
Ultrathin mesoporous Co3O4 nanosheets with excellent photo-/thermo-catalytic activity†
Yali
Zheng
,
Wenzhong
Wang
 *,
Dong
Jiang
,
Ling
Zhang
,
Xiaoman
Li
and
Zhong
Wang
*,
Dong
Jiang
,
Ling
Zhang
,
Xiaoman
Li
and
Zhong
Wang
State Key Laboratory of High Performance Ceramics and Superfine Microstructures, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, P. R. China. E-mail: wzwang@mail.sic.ac.cn
First published on 12th November 2015
Abstract
Ultrathin mesoporous Co3O4 nanosheets on a stainless steel mesh (SS–Co3O4) have been synthesized through an electrochemical deposition method. The SS–Co3O4 catalyst exhibits high photothermal performance, resulting in a considerable increase of temperature above the light-off temperature for VOC oxidation. Aerobic oxidations of propylene and propane under simulated sunlight were selected as probe reactions to explore the photo-/thermo-catalytic (PTC) activity. The results show that SS–Co3O4 with only 7 mg of Co3O4 grown on the substrate possesses superior PTC activity, which merely occurs around the directly grown SS–Co3O4 rather than the Co3O4 coated SS. The combined properties of SS–Co3O4 such as the ultrathin two-dimensional shape, foam-like porous inside architecture, and supported metal substrate could lead to the integrative advantages of the maximum utilization of photothermal conversion, more catalytically active sites, and enhanced amount and mobility of surface lattice oxygen species, resulting in excellent PTC activity of SS–Co3O4. This work provides insights into the rational design of monolith catalysts for the solar driven environmental purification and suggests new ideas toward the overall utilization of solar energy.
1. Introduction
The control of VOC emissions is very important for a wide range of applications. Catalytic oxidation using transition metal oxides is considered to be one of the most efficient routes. However, high operating temperature in this process means low energy efficiencies, and will compromise the long-term stability of catalysts and decrease the selectivity for the desired products.1 At present, the development of effective catalysts for complete oxidation of VOCs at low temperature is still a challenge to be solved.2In this regard, nature has already set a good example for us. UV-vis light of the solar spectrum with higher energy could be used to activate the photocatalytic (PC) reaction, while the infrared part can supply the needed endothermic energy for the thermocatalytic (TC) reaction.3 The PC conversion of solar energy to chemical energy is convenient and sustainable for chemical reactions because solar light is abundant and it can facilitate room temperature chemical transformations by generating electronically excited states in photocatalysts.4 Meanwhile, suitable catalysts can efficiently absorb light and release the absorbed light energy in the form of heat (photothermal effect), resulting in a considerable increase of temperature, which is just enough to stimulate the TC process. Therefore, integrating mild TC into PC should be promising for utilizing the solar energy and avoiding high operating temperature. Compared with sole PC and TC, photo-/thermo-catalysis (PTC) possesses the following advantages: (i) the processes are characterized by high energy efficiencies without input of thermal energy; (ii) the temperature on the catalyst surface rises instantaneously due to the photothermal effect,5 and the heat is confined on the surface of the catalyst (local temperature effect);6 (iii) the rate constant of the PTC process is much higher than a linear combination of TC and PC, which is attributed to the existence of a PTC synergetic effect;7–9 (iv) in some cases, PTC can effectively refrain the deactivation of the catalyst10 and increase the selectivity for the desired products. Recently, we proposed an integration of solar energy conversion with synergistic low-temperature catalysis in Ce1−xBixO2−δ nanorods, the coupled electronic and ionic conduction help improve the negative temperature effect and integrate synergistic low temperature catalysis into solar energy utilization.11
In most cases, the catalysts are used in the form of powder or supported on cordierite, which will not be helpful for the maximum utilization of the photothermal effect and will sacrifice the overall activity. Therefore, how to prepare supported catalysts with high efficiency for the photothermal utilization is particularly important, which is another issue to be considered. Metal substrate such as stainless steel mesh (SS) supported catalysts come to mind because they can realize much stronger photoabsorption abilities than traditional catalysts and display a strong photothermal effect. The use of metal substrates has a number of advantages. First, a high heating rate can be expected due to the low heat capacity of the metal substrate, it is much more easy to achieve the light-off temperature of the catalyst. Second, with good thermal conductivity and fast heat transfer properties, the metal substrate can effectively prevent the overheating induced deactivation of the catalyst. The catalyst can quickly achieve uniform temperature because of the high mass and heat transfer coefficient. Besides, research has shown that for stainless steel nets used as supports of dye-doped mesostructured films, the tiny grids of them could be regarded as laser resonant structures for the feedback of light.12 Taken together, the above effects can allow significant photothermal heating of the catalyst with surprisingly small amounts of optical energy input.13 Co3O4 has been reported to be most active in VOC catalytic combustion.14,15 As a certified photocatalyst and universal thermocatalyst, Co3O4 can be expected to show high PTC activity. Studies have shown that freestanding Co3O4 nanosheets on a conductive substrate could expose the maximum active surface, and the strong affinity between the support and the active materials could lead to good conductivity.16,17 Improved photothermal utilization and high-performance PTC activities can be expected for the Co3O4 catalysts grown on a metal substrate with a similar technique.
Specifically, herein we report ultrathin mesoporous Co3O4 nanosheets electrodeposited on a stainless steel mesh (SS–Co3O4), which have never been used directly as a catalyst in gas–solid phase catalytic oxidation. Aerobic oxidations of propylene (C3H6) and propane (C3H8) under simulated sunlight were selected as probe reactions to explore the PTC activity. The structural superiority of SS–Co3O4 and the actual contribution of the SS substrate have been integrally discussed. It was found that the excellent PTC activity of the SS–Co3O4 catalyst can be attributed to the maximum utilization of photothermal conversion, more catalytically active sites, and enhanced amount and mobility of surface lattice oxygen species. In addition, this work opens new avenues towards the design of more energy-efficient monolith catalysts for the solar driven environmental purification.
