Development of MgAl2O4-stabilized, Cu-doped, Fe2O3-based oxygen carriers for thermochemical water-splitting†
Received
26th August 2015
, Accepted 6th November 2015
First published on 9th November 2015
Abstract
The commercially dominating technology for hydrogen production (i.e. steam methane reforming) emits large quantities of CO2 into the atmosphere. On the other hand, emerging thermochemical water splitting cycles allow the production of a pure stream of H2 while simultaneously capturing CO2. From a thermodynamic point of view, the Fe2O3–Fe couple is arguably the most attractive candidate for thermochemical water splitting owing to its high H2 yield and H2 equilibrium partial pressure. However, the low reactivity of Fe2O3 with methane and the high activity of Fe for methane decomposition (leading to carbon deposition and in turn COx poisoning of the H2 stream) are major drawbacks. Here, we report the development of MgAl2O4-stabilized, Cu-modified, Fe2O3-based redox materials for thermochemical water-splitting that show a high reactivity towards CH4 and low rates of carbon deposition. To elucidate the effect of Cu doping on reducing significantly the rate of carbon deposition (while not affecting negatively the high redox activity of the material) extended X-ray absorption fine structure spectroscopy and energy dispersive X-ray spectroscopy was employed.
Introduction
An increasing global population in combination with the rapid industrial development of highly populous countries has resulted in a steep increase in the global electric power demand. Currently, fossil fuels have the largest share in the power generation mix (e.g. ∼68% in 2011) and will arguably remain a major energy carrier in the future.1,2 However, the combustion of fossil fuels is associated with the emission of large quantities of greenhouse gases, in particular carbon dioxide (CO2) into the atmosphere. The increasing concentration of greenhouse gases in the atmosphere is most likely linked to global warming and climate change.3 Thus, to mitigate climate change, there is an urgent need to reduce or at least stabilize anthropogenic CO2 emissions. In this regard, a transition to hydrogen (H2) as a clean energy carrier, could contribute appreciably to a reduction of CO2 emissions. However, the currently available technologies for the large scale production of H2, e.g. steam methane reforming and coal gasification, are very energy intensive and release large quantities of CO2 into the atmosphere.4 Thus, for H2 to become a major energy carrier, it must be produced in an efficient and ‘sustainable’ manner, i.e. if derived from hydrocarbon fuels, H2 must be produced in combination with carbon dioxide capture and storage (CCS).
In this context, thermochemical water-splitting is a promising process to produce high purity H2 (without the need of additional energy intensive H2 purification steps) while simultaneously capturing CO2.5 In the chemical looping based water-splitting cycle studied here, a hydrocarbon fuel is used to reduce an oxygen carrier. The reduced oxygen carrier is subsequently re-oxidized by steam to produce high-purity H2. Depending on thermodynamics and process configurations an additional air oxidation step is included after steam oxidation, allowing for an additional heat output. Iron oxide (Fe2O3) is a very attractive candidate for thermochemical water-splitting owing to its low cost, wide availability, minimal environmental impact and high equilibrium partial pressure of H2.6 The iron-oxide based thermochemical water-splitting cycle was originally referred to as the ‘steam-iron process’.7 A schematic diagram of the iron oxide mediated water-splitting process, using methane (CH4) as a fuel, is shown in Fig. 1.
 |
| | Fig. 1 Schematic diagram of the steam-iron process. | |
Depending on intrinsic kinetics and the ratio of CH4 to Fe2O3, the reduction of Fe2O3 with CH4 occurs via:
| | | CH4 + 3Fe2O3 → CO + 2H2 + 2Fe3O4, ΔH900 °C = 215.5 kJ mol−1 | (1a) |
| | | CH4 + Fe3O4 → CO + 2H2 + 3FeO, ΔH900 °C = 262.8 kJ mol−1 | (1b) |
| | | CH4 + FeO → CO + 2H2 + Fe, ΔH900 °C = 250.9 kJ mol−1 | (1c) |
or
| | | CH4 + 12Fe2O3 → CO2 + 2H2O + 8Fe3O4, ΔH900 °C = 145.1 kJ mol−1 | (2a) |
| | | CH4 + 4Fe3O4 → CO2 + 2H2O + 12FeO, ΔH900 °C = 334.3 kJ mol−1 | (2b) |
| | | CH4 + 4FeO → CO2 + 2H2O + 4Fe, ΔH900 °C = 286.6 kJ mol−1 | (2c) |
Subsequently, Fe is re-oxidized using steam to produce a pure stream of H2:
| | | 3Fe + 4H2O → Fe3O4 + 4H2, ΔH900 °C = −103.1 kJ mol−1 | (3) |
To close the cycle, Fe3O4 is oxidized back to Fe2O3 using air, viz.:
| | | 4Fe3O4 + O2 → 6Fe2O3, ΔH900 °C = −237.0 kJ mol−1 | (4) |
Addition of reactions (1b), (1c) and (3) yields the conventional steam reformation of CH4:
| | | CH4 + H2O → CO + 3H2, ΔH900 °C = 228.1 kJ mol−1 | (5) |
However, unlike in conventional steam reforming, in the iron oxide mediated process described above a pure stream of H2 is produced inherently. Moreover, the synthesis gas produced during the partial oxidation of CH4 (reaction (1a–c)) has a H2 to CO ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and is, thus, suitable for methanol or Fischer–Tropsch synthesis.8 Compared to steam methane reforming, the exergetic efficiency of the iron oxide mediated water-splitting process is promising,9 however, the poor cyclability of pure, i.e. unsupported Fe2O3 is currently a major drawback. It has been found that if reduced down to metallic Fe, unsupported Fe2O3 deactivates after only a few cycles, probably due to thermal sintering.10 To reduce its tendency for thermal sintering, Fe2O3 is commonly stabilized with a high Tammann temperature support, e.g. Al2O3, ZrO2, TiO2, SiO2 or MgAl2O4.11 Previous studies of the iron oxide mediated water-splitting process have largely focused on using CO or synthesis gas (CO + H2) as the reducing agents.12–17 The preference for CO (or synthesis gas) is due to the fact that carbon deposition via the Boudouard reaction (2CO → C + CO2) is thermodynamically limited at temperatures above 800 °C (Kp = ∼0.15 at 800 °C), whereas Fe catalyzes CH4 decomposition,18viz.:
:1 and is, thus, suitable for methanol or Fischer–Tropsch synthesis.8 Compared to steam methane reforming, the exergetic efficiency of the iron oxide mediated water-splitting process is promising,9 however, the poor cyclability of pure, i.e. unsupported Fe2O3 is currently a major drawback. It has been found that if reduced down to metallic Fe, unsupported Fe2O3 deactivates after only a few cycles, probably due to thermal sintering.10 To reduce its tendency for thermal sintering, Fe2O3 is commonly stabilized with a high Tammann temperature support, e.g. Al2O3, ZrO2, TiO2, SiO2 or MgAl2O4.11 Previous studies of the iron oxide mediated water-splitting process have largely focused on using CO or synthesis gas (CO + H2) as the reducing agents.12–17 The preference for CO (or synthesis gas) is due to the fact that carbon deposition via the Boudouard reaction (2CO → C + CO2) is thermodynamically limited at temperatures above 800 °C (Kp = ∼0.15 at 800 °C), whereas Fe catalyzes CH4 decomposition,18viz.:
| | | CH4(g) → C(s) + 2H2(g), ΔH900 °C = 58.5 kJ mol−1 | (6) |
Moreover, the oxygen carriers developed so far have shown a poor reactivity with CH4 when compared to H2 or CO.19,20 However, it should be noted that CO and synthesis gas are not primary fuels. Therefore, it is important to develop Fe2O3-based materials that have a high reactivity with CH4 and, at the same time, a low propensity for coke formation when using in-expensive CH4 as a fuel.
