhibited a Jsc of 17.49 mA cm−2, Voc of 0.71 V and FF of 0.63, yielding a PCE of 7.82%. It is obvious that the PCE of our graphene film-based PSCs is significantly lower to that of the control cell based on FTO. The major issues for our TCGFs based devices are relatively low Jsc and lower FF values as compared to the cell based on FTO. This might be due to the high Rs and poor optical transmittance of our graphene films. Although the PCE (0.62%) of our graphene electrode based device is far from that of the PSC fabricated with FTO, this efficiency value is higher than that achieved for previously published inorganic–organic hybrid solar cells24 or DSSCs27 in which graphene films act as the electron collection electrode. It should also be noted that the Voc value (0.71 V) and PCE achieved using our typical FTO based PSC is lower than recently reported values for standard cells using typical ITO or FTO transparent conducting electrodes.62–64 The perovskite precursor and deposition process we have used were chosen for their simplicity in deposition and under the conditions we followed typically yield PCEs of 7–9% with low Voc (0.7–0.8 V)65–67 which are consistent with our results using the standard transparent conducting electrodes. More importantly, here in this work, we demonstrate the feasibility of solution processed graphene films as alternatives to the traditional TCO electrodes in the state-of-the-art PSCs. We anticipate that significant improvement in the PCE can be made for this class of PV devices by enhancing the performance of the graphene films and/or using other solar cell architectures.
The J–V characteristics and device structures of the TCGFs based PSCs with and without graphene in the semiconducting oxide layers are illustrated in Fig. 6. The PV performances of these PSC devices have been summarized in Table 2. For comparison, the J–V curve and the corresponding energy level diagram of device 4 (TCGF based PSC without graphene in the semiconducting layer) is also plotted in Fig. 6a and a′, respectively. Since the work function of RGO is close to that of FTO, and lower than the conduction band of TiO2,25 it is reasonable to expect that the injected electrons at the TiO2 conduction band can be transferred to the graphene electrode without any barrier. Changes to the work function of gold chloride doped graphene have previously been shown to be minimal after thermal annealing, as has been done in this work.70
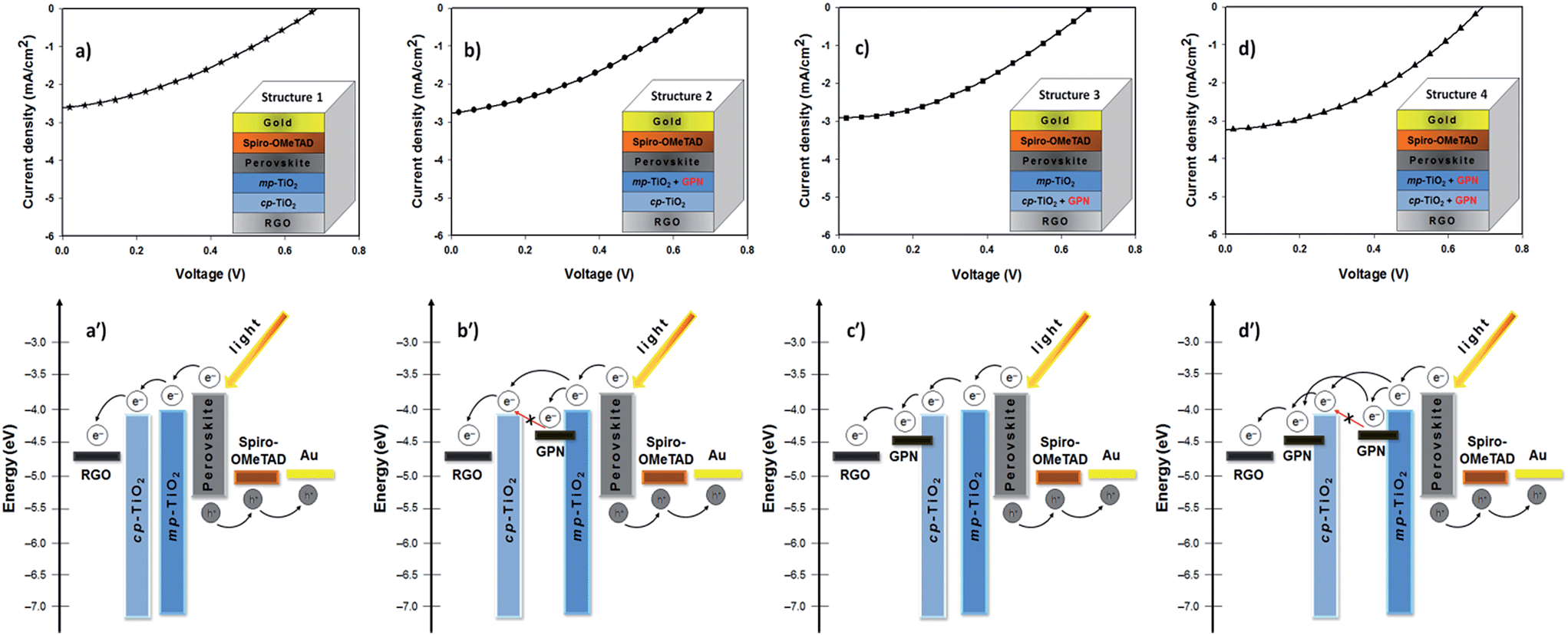 | ||
| Fig. 6 J–V curves (top) and the corresponding energy level diagrams (bottom) of TCGF film based PSCs with and without graphene in the semiconducting oxide layers. The device structures are shown in the insets. The abbreviations are as follows: RGO – reduced graphene oxide; graphene – GPN; mp-TiO2 – mesoporous TiO2; cp-TiO2 – compact TiO2. | ||
| Device | J sc (mA cm−2) | V oc (V) | FF | η (%) |
|---|---|---|---|---|
| Structure 1 | 2.55; 2.55 ± 0.03 | 0.690; 0.689 ± 0.001 | 0.35; 0.35 ± 0.01 | 0.62; 0.62 ± 0.00 |