2. Experimental
2.1 Catalyst preparation
A stainless steel mesh (SS, 200 mesh, 316 stainless steel, approximately 2 cm × 3 cm) was carefully cleaned with 2 M NaOH solution in an ultrasound bath for 30 min, and then rinsed with deionized water, acetone and absolute ethanol, respectively. SS–Co3O4 was synthesized through an electrochemical deposition method as previously reported.18 A standard three-electrode glass cell was used, consisting of the clean SS working electrode, a platinum plate counter electrode and a saturated calomel reference electrode (SCE). The green Co(OH)2 was electrodeposited upon SS in a 0.05 M Co(NO3)2·6H2O aqueous electrolyte using a CHI 660C electrochemical workstation (Shanghai Chenhua, China). The electrodeposition potential is −1.0 V (vs. SCE). After electrodeposition for 20 min, the green stainless steel mesh was carefully rinsed several times with deionized water and dried at 60 °C, followed by a calcination process at 250 °C for 2 h with a ramping rate of 1 °C min−1 to obtain Co3O4 nanosheets. On average, about 7 mg of Co3O4 nanosheets was grown on the 6 cm2 SS (Table S1†).2.2 Characterization
The purity and crystallinity of the SS–Co3O4 were measured by X-ray diffraction (XRD) using a Rigaku D/MAX 2250 V diffractometer with monochromatized Cu Kα radiation (λ = 0.15418 nm) under 40 kV, 100 mA and with the 2θ ranging from 10° to 80°. The morphologies and structures of the samples were characterized by scanning electron microscopy (SEM, Hitachi S-4800, Japan), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), selected area electron diffraction (SAED) (TecnaiG2 F20 S-Twin, accelerating voltage 200 kV), and N2 adsorption–desorption measurements using a V-sorb 2800P surface area analyzer. The specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method. The pore volume and pore size distribution plots were obtained by the Barrett–Joyner–Halenda (BJH) method. X-ray photoelectron spectroscopy (XPS) spectra were obtained by irradiating every sample with a 320 μm diameter spot of monochromated aluminum Kα X-rays at 1486.6 eV under ultrahigh vacuum conditions (performed on an ESCALAB 250, THERMO SCIENTIFIC Ltd.). Charging effects were corrected by adjusting the binding energy of C 1s to 284.6 eV. UV-vis diffuse reflectance spectra (DRS) of the samples were measured using a Hitachi UV-3010PC UV-vis spectrophotometer. Oxygen temperature-programmed desorption (O2-TPD) measurements were performed on a ChemiSorb 2750 instrument. Typically, 50 mg of the sample, placed in a glass tube, was pretreated by a He gas flow at 300 °C for 1 h. The adsorption of O2 was then performed in a 4 vol% O2/He gas flow for 1 h at room temperature. Afterward, the sample was heated to 900 °C at a rate of 10 °C min−1. The TPD signal was recorded by using a thermal conductivity detector. All the gas flow rates were set as 25 mL min−1.2.3 PTC oxidation of C3H6 and C3H8
The catalytic activities of all the samples were evaluated by the gas-phase aerobic oxidation of C3H6 (50 ppm) and C3H8 (50 ppm). The apparatus used was described in earlier papers.19 Experiments were performed in a gas-closed vitreous reactor (capacity 600 mL) with a quartz window, and a 500 mW cm−2 Xe lamp was lighted to simulate the optical excitation and thermal activation. Before the catalytic test, a thermocouple was placed on the catalyst to measure the temperature of the catalyst under the irradiation of the Xe lamp. During the oxidation process, a GC analyzer (GC 7900, Techcomp) equipped with two detective channels was used to detect the decrease of C3H6, C3H8 and the increase of CO2 simultaneously. The channel for C3H6 and C3H8 detection consists of a TM plot-Al2O3/S capillary column and a flame ionization detector (FID). Another channel for CO2 detection consists of a TDX-01, 80–100 mesh packed column followed by a methane conversion furnace and a FID.2.4 TC oxidation of C3H8
The TC activities of SS–Co3O4 for C3H8 oxidation were tested in a quartz tube reactor with an inner diameter of 10 mm at atmospheric pressure. The reagent gas consisted of 50 ppm C3H8 + 20% O2 + the balance made up of N2 at a flow rate of 535 mL min−1. 8 pieces of SS–Co3O4 were used as the catalyst. The reactants and products were analyzed online by using a gas chromatograph (GC) equipped with a TCD.3. Results and discussion
3.1 Structure and morphology analysis
SS–Co3O4 was synthesized through an electrochemical deposition method. Its XRD pattern was readily indexed to a pure spinel phase of Co3O4 (JCPDS card no. 65-3103, Fig. S1 in the ESI†). DTA/TG was performed to show the crystallization of Co3O4 (Fig. S2†). The morphology and size of the as-prepared SS–Co3O4 was estimated by SEM. Fig. 1 shows the SEM images of the SS–Co3O4 with different magnifications. It can be clearly seen that uniform Co3O4 nanosheets with high density are well aligned on SS, which are interconnected to each other.20 Especially concerning is that the thickness of every single Co3O4 nanosheet is estimated to be less than 10 nm (Fig. 1C and D). More notable from the high resolution SEM image is that the surface of every single Co3O4 nanosheet is full of voids, revealing that these nanosheets have foam-like porous inside architecture. These porous structures may be formed due to the dehydration and transformation of the crystalline structure during thermal treatment.17 Thus, most of the nanosheet surface is highly accessible by the gaseous pollutant when used as a catalyst for VOC disposal. | ||
| Fig. 1 SEM images of the Co3O4 nanosheet arrays supported on an SS substrate (SS–Co3O4). | ||
The structural feature of the SS–Co3O4 was further studied by TEM. Obviously, the ultrathin feature of the Co3O4 nanosheet is further verified by the typical wrinkled structure of nanosheets observed in Fig. 2A.21 Every ultrathin nanosheet consists of numerous interconnected nanoparticles with a size of ca. 10 nm forming a mesoporous structure (Fig. 2B and C). Furthermore, Fig. 3A and B prove that SS–Co3O4 has obvious porous foam-like inside architecture, which is well consistent with what we have observed from SEM. The lattice fringes in the HRTEM image (Fig. 2D) shows a well-defined crystalline structure with lattice spacings of 0.466 Å and 0.286 Å, corresponding to the value of the (111) and (202) planes of the Co3O4 phase. The SAED (inset of Fig. 2A and B) could be indexed to a polycrystalline pattern.
