
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereodivergent combined carbometalation/zinc homologation/C–C fragmentation reaction as an efficient tool to prepare acyclic allylic quaternary carbon stereocenters†
Sudipta Raha
Roy
,
Dorian
Didier
,
Amir
Kleiner
and
Ilan
Marek
*
The Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry and Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: chilanm@tx.technion.ac.il; Fax: +972-4-829-37-09; Tel: +972-4-829-37-09
First published on 24th May 2016
Abstract
A new strategy has been developed to construct enantiomerically enriched acyclic allylic quaternary carbon stereocenters in a single-pot operation through a combined carbometalation/zinc homologation/fragmentation sequence. Proper tuning of the reaction conditions enables the synthesis of the two enantiomers starting from a single enantiomer of the starting material.
Introduction
In the last few decades, numerous approaches to integrate multiple chemical steps in a single-pot operation1 have been described, offering reliable and powerful strategies for the synthesis of fine chemicals.2 In this context, the construction of several carbon–carbon (C–C) bonds with simultaneous control of newly formed asymmetric centers, including the formation of challenging quaternary carbon stereocenters,3 is of paramount importance for the development of complex molecular frameworks.4 Particularly interesting would be the formation of such stereocenters adjacent to allylic motifs4 as these sub-structures are abundant in biomolecules and natural products.5 Although several excellent and highly selective approaches have been reported for the construction of acyclic allylic carbon quaternary stereocenters,6–10 it becomes more intricate when the synthesis has to be performed in a single-pot operation.11 For instance, although copper catalysed asymmetric conjugate addition is probably the most well established transformation (Fig. 1, path A),12 asymmetric 1,4-addition to an extended conjugated system is more challenging as the 1,6-addition product is usually preferred over 1,4-addition (Fig. 1, path B).13 A more successful alternative approach to reach the same products is the asymmetric 1,4-addition of vinyl metal species to Michael acceptors.14 Although methods coupling asymmetric catalysis with fragmentation of strained building blocks have been reported recently, none could be used for the preparation of acyclic allylic carbon quaternary stereocenters.15 Herein, we would like to report our efforts to address this issue by performing a new tandem approach leading to the formation of two new carbon–carbon bonds in a single-pot operation, including the formation of the desired acyclic allylic quaternary carbon stereocenter (Fig. 1, Path C) from easily accessible starting materials.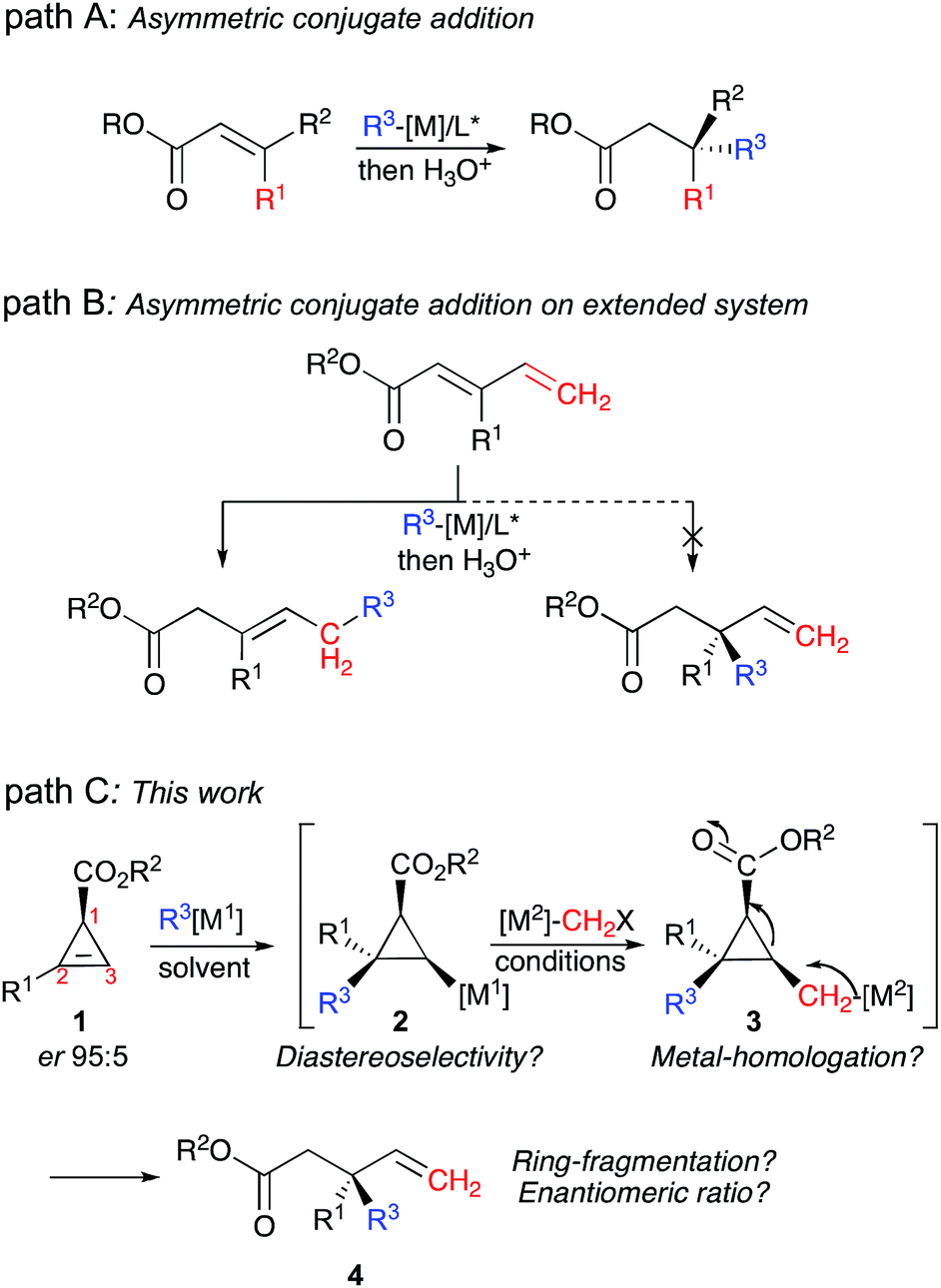 | ||
| Fig. 1 Approaches to construct allylic quaternary stereocenters. | ||
This combined reaction consists of the diastereoselective carbometalation reaction16 of enantiomerically enriched substituted cyclopropenyl esters 1, followed by a homologation reaction17 of the resulting cyclopropyl metal species 2 leading to a cyclopropylmethyl metal 3, which subsequently undergoes a carbon–carbon bond cleavage18 to give the enantiomerically enriched acyclic allylic quaternary carbon stereocenter 4 as described in Fig. 1, path C. Although this proposed sequence is very appealing, the unique formation of the linear product 4 directly from 1, promoted by a combination of organometallic species in high chemical yield and with a high enantiomeric ratio, requires a cascade of high-yielding events with perfect control of all the elementary steps. It should be noted that the enantiomerically enriched cyclopropenyl ester starting materials 1 are readily accessible through the asymmetric metal-catalyzed decomposition of diazoesters with alkynes.19 The enantiomeric ratio of the acyclic allylic quaternary stereocenter in 4 results from the diastereoselectivity of the carbometalation reaction.
