
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ALD SnO2 protective decoration enhances the durability of a Pt based electrocatalyst†
Catherine
Marichy
ab,
Giorgio
Ercolano
c,
Gianvito
Caputo‡
d,
Marc G.
Willinger
e,
Deborah
Jones
c,
Jacques
Rozière
c,
Nicola
Pinna
*d and
Sara
Cavaliere
*c
aLMI, CNRS UMR 5615, Université Lyon 1, 22 av. Gaston Berger – Bât. Berthollet, 69622 Cedex Villeurbanne, France
bDepartment of Chemistry, CICECO, University of Aveiro, 3810-193 Aveiro, Portugal
cInstitut Charles Gerhardt Montpellier, UMR CNRS 5253, Agrégats Interfaces Matériaux pour l'Energie, Université de Montpellier, 34095 Montpellier Cedex 5, France. E-mail: sara.cavaliere@umontpellier.fr
dHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Strasse 2, 12489 Berlin, Germany. E-mail: nicola.pinna@hu-berlin.de
eFritz Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, Faradayweg 4-6, 14195 Berlin, Germany
First published on 9th December 2015
Abstract
Electrospinning and atomic layer deposition (ALD) have been coupled to prepare functional hetero-structures with potential application in fuel cells. Electrocatalysts comprising platinum (Pt) nanoparticles dispersed onto electrospun carbon fibers were selectively decorated with tin dioxide (SnO2) using ALD. The presence of SnO2 led to a considerable enhancement of the catalyst durability during voltage cycling.
Introduction
Research on proton exchange membrane fuel cell (PEMFC) electrocatalysts has been mainly focused on understanding and improving their activity towards the kinetically limiting oxygen reduction reaction (ORR) at the cathode (and methanol/ethanol oxidation reaction at the direct alcohol fuel cell anode). Significant advances in this regard have been achieved by tuning the size and morphology of the Pt nanocatalyst and elaborating alloys.1 However, performance degradation over time has emerged as a critical aspect complementary to the electrocatalyst efficiency, and has been extensively studied.2 This issue, together with the low abundance of Pt in the Earth's crust and thus its cost, has a great impact on hindering the commercialization of PEMFC and the consequent development of a sustainable hydrogen-based economy. The two predominant and interacting processes affecting electrocatalyst lifetime are the corrosion of the carbon-based support and the agglomeration and detachment of the Pt nanoparticles leading to the loss of active surface area under specific cell operating conditions.At potential higher than 0.207 V vs. RHE (reversible hydrogen electrode), carbon is oxidized into CO2.3 At potentials encountered in normal PEMFC operation (0.6–0.9 V) the kinetics of the oxidation reaction are sluggish, while under dynamic processes, such as start-up and shut down, it is accelerated as a direct consequence of high potential values reached by the cathode, for example due to local fuel starvation4 (≥1.2 V). Furthermore, the presence of Pt particles accelerates the phenomenon catalyzing the reaction by providing highly reactive transient oxygen radicals.5 Carbon corrosion results in detachment/agglomeration of catalyst nanoparticles and a decrease in active surface area and performance, as well as in an increase in hydrophilicity and loss of porosity of the catalyst layer which negatively affect the mass-transport of gases and water management.2b Concerning the degradation of the nanocatalyst, it must be considered that under standard conditions platinum is thermodynamically unstable at pH values lower than 0 and at potentials greater than 1.0 V, and that the increase in potential and temperature is detrimental for the catalyst stability.6 Together with the detachment related to support oxidation, three other mechanisms are reported in literature:2e,7 Pt nanocrystal migration and coalescence, and Pt dissolution and re-precipitation on the support (electrochemical Ostwald ripening) or in the membrane (“Pt band”). Poisoning of the Pt surface with contaminants from the fuel and air, such as COx, NOx, SOx, H2S, NH3 and small organics, leads to the loss of electrocatalyst activity and fuel cell performance.8
Several strategies are employed to mitigate the degradation of PEMFC electrodes. To enhance the stability of the Pt based electrocatalyst, several alloys with noble and non-noble metals in binary and ternary catalysts have shown to be an effective route against dissolution and detachment.9 Use of core–shell nanoparticles and tuning of the catalyst morphology have also proved to be elegant approaches to improve the performance of Pt-based electrocatalysts.10 In order to avoid support corrosion, novel carbon nanostructured materials e.g. carbon nanotubes and nanofibers have been developed demonstrating improved stability.11 In this regard electrospinning has emerged as a straight-forward and readily up-scalable method to prepare 1D nanomaterials applicable in fuel cell electrodes.12 Another approach is to replace carbon by alternative materials such as transition metal oxides, e.g. TiO2 and SnO2, with intrinsic high electrochemical stability.13 The interaction of such oxides with platinum may also promote electrocatalytic activity through the so-called strong-metal–support-interaction.14 Nevertheless, the drawback of metal oxides in general, is their low electrical conductivity that can in some cases be overcome by doping15 or by using carbon based composite materials.16
In this work a promising composite electrocatalyst support material was elaborated based on electrically conducting carbon nanofibres (CFs) prepared by electrospinning and decorated with non-conducting stabilizing SnO2 using atomic layer deposition (ALD). In particular, the protecting tin oxide was deposited directly onto the Pt decorated CFs. This approach has been recently applied in heterogeneous catalysis, where porous Al2O3 ALD film stabilized Pd particles17 and in electrocatalysis where ZrO2 was deposited around Pt particles previously protected with an organic layer.18 Morphology, surface analysis, electrocatalytic properties towards ORR and the electrochemical degradation of such novel electrocatalysts were investigated. The addition of SnO2 permitted a remarkable increase of the catalyst durability without reducing Pt activity. So far ALD has been coupled to electrospinning to prepare a Pt electrocatalyst on fibers.19 Here ALD allows the protection of both the electrocatalyst and the support, while keeping intact their intrinsic catalytic activity and electrical conductivity, respectively. The unhindered activity may result from the very low and controlled amount of material deposited on the one hand, and the preferential growth of tin dioxide on the carbon fibers on the other hand.