 | ||
| Fig. 2 (A and B) TEM images and the insets in (A and B) are SAED patterns, (C and D) HRTEM images of the Co3O4 nanosheets scratched down from the SS substrate. | ||
 | ||
| Fig. 3 (A) N2 adsorption–desorption isotherms and (B) BJH desorption pore size distribution plots of the Co3O4 nanosheets scratched down from the SS substrate. | ||
The Co3O4 nanosheets were scratched down from the SS through ultrasonication to estimate their mesoporosity and textural properties. From Fig. 3A, a typical IV isotherm (according to the IUPAC classification) with a distinct H3 hysteresis loop22 in the pressure range of 0.6–1.0 P/P0 can be clearly observed, suggesting the presence of a mesoporous structure of Co3O4 nanosheets.23 The pore size distribution plot of the sample calculated by using the desorption isotherm via the BJH method (Fig. 3B) showed that the pore size of Co3O4 nanosheets is centered at around 6 nm, clarifying their mesoporosities. The BET specific surface area was measured to be 56.1 m2 g−1.
3.2 Photothermal effect
For PC, it is a prerequisite for the catalysts to efficiently absorb solar light, and the infrared absorption ability is quite important for efficient photothermal conversion. For comparison, Co3O4–O (Co3O4 nanoparticles, SI) was prepared through an oxalate combustion method. We measured the UV-vis-IR spectra of SS–Co3O4, Co3O4–O, and SS. Both SS–Co3O4 and Co3O4–O show strong absorption in the entire solar spectrum region with relatively weak absorption around 1000 nm (Fig. S3†), while the former shows higher absorption than the latter in the presence of an SS substrate.Before the catalytic tests, the solar thermal effect was measured. For black Co3O4–O and SS–Co3O4 with good vis-IR absorption (Fig. S3†), strong solar heating can be expected. The temperature evolution profiles of Co3O4–O, SS–Co3O4, and pure SS under the irradiation of the Xe lamp are shown in Fig. 4 and S6.† Impressively, the temperature of SS–Co3O4 quickly increases from room temperature to 110 °C in 60 s with the lamp on. When the temperature reaches a plateau, an equilibrium is established between the absorption of light energy and the energy dissipation from the catalyst to the surroundings.24 The plateau temperatures (Tp) of Co3O4–O and SS–Co3O4 are 181 °C and 175 °C, revealing their efficient photothermal conversion. By comparing the temperature evolutions of the three in detail (Fig. 4B), apparently, SS–Co3O4 shows a little higher heating rate in 60 s but a little lower Tp than Co3O4–O. In view of the inefficient photothermal conversion of the pure SS (Tp 108 °C) and the low amount of Co3O4 nanosheets (only 7 mg on each SS), it can be concluded that SS–Co3O4 with its unique composition and structure is promising for utilizing IR energy as a mild heat source.