One approach to reduce CH4 decomposition related carbon deposition is to limit the reduction of Fe2O3 to FeO, since FeO catalyzes only poorly reaction (6) when compared to Fe.21 However, this process modification reduces largely the yield of H2 expressed as mol H2 g−1 oxygen carrier, since the H2 yield for FeO is 3 times lower than for Fe. An alternative approach to suppress the formation of carbon during CH4 oxidation while at the same time ensuring a high H2 yield, is doping of Fe2O3 with a transition metal that is inactive for the dissociation of CH4, e.g. Cu, Pt or Pd.
In this context, Cu is an attractive candidate because of its low cost and high activation energy for the dissociative chemisorption of CH4 (compared to Pt and Pd).22 However, the main disadvantage of doping Fe2O3 with Cu is the possible formation of low Tammann temperature solid solutions, e.g. CuFe2O4 (Tammann temperature of only ∼400 °C).23 So far only Kang et al.24 have studied the redox characteristics of co-precipitated CuFe2O4, however, only five redox cycles were performed (CH4 reduction at 900 °C for 50 min followed by steam oxidation at 800 °C for 120 min). It was reported that compared to unsupported Fe3O4, CuFe2O4 exhibited fast reduction kinetics, low carbon deposition and high CO selectivity. Nonetheless, large fluctuations in the CH4 conversion (33–55 mol%) and H2 yield (6.7–11.3 mmol g−1) were observed over the 5 cycles tested (probably due to sintering). To stabilize the H2 yield, Kang et al.24 supported CuFe2O4 on ZrO2 or CeO2. However, this approach resulted in a reduction in the quantity of the active phase (CuFe2O4) to only 20 wt% CuFe2O4, a value that is arguably too small for practical applications. Nonetheless supported CuFe2O4 showed a higher CH4 conversion when compared to pure CuFe2O4. However, somewhat surprisingly the oxidation kinetics of the new oxygen carrier were reduced substantially, viz. 80 min were required to fully re-oxidize ZrO2 or CeO2 supported CuFe2O4 (1 g oxygen carrier and gas space velocity ∼4 min−1). In addition, the very low loading of CuFe2O4 in the material reduced the overall H2 yield to only 2 mmol g−1.
In this work, we demonstrate the potential of Cu-promoted, Fe2O3-based oxygen carriers for iron oxide mediated water-splitting. We were able to effectively stabilize CuO–Fe2O3 on MgAl2O4 using only 20 wt% support, thus, ensuring high H2 yields. The cyclic redox stability and water-splitting characteristics were determined and interpreted in light of the morphological and phase characteristics of the material. In order to understand the role of Cu in inhibiting carbon deposition, the reduced oxygen carriers were studied using extended X-ray absorption fine structure spectroscopy and energy dispersive X-ray spectroscopy. The materials developed here show very little coke deposition while at the same time maintaining high and stable H2 yields over many redox cycles.
Experimental
Oxygen carrier synthesis
MgAl2O4-stabilized, Cu-modified Fe2O3-based oxygen carriers were synthesized via a modified co-precipitation technique originally reported by He et al.25 The quantity of support required to stabilize Fe2O3 is typically in the range of 30–60 wt%.11 To maximize the quantity of Fe2O3 in the synthesized oxygen carriers, the quantity of Fe2O3 in the synthesized oxygen carriers was fixed to 70 wt% while the CuO content was varied from 0–30 wt%, the balance being MgAl2O4. In a typical synthesis, first appropriate amounts of Fe(NO3)3·9H2O, Cu(NO3)2·2.5H2O, Mg(NO3)2·6H2O and Al(NO3)3·9H2O were dissolved in 400 mL of deionized water (reverse osmosis, 15 MΩ cm). The molar ratio of Mg2+ to Al3+ was fixed to 1:2 while the total moles of cations were fixed to 0.3. Subsequently, 400 mL of an aqueous solution containing 24 g NaOH and 6 g Na2CO3 was added drop wise to the nitrate solution under continuous stirring. The pH of the resulting slurry was adjusted to 8.5 using NaOH. The mixture was kept at 80 °C for 15 h under total reflux. The resulting precipitate was filtered and washed several times with reverse osmosis water. The cake of the washed precipitate was dried in an oven at 70 °C for 24 h and subsequently calcined in a muffle furnace at 900 °C for 2 h using a temperature ramp of 5 °C min−1. The calcined oxygen carriers were crushed and sieved into the size range of 300–425 μm. The following nomenclature is used to describe the composition of the oxygen carriers: the symbols Fe, Cu and MgAl are followed by the weight percentage of Fe2O3, Cu and MgAl2O4 in the oxygen carrier, respectively. For example, Fe71Cu02MgAl27 is an oxygen carrier that contains 71 wt%, 2 wt% and 27 wt% of Fe2O3, Cu (after reduction of CuO) and MgAl2O4, respectively.
Characterization
The crystallinity and chemical composition of the fresh and cycled oxygen carriers was determined using a Bruker AXS D8 Advance X-ray diffractometer mounted with a Lynx eye super speed detector. The diffractometer was operated at 40 kV and 40 mA using CuKα radiation (λ = 1.5418 nm). Each sample was scanned within the range of 2θ = 20–80° using a step size of 0.0275° per second. X-ray diffractograms were further refined using the General Structure Analysis System (GSAS).26,27 Extended X-ray absorption fine structure (EXAFS) spectroscopy was performed at the Swiss-Norwegian beamline (SNBL, BM1B) of the European Synchrotron Radiation Facility (ESRF) to study the local structure of Fe and Cu in the reduced oxygen carriers. The oxygen carriers were mixed with cellulose (ratio 1:8) and pelletized. The pellets were scanned in transmission mode to acquire Fe and Cu K-edge EXAFS data. The acquired data were subsequently processed using the IFEFFIT package.28
N2 adsorption and desorption isotherms of the oxygen carriers were measured at −196 °C (Quantachrome NOVA 4000e analyser). Each sample was degassed for 3 h at 300 °C prior to the measurement. The surface area and pore volume were calculated using, respectively, the Brunauer et al.29 and Barrett et al.30 models. Scanning electron microscopy (Zeiss Gemini 1530 FEG) was applied to characterize the surface morphology of the fresh and cycled oxygen carriers. The average grain size was determined from electron micrographs using the ImageJ software package.31 The average grain size (and error bar) reported is based on the analysis of 50 individual grains. The elemental composition of the surface was mapped using energy dispersive X-ray (EDX) spectroscopy.
CH4-temperature programmed reduction was performed in a Mettler Toledo TGA/DSC 1 thermo-gravimetric analyzer (TGA). In a typical experiment, a small amount (∼30 mg) of the oxygen carrier was heated from room temperature to 1000 °C at a rate of 10 °C min−1 under a flow of 10 vol% CH4 in N2 (50 mL min−1) and kept at 1000 °C for 30 min. In all experiments, a constant N2 flow of 25 mL min−1 was used as purge flow over the micro-balance. Raman spectroscopy (Thermo Scientific DXR Raman microscope) was performed to characterize the carbon deposited on the reduced oxygen carriers. Raman spectra were collected by exciting the samples with 10% power of a 780 nm laser (24 mW total power) for 10 s at 1 cm−1 spectral resolution.