Results and discussion
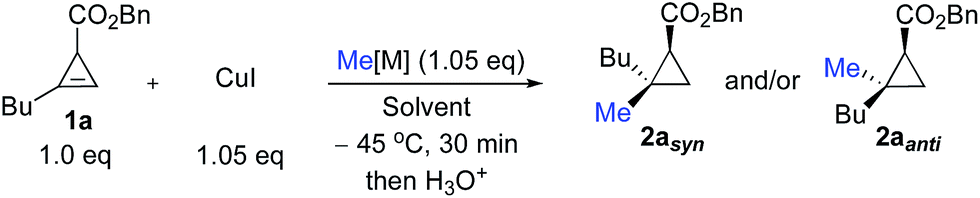
We started our research by investigating the diastereoselectivity of the carbometalation reaction of cyclopropenyl ester 1. In our synthetic plans, the presence of the ester was not only needed to achieve good diastereoselectivity for the carbometalation reaction, but was also a key element for successful ring-fragmentation of the newly formed donor–acceptor cyclopropylmethyl metal species 3.20Hence, to address the initial step of this sequence (transformation of 1 into 2), our model cyclopropenyl ester substrate (racemic 1a) was treated with various organocopper species in different solvents as described in Table 1. When 1a was treated at −45 °C in THF with 1.05 equivalents of MeCu, easily prepared by mixing MeLi and CuI in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, the carbometalated product 2aanti was rapidly obtained after hydrolysis (less than 30 minutes) with a very high anti-diastereoselectivity in good yield (Table 1, entry 1).21 The configuration of 2aanti was established by NOE experiments (see ESI†). The preferential formation of diastereomer 2aanti can only be possible if the carbometalation reaction is sterically driven. A coordinating solvent such as THF makes the organocopper species less prone to chelation by the ester and therefore it prefers to react on the re-face of the cyclopropene derivative 1a. If the assumption that solvent plays a crucial role in the control of the diastereoselectivity of the carbometalation step is correct, then a less Lewis basic solvent should favour addition on the si-face through coordination of the organometallic species with the ester. Indeed, when a slightly less coordinating solvent such as 2-methyl THF was used, the selectivity towards the formation of 2aanti dropped slightly (Table 1, entry 2). On further decreasing the Lewis basicity of the solvent (Table 1, entries 3 to 5, Et2O, hexane and toluene respectively), the tendency of the organometallic species to be coordinated by the ester group increases and therefore the isomer 2asyn could be prepared as the unique diastereoisomer (Table 1, entry 5). The relative configuration of 2asyn was established by comparing the NOE and 13C NMR experiments with 2aanti (see ESI†). The same trend was also found for the addition of organocopper species originating from Grignard reagent (prepared by mixing MeMgBr and CuI in a 1:1 ratio), and although the formation of the anti-addition product 2aanti in THF was less diastereoselective (Table 1, compare entries 1 and 6), it was still excellent for the formation of the syn-diastereoisomer 2asyn (Table 1, entry 8). It should be noted that diastereoselective carbometalation of cyclopropenyl esters such as 1a has already been described, but it was always achieved through variation of the organometallic species and never by simple variation of the solvent for a given organocopper entity.16