Results and discussion
The CFs comprise electrospun polyacrylonitrile fibers stabilized at 280 °C and graphitized under inert atmosphere at 1000 °C. The as-obtained fibers present an ID/IG ratio in Raman spectroscopy of 1.7 comparable to that of commercial carbon nanotubes.20 Investigation of the electrical properties using a four probe method demonstrates electrical conductivity comparable to that of carbon black Vulcan XC-72, a conventional electrocatalyst support.21Pt nanoparticles (NPs) were deposited onto the fibers using a microwave assisted polyol method.22 TEM images (Fig. 1) show a quite good Pt dispersion with a few aggregates. The size distribution (see Table SI.1 in ESI† and Fig. 1b) is centered at 2.3 ± 1.0 nm, while a domain size of 2.9 nm was determined by XRD. A small amount of SnO2 was deposited in a controlled manner using a non-aqueous sol–gel ALD approach.23 The number of ALD cycles was set for ∼1 nm thick SnO2 coating on Si reference wafer. No noticeable change on Pt nanoparticle distribution (Fig. 1 and Table SI.1†) was observed after deposition. It was previously demonstrated that the SnO2 deposition approach used leads to granular rather than continuous coating, especially on graphitized carbon.23,24 However, no clear evidence of SnO2 is visible in TEM images due to the very low amount of SnO2 deposited and the amorphous state of the oxide.22
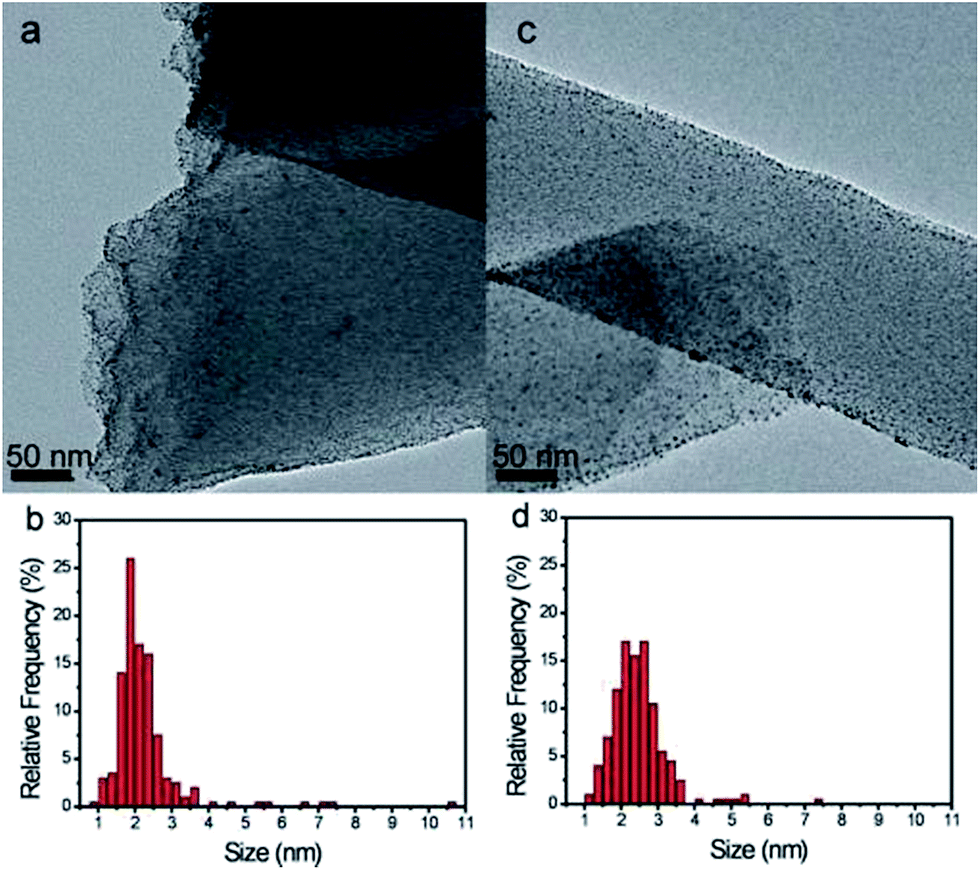 | ||
| Fig. 1 (a and c) TEM images and (b and d) particle distribution obtained from TEM analysis of (a and b) Pt/CFs, (c and d) SnO2/Pt/CFs before cyclic voltammetry. | ||
Indeed, the HRTEM images in Fig. 2 show that after ALD the Pt surface remains clean and comparable to uncoated CFs. Dark field STEM image recorded on coated samples (Fig. 2d) reveals tiny clusters (red arrows), almost not visible in bright field (Fig. 2c), dispersed onto the carbon, which could be attributed to SnO2.