Fixed bed measurements
The redox performance of the synthesized oxygen carriers was evaluated in a packed-bed reactor at 900 °C. A schematic diagram of the experimental set-up is given in Fig. S1 of the (ESI†). In a typical experiment, the oxygen carrier was reduced with 10 vol% CH4 in N2 (1.5 L min−1 at 25 °C and 1 bar). The reduced oxygen carrier was subsequently oxidized with 15 vol% steam in N2. The flow rate of the steam/N2 mixture was 2.83 L min−1 (at 200 °C and 1 bar). To close the cycle, the oxygen carrier was oxidized back to Fe2O3 with 5 vol% O2 in N2 using a flow rate of 1.97 L min−1 (at 25 °C and 1 bar). The duration of the CH4 reduction, steam oxidation and air oxidation segments was fixed to 15 min, 8 min and 5 min, respectively. The reactor was purged for 1 min with N2 (1.5 L min−1) after every segment. In total 15 cycles were performed. The moles of H2 produced during steam oxidation were calculated by integrating the molar flow rate of H2 produced with respect to time.
Results
Characterization of the freshly calcined oxygen carriers
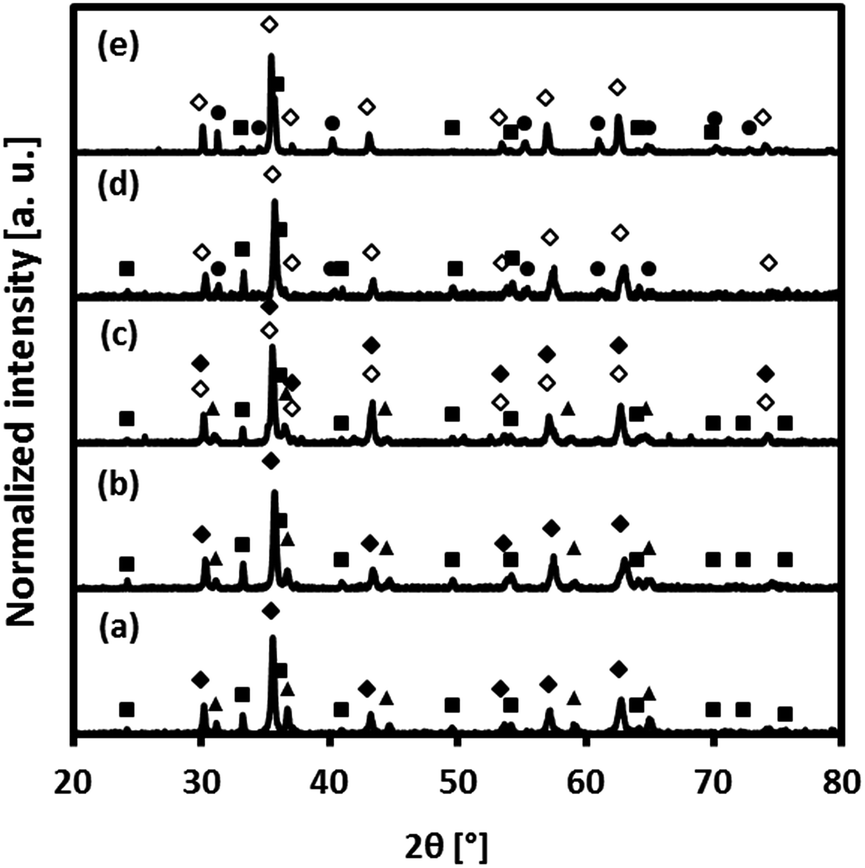
The composition of the freshly calcined oxygen carriers was determined using X-ray diffraction (Fig. 2). Hematite (Fe2O3) and MgAl2O4 were identified in fresh Fe70MgAl30. Rietveld refinement of the XRD data of Fe70MgAl30 gave ∼71.5 wt% Fe2O3 which is in good agreement with the theoretical value of 70 wt%. MgAl2O4-stabilized, Cu-modified Fe2O3-based oxygen carriers showed the formation of Fe2O3, CuFe2O4 and MgAl2O4. The X-ray diffractogram of fresh Fe71Cu02MgAl27 contained peaks due to Fe2O3 and MgAl2O4 only, probably because the quantity of Cu2+ phases (CuO or CuFe2O4) was below the detection limit of the equipment. Rietveld refinement‡ of the XRD data of Fe71Cu02MgAl27 yielded 70.9 wt% Fe2O3 and 29.1 wt% MgAl2O4, again in good agreement with the theoretically expected composition (70.0 wt% Fe2O3, 3.0 wt% CuO and 27.0 wt% MgAl2O4). The composition of Fe71Cu08MgAl21 was determined (Rietveld refinement) as 53.7 wt% Fe2O3, 27.0 wt% CuFe2O4 and 19.3 wt% MgAl2O4. Assuming that all CuO in Fe71Cu08MgAl21 forms CuFe2O4, ∼30 wt% CuFe2O4 would be expected theoretically in the calcined material. Interestingly, MgAl2O4 could not be observed in the diffractogram of Fe73Cu17MgAl10, indicating its presence in an amorphous form. The predicted composition‡ of Fe73Cu17MgAl10 (35.3 wt% Fe2O3 and 64.7 wt% CuFe2O4) was again in good agreement with its theoretical composition (30.0 wt% Fe2O3, 60.0 wt% CuFe2O4 and 10.0 wt% MgAl2O4, assuming the full solution of CuO into Fe2O3 forming CuFe2O4). Finally, only Bragg reflections corresponding to CuFe2O4 were identified in the diffractogram of fresh Fe75Cu25. Assuming a full solution of CuO into Fe2O3, 90.2 wt% CuFe2O4 and 9.8 wt% Fe2O3 would be expected for Fe75Cu25. Thus, our XRD measurements indicate that in Fe75Cu25, Fe2O3 may be present in an amorphous form.
 |
| | Fig. 2 X-ray diffractograms of the freshly calcined (i.e. unreacted) oxygen carriers: (a) Fe70MgAl30, (b) Fe71Cu02MgAl27, (c) Fe71Cu08MgAl21, (d) Fe73Cu17MgAl10 and (e) Fe75Cu25. The following compounds were identified: (■) Fe2O3, (▲) MgAl2O4 and (●) CuFe2O4. | |
Fig. S2 of the ESI† shows scanning electron micrographs of pure, i.e. unsupported Fe2O3, pure CuO and freshly calcined oxygen carriers. The surfaces of unsupported Fe2O3 and CuO are comprised of micrometre-sized polyhedrons (Fig. S2(g)† shows some sintering for unsupported CuO). On the other hand, the bimetallic oxygen carriers reveal different surface morphologies compared to unsupported Fe2O3 and CuO. The surfaces of Fe70MgAl30 and Fe71Cu02MgAl27 are comprised of nanometre-sized grains, as seen in Fig. S2(a) and (b),† respectively. The average size of the grains was determined as 88 ± 14 nm and 124 ± 16 nm for, respectively, Fe70MgAl30 and Fe71Cu02MgAl27. The surfaces of Fe71Cu08MgAl21 and Fe73Cu17MgAl10 showed some sintering and were, only in parts, comprised of small grains. The sintering tendency of the bi-metallic oxygen carriers (based on a visual inspection of the electron micrographs) was found to increase with increasing CuO content and can be explained by the low Tammann temperature of CuFe2O4 of only ∼400 °C.23 Indeed, Fig. S2(e)† shows that Fe75Cu25 (almost pure CuFe2O4) has a very smooth surface texture possibly due to severe thermal sintering during calcination.