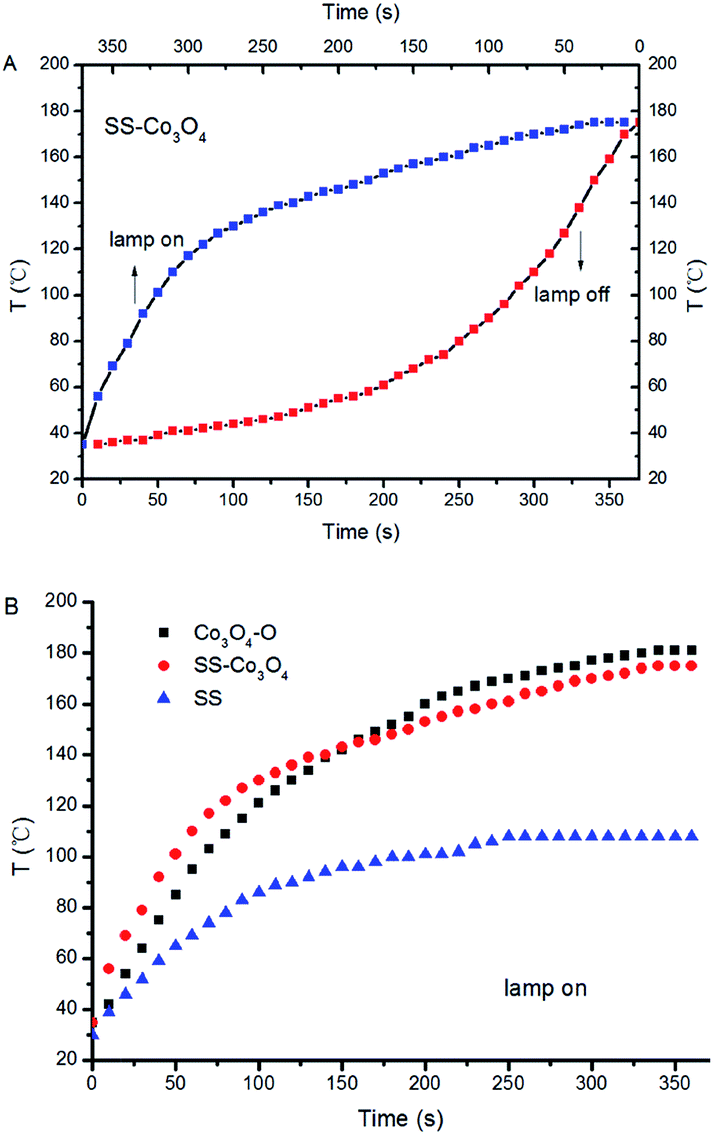 | ||
| Fig. 4 Temporal change of the temperature on the catalysts under the irradiation of the Xe lamp. | ||
3.3 PTC synergetic activity
The light-off temperature (Ts) for C3H8 oxidation is significantly higher than that for C3H6,25,26 which can be easily understood on the basis of the generally higher activity of alkenes compared to alkanes. The efficient PTC activity of SS–Co3O4 was investigated by evaluating the aerobic oxidation of C3H6 (50 ppm) and C3H8 (50 ppm) under the simulated solar irradiation of the Xe lamp. The C3H6 and C3H8 catalytic oxidation cannot proceed at room temperature without lighting. Besides, blank tests in the absence of any catalyst were performed (Fig. S7†). The photolysis of C3H6 contributed to less than 11% of C3H6 decrease under a Xe lamp, while changes in C3H8 content were not distinct, indicating the indispensable role of the catalyst. The catalytic test with just a piece of SS in the reactor was also conducted. Apparently, pure SS did not show any catalytic activity towards the target gas (Fig. S7†). The decrease in the concentration (Ct/C0) of the target gas (C3H6, C3H8) against the irradiation time over various catalysts is shown in Fig. 5A and B. A rapid decrease of both the C3H6 and C3H8 concentration was observed in the presence of a SS–Co3O4 catalyst. About 90% C3H6 oxidation was achieved within 5 min, while 72% C3H8 oxidation was realized at the same time. Along with the fact that there are only 7 mg of Co3O4 nanosheets grown on the 6 cm2 SS, it can be concluded that this novel SS–Co3O4 catalyst possesses superior PTC activity for both C3H6 and C3H8 oxidation. For comparison, we tested the catalytic activity of P25, TiO2, and Co3O4–O (20 mg each) under the same experimental conditions. Also, 3 pieces of SS–Co3O4 (approximately 20 mg Co3O4) were used as the catalyst. After the Xe lamp irradiation for 5 min, the C3H6 oxidation of Co3O4–O, P25 and TiO2 is only 17.7%, 63%, and 50.7%, respectively. Meanwhile, with the same amount of catalyst, 82% C3H8 oxidation was achieved within 5 min for 3 pieces of SS–Co3O4, while no obvious change in the C3H8 concentration can be seen for P25, TiO2, and Co3O4–O. In general, SS–Co3O4 exhibits far better PTC activity in comparison to the behavior of the other three. The PTC durability test of SS–Co3O4 under the simulated solar irradiation was conducted, with each cycle lasting for 15 min. Take the PTC oxidation of C3H8 as an example, as it is shown in Fig. S8,† despite some minor fluctuations, the C3H8 conversion ratio basically remains above 90%, indicating an excellent catalytic durability.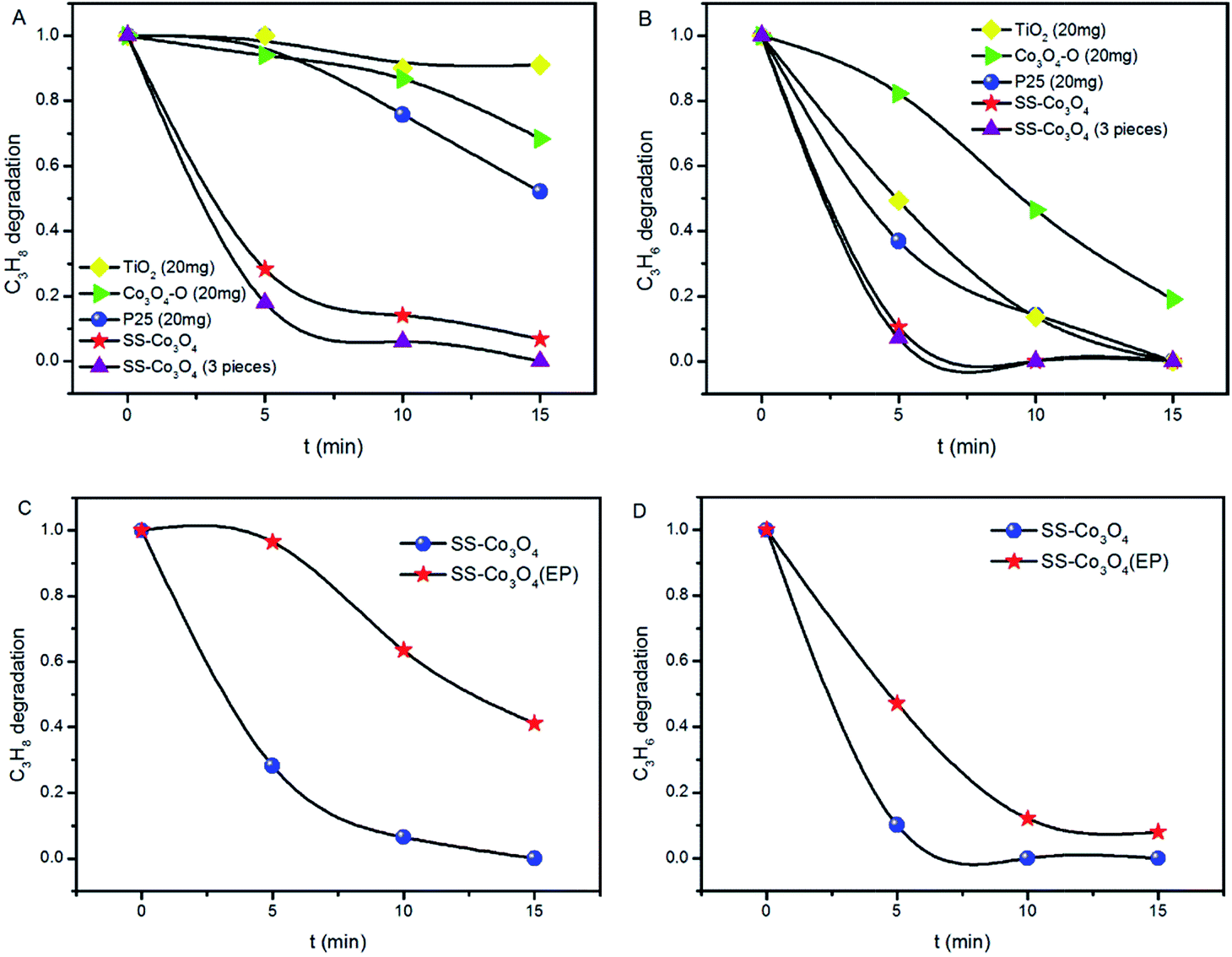 | ||
| Fig. 5 (A and B) C3H8 and C3H6 oxidation over SS–Co3O4, P25, TiO2, and Co3O4–O under simulated sunlight (Xe lamp). (C and D) C3H8 and C3H6 oxidation over SS–Co3O4 and SS–Co3O4 (EP) under simulated sunlight (Xe lamp). | ||