:1 ratio, the carbometalated product 2aanti was rapidly obtained after hydrolysis (less than 30 minutes) with a very high anti-diastereoselectivity in good yield (Table 1, entry 1).21 The configuration of 2aanti was established by NOE experiments (see ESI†). The preferential formation of diastereomer 2aanti can only be possible if the carbometalation reaction is sterically driven. A coordinating solvent such as THF makes the organocopper species less prone to chelation by the ester and therefore it prefers to react on the re-face of the cyclopropene derivative 1a. If the assumption that solvent plays a crucial role in the control of the diastereoselectivity of the carbometalation step is correct, then a less Lewis basic solvent should favour addition on the si-face through coordination of the organometallic species with the ester. Indeed, when a slightly less coordinating solvent such as 2-methyl THF was used, the selectivity towards the formation of 2aanti dropped slightly (Table 1, entry 2). On further decreasing the Lewis basicity of the solvent (Table 1, entries 3 to 5, Et2O, hexane and toluene respectively), the tendency of the organometallic species to be coordinated by the ester group increases and therefore the isomer 2asyn could be prepared as the unique diastereoisomer (Table 1, entry 5). The relative configuration of 2asyn was established by comparing the NOE and 13C NMR experiments with 2aanti (see ESI†). The same trend was also found for the addition of organocopper species originating from Grignard reagent (prepared by mixing MeMgBr and CuI in a 1:1 ratio), and although the formation of the anti-addition product 2aanti in THF was less diastereoselective (Table 1, compare entries 1 and 6), it was still excellent for the formation of the syn-diastereoisomer 2asyn (Table 1, entry 8). It should be noted that diastereoselective carbometalation of cyclopropenyl esters such as 1a has already been described, but it was always achieved through variation of the organometallic species and never by simple variation of the solvent for a given organocopper entity.16
|
|
|||
|---|---|---|---|
| Entry | Me[M] | Solvent | 2asyn/2aantia |
| a 1a was consumed completely and the ratio between products 2asyn and 2aanti was determined by GC-MS of the crude reaction mixture. Numbers in parentheses represent the isolated yields after purification by column chromatography. | |||
| 1 | MeLi | THF |
1:99 (75) |
| 2 | MeLi | 2-MeTHF | 6:94 |
| 3 | MeLi | Et2O | 89:11 |
| 4 | MeLi | Hexane | 97:3 |
| 5 | MeLi | C 6 H 5 Me |
99:1 (73) |
| 6 | MeMgBr | THF | 28:72 |
| 7 | MeMgBr | Et2O | 99:1 |
| 8 | MeMgBr | C 6 H 5 Me |
99:1 (72) |
Having access to both syn and anti diastereoisomers of the carbometalated products at will according to the nature of the solvent, we then turned our attention to the zinc homologation reaction of the racemic cyclopropyl copper species 2Cuanti originating from the carbometalation reaction of MeCu in THF (MeCu was formed from MeLi and CuI as described in Table 1, entry 1). For this purpose, CH2I2 and Et2Zn in a 1:1 ratio were added to the reaction mixture at −45 °C, and upon warming to −20 °C, within 2 hours, the homologated cyclopropylmethyl zinc derivative223 gave 4a as described in Fig. 2. Indeed, as cyclopropylcopper 2Cuanti does not react with CH2I2, the reaction between Et2Zn and CH2I2 occurs first leading to the in situ formation of the zinc carbenoid ICH2ZnEt.23 Then, 2Cuanti is homologated by the zinc carbenoid to generate the in situ reactive cyclopropylmethyl zinc (or copper) derivative 3 (see Fig. 1), which instantaneously undergoes a C–C bond fragmentation.24 Although the combined carbometalation/zinc homologation/fragmentation reaction proceeded smoothly according to our original plan to give 4a, the conversion of 2Cuanti into 4a was only moderate (2aanti/4a = 27/73 after hydrolysis).