The BET surface area and BJH pore volume of the calcined oxygen carriers obtained from N2 adsorption isotherms are summarized in Table 1. As expected from the electron micrographs, Fe70MgAl30 possessed the highest surface area and pore volume of all oxygen carriers synthesized. For the remaining oxygen carriers, a significant reduction in surface area and pore volume was observed for increasing Cu contents, most likely due to thermal sintering during calcination, as visualized in the electron micrographs shown in Fig. S2.†
Table 1 BET surface area and BJH pore volume of the calcined oxygen carriers
| Material |
BET surface area [m2 g−1] |
BJH pore volume [cm3 g−1] |
| Fe70MgAl30 |
12 |
0.11 |
| Fe71Cu02MgAl27 |
5 |
0.05 |
| Fe71Cu08MgAl21 |
2 |
0.02 |
| Fe73Cu17MgAl10 |
2 |
0.01 |
| Fe75Cu25 |
<1 |
<0.01 |
CH4-temperature programmed reduction
CH4-temperature programmed reduction (TPR) was performed to compare the reactivity of the oxygen carries synthesized with CH4 (Fig. 3, the CH4-TPR profiles of pure Fe2O3, CuO and CuFe2O4 are provided in Fig. S3†). The onset of reduction was defined as the temperature at which the oxygen carrier had lost 2% of the total weight loss. The reduction was considered to be completed once 98% of the total weight loss had been lost. Fig. S3† shows that pure CuO was reduced in a single step in the temperature range 480–900 °C. On the other hand, the reduction of pure Fe2O3 and CuFe2O4 started at ∼650 °C and ∼750 °C, respectively, and did not reach completion during the measurement time. Compared to unsupported Fe2O3, the reduction of MgAl2O4-supported Fe2O3 (Fe70MgAl30) started at a considerably lower temperature, viz. at ∼460 °C. For Fe70MgAl30, a three step reduction mechanism was identified in the CH4-TPR profile. The weight loss observed in the first step (temperature range 460–580 °C) agrees with the Fe2O3–Fe3O4 transition (see Fig. S4 of ESI†). Similarly, the weight losses recorded during the second and third steps correspond to the Fe3O4–FeO and FeO–Fe transitions, respectively. The reduction of Fe3O4 and FeO started at ∼620 °C and ∼800 °C, respectively. The increase in sample weight during the holding time (1000 °C) was due to carbon deposition via CH4 decomposition. The diffractogram of reduced Fe70MgAl30 (Fig. S5 of the ESI†) revealed the presence of Fe, MgAl2O4 and Fe3C.
 |
| | Fig. 3 CH4-TPR profiles of the synthesized oxygen carriers: ( ) Fe70MgAl30, ( ) Fe70MgAl30, ( ) Fe71Cu02MgAl27, ( ) Fe71Cu02MgAl27, ( ) Fe71Cu8MgAl21, ( ) Fe71Cu8MgAl21, ( ) Fe73Cu17MgAl10 and ( ) Fe73Cu17MgAl10 and ( ) Fe75Cu25. ) Fe75Cu25. | |
The reduction of Fe71Cu02MgAl27 and Fe71Cu08MgAl21 also occurred in three steps. The weight loss in the first step is in agreement with the CuO–Cu and Fe2O3–Fe3O4 transitions. The weight losses recorded in the second and third steps match well the theoretically predicted weight losses for the Fe3O4–FeO and FeO–Fe transitions. For Fe71Cu02MgAl27, the first, second and third reduction steps started at ∼480 °C, ∼690 °C and ∼800 °C, respectively. Interestingly, the weight increase of Fe71Cu02MgAl27 due to carbon deposition was appreciably lower compared to Fe70MgAl30. Indeed, peaks due to Fe3C could not be observed in the diffractogram of reduced (and further exposed to methane) Fe71Cu08MgAl21 (Fig. S5†), indicating that the presence of Cu inhibited notably carbon deposition. The reduction of Fe71Cu08MgAl21 started at ∼510 °C, with the second and third reduction steps occurring at ∼710 °C and ∼820 °C, respectively. For Fe71Cu08MgAl21, the tendency for carbon deposition was reduced further when compared to Fe70MgAl30 and Fe71Cu02MgAl27. Only Fe and Cu were identified in the diffractogram of reduced (and further exposed to methane) Fe71Cu08MgAl21.
The reduction of Fe73Cu17MgAl10 was found to occur in four steps. The weight losses recorded for the first, second, third and fourth step correspond to the CuFe2O4–Cu + Fe2O3, Fe2O3–Fe3O4, Fe3O4–FeO and FeO–Fe transitions, respectively. The reduction of CuFe2O4, Fe2O3, Fe3O4 and FeO in Fe73Cu17MgAl10 started at ∼580 °C, ∼660 °C, ∼760 °C and ∼890 °C, respectively. Interestingly, no increase in sample weight and, thus, no carbon deposition was observed for Fe73Cu17MgAl10. The diffractogram of the reduced Fe73Cu17MgAl10 showed peaks due to Fe and Cu only. The CH4-TPR profile of Fe75Cu25 revealed a single step reduction mechanism in the temperature range 650–930 °C. The weight loss recorded is in agreement with the reductions of CuFe2O4 to Cu + Fe2O3 and Fe2O3 to FeO. The diffractogram of reduced Fe75Cu25 showed peaks due to FeO, Fe and Cu, indicative of a very slow reduction of FeO to Fe in Fe75Cu25 (as confirmed in Fig. 3).