| Structure 2 | 2.77; 2.75 ± 0.02 | 0.684; 0.686 ± 0.002 | 0.36; 0.36 ± 0.00 | 0.66; 0.65 ± 0.01 |
| Structure 3 | 2.90; 2.85 ± 0.05 | 0.690; 0.695 ± 0.005 | 0.38; 0.38 ± 0.00 | 0.75; 0.74 ± 0.01 |
| Structure 4 | 3.05; 2.94 ± 0.11 | 0.687; 0.689 ± 0.002 | 0.38; 0.38 ± 0.01 | 0.81; 0.79 ± 0.02 |
On the other hand, the application of graphene in the semiconducting oxide layers should principally increase the efficiency of this class of solar cells due to enhanced charge transport.71 However, as shown in Fig. 6b, no significant improvement in the PV parameters for the PSC was observed after incorporating graphene into the mesoporous TiO2 layer only (structure 2). We hypothesize that these unchanged PV parameters are associated with the energy level alignment of TiO2 and graphene. In fact, the injected electrons from the excited perovskite sensitizer and/or mesoporous TiO2 into the graphene cannot be transferred to the conduction band of the compact TiO2 (Fig. 6b′) which results in incomplete electron transport within the networks.
Furthermore, the addition of graphene into the compact TiO2 layer of device (structure 3) exhibited some enhancement in the Jsc and FF parameters and displayed a PCE of 0.75%, as illustrated in Fig. 6c. These increased Jsc and FF values could be due to the suitable energy levels of graphene in the cell. The energy levels of graphene in the compact TiO2 layer can be ideal for this class of PSC (structure 3) as its work function sits between the TiO2 and graphene anode and so that the electrons transfer stepwise from the perovskite to the graphene anode without an energy barrier (see Fig. 6c′). Here, graphene, which was incorporated into the compact TiO2 layer, acts as a bridge between TiO2 and graphene anode. In the energy diagram, it is reasonable to assume that the work function of RGO (graphene anode; used as a transparent conductive film in the PSC) is higher than that of the graphene used in the semiconducting oxide layers because the extent of reduction in the electrode is relatively high.
Structure 4 showed a promising improvement in the power conversion efficiency (0.81%) (Fig. 6d). In particular, the Jsc and FF values of structure 4 increased to 3.04 mA cm−2 and 0.38, respectively, after incorporating graphene structures into both the compact TiO2 and mesoporous TiO2 layers. The improvement in these parameters (Jsc and FF) can be ascribed to the fact that the conductive graphene in the cells enhances the charge transport rate and suppresses the charge recombination. Moreover, it is reasonable to expect that the presence of graphene in both the compact and mesoporous TiO2 layers provides a thermodynamically favorable energy transfer path and potentially offers an extra graphene to graphene conduction path both of which enable successful charge collection and hence higher PCE (see Fig. 6d′). A detailed investigation on the effect of carbonaceous materials in the TiO2 photoanodes of PSCs is ongoing research in our group.