 | ||
| Fig. 2 (a and b) HRTEM images of Pt/CFs and SnO2/Pt/CFs before cyclic voltammetry. (c and d) STEM images of SnO2/Pt/CFs in bright and dark field. | ||
XPS analyses were performed on SnO2-coated samples to confirm the presence of the metal oxide and the Pt loading. Table SI.2† reports the weight percentage of Pt, Sn, O and C in the sample. In the high resolution scan across the Sn 3d region of Fig. 3a the two Sn 3d5/2 peaks at 486.9 eV and 495.3 eV confirm the presence of tin in an oxidized state.23,25 The calculated stoichiometric ratio Sn/O in our samples, utilizing the O 1s peak at 530.7 eV attributed to the metal oxide, is 0.54, in good agreement with value reported in the literature15a and confirms the presence of tin in the oxidation state (IV).26 Platinum, tin, oxygen and carbon were also observed in the survey spectrum. Looking at the high resolution scan across the Pt 4f region (Fig. 3b) mostly Pt in 0 oxidation state is detected. It is worthy to note a very small shift of the two Pt 4f5/2 and Pt 4f7/2 peaks to lower binding energy compared to Pt/CFs, which can be attributed to a slight increase of the local electron density due to charge transfer from SnO2 to Pt resulting from strong metal–metal oxide interaction.27
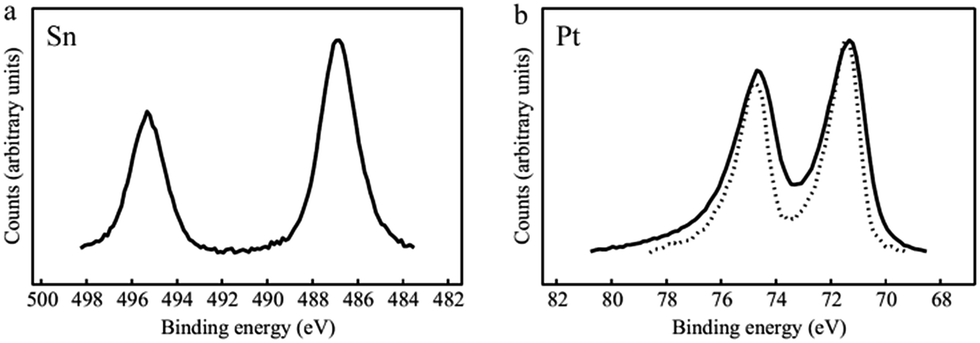 | ||
| Fig. 3 XPS spectra of SnO2/Pt/CFs (full line) and Pt/CFs (dotted line). High resolution scans across (a) the Sn 3d and (b) Pt 4f edges, respectively. | ||
In order to evaluate the impact of the presence of tin oxide on the electrocatalyst accessibility and performance, cyclic voltammetry (CV) in a N2 saturated 0.5 M aqueous H2SO4 was performed and is presented in Fig. 4a. Similar profiles are observed for Pt/CFs with and without SnO2. Both electrocatalysts clearly showed H adsorption and desorption peaks at 0.05–0.3 V (ref. 28) as well as PtO formation and reduction peaks at 0.7–0.9 V. The Pt electrochemically active surface area (ECSA) was determined from H adsorption and desorption: no significant change of ECSA is noted between Pt/CFs (27 m2 gPt−1) and SnO2-decorated Pt/CFs (29 m2 gPt−1). Similar and comparable values have been previously reported for other carbon nanofiber based Pt catalysts with similar loading.11c,29 Thus, the presence of SnO2 does not seem to influence the overall electrochemical behavior of the samples, proving that the coating does not prevent reacting species from reaching the catalyst. One can therefore conclude that the deposited SnO2 particles does not cover the Pt NPs, but must be preferentially deposited onto the carbon support. This is also supported by the fact that a retarded nucleation process on noble metal substrates was already demonstrated in the literature.30 Indeed an onset of deposition was noted on a Pt support;30a in the case of HfO2 ALD from HfCl4 and H2O, 30–50 cycles are required to initiate the growth.30c This behavior has been attributed to the relative inertness of noble metals toward hydroxylation that is required for the creation of anchoring sites.30b No oxide is present to initiate the growth and different mechanisms that on oxide or silicon may occur at the early stage, e.g. it was shown that Pt surface provides a different bonding environment for ALD HfO2.31 Furthermore a significant lattice mismatch between Pt and metal oxide may also restrict the nucleation of grains leading to a nucleation delay.30b Considering the growth rate and the low number of cycles performed here (20 cycles), no real growth on the Pt NP surface may occur as the offset may have not been reached, even though deposition of few SnO2 clusters on the metal particles cannot be completely excluded. On the other hand, the functional groups available on the surface of carbon fibers promote the nucleation of SnO2 even after only few cycles.32 Therefore, SnO2 should grow preferentially from oxygen functional groups present onto the carbon substrate in proximity to the Pt catalyst nanoparticles, permitting a strong metal–metal oxide interaction without significant shielding effect. A schematic representation of the electrocatalyst surface is shown in Fig. 5.