Redox performance of the synthesized oxygen carriers
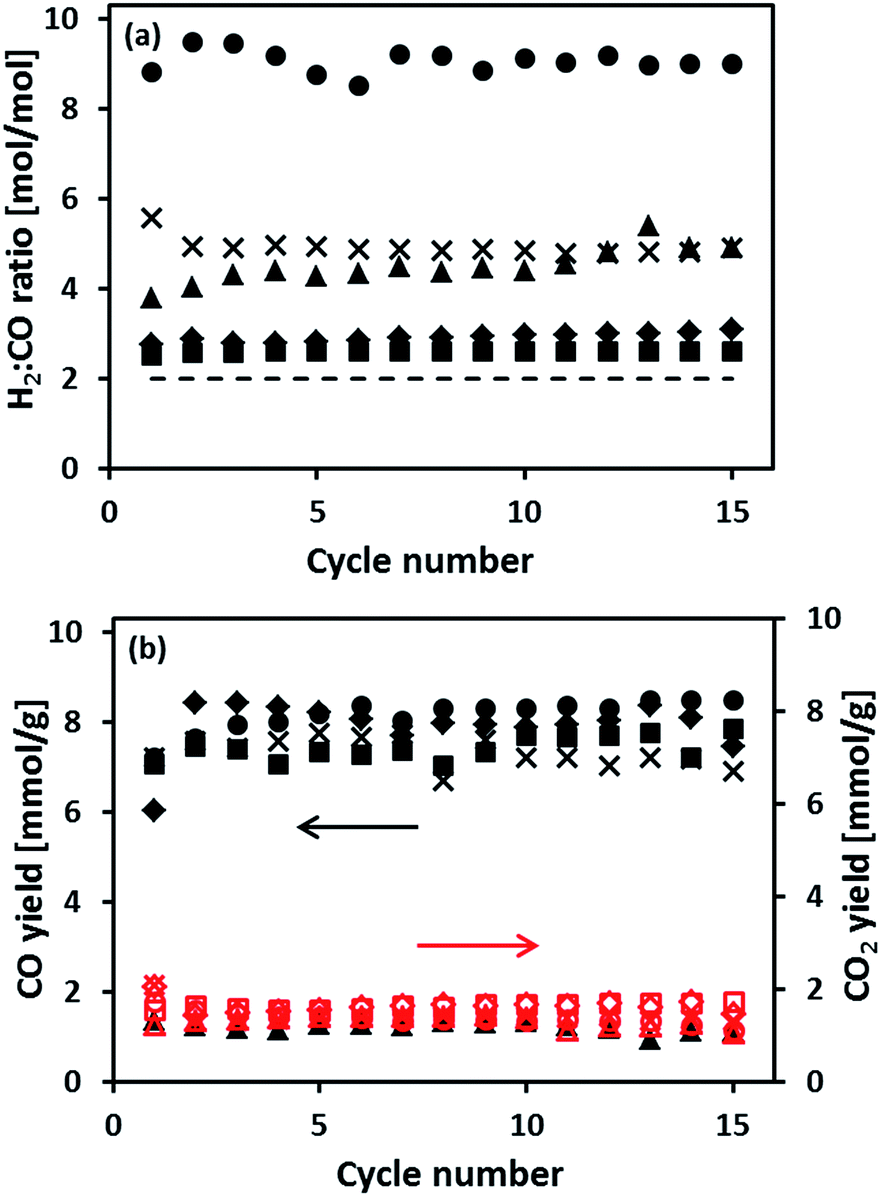
The cyclic redox performance of the synthesized oxygen carriers was assessed in a fixed bed reactor at 900 °C (reduction in 10 vol% CH4 in N2). The yield of H2 during steam oxidation is plotted as a function of cycle number in Fig. 4. Fig. 5(a and b) plot, respectively, the molar ratio of H2 to CO in the synthesis gas and the moles of COx produced during the oxidation of CH4 as a function of cycle number. We observe that the H2 yield of Fe70MgAl30 was ∼3.5 times higher than the theoretically expected value (11.6 mmol H2 goxygen carrier−1 assuming a full reduction to Fe) with some fluctuations during the first 10 cycles. For Fe71Cu02MgAl27, Fe71Cu08MgAl21 and Fe73Cu17MgAl10 a stable H2 yield of ∼16.2 mmol H2 goxygen carrier−1, ∼14.2 mmol H2 goxygen carrier−1 and ∼13.6 mmol H2 goxygen carrier−1, respectively, was obtained over the 15 redox cycles tested. Finally, the reference material Fe75Cu25 showed a stable, but low H2 yield of ∼4.2 mmol H2 goxygen carrier−1, which is only ∼36% of the theoretically expected value.
 |
| | Fig. 4 H2 yield during re-oxidation using 15 vol% steam in N2. The experiments were performed in a packed bed at 900 °C and the duration of the steam oxidation reaction was fixed to 8 min: (●) Fe70MgAl30, (×) Fe71Cu02MgAl27, (■) Fe71Cu08MgAl21, (♦) Fe73Cu17MgAl10 and (▲) Fe75Cu25. The dashed line corresponds to the theoretical H2 yield of 11.6 mmol H2 g−1 oxygen carrier for an oxygen carrier containing 70 wt% Fe2O3. | |
 |
| | Fig. 5 (a) H2 to CO ratio and (b) moles of CO (closed black symbols) and CO2 (open red symbols) produced during reduction in 10 vol% CH4 in N2. The experiments were performed in a packed bed at 900 °C. The duration of the reduction reaction was fixed to 15 min: (●) Fe70MgAl30, (×) Fe71Cu02MgAl27, (■) Fe71Cu08MgAl21, (♦) Fe73Cu17MgAl10 and (▲) Fe75Cu25. The dashed line corresponds to the theoretical H2 to CO ratio of 2:1 assuming the partial oxidation of CH4 to H2 and CO. | |
Turning now to the production of a synthesis gas during the reduction of the oxygen carriers with methane, Fig. 5(a) shows that the H2 to CO ratio for Fe70MgAl30 fluctuated somewhat during the first 10 cycles. From the eleventh cycle onwards, the H2 to CO ratio stabilized at ∼9:1. On the other hand, Cu modified oxygen carriers showed a significantly lower H2 to CO ratio. For Fe71Cu03MgAl27, the H2 to CO ratio was ∼6:1 in the first cycle which decreased to ∼5:1 in the fifteenth cycle. In cases of Fe71Cu08MgAl21 and Fe73Cu17MgAl10 the H2 to CO ratio remained stable at ∼3:1 over repeated redox cycles. Finally, for the reference material Fe75Cu25 a synthesis gas with a H2 to CO ratio of ∼4:1 was obtained in the first cycle. However, the H2 to CO ratio gradually increased, reaching ∼5:1 in the fifteenth cycle. Among the synthesized oxygen carriers, Fe75Cu25 possessed the lowest CO yield of only ∼1.2 mmol CO goxygen carrier−1, see Fig. 5(b). On the other hand, the CO yield of the MgAl2O4-stabilized oxygen carriers was 6–7 times higher than that of unsupported Fe75Cu25. It is also worth noting that the CO2 yield of all oxygen carriers tested was ∼1.5 mmol CO2 goxygen carrier−1, independent whether Fe2O3 was supported or promoted with Cu.
Carbon deposition
Carbon deposition via CH4 decomposition during reduction reduces the purity of the H2 produced during steam oxidation due to the simultaneous formation of COx. Fig. 6 plots the moles of carbon deposited during CH4 reduction as a function of cycle number. The quantity of carbon deposited was calculated from the moles of COx produced during re-oxidation (the mole fraction of COx in the product gas is plotted in Fig. S6† as a function of cycle number). The activity for CH4 decomposition over the 15 redox cycles tested decreased in the following order: Fe70MgAl30 > Fe71Cu02MgAl27 > Fe71Cu08MgAl21 > Fe73Cu17MgAl10 > Fe75Cu25. The average quantity of carbon deposited on the oxygen carriers over 15 redox cycles was 28.3 mmol g−1, 3.7 mmol g−1, 2.7 mmol g−1, 1.8 mmol g−1 and 0.4 mmol g−1 for Fe70MgAl30, Fe71Cu02MgAl27, Fe71Cu08MgAl21, Fe73Cu17MgAl10 and Fe75Cu25, respectively.
 |
| | Fig. 6 Carbon deposition during CH4 reduction. The experiments were performed at 900 °C in a packed bed: (●) Fe70MgAl30, (×) Fe71Cu02MgAl27, (■) Fe71Cu08MgAl21, (♦) Fe73Cu17MgAl10 and (▲) Fe75Cu25. | |
To understand better the inhibiting effect of Cu for carbon deposition, reduced Fe70MgAl30 and Fe73Cu17MgAl10 were studied using EXAFS spectroscopy. The acquired Fe and Cu K-edge EXAFS functions of the reduced materials are plotted in Fig. 7(a and b), respectively. The EXAFS functions of the reference Fe and Cu foils are also provided in Fig. 7. The Fe K-edge EXAFS functions (k2-weighted) of both oxygen carriers examined are similar to the reference Fe foil. Fitting of the first shell around the central Fe atom confirms that the experimentally determined Fe–Fe distances in the reduced oxygen carriers (Table S1†) are similar to the Fe–Fe distances in the reference Fe foil. A slightly higher Debye–Waller factor (σ2), which indicates disorder, was determined for the reduced oxygen carriers when compared to the reference Fe foil. Similarly, the k2-weighted Cu K-edge EXAFS function of reduced Fe73Cu17MgAl10 resembles well the EXAFS function of the reference Cu foil. Fitting of the first shell around the central Cu atom yielded similar Cu–Cu distances as in the reference Cu foil (Table S2†). The Debye–Waller factor of reduced Fe73Cu17MgAl10 was again higher when compared to the Cu foil. Importantly, the acquired Fe and Cu K-edge EXAFS functions of the reduced oxygen carriers confirmed that Fe and Cu did not form an alloy. The surface of reduced Fe73Cu17MgAl10 was also probed via EDX spectroscopy. The Fe L-edge EDX spectrum of reduced Fe73Cu17MgAl10 (Fig. 8(b)) indicates that Fe is present on the entire surface of the reduced material. However, the Cu L-edge EDX spectrum (Fig. 8(c)) and the contrast in Fig. 8(b) suggest that Cu covers Fe only partially on the surface of reduced Fe73Cu17MgAl10.