The external quantum efficiency (EQE) is an important parameter for evaluating the performance of solar cells. PSC devices (structure 4 in Fig. 6) with TCGF and graphene in mesoporous and compact TiO2 were chosen for EQE analysis. For comparison, the EQE characteristic of the conventional PSC fabricated on FTO electrode without graphene was also investigated. Fig. 7 compares the obtained EQE spectra. Both cells show a broad EQE peak across the visible region, typical for PSCs.66 The cell fabricated with graphene (structure 4 in Fig. 6) shows a similar shape to the FTO-electrode based PSC (see Fig. 7 inset) but much lower EQE value, showing that the difference is wavelength independent which indicates that the use of graphene film did not alter the internal mechanism of the PSC. The lower EQE value of TCGF based cell is expected when considering the low PCE obtained, as discussed previously. Moreover, the stability of these two PSCs, namely FTO-based and TCGF-based, was investigated for 60 h and the results are plotted in Fig. S6.† The degradation rate of TCGF based cell was very similar to that of an FTO-based device.
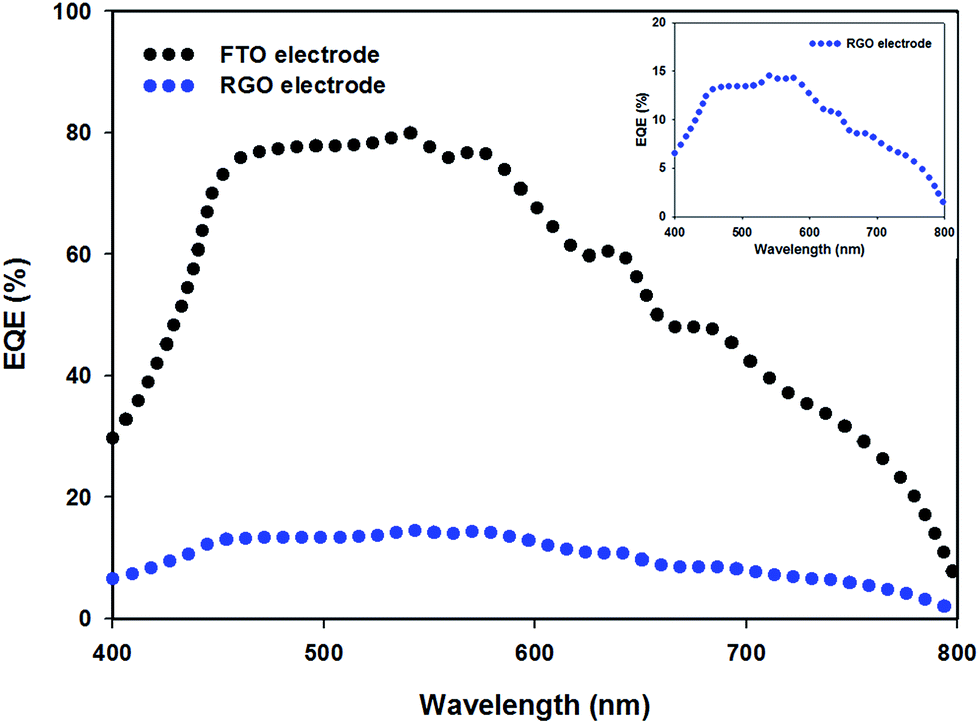 | ||
| Fig. 7 EQE spectra of FTO electrode (black dots) and TCGF (blue dots, structure 4 (from Fig. 6 and Table 2)) based PSCs. Inset shows the expanded EQE spectrum of RGO electrode based PSC. | ||
The initial reported PCE of PSCs was relatively low but has increased rapidly in just a few years. It is anticipated that PCE of TCGF in PSCs will show a similar rapid improvement as they have in other solar cell architectures.19 A promising result is that the observed Voc for all devices fabricated with TCGF films were similar to that of FTO electrodes based cells, indicating that the energy bands of graphene are suitable for application in PSCs, supporting theoretical predictions.32 Therefore, our results demonstrate that the use of graphene films as the electron transporting transparent conducting electrode in the PSCs is viable. The two key areas for research are the improvement in Rs with high transmittance and the creation of flexible PSCs using TCGFs. Further modification of the reduction of GO to increase flake size could produce graphene films with better performance for PSCs without increasing manufacturing cost.