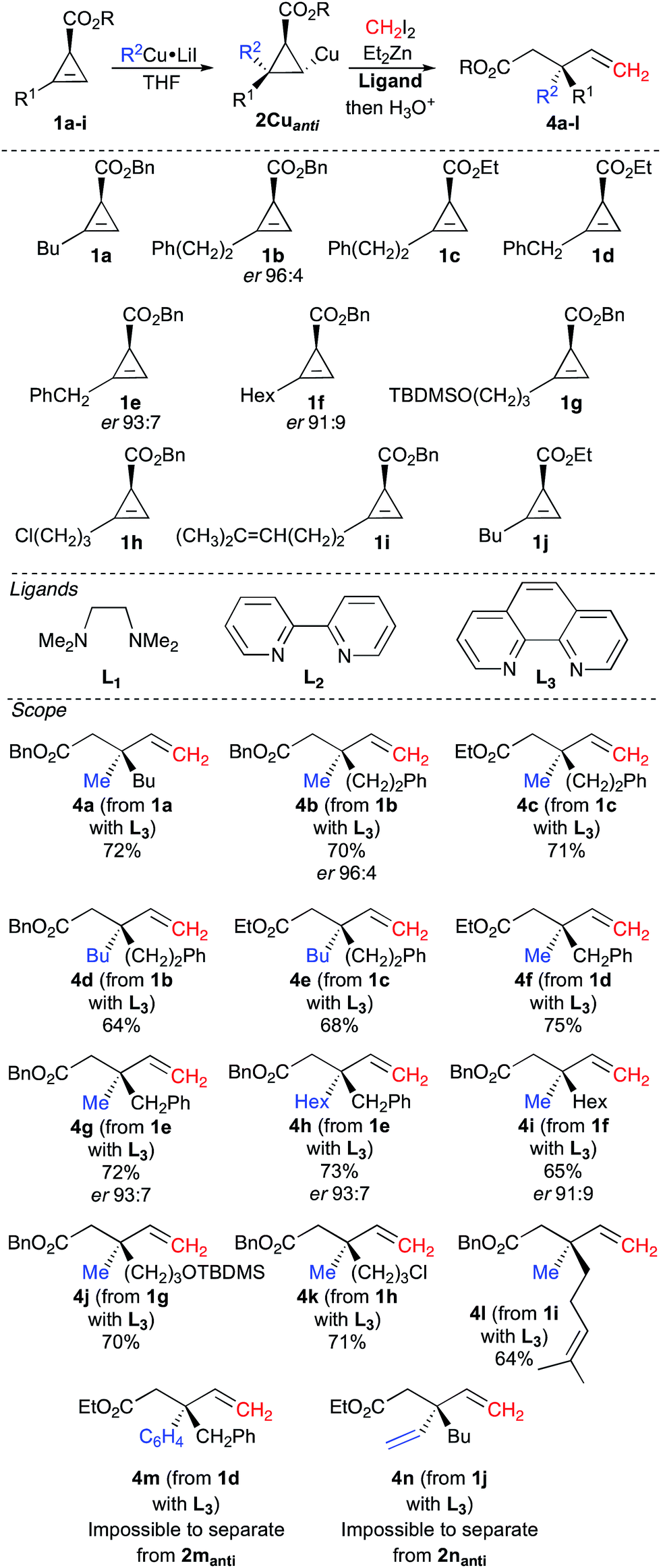 | ||
| Fig. 2 Combined carbometalation/zinc homologation/fragmentation reaction of cyclopropenyl ester 1. | ||
To improve the conversion, it was then anticipated that increasing the nucleophilicity of the carbon atom bearing the organocopper species should increase its reactivity towards the ambiphilic zinc carbenoid species EtZnCH2I.
To this end, various donor ligands were added to the reaction mixture to improve the reactivity of the cyclopropyl copper species (see ligands in Fig. 2).25 Indeed, we were pleased to observe that the addition of TMEDA (L1) or 2-2′-bipyridine (L2) as ligand improved the conversion of the desired product 4a (2aanti/4a = 12/88 and 13/87 respectively after hydrolysis).
The best conversion could be reached when phenanthroline (L3) was added to 2Cuanti, as the ratio became excellent (2aanti/4a = 6/94 after hydrolysis) and the final product 4a could be isolated in 72% yield. This sequence of carbocupration/zinc homologation/C–C bond cleavage has been generalized to different functionalized substrates (see scope in Fig. 2) and in all cases, the reactions proceed smoothly for the one-pot transformation of 1 into 4.
Both cyclopropenyl esters (–OEt and –OBn) undergo this combined transformation (compare 4b to 4c, 4d to 4e and 4f to 4g in Fig. 2), but it should be noted that the products resulting from the reaction with the benzyl ester (4b, 4d, 4g) are easier to purify by column chromatography from the remaining carbometalated products after hydrolysis (2anti) than the products possessing the ethyl ester (4c, 4e, 4f). These combined reactions are not restricted to the introduction of a methyl group, as various other alkyl groups could be added in good yields by simply changing the nature of the starting organocopper reagent (compare 4b to 4d, 4f to 4h and 4g to 4h, Fig. 2). The reaction of cyclopropenyl ester 1d with phenylcopper also proceeds well, but we were unable to separate the desired product 4m from the carbometalated product 2manti after hydrolysis. Similarly, when we treated a vinylcopper derivative (prepared from ethenylmagnesium bromide and CuI in a 1:1 ratio), as a representative example of an sp2 organometallic species, with cyclopropenyl ester 1j, the reaction proceeds smoothly but again, we were unable to separate the desired product 4n from the hydrolysed carbometalated product 2nanti. It is worth mentioning that this sequence of vinyl cupration/zinc homologation/fragmentation opens new avenues for the preparation of skipped dienes possessing a quaternary carbon stereocenter. Notably, the presence of other functional groups in the original alkyl chain of the cyclopropenyl ester is well tolerated (formation of 4j–l, Fig. 2). Having established an easy protocol for the preparation of racemic 4 from these very simple starting materials, the synthesis of enantiomerically enriched cyclopropenyl esters 1b (R1 = Ph(CH2)2), 1e (R1 = PhCH2) and 1f (R1 = Hex) was easily achieved in good enantiomeric ratio (1ber 96:4, 1eer 93:7 and 1fer 91:9) through cyclopropenation of the terminal alkyne with benzyl diazoacetate and Rh2(OAc)(R,R-DPTI)3 (DPTI = diphenyltriflylimidazolidinone) as catalyst.26 Interestingly, the enantiomeric ratios of these cyclopropenyl benzyl esters are lower than the ones obtained for cyclopropenyl ethyl esters (ethyl diazoacetate generally leads to enantiomeric ratios in the range of 97:3), but for the sake of simplicity, we decided to illustrate our concept with starting materials that would require a minimum number of chemical steps. When the sequence of carbocupration, zinc homologation and C–C bond fragmentation was performed on these enantiomerically enriched cyclopropenyl esters, enantioenriched acyclic allylic molecular architectures (4b,g,h,i) possessing a quaternary carbon stereocenter were easily obtained through the creation of two C–C bonds in a single-pot operation. HPLC analyses (see ESI†) show that the chiral information at the carbon atom connected to the ester moiety in 1b,e and f was quantitatively transferred to the final products 4b (er 96:4), 4g and 4h (er 93:7) and 4i (er 91:1), Fig. 2. It was therefore clear that the quaternary stereocenter was at no risk of epimerization through the whole process, and one could prepare the expected products with the same enantiomeric ratio as in the starting materials.