 |
| | Fig. 7 (a) Fe K-edge EXAFS functions (k2-weighted) of ( ) Fe foil, ( ) Fe foil, ( ) reduced Fe70MgAl30 and ( ) reduced Fe70MgAl30 and ( ) reduced Fe73Cu17MgAl10. (b) Cu K-edge EXAFS functions (k2-weighted) of ( ) reduced Fe73Cu17MgAl10. (b) Cu K-edge EXAFS functions (k2-weighted) of ( ) Cu foil and ( ) Cu foil and ( ) reduced Fe73Cu17MgAl10. ) reduced Fe73Cu17MgAl10. | |
 |
| | Fig. 8 (a) Scanning electron micrograph of reduced Fe73Cu17MgAl10, (b) Fe L-edge, (c) Cu L-edge and (d) Fe and Cu L-edge EDX spectra. | |
Characterisation of the cycled oxygen carriers
X-ray diffractograms, scanning electron micrographs and N2 physisorption isotherms of the cycled oxygen carriers (oxidized form) were acquired to probe compositional and morphological changes over repeated redox cycles. Fig. 9 shows that, compared to the freshly calcined materials, additional crystalline phases appeared in the cycled samples. For example, magnesium aluminium iron oxide (MgAl0.6Fe1.4O4) was identified in the diffractograms of cycled Fe70MgAl30, Fe71Cu02MgAl27 and Fe71Cu08MgAl21. Owing to the formation of the MgAl0.6Fe1.4O4 spinel, some amorphous Al2O3 might also be present in the cycled materials. Importantly, the formation of MgAl0.6Fe1.4O4 indicates that MgAl2O4 is not inert as it has been assumed generally in previous studies.17,32,33 Cycled Fe73Cu17MgAl10 was found to be comprised mainly of CuFe2O4 and Fe2O3, however, weak Bragg reflections due to delafossite, CuFeO2, were also identified. Intriguingly, MgAl2O4 could not be observed in cycled Fe73Cu17MgAl10. Finally, cycled Fe75Cu25 contained CuFe2O4, CuFeO2 and Fe2O3.
 |
| | Fig. 9 X-ray diffractograms of the cycled materials: (a) Fe70MgAl30, (b) Fe71Cu02MgAl27, (c) Fe71Cu08MgAl21, (d) Fe73Cu17MgAl10 and (e) Fe75Cu25. The following compounds were identified: (■) Fe2O3, (♦) MgAl0.6Fe1.4O4, (▲) MgAl2O4, (◊) CuFe2O4 and (●) CuFeO2. | |
In addition, also the morphology of the materials changed over multiple redox cycles as confirmed by electron microscopy (Fig. S7†) and N2 physisorption (Table 2). For example, the electron micrographs of cycled Fe70MgAl30 (Fig. S7(a)†) and Fe71Cu02MgAl27 (Fig. S7(b)†) show that the nanometre-sized grains, originally present in freshly calcined Fe70MgAl30 and Fe71Cu02MgAl27, had sintered, viz. the average grain size of cycled Fe70MgAl30 (135 ± 20 nm) and Fe71Cu02MgAl27 (210 ± 29 nm) increased by a factor of 1.5. This observation is in line with the significantly reduced BET surface area and BJH pore volume of cycled Fe70MgAl30 and Fe71Cu02MgAl27 (Table 2). The surface morphology of cycled Fe71Cu08MgAl21 (Fig. S7(c)†) and Fe73Cu17MgAl10 (Fig. S7(d)†) was also altered during repeated redox cycles. Cycled Fe71Cu08MgAl21 and Fe73Cu17MgAl10 comprised of large grains that were often fused together. The average grain size was 198 ± 54 nm and 172 ± 41 nm for Fe71Cu08MgAl21 and Fe73Cu17MgAl10, respectively. However, comparing the data listed in Tables 1 and 2, the surface area and pore volume of Fe71Cu08MgAl21 did not change appreciably over 15 redox cycles. On the other hand, the surface area of Fe73Cu17MgAl10 decreased from 2 m2 g−1 to 1 m2 g−1. Finally, the electron micrograph of cycled Fe75Cu25 (Fig. S7(e)†) shows a surface comprised of well-defined edges and steps. This change in morphology of Fe75Cu25 was accompanied by an increase in surface area (from <1 m2 g−1 to 3 m2 g−1).
Table 2 BET surface area and BJH pore volume of the cycled materials
| Material |
BET surface area [m2 g−1] |
BJH pore volume [cm3 g−1] |
| Fe70MgAl30 |
2 |
0.02 |
| Fe71Cu02MgAl27 |
2 |
0.01 |
| Fe71Cu08MgAl21 |
2 |
0.01 |
| Fe73Cu17MgAl10 |
1 |
0.01 |
| Fe75Cu25 |
3 |
0.01 |
Discussion
Reduction characteristics of the synthesized oxygen carriers
CH4-TPR measurements of the synthesized oxygen carriers (Fig. 3) show that supporting Fe2O3 on MgAl2O4 shifts its reduction to considerably lower temperatures (compared to unsupported Fe2O3). With regard to the lower reduction temperature of MgAl2O4-supported Fe2O3, it is conceivable that the MgAl2O4 support influences the reducibility of Fe2O3 by modifying CH4 chemisorption and charge transport characteristics of the oxygen carrier. Cui et al.34 studied the influence of α-Al2O3 and MgAl2O4 supports on the dissociative chemisorption of CH4 on Ni. Using CH4 pulse experiments, Cui et al.34 demonstrated that the MgAl2O4 support lowered the activation energy of CH4 dissociation on Ni sites when compared to α-Al2O3 supported and unsupported Ni. The effect of the support on the reduction temperature of Fe2O3 was also studied by Galinsky et al.35 who reported that the reduction temperature of La0.8Sr0.2FeO3-supported Fe2O3 is roughly 250 °C lower than for TiO2-supported Fe2O3. The high reactivity of La0.8Sr0.2FeO3-supported Fe2O3 was attributed to a faster solid state O2− and electron/hole transport from the bulk to the particle surface. On the other hand, the addition of Cu shifts the reduction of Fe2O3 to higher temperatures, viz. the reduction temperature of Fe2O3 increased in the following order: Fe70MgAl30 < Fe71Cu02MgAl27 ≈ Fe71Cu08MgAl21 ≈ Fe73Cu17MgAl10 < Fe75Cu25. CH4-TPR profiles of unsupported Fe2O3 and CuFe2O4 showed that CuFe2O4 and Fe2O3 were reduced at ∼750 °C and ∼650 °C, respectively. Therefore, the shift to higher reduction temperatures for Cu containing materials may be explained by the formation of CuFe2O4 in the oxygen carriers. The CH4-TPR profiles also revealed that metallic Cu formed through the reduction of CuFe2O4, viz.| | | 3CuFe2O4 + 4CH4 → 4CO + 8H2 + 3Cu + 2Fe3O4 | (7) |
decreases slightly the (apparent) rate of reduction of Fe3O4 (to FeO) and FeO (to Fe). These observations are in line with the fact that the activation of CH4 (CH4,s ↔ CH3,s + Hs) on Fe is exothermic (activation energy = −62.8 kJ mol−1) but endothermic on Cu (activation energy = 123.1 kJ mol−1).36 As a consequence, the formation of Cu decreases the (apparent) rate of reduction of Fe3O4 and FeO with CH4.