Conclusions
Herein, we demonstrate the feasibility of transparent conductive graphene films (TCGFs) formed by solution processing as alternatives to the conventional transparent conducting oxide (TCO) electrodes in PSC devices. The TCGFs were prepared by using a low-temperature annealing process as well as chemical post-treatments. By using an optimal balance of Rs and transparency of the graphene films, a maximum PCE of 0.62% was obtained. By incorporating graphene structures into both compact TiO2 and mesoporous TiO2 layers of the PSCs, the PCE was further improved to 0.81%. Further PCE enhancement is expected in this class of solar cells by applying high-quality graphene films with improved electrical conductivity and high transparency. Finally, we anticipate that the current work will open new avenues for the development of graphene materials in perovskite based solar cells.Experimental
Materials
Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich and used without further purification. Methylammonium iodide (CH3NH3I), TiO2 paste (18NR-T) and tris(1-(pyridin-2-yl)-1H-pyrazol)cobalt(III)tris(hexafluorophosphate) (FK102 Co(III) PF6) salt were purchased from Dyesol. (2,2′,7,7′-Tetrakis-(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene) (Spiro-OMeTAD) was obtained from Solaronix.Preparation of graphene films
Graphite oxide was prepared via the oxidation of natural graphite according to an improved Hummers method.34 In brief, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of concentrated sulfuric acid (95–98% H2SO4) and phosphoric acid (85% H3PO4) (240:27 mL) was kept in the cold room (3–5 °C) until it was added to a mixture of graphite flakes (2 g) and potassium permanganate (99% KMnO4) (12 g). Then the oxidation process was carried out by stirring at 50 °C for 12 h. Upon completion, the reaction was cooled down to room temperature and poured onto ice (approximately 300 mL) with 30% hydrogen peroxide (H2O2) (2 mL). The mixture was then washed with distilled (DI) water, 30% hydrochloric acid (HCl) and ethanol (2 times). For each sequential wash, the product was centrifuged at 4400 rpm for 3 h and the supernatant decanted away. The obtained light brown sample was then vacuum-dried overnight at room temperature.
:1 (v/v) mixture of concentrated sulfuric acid (95–98% H2SO4) and phosphoric acid (85% H3PO4) (240:27 mL) was kept in the cold room (3–5 °C) until it was added to a mixture of graphite flakes (2 g) and potassium permanganate (99% KMnO4) (12 g). Then the oxidation process was carried out by stirring at 50 °C for 12 h. Upon completion, the reaction was cooled down to room temperature and poured onto ice (approximately 300 mL) with 30% hydrogen peroxide (H2O2) (2 mL). The mixture was then washed with distilled (DI) water, 30% hydrochloric acid (HCl) and ethanol (2 times). For each sequential wash, the product was centrifuged at 4400 rpm for 3 h and the supernatant decanted away. The obtained light brown sample was then vacuum-dried overnight at room temperature.
The as-prepared graphite oxide was exfoliated in water (1 mg mL−1) by bath ultrasonication (Elma, Germany) for 40 min in the presence of SDBS (1 wt% in the solution). The obtained homogenous dispersion was named “GO–SDBS solution”. The GO–SDBS colloidal dispersion (10 mL) was chemically reduced by hydrazine monohydrate solution (40 μL, 64–65% N2H4·H2O) and ammonium hydroxide solution (120 μL, 30% NH3·H2O).44 The chemical reduction was performed in an oil bath at 100 °C overnight. The resultant solution (termed as “CRGO–SDBS”) was then diluted with DI water to obtain the final concentration of CRGO–SDBS (0.16 mg L−1). The diluted solution was further used to prepare the transparent films. For comparison, the chemical reduction of GO was performed in the absence of SDBS and the resultant solution was named CRGO-only.