In contrast to any typical enantioselective synthesis where the independent formation of the two enantiomers of a product can only be achieved by changing the chirality of the ligand (or substrate) associated with the enantioselective transformation,27 the diastereodivergent carbometalation of 1, controlled by the nature of the solvent as described in Table 1, should therefore lead to the enantiodivergent synthesis of 4 from the same enantiomerically enriched starting material 1 (as summarized in Fig. 3).
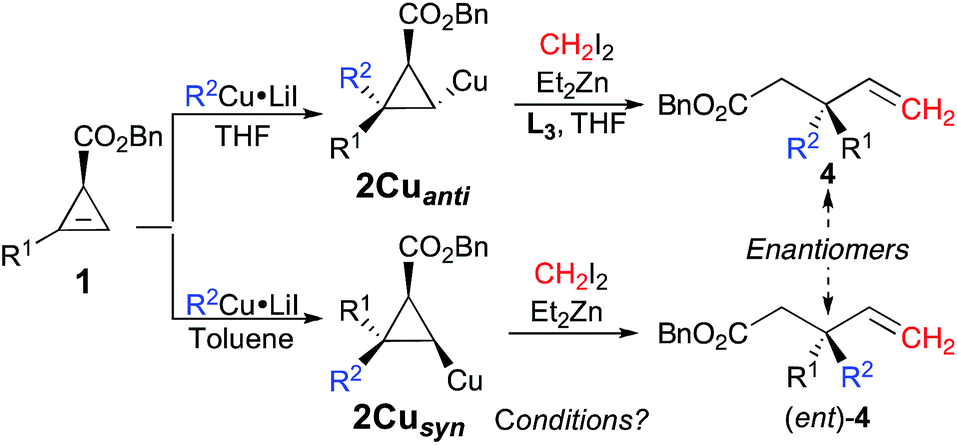 | ||
| Fig. 3 Diastereodivergent combined carbometalation/zinc homologation/fragmentation en route to enantioenriched allylic quaternary carbon stereocenters. | ||
Although the best ligand to promote the zinc homologation of 2Cuanti was phenanthroline L3, it was unclear that the same ligand would be optimal when the first step was performed in a non-polar solvent. To reach this new goal, we had to again optimise the reaction sequence for the formation of the desired products 4 from the syn-diastereomer 2Cusyn in the presence of a non-polar solvent (such as toluene, see Table 1, entries 5 and 8). It was first rapidly found that the zinc homologation for the transformation of 2Cusyn into 4a was more efficient when the initial organocopper species was prepared from an organolithium (R2Cu·LiI, Table 1, entry 5) instead of an organomagnesium species (R2Cu·MgX2, Table 1, entry 7). After extensive experiments, we were pleased to find that the zinc homologation could now be best performed in the presence of a non-nucleophilic potassium tert-butoxide ligand (for all other ligands tested, see ESI†) with addition of THF as co-solvent. It is important to note that the reaction performed under identical conditions but in the absence of the potassium tert-butoxide ligand results in very poor conversion, which underlines the effect of this particular ligand on the nucleophilicity of the metalated carbon center stabilized by the chelating ester. Once the best experimental conditions were in hand, various allylic esters 4 possessing quaternary carbon stereocenters were prepared in a single-pot operation from cyclopropenyl esters 1 as illustrated in Fig. 4.