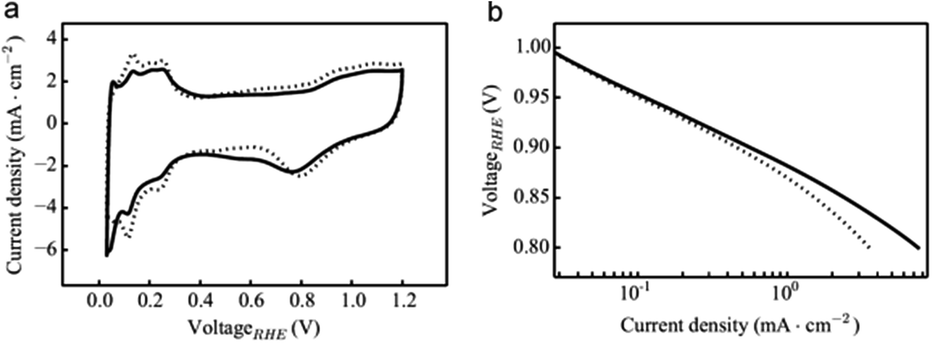 | ||
| Fig. 4 (a) Cyclic voltammetry at 50 mV s−1 in a 0.5 M H2SO4 solution saturated with N2. (b) ORR Tafel plot for uncoated (dotted line) and SnO2-decorated carbon fibers (full line). | ||
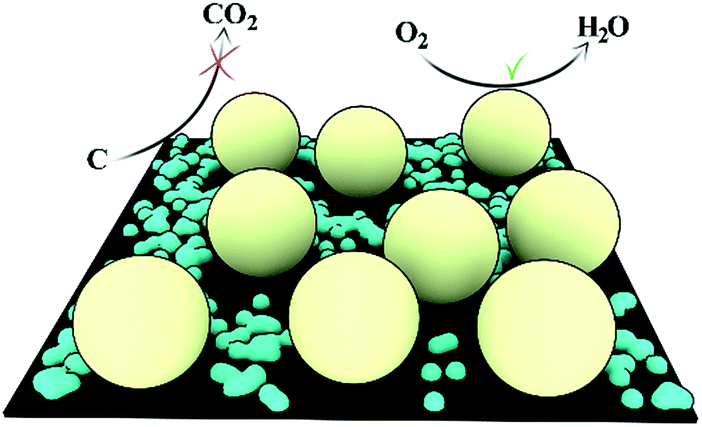 | ||
| Fig. 5 Schematic representation of the SnO2(blue)/Pt(yellow)/CFs (black) surface. | ||
To confirm that the electrocatalyst activity is not hindered by the presence of tin dioxide, ORR was performed in an O2 saturated 0.5 M aqueous H2SO4 at different rotating electrode speeds (Fig. SI.2†). No loss of the performance was noted in the presence of the metal oxide as shown by the ORR mass activities (Table SI.3†). The calculated number of electrons involved in the oxygen reduction mechanism using the Koutecky–Levich analysis is ∼4 for both electrocatalysts as expected for Pt, suggesting a mechanism that does not involve production of H2O2.33 Furthermore, the corresponding kinetic currents involved were calculated and are reported in the Tafel plot in Fig. 4b. Even though at low current density similar kinetic currents are noted for unprotected Pt/CFs and SnO2/Pt/CFs, a significant decrease in potential is observed for Pt/CFs compared to the SnO2 decorated electrocatalyst. This suggests a beneficial effect of SnO2-coated fibers at higher potential values, which we attribute to the proximity of platinum to the metal oxide and its promoting catalytic effect (Fig. 5).
In order to evaluate the durability of the electrocatalyst, an accelerated degradation protocol was applied by cycling from 0.6 to 1.2 V at 100 mV s−1 for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and monitoring the electrochemical surface area decrease every 100th cycle performing a CV from 0.05 to 1.2 V at 50 mV s−1. Fig. 6 presents the ECSA as a function of the cycle number and shows a reduced ECSA loss for the SnO2-decorated catalysts compared to pristine Pt/CFs electrocatalysts. In a previous study, the significant ECSA loss and thus catalyst degradation over cycling has been defined as the major issue for electrospun CFs.21 SnO2-decorated Pt/CFs samples maintained an active surface area around 20 m2 gPt−1 by the end of the cycling, while the loss of Pt active surface on unprotected carbon fibers was much more significant. The loss is halved with addition of SnO2 as reported in Table SI.4† and thus a positive influence of SnO2 decoration with respect to degradation over cycling is clearly demonstrated. This result is in agreement with the high electrochemical stability reported in the literature of the metal oxide, used either as additive or as replacement for conventional carbon-based electrocatalyst supports.15a,34
000 cycles and monitoring the electrochemical surface area decrease every 100th cycle performing a CV from 0.05 to 1.2 V at 50 mV s−1. Fig. 6 presents the ECSA as a function of the cycle number and shows a reduced ECSA loss for the SnO2-decorated catalysts compared to pristine Pt/CFs electrocatalysts. In a previous study, the significant ECSA loss and thus catalyst degradation over cycling has been defined as the major issue for electrospun CFs.21 SnO2-decorated Pt/CFs samples maintained an active surface area around 20 m2 gPt−1 by the end of the cycling, while the loss of Pt active surface on unprotected carbon fibers was much more significant. The loss is halved with addition of SnO2 as reported in Table SI.4† and thus a positive influence of SnO2 decoration with respect to degradation over cycling is clearly demonstrated. This result is in agreement with the high electrochemical stability reported in the literature of the metal oxide, used either as additive or as replacement for conventional carbon-based electrocatalyst supports.15a,34
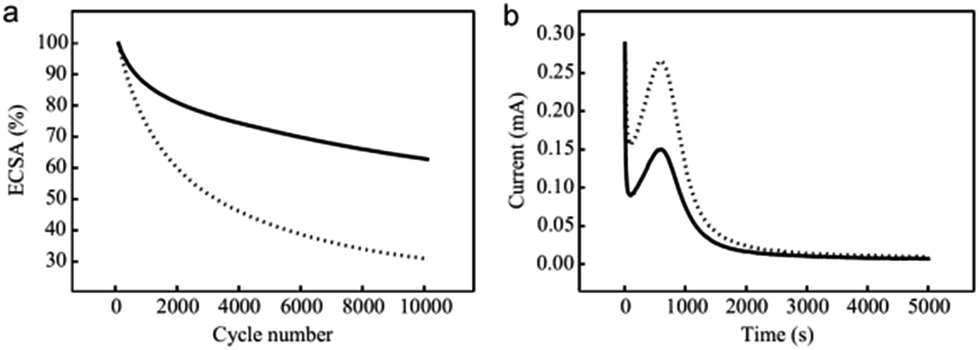 | ||
| Fig. 6 (a) ECSA of Pt/CFs (dotted line) and SnO2/Pt/CFs (full line) calculated from cyclic voltammetry at 50 mV s−1 in a N2 saturated 0.5 M H2SO4 solution every 100 cycles performed at 100 mV s−1. (b) Oxidation/corrosion characteristics for Pt/CFs (dotted line) and SnO2/Pt/CFs (full line) at 1.4 V/RHE. | ||
To further confirm the protecting role of the tin oxide component on the Pt/CFs, an accelerated corrosion test was performed in conditions providing information on degradation under fuel starvation and reverse current at the cathode side (1.4 V/RHE, 80 °C, 0.5 M H2SO4, 2 h). The corrosion is quantified in terms of loss of material by the effect of electrochemical oxidation.35 For carbon based materials the corrosion reaction is well known:
| C + H2O → CO2 + 4H+ + 4e−ref. 3b |
From integration of the oxidation currents over time (oxidation charge) it is possible to estimate the carbon loss into CO2 from the Faraday law. Fig. 6b presents the currents obtained versus time for ALD SnO2 protected and unprotected samples. It is evident that the oxidation charge is lower for the former (3.2 C mg−1, carbon loss 12.6%) than for the pristine CFs (5.3 C mg−1, carbon loss 16.2%). This accelerated test clearly proves that the metal oxide is able to cover and partially protect the carbon support from corrosion (see Fig. 5).
