
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtmospheric-pressure ionization and fragmentation of peptides by solution-cathode glow discharge†
Andrew J.
Schwartz
a,
Jacob T.
Shelley‡
*b,
Courtney L.
Walton
b,
Kelsey L.
Williams
b and
Gary M.
Hieftje
a
aDepartment of Chemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: jshelley@kent.edu; Tel: +1-330-672-2986
bDepartment of Chemistry and Biochemistry, Kent State University, Kent, OH 44242, USA
First published on 27th June 2016
Abstract
Modern “-omics” (e.g., proteomics, glycomics, metabolomics, etc.) analyses rely heavily on electrospray ionization and tandem mass spectrometry to determine the structural identity of target species. Unfortunately, these methods are limited to specialized mass spectrometry instrumentation. Here, a novel approach is described that enables ionization and controlled, tunable fragmentation of peptides at atmospheric pressure. In the new source, a direct-current plasma is sustained between a tapered metal rod and a flowing sample-containing solution. As the liquid stream contacts the electrical discharge, peptides from the solution are volatilized, ionized, and fragmented. At high discharge currents (e.g., 70 mA), electrospray-like spectra are observed, dominated by singly and doubly protonated molecular ions. At lower currents (35 mA), many peptides exhibit extensive fragmentation, with a-, b-, c-, x-, and y-type ion series present as well as complex fragments, such as d-type ions, not previously observed with atmospheric-pressure dissociation. Though the mechanism of fragmentation is currently unclear, observations indicate it could result from the interaction of peptides with gas-phase radicals or ultraviolet radiation generated within the plasma.
Introduction
Over the last thirty years, mass spectrometry (MS) has become an essential tool in the identification and characterization of proteins and peptides. Since biological activity of peptides and proteins is dictated by their amino-acid sequences, methods that can systematically sever bonds along a peptide backbone and detect the resulting fragments are central to the underlying goal of proteomics—the study of gene and cellular function at the protein/peptide level. The pervasiveness of MS in proteomic analysis is thereby attributable to the capability of tandem mass spectrometry (MS/MS or MSn) to provide a rapid, sensitive means to elucidate biopolymer sequences as well as post-translational modifications (PTMs).Though many MSMS methods are used in proteomics the most common employ collision-induced dissociation (CID).1 In CID, a peptide is activated by multiple collisions with neutral, non-reactive gases at reduced pressure. At these lower pressures, ions can be easily accelerated to kinetic energies in excess of the covalent bond dissociation energies. Redistribution of the energy from collisions into internal vibrational modes ultimately results in dissociation of the weakest chemical bonds along the amino-acid chain, most commonly resulting in formation of b- and y-type fragments. Though widely utilized, CID suffers from two major weaknesses: incomplete sequence coverage and the inability to preserve labile modifications. Since CID fragmentation is initiated by conversion of translational energy into internal energy, fragmentation of larger biomolecules becomes increasingly difficult as a result of increased degrees of freedom through which energy can be distributed; complete sequence coverage is attainable only for small (<3 kDa) peptides.2–5 Vibrational activation is similarly disadvantageous for identification of labile PTMs, such as phosphorylation or glycosylation, which alter the biological function of the molecule. Attached by chemical bonds weaker than those of the peptide backbone, PTMs are often lost prior to cleavage of peptide bonds when vibrational excitation occurs.6
Methods that induce fragmentation by transfer of an electron to the peptide, either from low-energy free electrons (electron-capture dissociation, ECD)5 or reagent anions (electron-transfer dissociation, ETD)3 have also become common. In either ECD or ETD, the exothermic neutralization reaction of an electron with a multiply charged peptide initiates a series of reactions that typically end in generation of complementary c- and z-type fragments with minor abundances of a- and y-type ions.7 Unlike CID, ECD and ETD result in rapid (i.e. vertical) electronic transitions resulting in little vibrational activation of the peptide; as a result, weakly bound PTMs are maintained during the ECD/ETD process. Further, because electron capture is equally probable to occur at any residue along a peptide backbone, sequence coverage is independent of peptide length and generally superior to CID. The largest drawbacks to ECD and ETD are that they induce charge reduction upon fragmentation, so only multiply charged peptides and proteins can be subjected to ECD/ETD, and that stearic hindrance prevents the fragmentation reactions from occurring at proline residues.3,5,7