To further emphasize the structural superiority of SS–Co3O4 and to identify the possible contribution of the metal substrate, Co3O4–O was deposited on SS with the same area using electrophoresis technology. The resulting sample was denoted as SS–Co3O4 (EP) and was used to test the PTC activity. From Fig. 5C and D, about 3.5% C3H8 oxidation and 52.9% C3H6 oxidation of SS–Co3O4 (EP) are achieved within 5 min, which is much lower than that of the SS–Co3O4 catalyst. Integrating the behavior of SS, SS–Co3O4, SS–Co3O4 (EP) and Co3O4–O, it can be derived that (1) the excellent PTC activity merely occurs around the directly grown SS–Co3O4 rather than the Co3O4 coated SS; (2) a synergetic interaction between the SS substrate and the Co3O4 nanosheets has enabled the superior performance. Detailed analysis will be in the Mechanism section.
We tested the catalytic activity of the SS–Co3O4 under the infrared irradiation of the Xe lamp by using a cutoff filter to filter out the wavelengths below 690 nm, and also under the irradiation of a UV lamp (15 W). It can be seen from Fig. 6A and B that SS–Co3O4 exhibits efficient infrared-light driven catalytic activity for the oxidation of target gas. Specifically, about 39.2% C3H8 oxidation and 60% C3H6 oxidation were achieved within 5 min, which accounted for nearly 60% of the catalytic activities under the full solar spectrum Xe lamp irradiation. The red dots in Fig. 6A and B show results using a UV lamp as a light source. The UV part could only show a slight contribution, giving an oxidation ratio of 17.3%, 22.5% within 5 min for C3H6 and C3H8, respectively, with the results remaining almost unchanged for the next 10 min. Moreover, as the oxidation of C3H8 was much more difficult than that of C3H6, C3H8 was chosen in the individual TC oxidation tests to determine the temperature at which the catalytic activity of SS–Co3O4 starts (Ts). Impressively, C3H8 oxidation starts at around 100 °C (Ts, Fig. 6C), which is much lower than the corresponding Tp (175 °C) of SS–Co3O4 under the Xe lamp irradiation. Thus, the high Tp induced by efficient photothermal conversion is enough to trigger off TC C3H8 oxidation for SS–Co3O4 in the Xe lamp irradiation experiments. Hou et al.24 demonstrated a novel Ce ion substituted OMS-2 catalyst, which can efficiently transform the absorbed solar energy into thermal energy, and a mechanism of the solar light driven TC was proposed. For the SS–Co3O4 catalyst in this study, not only the solar light driven TC existed, but also PC oxidation was present with the Xe lamp on. Recent studies7,27 have shown that a synergetic effect between the PC and the TC oxidation does exist, resulting in excellent PTC oxidation activity.