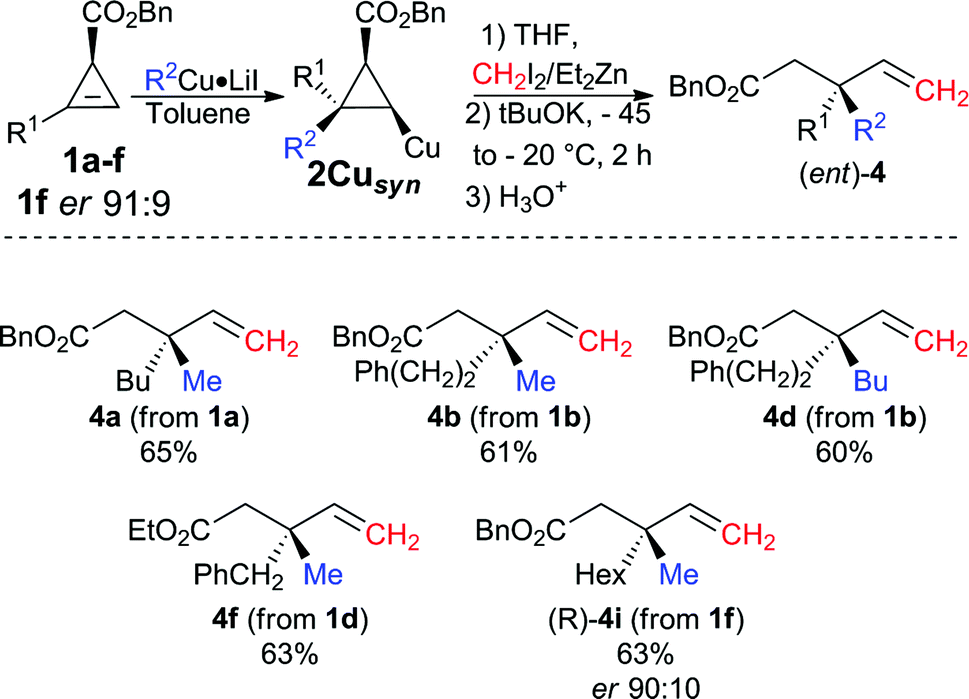 | ||
| Fig. 4 Combined carbometalation/zinc homologation/fragmentation reactions for the preparation of the opposite enantiomer. | ||
The scope of this new reaction sequence of syn-diastereoselective carbometalation, zinc homologation and selective C–C bond cleavage is equally good albeit with slightly lower yields for the final products 4. To illustrate the potential of this diastereodivergent approach, both enantiomers of 4i were prepared from the same cyclopropenyl ester enantiomer 1f (R1 = Hex, er 91:9). When the carbocupration of (S)-1f was performed with MeCu·LiI in THF followed by the subsequent addition of CH2I2, Et2Zn and phenanthroline (L3) as ligand, the selective C–C bond cleavage led to the corresponding allylic (S)-4i with an enantiomeric ratio of 91:9 in 65% yield (Fig. 5).
 | ||
| Fig. 5 Diastereodivergent strategy for the preparation of enantioenriched 4. | ||
On the other hand, if the same cyclopropenyl ester (S)-1f is added to the organocopper MeCu·LiI in toluene followed by subsequent zinc homologation in the presence of potassium tert-butoxide as ligand, the selective C–C bond cleavage provides the allylic (R)-4i in similar yield (63%) with almost the same enantiomeric ratio (er 90:10) (Fig. 5).
Conclusions
A new sequence of diastereoselective carbometalation/zinc homologation/C–C bond cleavage allows the easy transformation of enantiomerically enriched cyclopropenyl esters into acyclic allylic moieties bearing challenging quaternary carbon stereocenters in a single-pot reaction through the formation of two new C–C bonds. As the carbometalation reaction may lead to two different diastereoisomers according to the nature of the solvent, this strategy paves the way to the diastereodivergent synthesis of both enantiomers of 4 at will.Acknowledgements
This research is supported by the European Research Council under the European Community's Seventh Framework Program (ERC grant agreement no. 338912) and by the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities (140/12). S. R. R. acknowledges the Planning and Budgeting Committee (PBC) for financial assistance. I. M. is holder of the Sir Michael and Lady Sobell Academic Chair.Notes and references
- For recent reviews, see: (a) H. Pellissier, Tetrahedron, 2013, 69, 7171 CrossRef CAS; (b) H. Pellissier, Chem. Rev., 2013, 113, 442 CrossRef CAS PubMed; (c) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (d) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC; (e) H. Clavier and H. Pellissier, Adv. Synth. Catal., 2012, 354, 3347 CrossRef CAS; (f) C. De Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969 RSC; (g) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS; (h) Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 11778 CrossRef CAS PubMed; (i) S. Piovesana, D. M. Scarpino Schietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216 CrossRef CAS PubMed; (j) L. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef CAS PubMed; (k) M. Ruiz, P. Lopez-Alvarado, G. Giorgi and J. C. Menendez, Chem. Soc. Rev., 2011, 40, 3445 RSC; (l) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS PubMed.
- R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253 CrossRef CAS.
- For recent reviews on the formation of quaternary carbon stereogenic centers, see: (a) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed; (b) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed; (c) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (d) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (e) M. Bella and T. Casperi, Synthesis, 2009, 1583 CrossRef CAS; (f) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (g) I. Marek and G. Sklute, Chem. Commun., 2007, 43, 1683 RSC; (h) B. M. Trost and C. Jiang, Synthesis, 2006, 369 CrossRef CAS; (i) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473 CrossRef CAS; (j) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed; (k) I. Denissova and L. Barriault, Tetrahedron, 2003, 59, 10105 CrossRef CAS; (l) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (m) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef.
- (a) G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175 CrossRef CAS PubMed; (b) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (c) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC; (d) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404 CrossRef CAS PubMed; (e) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed; (f) B. M. Trost, Science, 1991, 254, 1471 CAS; (g) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS PubMed; (h) I. Marek, Chem.–Eur. J., 2008, 14, 7460 CrossRef CAS PubMed.