Other limitations exist that are common among CID, ETD, and ECD. Most prevalent is the necessity for expensive, complex instrumentation. In the case of CID, an instrument capable of extracting ions within a desired mass-to-charge (m/z) range in conjunction with a collision cell is required (e.g., triple quadruple, ion trap, etc.). Electron-transfer dissociation presents additional complexity as the instrument (most commonly an ion trap) must be equipped with a secondary ionization source and associated ion optics for generation of reagent anions. Further, the instrument must be able to combine and trap precursor cations with anionic reagents, terminate the reaction, and detect product fragments. Thus, this approach requires precise control of DC, AC, and radiofrequency electronics as well as the relative timing between them. The most complicated method of fragmentation is ECD, where precursor ions must be immersed in a high number density of low-energy, free electrons. Because free electrons cannot be efficiently retained at low energies within radiofrequency fields of quadrupolar ion traps, ECD remains a technique that is limited to the static magnetic field provided by Fourier transform ion cyclotron resonance mass spectrometers, the most expensive type of mass analyzer. All of the aforementioned methods are further complicated by reduced-pressure requirements. At atmospheric pressure, the mean free path is short (<100 nm)8 and frequent collisions lead to complications for the aforementioned dissociation pathways. In CID, rapid collisional cooling at atmospheric pressure prevents precursors from acquiring enough internal energy for dissociation to occur. For ETD and ECD, trapping efficiency declines at atmospheric pressure while reaction of atmospheric gases with reagent anions or free electrons further diminishes fragmentation efficiency.
Despite the advances made in development of reduced-pressure fragmentation methods, little has been done to address the instrumental complexity required to utilize them. Much of the complexity in reduced-pressure approaches could be eliminated by controllably fragmenting peptides within the ion source, at atmospheric pressure, obviating the requirement of tandem MS for structural elucidation. This approach reduces instrument cost and complexity, and enables structural characterization with a wider variety of instruments (e.g., single quadrupole, sector field, time-of-flight, etc.). Furthermore, each stage of ion isolation and fragmentation in tandem-MS can result in significant ion losses (50–90%);9 in contrast, atmospheric-pressure fragmentation approaches exhibit higher ionization and transport efficiencies. To date, reported methods of atmospheric-pressure peptide fragmentation have utilized an electrospray-ionization (ESI) source to generate gas-phase peptide cations. These cations are directed to a secondary source, either a photoionization lamp10,11 or corona discharge,2,12,13 that promotes dissociation by a variety of mechanisms. For photoionization,10,11 ultraviolet (UV) radiation produces free electrons that promote dissociation by an ECD-like mechanism. Generation of free electrons by a corona discharge has also been found to lead to ECD-like dissociation,12 though electrochemical ionization13 and reaction with gas-phase radicals2 have also been reported. While these earlier methods did achieve fragmentation at atmospheric pressure, a combination of two sources, ESI and a secondary source for fragmentation, were required. A single source that can provide ionization and controlled fragmentation would be more desirable, and further reduce instrument complexity.
Here, we introduce an atmospheric-pressure ionization source, the solution-cathode glow discharge (SCGD), capable of ionizing and controllably fragmenting peptides directly from bulk solution. Originally used as an atomization and excitation source for atomic emission spectrometry,14–16 the SCGD is a compact, direct-current plasma formed between a tapered metal anode and flowing liquid cathode. As the liquid stream contacts the plasma, peptides within the solution are vaporized, ionized, and fragmented by interaction with the discharge. Unlike earlier atmospheric-pressure fragmentation methods, ionization and fragmentation are consolidated into a single source and peptide fragmentation is controllably tunable by simple adjustment of electrical current in the discharge. Counterintuitively, higher currents produce intact molecular ions and more ESI-like spectra, while lower currents yield extensive fragmentation to form a-, b-, c-, x-, y- and even d-type ion series. This new tunable ionization source for biological mass spectrometry provides improved sequence coverage over CID, and offers a simple ionization/fragmentation platform adaptable to virtually any atmospheric-pressure inlet MS.