The glass substrates (25 mm × 25 mm) were cleaned by detergent (Pyroneg) followed by washing with acetone, ethanol and Milli-Q water under ultrasonication for 10 min each and subsequently dried with a stream of nitrogen gas. The cleaned glass substrates were pretreated with 3-aminopropyl-triethoxysilane (APTES) (3% in toluene) to improve the surface functionalities of the substrates.24,39,56 Transparent graphene films were prepared on mixed cellulose ester (MCE) membranes (0.45 μm HAWP, Millipore) through the vacuum filtration of CRGO–SDBS solution.44 The transparency of the films was controlled by varying the effective filtration volume of solutions. The filtered films (CRGO–SDBS/MCE membrane) were subsequently pressed against the APTES-modified glass surface with the graphene side in contact with the substrate. The substrates where then firmly clamped in place at room temperature for 2 days to completely adhere the CRGO–SDBS film to the substrate. The MCE membranes were dissolved in an acetone bath to leave CRGO–SDBS film on the substrate. The obtained CRGO–SDBS films were then rinsed with methanol and dried by blowing nitrogen. To further improve the electrical conductivity of the films, the as-produced CRGO–SDBS films were thermally reduced in a tube furnace at low temperature (400 °C) for 1 h. The annealing and cooling processes were performed under the protection of an Ar and H2 (20:1) atmosphere. The obtained films are named “RGO–SDBS”. To remove the residual SDBS surfactant from the films, the RGO–SDBS films were then immersed in concentrated nitric acid (HNO3, 68%) solution for 3 h and rinsed thoroughly with DI water, and dried at 100 °C for 1 h in a hot oven. After the application of the HNO3-treatment, the samples are called “RGO films”. The AuNPs were then deposited onto the RGO films by dynamic spin coating of 0.5 mM HAuCl4 in nitromethane, and finally dried completely at 200 °C overnight. The prepared films are named “AuNPs–RGO films” and have been used to fabricate the PSC devices.
Fabrication of PSC devices
PSC devices with the structure of graphene anode/compact TiO2/mesoporous TiO2/CH3NH3PbI3−xClx/Spiro-OMeTAD/Au were fabricated according to the following procedure. The fabrication process of PSCs has been reported elsewhere.12,14 A thin compact TiO2 layer was spin-coated onto the previously prepared graphene film and/or cleaned FTO electrode (∼12 Ω sq−1, Solaronix TCO30-8) substrate at a rotation speed of 2000 rpm for 20 s using 0.2 M titanium diisopropoxide bis(acetylacetonate) (75 wt% in isopropanol, Aldrich) in 1-butanol solution, followed by heating at 125 °C for 5 min. The same process was repeated twice with the above solution, followed by drying at 125 °C for 5 min and sintering at 500 °C for 1 h. For the preparation of the graphene incorporated compact TiO2 layer, GO–ethanol solution (1 mg mL−1) was added into the titanium diisopropoxide bis(acetylacetonate) in 1-butanol solution. The concentration of the GO in the composite was calculated to be 0.6 wt%. After cooling to room temperature, a thick mesoporous TiO2 layer was deposited onto the compact TiO2 layer by spin coating a solution of TiO2 paste (Dyesol 18NR-T) in a 2:7 weight ratio to ethanol at 4000 rpm for 30 s. After drying at 125 °C for 5 min, the films were sintered at 500 °C for 1 h. The mesoporous TiO2 deposited films were then immersed in 40 mM aqueous TiCl4 (Aldrich) solution at 70 °C for 30 min, which was again annealed at 500 °C for 30 min. Similarly, to prepare the graphene/mesoporous TiO2 layer, the GO-ethanol solution was also added into the diluted TiO2 paste solution and the concentration of the GO in the composite was controlled to be 0.2 wt%. The GO in the compact and/or mesoporous TiO2 layers can simply be converted to graphene during the annealing processes. Moreover, during the deposition of the compact and mesoporous layers on the transparent electrodes, Parafilm® M seal was rolled onto one side of the TCGFs to protect the graphene anode contact. After the completion of all annealing processes at 500 °C, conductive adhesive tape was carefully applied onto the graphene anode to serve as electrical contact. Notably, we measured the Rs of the graphene films before and after annealing at 500 °C for 1 h as this thermal annealing process was done after the deposition of TiO2 layers and no significant changes in the Rs were observed. Particularly, the Rs of HNO3 and HAuCl4 treated RGO films before and after thermal treatment at 500 °C were measured to be 4.08 ± 0.04 kΩ sq−1 and 4.21 ± 0.12 kΩ sq−1, respectively. It should also be noted that for the fabrication of PSC devices with graphene structures, the thermal annealing processes at more than 400 °C were carried out under the protection of Ar to protect graphene from the mild oxidation.