Finally, TEM studies were performed on the 5% Pt loaded CF catalysts with and without SnO2 after CV prolonged cycling (Fig. 7a and c). It is evident that the amount of Pt remaining on the fibers after 10000 cycles is much higher on the SnO2 coated CFs (Fig. 7b) than on the non-protected CFs (Fig. 7a). A better dispersion of the particles is also observed in the presence of the oxide. For both samples, growth of Pt nanoparticles is noted with a size distribution (Table SI.1†) increasing from ∼2 nm before to either 7 nm or 4.9 nm after cycling for Pt/CFs and SnO2/Pt/CFs, respectively, indicating that Ostwald ripening is almost suppressed by SnO2 surrounding the noble metal particles (Fig. 5). It must be also noted that a narrower particle distribution is obtained in the presence of the metal oxide (Fig. 7b and d). Therefore the improved durability is probably related not only to the protection of carbon from corrosion but also to a better stability, adhesion and confinement onto CFs of Pt NPs in presence of SnO2.
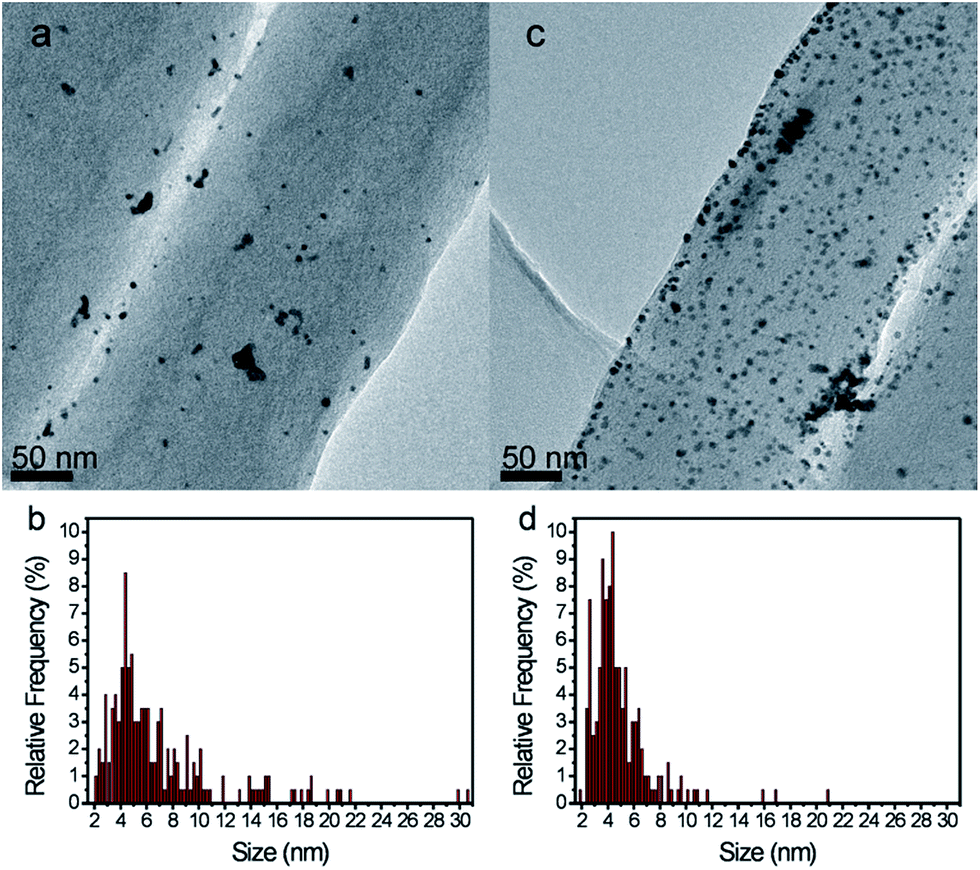 | ||
| Fig. 7 TEM images (a and c) and (b and d) particle distribution from TEM analysis of (a and b) Pt5%–CFs and (c and d) SnO2–Pt5%–CFs after 10000 potential cycles. | ||
Experimental
Polyacrylonitrile (PAN, Mw = 150000, Sigma Aldrich) based carbon fibers were prepared combining electrospinning with thermal treatment as already reported.21 8% (w/w) PAN solution in N,N-dimethylformamide (DMF, 98%, Sigma Aldrich) was feed to a metallic needle via a syringe pump with a flow rate of 1 ml h−1. An electric field of 15 kV was applied between the needle and a metallic rotating drum collector (from Linari Engineering) placed 10 cm away. As-prepared PAN fibers were collected in the form of non-woven mats. Fiber carbonization was realized by a two-step thermal treatment. First an oxidative stabilization was performed at 280 °C for 1 h followed by a carbonization for 1 h under N2 at 1000 °C using a heating ramp of 1 °C min−1.