Experimental section
Chemicals and reagents
Water, methanol, and trifluoroacetic acid (TFA) used as solvents and additives throughout these experiments were obtained from Fisher Scientific (Optima LC/MS grade). The modified peptide renin substrate I (97%), human angiotensin I (90%), human angiotensin II (93%), bradykinin (98%), substance P (95%), Met5-enkephalin (95%), and [D-Ala2,D-Leu5]-enkephalin (95%) were all purchased as acetate salts from Sigma-Aldrich (St. Louis, MO). Biotinylated, phosphorylated SAMS peptide was obtained from Enzo Life Sciences (Farmingdale, NY). Support solutions for the SCGD were prepared by dilution of concentrated, trace metal grade hydrochloric acid (J.T. Baker, Center Valley, PA) with deionized water of 18.2-MΩ resistivity, prepared in-house with a mixed-bed ion-exchange deionization unit.SCGD cell, sample introduction, and ion sampling
The present study employs a compact SCGD cell similar to those described by Doroski et al.17 and Shekhar et al.18 In this design (cf. Fig. S1†), a support solution (0.2 M HCl with 6% methanol) was delivered by a Gilson Minipuls 3 peristaltic pump (Middletown, WI) at a rate of 1.70 mL min−1 to a vertically oriented glass capillary (0.38 mm ID, 1.1 mm OD). This solution composition was chosen because it was found to produce higher ion signals for peptides than did more typical SCGD support solutions (e.g. 0.1 M HCl). Sheathing the capillary was a graphite tube of 1.40 mm ID and 4.53 mm OD, tapered to a point, with a contact angle of 15°, nearest the tip of the glass inlet capillary. The graphite tube and capillary were mounted within a polytetrafluoroethylene (PTFE) basin of approximately 20 mL volume. A stainless-steel screw, mounted through the side of the basin, served to electrically ground the graphite tube. Liquid overflow from the tip of the capillary, positioned 3 mm above the top of graphite tube, creates a connection between electrical ground and the incoming liquid cathode. Mounted directly above the glass capillary was a tungsten rod (3.18 mm diameter, 25.42 mm length) which served as the anode for the discharge. The tip closest to the solution-inlet capillary was tapered at a contact angle of 15° to provide a pointed structure that results in a more stable discharge. When a high potential (≥1000 V) is applied to the tungsten rod from a direct-current power supply (Kepco, Flushing, NY, BHK 2000-0.1 MG), electrical breakdown occurs in the ambient air between the rod and the solution. This results in the formation of a plasma that is sustained between the high-voltage anode and grounded solution cathode. To prevent the plasma from devolving into an electrical arc, a 1.25 kΩ ballast resistor is inserted between the high-voltage power supply and anode. Any solution that is not vaporized by the plasma drains into the PTFE basin, from which it is removed through a waste outlet. The discharge was operated in constant-current mode, with currents ranging from 35 to 90 mA. A 5 mm inter-electrode gap (distance between the tungsten rod and grounded solution) was used for all experiments.Peptide solutions were introduced into the SCGD via flow injection, as shown in Fig. 1. Here, a carrier stream of 50 μL min−1 deionized water (18.2 MΩ specific resistance) was provided by a syringe pump (Model Fusion 100T, Chemyx, Inc., Stafford, TX) to a Rheodyne 6-port injection valve (Model MXT715-000, Oak Harbor, WA), outfitted with a 5 μL sample loop. Sample solutions exiting the valve passed into a fused-silica capillary (50 μm ID, 150 μm OD). Fed through a T-connector and into the inlet capillary of the SCGD cell, the fused-silica capillary was terminated 1 mm below the surface of the support solution overflow. This nested-capillary arrangement prevents sample injections from mixing with the support flow until the very tip of the SCGD inlet capillary, and thereby reduces void volume and dilution experienced by sample injections.