For the preparation of CH3NH3PbI3−xClx perovskite, a 1:3 molar ratio of PbCl2:CH3NH3I was mixed in anhydrous N,N-dimethylformamide (DMF) (99.8% Aldrich), with a concentration of 0.73 M and 2.2 M, respectively. The mixture was stirred at room temperature for at least 6 h before spin coating (100 μL of the solution) onto the mesoporous layers at 2500 rpm for 30 s in air and then heated at 100 °C for 1 h. The deposition process of the perovskite was carried out in controlled humidity under 35%.
The HTM (120 μL of the prepared solution) was then deposited onto the perovskite layer by spin coating at 4000 rpm for 30 s in a nitrogen-filled glovebox. The HTM was prepared by dissolving 72.3 mg Spiro-OMeTAD, 28.8 μL 4-tert-butylpyridine (tBP), 17.5 μL of a stock solution of 520 mg mL−1 lithium bis(trifluoromethylsulphonyl)imide (Li-TFSI) in acetonitrile and 29 μL of a stock solution of 300 mg mL−1 FK102 Co(III) PF6 salt in acetonitrile, in 1 mL chlorobenzene. Finally, 60 nm gold electrodes were deposited on top of devices by thermal evaporation at a rate of 1 Å s−1 under a high vacuum (∼10−6 bar) through a shadow mask.
Measurement and characterizations
AFM images were acquired in air using a Bruker Dimension FastScan AFM with Nanoscope V controller, operating in tapping mode. Silicon cantilevers (MikroMasch) with a fundamental resonance frequency of between 300 and 400 kHz were used. Images were obtained using a scan rate of 1 Hz with the set point, amplitude, and feedback control parameters optimized manually for each sample. The images presented have been flattened using NanoScope Analysis v1.4 software. SEM images were obtained using an Inspect F50 SEM (FEI) with accelerating voltage of 20 kV. EDX analysis was completed on the same system with Team EDS Octane Pro (EDAX) attachment. ATR-FTIR spectra were acquired over a wavenumber range of 4000–650 cm−1 in transmission mode using a Frontier FTIR spectrometer (Perkin Elmer, USA) with a germanium crystal. The elemental compositions of the samples were characterized at binding energies ranging from 0 eV to 1200 eV using a XPS, Leybold Heraeus LHS-10 with a SPECS XR-50 dual anode source operating at 250 W. A Mg-Kα source, which has energy of 1253.6 eV, was used for the XPS analysis. Curve fitting of the C 1s in XPS spectra was done using peak fitting software “Fityk”.72 High resolution XPS of the C 1s were collected with a step size of 0.1 eV and the presented spectra are an average of 5 collections. Raman spectroscopy was performed on LabRAM HR Evolution spectrometer (Horiba Jobin Yvon, Japan). Raman spectra were collected using a 532 nm laser (mpc 3000) as the excitation source. A 50× objective was used with a confocal hole size of 100 μm. Thermal decomposition of SDBS was performed using a thermal gravimetric analyser (TA Instruments TGA 2950 Thermogravimetric Analyzer, USA) under a flow of nitrogen at a rate of at 20.0 mL min−1. The transmittances of the films on glass slides were determined using a Varian Cary 50G UV-vis spectrophotometer at wavelengths ranging from 400 to 1000 nm. Sheet resistance measurements were performed on the same films using a four point probe technique (KeithLink Technology Co., Ltd. Taiwan). The J–V curves were measured using a Keithley 2400 SMU instrument and recorded using a custom LabView Virtual Instrument program. A standard silicon test cell with NIST-traceable certification was used to calibrate the power density as 100 mW cm−2 at the sample plane of the collimated xenon-arc light source, which was passed through an AM 1.5G filter. The active area of each device was 0.075 cm2. The J–V curves were obtained in the air in reverse-scan direction from 1 V to −1 V. EQE measurements as a function of wavelength ranging from 400 nm to 800 nm were taken by passing chopped light from a xenon source through a monochromator and onto the devices.Acknowledgements
The support of the Australian Research Council Discovery Program (DP130101714) is gratefully acknowledged. Munkhbayar Batmunkh acknowledges International Postgraduate Research Scholarship (IPRS) and Australian Postgraduate Award (APA) for their financial support during his study in Australia. We acknowledge the use of South Australian node of the Australian Microscopy & Microanalysis Research Facility (AMMRF) at Flinders University. The authors would also like to thank Meisam V. Kiamahalleh of School of Chemical Engineering at the University of Adelaide for graphic design.Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- T. Saga, NPG Asia Mater., 2010, 2, 96–102 CrossRef.