CFs were loaded at 20% (w/w) with Pt NPs synthesized by a microwave assisted polyol synthesis.15a Thermogravimetric analysis was performed in air up to 800 °C (10 °C min−1) using a Netzsch TG 439 thermobalance to determine the Pt loading on Pt/CF electrospun fibers.
After Pt deposition, the differently heat-treated CFs were coated with SnO2 by nonaqueous ALD.23 Based on approaches used for CNT coating,36 Pt/CFs were contained during the deposition inside of a “tea bag” made of Kevlar mat. Tin tert-butoxide and acetic acid were used as metal and oxygen source, respectively.23 Depositions took place in exposure mode home-made reactor at 175 °C and 20 ALD cycles were performed.
Structural characterization was performed by X-ray diffraction on a PANAlytical X'pert powder diffractometer equipped with CuKα radiation (λ = 1.542 Å). The Pt crystallite size was estimated by the Scherrer equation using the Pt (111) diffraction line at 39.7°.
Bright field TEM images were acquired on a Philips CM 200 LaB6 microscope operated at 200 kV. Phase contrast and STEM images were acquired on a Jeol-ARM200F equipped with a cold field emission gun and operated at 200 kV.
X-ray photoelectron spectroscopy was performed using a Physical Electronics PHI 5700 spectrometer with a non-monochromatic Mg Kα radiation (300 W, 15 kV, hν = 1253.6 eV) as excitation source. Spectra were recorded at a 45° take-off angle by a concentric hemispherical analyzer operating in the constant pass energy mode at 25.9 eV, using a 720 μm diameter analysis area. Core level C 1s, O 1s, Pt 4f and Sn 3d signals were recorded. Powdered solids were mounted on a sample holder without adhesive tape and kept overnight in a high vacuum chamber before they were transferred inside the analysis chamber of the spectrometer. Each region was scanned with several sweeps until a good signal to noise ratio was observed. The pressure in the analysis chamber was maintained below 10−7 Pa. PHI ACCESS ESCA-V6.0 F and Multipak 8.2b software packages were used for acquisition and data analysis. A Shirley-type background was subtracted from the signals. Recorded spectra were always fitted using Gauss–Lorentz curves in order to determine the binding energy of the different element core levels more accurately. The accuracy of the binding energy (BE) values was within ±0.1 eV. Electrochemical analyses were carried out in a three-electrode cell comprising a gold (chronoamperometry) or glassy carbon (other voltammetric characterizations) rotating disk electrode (RDE) (working electrode geometric area 0.196 cm2), a reversible hydrogen electrode (RHE, reference electrode) and a platinum wire (counter electrode), and a Pine bipotentiostat model AFCBP1. All the potential values are referred to the RHE. Catalyst ink was prepared by mixing 10 mg Pt/CF catalyst with 108.5 μl of 5% (w/w) aqueous/alcoholic solution of Nafion, 300 μl of ethanol and 30 μl of DI water. After 20 min of sonication, 7 μl of ink was deposited onto the electrodes and dried for 10 min to obtain a total Pt loading of 32 μg on the RDE tip. CV was performed in 0.5 M H2SO4 solution saturated with either N2 or O2 on glassy carbon electrode after deposition of the electrocatalyst. In particular ECSA determination was performed by CV in 0.5 M H2SO4 solution saturated with N2 cycling between 0.05 and 1.2 V (vs. RHE) at a speed of 50 mV s−1 while ORR tests were performed by linear sweep voltammetry in 0.5 M H2SO4 saturated with O2 between 0.2 and 1.2 V (vs. RHE) at a scan speed of 20 mV s−1 and RDE rotating speeds of 400, 900, 1600 and 2500 RPM.
Cyclic voltammetry based accelerated degradation tests were performed by cycling between 0.6 and 1.2 V at 100 mV s−1 for 10000 cycles measuring the ECSA every 100th cycle at 50 mV s−1.
Potentiostatic accelerated stress tests were performed by holding the gold working electrode potential at 1.4 V vs. RHE for 2 h in 0.5 M H2SO4 at 80 °C. Pt/CFs before and after SnO2 ALD were characterized by TEM.