Reactivity measurements
The molar H2 to CO ratio of the synthesis gas produced during the reduction of iron oxide was higher than the theoretically expected value of 2:1. The average molar H2 to CO ratio (over 15 redox cycles) increased in the following order: Fe73Cu17MgAl10 (3:1) ≈ Fe71Cu08MgAl21 (3:1) < Fe71Cu02MgAl27 (5:1) ≈ Fe75Cu25 (5:1) < Fe70MgAl30 (9:1). Fig. 5 indicates that all synthesized oxygen carriers show similar CO2 yields during CH4 oxidation. In contrast, the CO yields of all MgAl2O4-stabilized materials were similar and significantly higher than for unsupported Fe75Cu25. Thus, the high H2 to CO ratio of MgAl2O4-stabilized materials can be explained only by the deposition of carbon during the reduction step. Previously, Neal et al.37 have observed four regimes during CH4 reduction of Fe2O3-based oxygen carriers viz. (i) full oxidation of CH4 to CO2, (ii) competing full oxidation to CO2 and partial oxidation to CO, (iii) partial oxidation of CH4 to CO and (iv) CH4 decomposition. For MgAl2O4-supported materials, the reaction between CH4 and the oxygen carriers synthesized was sufficiently fast to reach regime (iv) within 15 min, resulting in carbon deposition which in turn led to a high H2 to CO ratio. Galinsky et al.20 studied the effect of three different supports, viz. Ca0.8Sr0.2Ti0.8Ni0.2O3, CeO2 and MgAl2O4, on the methane conversion characteristics of Fe2O3 and found that MgAl2O4 has the highest activity for CH4 decomposition. Among the materials tested here, Fe75Cu25 has the lowest reactivity with CH4 probably due to the absence of a MgAl2O4 support (see Fig. 3). As a consequence, the reduction of Fe70Cu25 was still in regime (ii) after 15 min. Thus, for Fe75Cu25 the high H2 to CO ratio is due to a high CO2 mole fraction (but low CO mole fraction) in the off gas.
Turning now to the steam oxidation step, the high propensity of Fe70MgAl30 for coke formation led to a substantial COx contamination of the H2 produced according to:
| | | C + H2O → CO + H2, ΔH900 °C = 169.6 kJ mol−1 | (8) |
| | | C + 2H2O → CO2 + 2H2, ΔH900 °C = 136.9 kJ mol−1 | (9) |
Owing to severe coke deposition, the H2 yield of Fe70MgAl30 was 3.5 times higher than the theoretically expected value. For Cu-modified materials, the quantity of carbon deposited decreased with increasing Cu content. For example, for Fe71Cu02MgAl27, Fe71Cu08MgAl21 and Fe73Cu17MgAl10 the H2 yield was, respectively, only 1.4 times, 1.2 times and 1.1 times higher than the theoretically expected value, indicating that Cu significantly reduced coke deposition. Finally, the low H2 yield of only ∼32% of the theoretically expected value for Fe75Cu25 is due to the incomplete reduction of FeO to Fe with CH4. The yield of H2 obtained through the oxidation of FeO is 3 times lower than that from Fe. For all Cu modified materials, the COx content in the hydrogen produced during steam oxidation is >5 vol% (Fig. S6†), however, with easily-implementable process modifications the purity of the H2 produced can be increased further. Fig. S8† plots the composition of the effluent gas during steam oxidation (15th cycle) for Fe70MgAl30 and Fe71Cu08MgAl21. For Fe70MgAl30, CO is produced in large quantities during the entire steam oxidation step. On the other hand, for Fe71Cu08MgAl21 the H2 produced after the initial 30 s is essentially COx free. Thus, by a simple switching operation, high purity hydrogen can be produced with Fe71Cu08MgAl21.
Influence of Cu on CH4 decomposition
CH4-TPR (Fig. 3) and reactive measurements (Fig. 6) confirm that the addition of Cu to MgAl2O4-stabilized, Fe2O3-based oxygen carriers improved appreciably the materials' resistance to carbon deposition via CH4 decomposition. The propensity of the oxygen carriers to carbon deposition decreased with increasing Cu content. CH4-TPR in combination with Raman spectroscopy (Fig. S9†) showed that the decomposition of CH4 started once Fe2O3 was fully reduced to metallic iron. Previous studies of Bengaard et al.38 and Besenbacher et al.39 reported that (i) the dissociation of CH4 molecules (CH4,s ↔ Cs + 4Hs) on a metal surface and (ii) the nucleation of graphite on steps and edges play a key role in carbon formation. The reaction enthalpy for CH4 dissociation over Fe is exothermic (−216.9 kJ mol−1) but endothermic (251.2 kJ mol−1) for Cu.36 Our EXAFS (Fig. 7) and EDX (Fig. 8) measurements confirm that in the reduced oxygen carriers Fe and Cu do not form a solid solution, in line with an experimentally determined Fe–Cu equilibrium phase diagram (using thermal analysis and electron microscopy).40 Owing to the phase separation of Cu and Fe a sharp interphase on the surface of the reduced oxygen carriers can be expected, as illustrated in Fig. 10. We speculate that the, at least partial, covering of surface Fe (largely steps) with Cu decreases the rate of CH4 dissociation, which in turn reduces carbon deposition. Furthermore, our preliminary kinetic data show that the rate of oxidation of Fe with steam is not significantly affected by the presence of Cu. The fact that Fe and Cu occur in separate phases in the reduced state ensures that Cu-promoted Fe2O3 maintains the excellent redox characteristic of Fe2O3 such as a high H2 yield and reaction kinetics during the steam oxidation step.
 |
| | Fig. 10 A schematic representation of carbon deposition on (a) MgAl2O4-stabilized Fe2O3 and (b) Cu-modified, MgAl2O4-stabilized Fe2O3. | |
Conclusions
In this work, we have demonstrated that MgAl2O4-stabilized, CuO–Fe2O3-based oxygen carriers possess a high and stable H2 yield over many redox cycles while maintaining a low propensity for coke deposition. CH4-temperature programmed reduction experiments revealed that the propensity for coke formation decreased with increasing Cu content. Furthermore, Cu shifted the reduction of Fe3O4 and FeO to slightly higher temperatures (using CH4). At 900 °C (using 10 vol% CH4 in N2) MgAl2O4-stabilized Fe2O3 produced a synthesis gas with a high H2 to CO ratio of 9:1 owing to substantial carbon deposition via CH4 decomposition. However, increasing the Cu content in the oxygen carrier decreased the H2 to CO ratio (compared to MgAl2O4-stabilized Fe2O3). On the other hand, unsupported Cu-modified Fe2O3 showed low H2 yields reaching only ∼32% of the theoretically expected value. This was due to the incomplete reduction of FeO to Fe using CH4. During steam oxidation, MgAl2O4-stabilized, Cu-modified, Fe2O3-based oxygen carriers exhibited H2 yields close to the theoretically expected value owing to a low CH4 decomposition activity for these materials. Extended X-ray absorption fine structure spectroscopy and energy dispersive X-day spectroscopy confirmed that Fe and Cu did not form an alloy in the reduced oxygen carrier. The high resistance to carbon deposition of Cu modified oxygen carriers was, therefore, attributed to the (partial) coverage of surface Fe with Cu. Owing to their low tendency for coke deposition Cu-modified, MgAl2O4-stabilized Fe2O3 based oxygen carrier, produced hydrogen with very little COx contamination. The oxygen carrier that contained 71 wt% Fe2O3, 8 wt% Cu and 21 wt% MgAl2O4 was identified as a highly promising material for CH4-based water-splitting cycles for H2 production.