- M. Bosi and C. Pelosi, Prog. Photovoltaics, 2007, 15, 51–68 CAS.
- N.-G. Park, J. Phys. Chem. Lett., 2013, 4, 2423–2429 CrossRef CAS.
- G. Hodes, Science, 2013, 342, 317–318 CrossRef CAS PubMed.
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013–4030 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- G. E. Eperon, V. M. Burlakov, P. Docampo, A. Goriely and H. J. Snaith, Adv. Funct. Mater., 2014, 24, 151–157 CrossRef CAS.
- X. Chen, S. Yang, Y. C. Zheng, Y. Chen, Y. Hou, X. H. Yang and H. G. Yang, Adv. Sci., 2015, 2, 1500105 Search PubMed.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 CrossRef CAS PubMed.
- D. S. Hecht, L. Hu and G. Irvin, Adv. Mater., 2011, 23, 1482–1513 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- Z. Yin, J. Zhu, Q. He, X. Cao, C. Tan, H. Chen, Q. Yan and H. Zhang, Adv. Energy Mater., 2014, 4, 1300574 Search PubMed.
- H. Chang and H. Wu, Adv. Funct. Mater., 2013, 23, 1984–1997 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- M. Hösel, D. Angmo, R. R. Søndergaard, G. A. dos Reis Benatto, J. E. Carlé, M. Jørgensen and F. C. Krebs, Adv. Sci., 2014, 1, 1400002 Search PubMed.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS PubMed.
- Z. Yin, S. Sun, T. Salim, S. Wu, X. Huang, Q. He, Y. M. Lam and H. Zhang, ACS Nano, 2010, 4, 5263–5268 CrossRef CAS PubMed.
- E. Kymakis, K. Savva, M. M. Stylianakis, C. Fotakis and E. Stratakis, Adv. Funct. Mater., 2013, 23, 2742–2749 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- P. You, Z. Liu, Q. Tai, S. Liu and F. Yan, Adv. Mater., 2015, 27, 3632–3638 CrossRef CAS PubMed.
- F. Lang, M. A. Gluba, S. Albrecht, J. Rappich, L. Korte, B. Rech and N. H. Nickel, J. Phys. Chem. Lett., 2015, 6, 2745–2750 CrossRef CAS PubMed.
- Z. Liu, S. P. Lau and F. Yan, Chem. Soc. Rev., 2015, 44, 5638–5679 RSC.
- M. Batmunkh, C. J. Shearer, M. J. Biggs and J. G. Shapter, J. Mater. Chem. A, 2015, 3, 9020–9031 CAS.
- G. Volonakis and F. Giustino, J. Phys. Chem. Lett., 2015, 6, 2496–2502 CrossRef CAS PubMed.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS PubMed.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 Search PubMed.
- S. J. Wang, Y. Geng, Q. Zheng and J.-K. Kim, Carbon, 2010, 48, 1815–1823 CrossRef CAS.
- H. Chang, G. Wang, A. Yang, X. Tao, X. Liu, Y. Shen and Z. Zheng, Adv. Funct. Mater., 2010, 20, 2893–2902 CrossRef CAS.
- Q. Zheng, Z. Li, J. Yang and J.-K. Kim, Prog. Mater. Sci., 2014, 64, 200–247 CrossRef CAS.
- C. J. Shearer, A. Cherevan and D. Eder, Adv. Mater., 2014, 26, 2295–2318 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- M. S. Kang, K. T. Kim, J. U. Lee and W. H. Jo, J. Mater. Chem. C, 2013, 1, 1870–1875 RSC.
- H.-Z. Geng, K. K. Kim, K. P. So, Y. S. Lee, Y. Chang and Y. H. Lee, J. Am. Chem. Soc., 2007, 129, 7758–7759 CrossRef CAS PubMed.
- A. K. K. Kyaw, H. Tantang, T. Wu, L. Ke, C. Peh, Z. H. Huang, X. T. Zeng, H. V. Demir, Q. Zhang and X. W. Sun, Appl. Phys. Lett., 2011, 99, 021107 CrossRef.
- S. Kim, J. Yim, X. Wang, D. D. C. Bradley, S. Lee and J. C. deMello, Adv. Funct. Mater., 2010, 20, 2310–2316 CrossRef CAS.