Conclusions
By combining original synthetic approaches, i.e. electrospinning, microwave-assisted synthesis and atomic layer deposition, a novel electrocatalyst was elaborated with significantly improved stability over cycling. The addition of SnO2 permitted a remarkable increase of the catalyst durability and even a slight increase in the Pt activity. Regarding the different characterizations presented in this work, it can be safely concluded that ALD of SnO2 allows the protection of both the electrocatalyst and the support, while keeping intact their intrinsic catalytic activity and electrical conductivity, respectively. Lower corrosion of the carbon support, moderate aggregation of Pt particles and improved stability, adhesion and confinement on CFs of Pt NPs were observed when the electrocatalyst is decorated with tin oxide. The protection of Pt/CFs with metal oxide halves the degradation of the electrocatalyst over prolonged electrochemical cycling. Furthermore unhindered activity of Pt was observed suggesting no growth on the noble metal and preferential grow of tin dioxide on the carbon fibers although metal – metal oxide interaction seems to occur due to the proximity between these CF-supported components. It must be stressed that the successful selective SnO2 decoration is only based on the difference in reactivity and density of anchoring sites of the Pt and CFs, without need of a protective organic layer around Pt, for example. Finally, this work certainly paves the way to the elaboration of modified carbon-based electrodes for fuel cells and related energy applications.Acknowledgements
The authors acknowledge R. M. Silva and I. Jiménez-Morales for their experimental support. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n. 306682.Notes and references
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- (a) R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed; (b) L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J.-C. Perrin, E. Moukheiber, A. ElKaddouri, G. de Moor, C. Bas, L. Flandin and N. Caqué, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560 CrossRef CAS; (c) J. C. Meier, C. Galeano, I. Katsounaros, A. A. Topalov, A. Kostka, F. Schüth and K. J. J. Mayrhofer, ACS Catal., 2012, 2, 832–843 CrossRef CAS; (d) Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558–566 CrossRef CAS; (e) Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS; (f) S. Zhang, X.-Z. Yuan, J. N. C. Hin, H. Wang, K. A. Friedrich and M. Schulze, J. Power Sources, 2009, 194, 588–600 CrossRef CAS.
- (a) F. N. Büchi, M. Inanba and T. J. Schmidt, Polymer Electrolyte Fuel Cell Durability, Springer-Verlag, New York, 2009 Search PubMed; (b) S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, J. Power Sources, 2008, 176, 444–451 CrossRef CAS; (c) Y. Yu, H. Li, H. Wang, X.-Z. Yuan, G. Wang and M. Pan, J. Power Sources, 2012, 205, 10–23 CrossRef CAS.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS.
- (a) M. Cai, M. S. Ruthkosky, B. Merzougui, S. Swathirajan, M. P. Balogh and S. H. Oh, J. Power Sources, 2006, 160, 977–986 CrossRef CAS; (b) J. Willsau and J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem., 1984, 161, 93–101 CrossRef CAS; (c) K. Kinoshita and J. A. S. Bett, Carbon, 1974, 12, 525–533 CrossRef CAS.
- B. M. J. N. Pourbaix, J. van Muylder and N. de Zoubov, Platinum Metals Review, 1959, 3, 47–53 Search PubMed.
- (a) P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef; (b) K. G. Gallagher, R. M. Darling and T. F. Fuller, in Handbook of Fuel Cells: Advances in Electrocatalysis, Materials, Diagnostics and Durability, ed. W. Vielstich, H. Yokokawa and H. A. Gasteiger, John Wiley and Sons, Chichester, 2003, vol. 6, pp. 819–829 Search PubMed; (c) B. Vion-Dury, M. Chatenet, L. Guétaz and F. Maillard, ECS Trans., 2011, 41, 697–708 CrossRef CAS.
- (a) O. Yamazaki, Y. Oomori, H. Shintaku and T. Tabata, ECS Trans., 2007, 11, 287–295 CrossRef CAS; (b) X. Cheng, Z. Shi, N. Glass, L. Zhang, J. Zhang, D. Song, Z.-S. Liu, H. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS.
- (a) S. Chen, H. A. Gasteiger, K. Hayakawa, T. Tada and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, A82–A97 CrossRef CAS; (b) H. R. Colón-Mercado, H. Kim and B. N. Popov, Electrochem. Commun., 2004, 6, 795–799 CrossRef; (c) J. Xi, J. Wang, L. Yu, X. Qiu and L. Chen, Chem. Commun., 2007, 1656–1658 RSC; (d) J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- M. Cao, D. Wu and R. Cao, ChemCatChem, 2014, 6, 26–45 CrossRef CAS.
- (a) Z. Lin, L. Ji, W. E. Krause and X. Zhang, J. Power Sources, 2010, 195, 5520–5526 CrossRef CAS; (b) S. Park, Y. Shao, R. Kou, V. V. Viswanathan, S. A. Towne, P. C. Rieke, J. Liu, Y. Lin and Y. Wang, J. Electrochem. Soc., 2011, 158, B297–B302 CrossRef CAS; (c) D. Sebastián, A. G. Ruíz, I. Suelves, R. Moliner, M. J. Lázaro, V. Baglio, A. Stassi and A. S. Aricò, Appl. Catal., B, 2012, 115–116, 269–275 CrossRef.
- (a) S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Rozière, Energy Environ. Sci., 2011, 4, 4761 RSC; (b) V. Thavasi, G. Singh and S. Ramakrishna, Energy Environ. Sci., 2008, 1, 205–221 RSC.
- (a) Y. Shao, J. Liu, Y. Wang and Y. Lin, J. Mater. Chem., 2009, 19, 46–59 RSC; (b) Y.-J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed; (c) I. Savych, S. Subianto, Y. Nabil, S. Cavaliere, D. Jones and J. Roziere, Phys. Chem. Chem. Phys., 2015, 17, 16970–16976 RSC.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- (a) S. Cavaliere, S. Subianto, I. Savych, M. Tillard, D. J. Jones and J. Rozière, J. Phys. Chem. C, 2013, 117, 18298–18307 CrossRef CAS; (b) N. R. Elezović, B. M. Babić, V. R. Radmilović and N. V. Krstajić, J. Electrochem. Soc., 2013, 160, F1151–F1158 CrossRef; (c) A. Masao, S. Noda, F. Takasaki, K. Ito and K. Sasaki, Electrochem. Solid-State Lett., 2009, 12, B119–B122 CrossRef CAS.