- (a) K.-i. Takao, K. Tsunoda, T. Kurisu, A. Sakama, Y. Nishimura, K. Yoshida and K.-i. Tadano, Org. Lett., 2015, 17, 756 CrossRef CAS PubMed; (b) L.-Q. Wang, Y.-G. Chen, J.-J. Xu, Y. Liu, X.-M. Li and Y. Zhao, Chem. Biodiversity, 2008, 5, 1879 CrossRef CAS PubMed; (c) B. Biswas and R. V. Venkateswaran, Synth. Commun., 2005, 35, 1769 CrossRef CAS; (d) Y. Kita, A. Furukawa, J. Futamura, K. Ueda, Y. Sawama, H. Hamamoto and H. Fujioka, J. Org. Chem., 2001, 66, 8779 CrossRef CAS PubMed; (e) M. Takahashi, Y. Shioura, T. Murakami and K. Ogasawara, Tetrahedron: Asymmetry, 1997, 8, 1235 CrossRef CAS.
- (a) M. Raducan, R. Alam and K. J. Szabό, Angew. Chem., Int. Ed., 2012, 51, 7263 CrossRef PubMed; (b) Y. Yatsumonji, T. Nishimura, A. Tsubouchi, K. Noguchi and T. Takeda, Chem.–Eur. J., 2009, 15, 2680 CrossRef CAS PubMed; (c) G. Dunet, P. Meyer and P. Knochel, Org. Lett., 2008, 10, 117 CrossRef CAS PubMed; (d) H. Ren, G. Dunet, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2007, 129, 5376 CrossRef CAS PubMed; (e) L. F. Tietze, T. Knizel and S. Schmatz, J. Am. Chem. Soc., 2006, 128, 11483 CrossRef CAS PubMed; (f) M. Yasuda, K. Hirata, A. Nishino, A. Yamamoto and A. Baba, J. Am. Chem. Soc., 2002, 124, 13442 CrossRef CAS PubMed.
- (a) J.-N. Heo, G. C. Micalizio and W. R. Roush, Org. Lett., 2003, 5, 1693 CrossRef CAS PubMed; (b) J. W. J. Kennedy and D. G. Hall, J. Am. Chem. Soc., 2002, 124, 898 CrossRef CAS PubMed; (c) S. E. Denmark and J. Fu, Org. Lett., 2002, 4, 1951 CrossRef CAS PubMed; (d) S. E. Denmark and J. Fu, J. Am. Chem. Soc., 2001, 123, 9488 CrossRef CAS PubMed.
- (a) R. M. Maksymowicz, A. J. Bissette and S. P. Fletcher, Chem.–Eur. J., 2015, 21, 5668 CrossRef CAS PubMed; (b) K. Hojoh, Y. Shido, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2014, 53, 4954 CrossRef CAS PubMed; (c) F. Gao, K. P. McGrath, Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 14315 CrossRef CAS PubMed; (d) C. A. Luchaco-Cullis, H. Mizutani, K. E. Murphy and A. H. Hoveyda, Angew. Chem., Int. Ed., 2001, 40, 1456 CrossRef CAS.
- (a) F. Nowrouzi, A. N. Thadani and R. A. Batey, Org. Lett., 2009, 11, 2631 CrossRef CAS PubMed; (b) U. Schneider, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13284 Search PubMed; (c) L. Carosi and D. G. Hall, Angew. Chem., Int. Ed., 2007, 46, 5913 CrossRef CAS PubMed; (d) S. R. Denmark, J. Fu and M. J. Lawler, J. Org. Chem., 2006, 71, 1523 CrossRef CAS PubMed; (e) S. R. Denmark, J. Fu, D. M. Coe, X. Su, N. E. Pratt and B. D. Griedel, J. Org. Chem., 2006, 71, 1513 CrossRef CAS PubMed; (f) R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8910 CrossRef CAS PubMed.
- (a) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabό, J. Am. Chem. Soc., 2015, 137, 11262 CrossRef CAS PubMed; (b) J. L.-Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 10992 CrossRef CAS PubMed.
- M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed.
- (a) D. Müller, M. Tissot and A. Alexakis, Org. Lett., 2011, 13, 3040 CrossRef PubMed; (b) Q. Naeemi, T. Robert, D. Kranz, J. Velder and H.-G. Schmalz, Tetrahedron: Asymmetry, 2011, 22, 887 CrossRef CAS; (c) T. Robert, J. Velder and H.-G. Schmalz, Angew. Chem., Int. Ed., 2008, 47, 7718 CrossRef CAS PubMed; (d) M. Vuagnoux-d'Augustin and A. Alexakis, Chem.–Eur. J., 2007, 13, 9647 CrossRef PubMed; (e) K. H. Ahn, R. B. Klassen and S. J. Lippard, Organometallics, 1990, 9, 3178 CrossRef CAS.
- (a) T. den Hartog, Y. Huang, M. Fananas-Mastral, A. Meuwese, A. Rudolph, M. Perez, A. J. Minnaard and B. L. Feringa, ACS Catal., 2015, 5, 560 CrossRef; (b) N. Yoshikai, T. Yamashita and E. Nakamura, Angew. Chem., Int. Ed., 2005, 44, 4721 CrossRef CAS PubMed; (c) S. Mori, M. Uerdingen, N. Kraus and K. Morokuma, Angew. Chem., Int. Ed., 2005, 44, 4715 CrossRef CAS PubMed; (d) Y. Yamamoto, S. Yamamoto, H. Yatagai, Y. Ishihara and K. Maruyama, J. Org. Chem., 1982, 47, 119 CrossRef CAS.