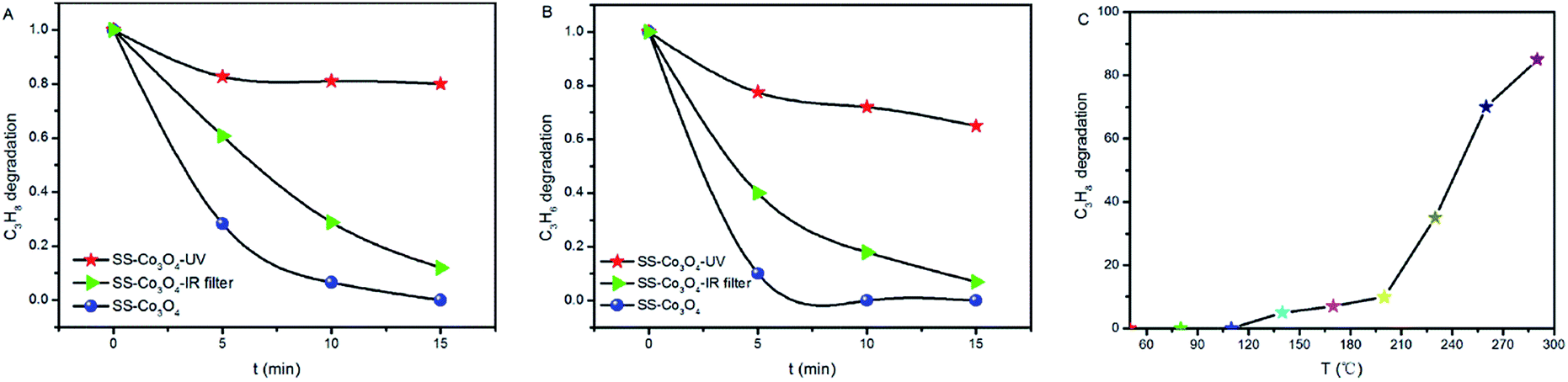 | ||
| Fig. 6 (A and B) C3H8 and C3H6 oxidation over SS–Co3O4 under simulated sunlight, UV light, and infrared light. (C) Thermocatalytic performance of SS–Co3O4 for C3H8 oxidation. | ||
3.4 Mechanism
The question is why the SS–Co3O4 with small amounts of Co3O4 on the SS substrate exhibits highly efficient PTC activity for the catalytic oxidation of light hydrocarbons. In this section, the origins of the excellent PTC activity of Co3O4 are discussed in detail.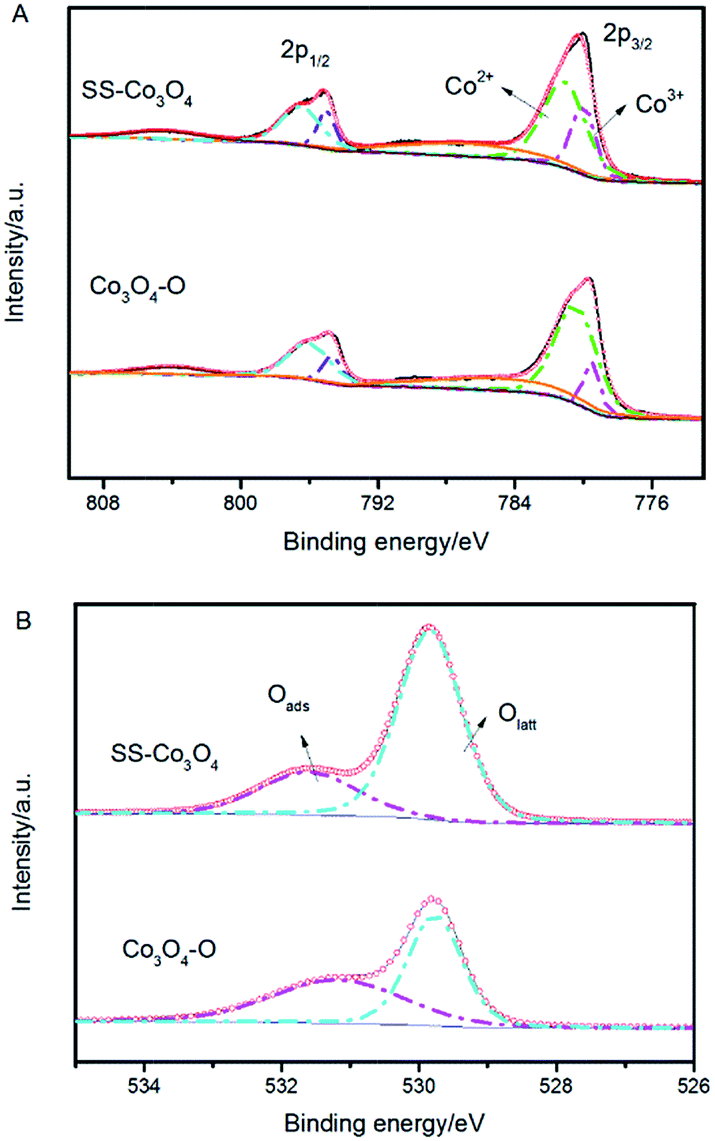 | ||
| Fig. 7 (A) Co 2p XPS spectra and (B) O 1s XPS spectra of SS–Co3O4 and Co3O4–O. | ||
| Sample | % area | |||
|---|---|---|---|---|
| Co 2p3/2 | O 1s | |||
| Co3+ | Co2+ | Olatt | Oads | |
| SS–Co3O4 | 20.4 | 46.2 | 73.8 | 26.2 |
| Co3O4–O | 11.9 | 54.7 | 50.9 | 49.1 |
Co3+ is generally considered to be the catalytic active site of Co3O4. The relative surface content of Co3+ cations played a very important role in the catalytic activities of Co3O4.32 A breakthrough on Co3O4 for catalytic CO oxidation showing that Co3O4 nanorods with four {110} surface planes exhibit a much higher catalytic activity for CO oxidation and larger resistance to deactivation by water than Co3O4 nanoparticles was reported by Xie et al.33 The high catalytic activity of Co3O4 {110} planes is attributed to the higher concentration of Co3+ cations (correspondingly fewer Co2+ cations) than other crystal planes, since only Co3+ cations surrounded by Co2+ ions are active for catalytic oxidation of CO.34 It has also been observed that the variation of the catalytic activity of Co3O4/ZSM-5 catalysts for propane oxidation parallels both the relative content of surface Co3+ and the reducibility of Co3+.30 Hence, the obtained ultrahigh PTC activity of the SS–Co3O4 was in part because of the abundance of active Co3+ on the surface.