- (a) A. Bauer, C. Song, A. Ignaszak, R. Hui, J. Zhang, L. Chevallier, D. Jones and J. Rozière, Electrochim. Acta, 2010, 55, 8365–8370 CrossRef CAS; (b) K. Miyazaki, K.-I. Kawakita, T. Abe, T. Fukutsuka, K. Kojima and Z. Ogumi, J. Mater. Chem., 2011, 21, 1913–1917 RSC.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- N. Cheng, M. N. Banis, J. Liu, A. Riese, X. Li, R. Li, S. Ye, S. Knights and X. Sun, Adv. Mater., 2015, 27, 277–281 CrossRef CAS PubMed.
- Q. Du, J. Wu and H. Yang, ACS Catal., 2014, 4, 144–151 CrossRef CAS.
- J.-P. Tessonnier, D. Rosenthal, T. W. Hansen, C. Hess, M. E. Schuster, R. Blume, F. Girgsdies, N. Pfänder, O. Timpe, D. S. Su and R. Schlögl, Carbon, 2009, 47, 1779–1798 CrossRef CAS.
- I. Savych, J. Bernard d'Arbigny, S. Subianto, S. Cavaliere, D. J. Jones and J. Rozière, J. Power Sources, 2014, 257, 147–155 CrossRef CAS.
- Y. Lin, S. Zhang, S. Yan and G. Liu, Electrochim. Acta, 2012, 66, 1–6 CrossRef CAS.
- C. Marichy, N. Donato, M.-G. Willinger, M. Latino, D. Karpinsky, S.-H. Yu, G. Neri and N. Pinna, Adv. Funct. Mater., 2011, 21, 658–666 CrossRef CAS.
- C. Marichy, P. A. Russo, N. Donato, M. Latino, G. Neri and N. Pinna, J. Phys. Chem., 2013, 117, 19729–19739 CAS.
- L. Jie and X. Chao, J. Non-Cryst. Solids, 1990, 119, 37–40 CrossRef.
- J.-M. Themlin, M. Chtaïb, L. Henrard, P. Lambin, J. Darville and J.-M. Gilles, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 2460–2466 CrossRef CAS.
- (a) A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Phys. Chem. C, 2011, 115, 20153–20159 CrossRef CAS; (b) G. Samjeske, S.-I. Nagamatsu, S. Takao, K. Nagasawa, Y. Imaizumi, O. Sekizawa, T. Yamamoto, Y. Uemura, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2013, 15, 17208–17218 RSC.
- G. Jerkiewicz, Electrocatalysis, 2010, 1, 179–199 CrossRef CAS.
- E. Yli-Rantala, A. Pasanen, P. Kauranen, V. Ruiz, M. Borghei, E. Kauppinen, A. Oyarce, G. Lindbergh, C. Lagergren, M. Darab, S. Sunde, M. Thomassen, S. Ma-Andersen and E. Skou, Fuel Cells, 2011, 11, 715–725 CrossRef CAS.
- (a) A. J. Niskanen, T. Ylinen-Hinkka, M. Pusa, S. Kulmala and S. Franssila, Thin Solid Films, 2010, 519, 430–433 CrossRef CAS; (b) K. Kukli, M. Ritala, T. Pilvi, T. Aaltonen, J. Aarik, M. Lautala and M. Leskelä, Mater. Sci. Eng. B, 2005, 118, 112–116 CrossRef; (c) K. Kukli, T. Aaltonen, J. Aarik, J. Lu, M. Ritala, S. Ferrari, A. Hårsta and M. Leskelä, J. Electrochem. Soc., 2005, 152, F75 CrossRef CAS.
- C. L. Platt, N. Li, K. Li and T. M. Klein, Thin Solid Films, 2010, 518, 4081–4086 CrossRef CAS.
- C. Marichy, J.-P. Tessonnier, M. C. Ferro, K.-H. Lee, R. Schlögl, N. Pinna and M.-G. Willinger, J. Mater. Chem., 2012, 22, 7323–7330 RSC.
- (a) H. S. Wroblowa, P. Yen Chi and G. Razumney, J. Electroanal. Chem. Interfacial Electrochem., 1976, 69, 195–201 CrossRef CAS; (b) N. M. Marković, T. J. Schmidt, V. Stamenković and P. N. Ross, Fuel Cells, 2001, 1, 105–116 CrossRef.
- (a) A. Rabis, E. Fabbri, A. Foelske, M. Horisberger, R. Kötz and T. J. Schmidt, ECS Trans., 2013, 50, 9–17 CrossRef; (b) K. Kanda, Z. Noda, Y. Nagamatsu, T. Higashi, S. Taniguchi, S. M. Lyth, A. Hayashi and K. Sasaki, ECS Electrochem. Lett., 2014, 3, F15–F18 CrossRef CAS; (c) P. Zhang, S.-Y. Huang and B. N. Popov, J. Electrochem. Soc., 2010, 157, B1163–B1172 CrossRef CAS.
- P. L. Antonucci, F. Romeo, M. Minutoli, E. Alderucci and N. Giordano, Carbon, 1988, 26, 197–203 CrossRef CAS.
- C. K. Devine, C. J. Oldham, J. S. Jur, B. Gong and G. N. Parsons, Langmuir, 2012, 27, 14497–14507 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Pt particle size distribution, ORR mass activities and ECSA loss after accelerated stress test. See DOI: 10.1039/c5ta08432f |
| ‡ Current address: Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. |
| This journal is © The Royal Society of Chemistry 2016 |