- A. Kasry, M. A. Kuroda, G. J. Martyna, G. S. Tulevski and A. A. Bol, ACS Nano, 2010, 4, 3839–3844 CrossRef CAS PubMed.
- L. J. Larsen, C. J. Shearer, A. V. Ellis and J. G. Shapter, RSC Adv., 2015, 5, 38851–38858 RSC.
- R. Jackson, B. Domercq, R. Jain, B. Kippelen and S. Graham, Adv. Funct. Mater., 2008, 18, 2548–2554 CrossRef CAS.
- Q. W. Li, Y. Li, X. F. Zhang, S. B. Chikkannanavar, Y. H. Zhao, A. M. Dangelewicz, L. X. Zheng, S. K. Doorn, Q. X. Jia, D. E. Peterson, P. N. Arendt and Y. T. Zhu, Adv. Mater., 2007, 19, 3358–3363 CrossRef CAS.
- B.-S. Kong, J. Geng and H.-T. Jung, Chem. Commun., 2009, 2174–2176 RSC.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- Y. Xu, G. Long, L. Huang, Y. Huang, X. Wan, Y. Ma and Y. Chen, Carbon, 2010, 48, 3308–3311 CrossRef CAS.
- Q. B. Zheng, M. M. Gudarzi, S. J. Wang, Y. Geng, Z. Li and J.-K. Kim, Carbon, 2011, 49, 2905–2916 CrossRef CAS.
- J. Geng, L. Liu, S. B. Yang, S.-C. Youn, D. W. Kim, J.-S. Lee, J.-K. Choi and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 14433–14440 CAS.
- L. Zhao, L. Zhao, Y. Xu, T. Qiu, L. Zhi and G. Shi, Electrochim. Acta, 2009, 55, 491–497 CrossRef CAS.
- J. Wu, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 263302 CrossRef.
- X. Wang, L. Zhi, N. Tsao, Ž. Tomović, J. Li and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 2990–2992 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- H.-S. Ko, J.-W. Lee and N.-G. Park, J. Mater. Chem. A, 2015, 3, 8808–8815 CAS.
- Y. Liu, Q. Chen, H.-S. Duan, H. Zhou, Y. Yang, H. Chen, S. Luo, T.-B. Song, L. Dou, Z. Hong and Y. Yang, J. Mater. Chem. A, 2015, 3, 11940–11947 CAS.
- P. Qin, M. Paulose, M. I. Dar, T. Moehl, N. Arora, P. Gao, O. K. Varghese, M. Grätzel and M. K. Nazeeruddin, Small, 2015, 11, 5533–5539 CrossRef CAS PubMed.
- F. Di Giacomo, V. Zardetto, A. D'Epifanio, S. Pescetelli, F. Matteocci, S. Razza, A. Di Carlo, S. Licoccia, W. M. M. Kessels, M. Creatore and T. M. Brown, Adv. Energy Mater., 2015, 5, 1401808 Search PubMed.
- Y. Shi, Y. Xing, Y. Li, Q. Dong, K. Wang, Y. Du, X. Bai, S. Wang, Z. Chen and T. Ma, J. Phys. Chem. C, 2015, 119, 15868–15873 CAS.
- M. Lyu, J.-H. Yun, R. Ahmed, D. Elkington, Q. Wang, M. Zhang, H. Wang, P. Dastoor and L. Wang, J. Colloid Interface Sci., 2015, 453, 9–14 CrossRef CAS PubMed.
- M. Batmunkh, M. J. Biggs and J. G. Shapter, Adv. Sci., 2015, 2, 1400025 Search PubMed.
- M. Batmunkh, M. J. Biggs and J. G. Shapter, Small, 2015, 11, 2963–2989 CrossRef CAS PubMed.
- K. C. Kwon, B. J. Kim, J.-L. Lee and S. Y. Kim, J. Mater. Chem. C, 2013, 1, 2463–2469 RSC.
- J. T.-W. Wang, J. M. Ball, E. M. Barea, A. Abate, J. A. Alexander-Webber, J. Huang, M. Saliba, I. Mora-Sero, J. Bisquert, H. J. Snaith and R. J. Nicholas, Nano Lett., 2014, 14, 724–730 CrossRef CAS PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta08996d |
| This journal is © The Royal Society of Chemistry 2016 |