- (a) K. P. McGrath and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 53, 1910 CrossRef CAS PubMed; (b) D. Müller and A. Alexakis, Chem. Commun., 2012, 48, 12037 RSC.
- (a) L. Souillart and N. Cramer, Chem. Sci., 2014, 5, 837 RSC; (b) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed; (c) T. Seiser and N. Cramer, Angew. Chem., Int. Ed., 2008, 47, 9294 CrossRef CAS PubMed; (d) T. Matsuda, M. Shigeno and M. Murakami, J. Am. Chem. Soc., 2007, 129, 12086 CrossRef CAS PubMed.
- D. Didier, P.-O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038 CrossRef CAS PubMed.
- (a) R. Vabre, B. Island, C. J. Diehl, P. R. Schreiner and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 9996 CrossRef CAS PubMed; (b) T. Mejuch, M. Botoshansky and I. Marek, Org. Lett., 2011, 13, 3604 CrossRef CAS PubMed; (c) B. Dutta, N. Gilboa and I. Marek, J. Am. Chem. Soc., 2010, 132, 5588 CrossRef CAS PubMed; (d) G. Sklute and I. Marek, J. Am. Chem. Soc., 2006, 128, 4642 CrossRef CAS PubMed; (e) A. Abramovitch, J. P. Varghese and I. Marek, Org. Lett., 2004, 6, 621 CrossRef CAS PubMed; (f) G. Sklute, D. Amsallem, A. Shabli, J. P. Varghese and I. Marek, J. Am. Chem. Soc., 2003, 125, 11776 CrossRef CAS PubMed.
- For recent reviews on C–C cleavage, see: (a) I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 CAS; (b) K. Ruhland, Eur. J. Org. Chem., 2012, 2683 CrossRef CAS; (c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (d) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (e) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (f) T. Satoh and M. Miura, Top. Organomet. Chem., 2005, 14, 1 CAS; (g) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (h) M. E. Van Der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (i) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef.
- For recent examples of enantioselective cyclopropenylation of alkynes, see: (a) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS PubMed; (b) M. Uehara, H. Suematsu, Y. Yasutomi and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 170 CrossRef CAS PubMed; (c) X. Cui, X. Xu, H. Lu, S. Zhu, L. Wojitas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 3304 CrossRef CAS PubMed; (d) K. Goto, K. Takeda, N. Shiamda, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803 CrossRef PubMed; (e) J. F. Briones and H. M. L. Davies, Tetrahedron, 2011, 67, 4313 CrossRef CAS; (f) J. F. Briones, J. H. Hansen, K. I. Hardcastle, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 17211 CrossRef CAS PubMed; (g) M. P. Doyle, W. R. Winchester, J. A. A. Hoorn, V. Lynch, S. H. Simonsen and R. Ghosh, J. Am. Chem. Soc., 1993, 115, 9968 CrossRef CAS.
- (a) T. F. Schneider, J. Kaschel and D. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (b) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (c) M. Yu and B. L. Pagenkopf, Tetrahedron, 2005, 61, 321 CrossRef CAS; (d) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed.
- D. Müller and I. Marek, Chem. Soc. Rev., 2016 10.1039/c5cs00897b.
- (a) M. Pasco, N. Gilboa, T. Mejuch and I. Marek, Organometallics, 2013, 32, 942 CrossRef CAS; (b) I. Marek, Tetrahedron, 2002, 58, 9463 CrossRef CAS; (c) P. Knochel, N. Jeong, M. J. Rozema and M. C. P. Yeh, J. Am. Chem. Soc., 1989, 111, 6474 CrossRef CAS; (d) P. Knochel, T.-S. Chou, H. G. Chen, M. C. P. Yeh and M. J. Rozema, J. Org. Chem., 1989, 54, 5202 CrossRef CAS; (e) A. R. Sidduri and P. Knochel, J. Am. Chem. Soc., 1992, 114, 7579 CrossRef CAS; (f) A. R. Sidduri, M. J. Rozema and P. Knochel, J. Org. Chem., 1993, 58, 2694 CrossRef CAS.
- (a) A. Voituriez, L. E. Zimmer and A. B. Charette, J. Org. Chem., 2010, 75, 1244 CrossRef CAS PubMed; (b) J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron Lett., 1966, 7, 3353 CrossRef.
- (a) M. Simaan, P.-O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345 CrossRef CAS PubMed; (b) P. O. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333 CrossRef CAS PubMed.
- (a) E. Nakamura and S. Mori, Angew. Chem., Int. Ed., 2000, 39, 3750 CrossRef CAS; (b) M. D. Janssen, M. A. Corsten, A. L. Spek, D. M. Grove and G. van Koten, Organometallics, 1996, 15, 2810 CrossRef CAS.
- Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8916 CrossRef CAS PubMed.
- Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, vol. I–III, Suppl. I–II Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c6sc02191c |
| This journal is © The Royal Society of Chemistry 2016 |