Acknowledgements
We are grateful to the Swiss National Science Foundation (SNF) (Project 406640_13670011) and the Swiss Federal Office of Energy (SFOE) (Projects SI/500652 and SI/500653) for financial support. We also thank Mrs Lydia Zender for her help with the XRD analysis and the Scientific Centre for Optical and Electron Microscopy (ScopeM) for providing access to and training on scanning electron microscopes. We acknowledge SNBL and ESRF for the provision of beamtime.
Notes and references
- Key World Energy Statistics 2013, 2013.
- Energy Technology Perspectives 2014: Harnessing Electricity's Potential, 2014.
-
B. Metz, Intergovernmental Panel on Climate Change. Working Group III, IPCC special report on carbon dioxide capture and storage, Cambridge University Press, for the Intergovernmental Panel on Climate Change, Cambridge, 2005 Search PubMed.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 CAS.
- M. A. Rosen, Energy, 2010, 35, 1068–1076 CrossRef CAS.
- R. D. Solunke and G. Veser, Ind. Eng. Chem. Res., 2010, 49, 11037–11044 CrossRef CAS.
-
A. Messerschmitt, Germany Pat., 266863, 1911.
- K. S. Go, S. R. Son, S. D. Kim, K. S. Kang and C. S. Park, Int. J. Hydrogen Energy, 2009, 34, 1301–1309 CrossRef CAS.
- J. P. E. Cleeton, C. D. Bohn, C. R. Muller, J. S. Dennis and S. A. Scott, Int. J. Hydrogen Energy, 2009, 34, 1–12 CrossRef CAS.
- C. D. Bohn, C. R. Muller, J. P. Cleeton, A. N. Hayhurst, J. F. Davidson, S. A. Scott and J. S. Dennis, Ind. Eng. Chem. Res., 2008, 47, 7623–7630 CrossRef CAS.
- J. Adanez, A. Abad, F. Garcia-Labiano, P. Gayan and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef CAS.
- C. D. Bohn, J. P. Cleeton, C. R. Muller, S. Y. Chuang, S. A. Scott and J. S. Dennis, Energy Fuels, 2010, 24, 4025–4033 CrossRef CAS.
- A. M. Kierzkowska, C. D. Bohn, S. A. Scott, J. P. Cleeton, J. S. Dennis and C. R. Muller, Ind. Eng. Chem. Res., 2010, 49, 5383–5391 CrossRef CAS.
- P. Datta, L. K. Rihko-Struckmann and K. Sundmacher, Mater. Chem. Phys., 2011, 129, 1089–1095 CrossRef CAS.
- V. Galvita and K. Sundmacher, Chem. Eng. J., 2007, 134, 168–174 CrossRef CAS.
- A. Murugan, A. Thursfield and I. S. Metcalfe, Energy Environ. Sci., 2011, 4, 4639–4649 CAS.
- S. Z. Wang, G. X. Wang, F. Jiang, M. Luo and H. Y. Li, Energy Environ. Sci., 2010, 3, 1353–1360 CAS.
- Y. Nishiyam and Y. Tamai, J. Catal., 1974, 33, 98–107 CrossRef.
- T. Kodama, H. Ohtake, S. Matsumoto, A. Aoki, T. Shimizu and Y. Kitayama, Energy, 2000, 25, 411–425 CrossRef CAS.
- N. L. Galinsky, A. Shafiefarhood, Y. G. Chen, L. Neal and F. X. Li, Appl. Catal., B, 2015, 164, 371–379 CrossRef CAS.
- X. Zhu, Y. G. Wei, H. Wang and K. Z. Li, Int. J. Hydrogen Energy, 2013, 38, 4492–4501 CrossRef CAS.
- M. S. Liao and Q. E. Zhang, J. Mol. Catal. A: Chem., 1998, 136, 185–194 CrossRef CAS.
-
I. Barin, O. Knacke and O. Kubaschewski, Thermochemical properties of inorganic substances: supplement, Springer-Verlag, Berlin, New York, 1977 Search PubMed.
- K. S. Kang, C. H. Kim, W. C. Cho, K. K. Bae, S. W. Woo and C. S. Park, Int. J. Hydrogen Energy, 2008, 33, 4560–4568 CrossRef CAS.
- L. He, H. Berntsen, E. Ochoa-Fernandez, J. C. Walmsley, E. A. Blekkan and D. Chen, Top. Catal., 2009, 52, 206–217 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
-
A. C. Larson and R. B. V. Dreele, General Structure Analysis System (GSAS), 2004 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- W. Liu, M. Ismail, M. T. Dunstan, W. Hu, Z. Zhang, P. S. Fennell, S. A. Scott and J. S. Dennis, RSC Adv., 2015, 5, 1759–1771 RSC.
- M. Rydén and M. Arjmand, Int. J. Hydrogen Energy, 2012, 37, 4843–4854 CrossRef.
- Y. H. Cui, H. Y. Xu, Q. J. Ge, Y. Z. Wang, S. F. Hou and W. Z. Li, Chin. J. Catal., 2006, 27, 479–484 CrossRef CAS.
- N. L. Galinsky, Y. Huang, A. Shafiefarhood and F. X. Li, ACS Sustainable Chem. Eng., 2013, 1, 364–373 CrossRef CAS.
- M. J. Hei, H. B. Chen, J. Yi, Y. J. Lin, Y. Z. Lin, G. Wei and D. W. Liao, Surf. Sci., 1998, 417, 82–96 CrossRef CAS.
- L. M. Neal, A. Shafiefarhood and F. X. Li, ACS Catal., 2014, 4, 3560–3569 CrossRef CAS.
- H. S. Bengaard, J. K. Norskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Norskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS PubMed.
- S. E. Amara, A. Belhadj, R. Kesri and S. Hamar-Thibault, Z. Metallkd., 1999, 90, 116–123 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta06753g |
| ‡ In calcined materials, only MgAl2O4 or CuFe2O4 was present in an amorphous phase. Thus, using Rietveld refinement, the composition of the samples was estimated on a MgAl2O4- or CuFe2O4-free basis and compared with the theoretically expected values. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 



 ) Fe70MgAl30, (
) Fe70MgAl30, ( ) Fe71Cu02MgAl27, (
) Fe71Cu02MgAl27, ( ) Fe71Cu8MgAl21, (
) Fe71Cu8MgAl21, ( ) Fe73Cu17MgAl10 and (
) Fe73Cu17MgAl10 and ( ) Fe75Cu25.
) Fe75Cu25.


 ) Fe foil, (
) Fe foil, ( ) reduced Fe70MgAl30 and (
) reduced Fe70MgAl30 and ( ) reduced Fe73Cu17MgAl10. (b) Cu K-edge EXAFS functions (k2-weighted) of (
) reduced Fe73Cu17MgAl10. (b) Cu K-edge EXAFS functions (k2-weighted) of ( ) Cu foil and (
) Cu foil and ( ) reduced Fe73Cu17MgAl10.
) reduced Fe73Cu17MgAl10.