(1) 100–200 °C: desorption of surface adsorbed peroxy species O2− (ad).
(2) 200–400 °C: desorption of surface adsorbed monatomic species O− (ad).
(3) 400–700 °C: desorption of surface lattice oxygen O2− (ad/lattice), and
(4) Beyond 700 °C: desorption of bulk lattice oxygen.
From Fig. 8, it can be seen that the Co3O4 nanosheets scratched down from the SS foam (red line) did not show an obvious O2− (ad) desorption peak in the temperature range of 100–200 °C, where the Co3O4–O sample (black line) revealed the distinct O2− (ad) desorption peak. The O− (ad) desorption peak of the two samples was located almost at the same temperature. Notably, for the O2− (ad/lattice) desorption, SS–Co3O4 revealed a much lower temperature and much wider range, indicating that the desorption amount and the mobility of surface lattice oxygen species of the SS–Co3O4 catalyst are greatly enhanced in comparison with Co3O4–O nanoparticles. It has been widely accepted that VOC oxidation over metal oxides follows a mechanism developed by Mars–van and Krevelen involving the participation of a lattice oxygen by a redox cycle.39 Lattice oxygen can be both the adsorption site and active site of the catalyst for VOC catalytic oxidation.40 In the research of Finocchio,26 the experimental data strongly support a typical Mars–van Krevelen type mechanism for C3H8 and C3H6 catalytic combustion over Co3O4 catalysts where the oxidizing center is, accordingly, Co3+ and the active oxygen species is nucleophilic O2− (ad/lattice) at the oxidized surface. For the combustion of methane over the cobalt oxide catalyst, it also follows a Mars–van Krevelen mechanism and depends on the fast migration of oxygen ions through the lattice of cobalt oxide.41 Herein, the amount and mobility of surface lattice oxygen is a significant factor to sustain the much higher catalytic activity of the SS–Co3O4.
 | ||
| Fig. 8 O2-TPD profiles of the Co3O4–O nanoparticles and the Co3O4 nanosheets scratched down from the SS substrate. | ||
On the basis of the above discussion of a series of critical factors for the PTC oxidation process, it can be concluded that the combined properties of SS–Co3O4 such as the ultrathin two-dimensional shape, foam-like porous inside architecture, and supported metal substrate could lead to integrative advantages of the maximum utilization of photothermal conversion, more catalytic active sites, and enhanced amount and mobility of surface lattice oxygen species, resulting in the excellent PTC activity of the SS–Co3O4 catalyst.
4. Conclusions
In summary, ultrathin mesoporous Co3O4 nanosheets on a stainless steel mesh (SS–Co3O4) have been synthesized through an electrochemical deposition method. The SS–Co3O4 catalyst exhibits strong absorption in the entire solar spectrum and shows a strong solar heating effect, resulting in a considerable increase of temperature above the light-off temperature for VOC oxidation. Aerobic oxidations of C3H6 and C3H8 under simulated sunlight, UV light, and infrared light irradiation were performed. This novel SS–Co3O4 catalyst with only 7 mg of Co3O4 grown possesses superior PTC activity, which is much better than the that of P25, TiO2, and Co3O4 nanoparticles. The structural superiority of SS–Co3O4 and the actual contribution of the metal substrate to the PTC process have been integrally discussed. The C3H6 and C3H8 oxidation over SS–Co3O4 follows the Mars–van Krevelen redox mechanism. The excellent PTC activity of the SS–Co3O4 catalyst can be attributed to the maximum utilization of photothermal conversion, more catalytically active sites, and enhanced amount and mobility of surface lattice oxygen species. On the basis of this study, a brand new strategy for catalyst design toward the full use of both the light and heat of solar energy is suggested. In addition, the system of metal substrate supported ultrathin cobalt oxide nanosheets could be of interest as a monolith catalyst in many practical applications.Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (2013CB933200) and National Natural Science Foundation of China (51272269, 51272303, and 51472260).Notes and references
- P. Christopher, H. L. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- B. Bai and J. Li, ACS Catal., 2014, 4, 2753–2762 CrossRef CAS.
- J. H. W. Prengle and W. E. Wentworth, Solar Photo-Thermal Catalytic Reactions to Produce High Value Chemicals, University of Houston, 1992 Search PubMed.
- T. T. Trinh, R. Sato, M. Sakamoto, Y. Fujiyoshi, M. Haruta, H. Kurata and T. Teranishi, Nanoscale, 2015, 7, 12435–12444 RSC.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007 CAS.
- Y. H. Qu, F. Liu, Y. Wei, C. L. Gu, L. H. Zhang and Y. Liu, Appl. Surf. Sci., 2015, 343, 207–211 CrossRef CAS.
- W. Xie, Y. Z. Li, W. Q. Shi, L. Zhao, X. J. Zhao, P. F. Fang, F. Zheng and S. J. Wang, Chem. Eng. J., 2012, 213, 218–224 CrossRef CAS.
- Y. Li, Q. Sun, M. Kong, W. Shi, J. Huang, J. Tang and X. Zhao, J. Phys. Chem. C, 2011, 115, 14050–14057 CAS.
- J. L. Falconer and K. A. Magrini-Bair, J. Catal., 1998, 179, 171–178 CrossRef CAS.
- Y. Zheng, W. Wang, D. Jiang and L. Zhang, Chem. Eng. J., 2016, 284, 21–27 CrossRef CAS.
- D. Jiang, W. Wang, E. Gao, L. Zhang and S. Sun, J. Phys. Chem. C, 2013, 117, 24242–24249 CAS.
- H. Yang, G. Zhu, D. Zhang, D. Xu and S. Qiu, Microporous Mesoporous Mater., 2007, 102, 95–100 CrossRef CAS.
- J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Nano Lett., 2009, 9, 4417–4423 CrossRef CAS PubMed.
- M. Haneda, Y. Kintaichi, N. Bion and H. Hamada, Appl. Catal., B, 2003, 46, 473–482 CrossRef CAS.
- L. F. Liotta, H. Wu, G. Pantaleo and A. M. Venezia, Catal. Sci. Technol., 2013, 3, 3085 CAS.
- Y. Fan, N. Zhang, L. Zhang, H. Shao, J. Wang, J. Zhang and C. Cao, J. Electrochem. Soc., 2013, 160, F218–F223 CrossRef CAS.
- Y. Fan, H. Shao, J. Wang, L. Liu, J. Zhang and C. Cao, Chem. Commun., 2011, 47, 3469–3471 RSC.
- C. Yuan, L. Yang, L. Hou, L. Shen, X. Zhang and X. W. Lou, Energy Environ. Sci., 2012, 5, 7883 CAS.
- D. Jiang, W. Wang, S. Sun, L. Zhang and Y. Zheng, ACS Catal., 2015, 5, 613–621 CrossRef CAS.
- C. Cheng, G. Zhou, J. Du, H. Zhang, D. Guo, Q. Li, W. Wei and L. Chen, New J. Chem., 2014, 38, 2250–2253 RSC.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- M. Roy, S. Ghosh and M. K. Naskar, Phys. Chem. Chem. Phys., 2015, 17, 10160–10169 RSC.
- C. Santra, S. Rahman, S. Bojja, O. O. James, D. Sen, S. Maity, A. K. Mohanty, S. Mazumder and B. Chowdhury, Catal. Sci. Technol., 2013, 3, 360–370 CAS.
- J. Hou, Y. Li, M. Mao, Y. Yue, G. N. Greaves and X. Zhao, Nanoscale, 2015, 7, 2633–2640 RSC.
- A. C. Gluhoi, N. Bogdanchikova and B. E. Nieuwenhuys, Catal. Today, 2006, 113, 178–181 CrossRef CAS.
- E. Finocchio, G. Busca, V. Lorenzelli and V. S. Escribanob, J. Chem. Soc., Faraday Trans., 1996, 9, 1587–1593 RSC.
- S. Kohtani, K. Yoshida, T. Maekawa, A. Iwase, A. Kudo, H. Miyabe and R. Nakagaki, Phys. Chem. Chem. Phys., 2008, 10, 2986–2992 RSC.
- Y. Fan, G. Shao, Z. Ma, G. Wang, H. Shao and S. Yan, Part. Part. Syst. Charact., 2014, 31, 1079–1083 CrossRef CAS.
- Q. Shen, Z. Chen, X. Huang, M. Liu and G. Zhao, Environ. Sci. Technol., 2015, 49, 5828–5835 CrossRef CAS PubMed.
- Z. Zhu, G. Lu, Z. Zhang, Y. Guo, Y. Guo and Y. Wang, ACS Catal., 2013, 1154–1164 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- Y. Sun, P. Lv, J. Y. Yang, L. He, J. C. Nie, X. Liu and Y. Li, Chem. Commun., 2011, 47, 11279–11281 RSC.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- M. Zhou, L. Cai, M. Bajdich, M. García-Melchor, H. Li, J. He, J. Wilcox, W. Wu, A. Vojvodic and X. Zheng, ACS Catal., 2015, 5, 4485–4491 CrossRef CAS.
- A. Bielański and J. Haber, Catal. Rev., 1979, 19, 1–41 Search PubMed.
- C. S. Deng, B. Li, L. H. Dong, F. Y. Zhang, M. G. Fan, G. Z. Jin, J. B. Gao, L. L. Gao, Z. G. Fei and X. Zhou, Phys. Chem. Chem. Phys., 2015, 17, 16092–16109 RSC.
- K. B. Sravan Kumar and P. A. Deshpande, J. Phys. Chem. C, 2015, 119, 8692–8702 CAS.
- W. Song, A. S. Poyraz, Y. Meng, Z. Ren, S.-Y. Chen and S. L. Suib, Chem. Mater., 2014, 26, 4629–4639 CrossRef CAS.
- B. Solsona, T. E. Davies, T. Garcia, I. Vázquez, A. Dejoz and S. H. Taylor, Appl. Catal., B, 2008, 84, 176–184 CrossRef CAS.
- H. Sun, Z. Liu, S. Chen and X. Quan, Chem. Eng. J., 2015, 270, 58–65 CrossRef CAS.
- N. Bahlawane, Appl. Catal., B, 2006, 67, 168–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Co3O4–O preparation, electrophoresis of SS–Co3O4 (EP), electrochemical analysis, and TG/DTA analysis. Fig. S1–S10 display the XRD patterns, UV-vis-IR spectra, TEM images, N2 adsorption–desorption isotherm, pore size distributions, temporal change of the temperature under Xe irradiation, blank experiments for C3H6 and C3H8 oxidation, consecutive cyclic tests and current–time curves of the samples. See DOI: 10.1039/c5ta07617j |
| This journal is © The Royal Society of Chemistry 2016 |