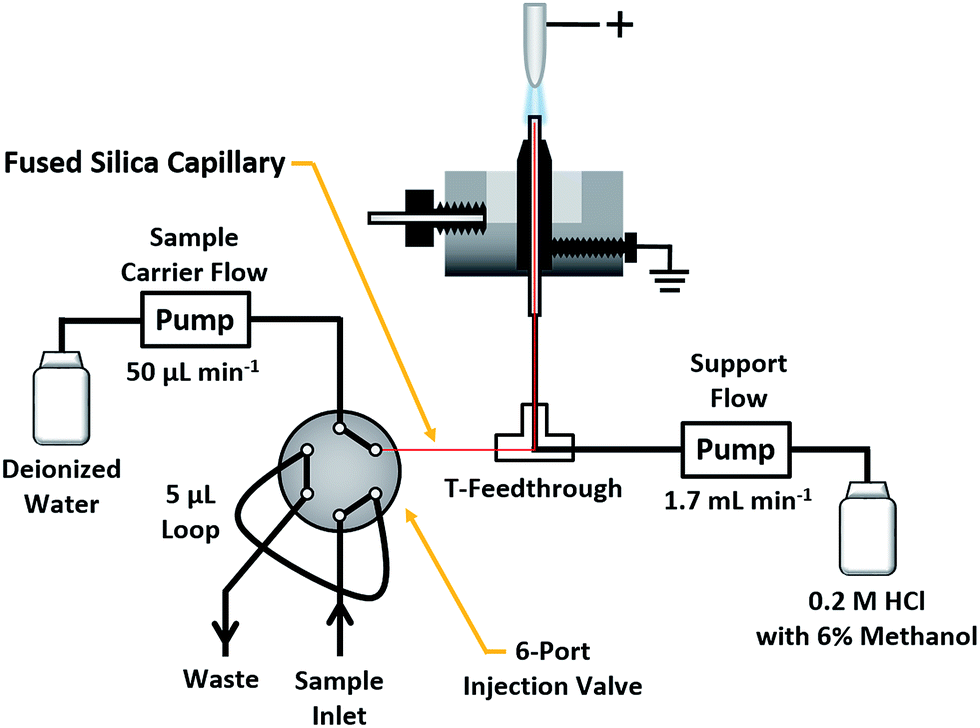 | ||
| Fig. 1 Schematic of the SCGD design and flow-injection arrangement used to introduce peptide solutions into the discharge. | ||
Gas-phase ions produced by the SCGD were sampled into the reduced-pressure environment of the MS through a locally fabricated extended capillary interface and detected by a high-resolution Orbitrap MS (Exactive Plus, Thermo Scientific, Bremen, Germany). The custom inlet capillary consisted of a stainless-steel tube (0.5 mm ID, 1.6 mm OD) that extended 4 cm beyond the conventional capillary inlet of the MS. The capillary was heated to 320 °C with the instrument's standard inlet heater and controlled/monitored through the instrument-control software. The use of thermal insulation around the exposed part of the capillary was found to have no impact on mass spectra or ion signals and, as such, was not used for these studies. In addition, no gas flow, apart from the vacuum pull within the first stage of the MS, was used to direct ions from the SCGD to the inlet. For all experiments, the stainless-steel capillary was positioned 3 to 6 mm below the SCGD plasma, at a distance of 3 to 5 mm from the SCGD sample inlet capillary (cf. Fig. S2†).
Mass spectra in the text represent the background-subtracted average (approximately 10 scans) of the mass-spectral signal acquired over a single flow-injection peak for a given peptide. Experimental details pertaining to the ESI-tandem mass spectra collected for comparison to those of SCGD-MS are included within the ESI.†
Results
Initial evaluation of the SCGD as a biomolecular ionization source was performed with the modified peptide renin substrate I, which contains an N-terminal succinylation (Suc) and a C-terminal aminomethyl coumarin (AMC). This peptide has the sequence Suc-RPFHLLVY-AMC and a molecular weight (MW) of 1300.485 Da. Fig. 2a shows the mass spectrum of renin substrate I obtained with the SCGD; the discharge was operated at 70 mA, a current commonly used for earlier atomic-emission studies.19 In this mode, both the protonated and doubly protonated molecular ions (MH+ and MH2+, respectively) are readily detected. Presence of the doubly charged molecular ion suggests that an electrospray-like ionization process is occurring. In ESI, a high electric field generates droplets that contain analyte, solvent, and an excess number of charges, most commonly protons. As these droplets desolvate, only the analyte species, now with one or more charges, remain.20 In contrast, gas-phase chemical ionization, such as interaction with proton donors or charge-transfer reagents (e.g. H3O+ or O2+), is unlikely to produce multiply charged species as it necessitates two sequential reactions to occur, with the second requiring two species charged with the same polarity to come into sufficient proximity for reaction. From Coulomb's law, the ions would need to have energies of at least 1.4 eV to overcome the repulsive force. Even at the elevated ion temperatures in the SCGD, with a maximum of 5000 K, the two reactants would have average energies of 0.43 eV, which is insufficient to overcome the coulombic barrier, let alone the activation energy of the reaction. Use of atmospheric-pressure chemical ionization (APCI) for the ionization of peptides rarely produces singly charged ions21 and, even then, does not produced multiply protonated analyte ions.22,23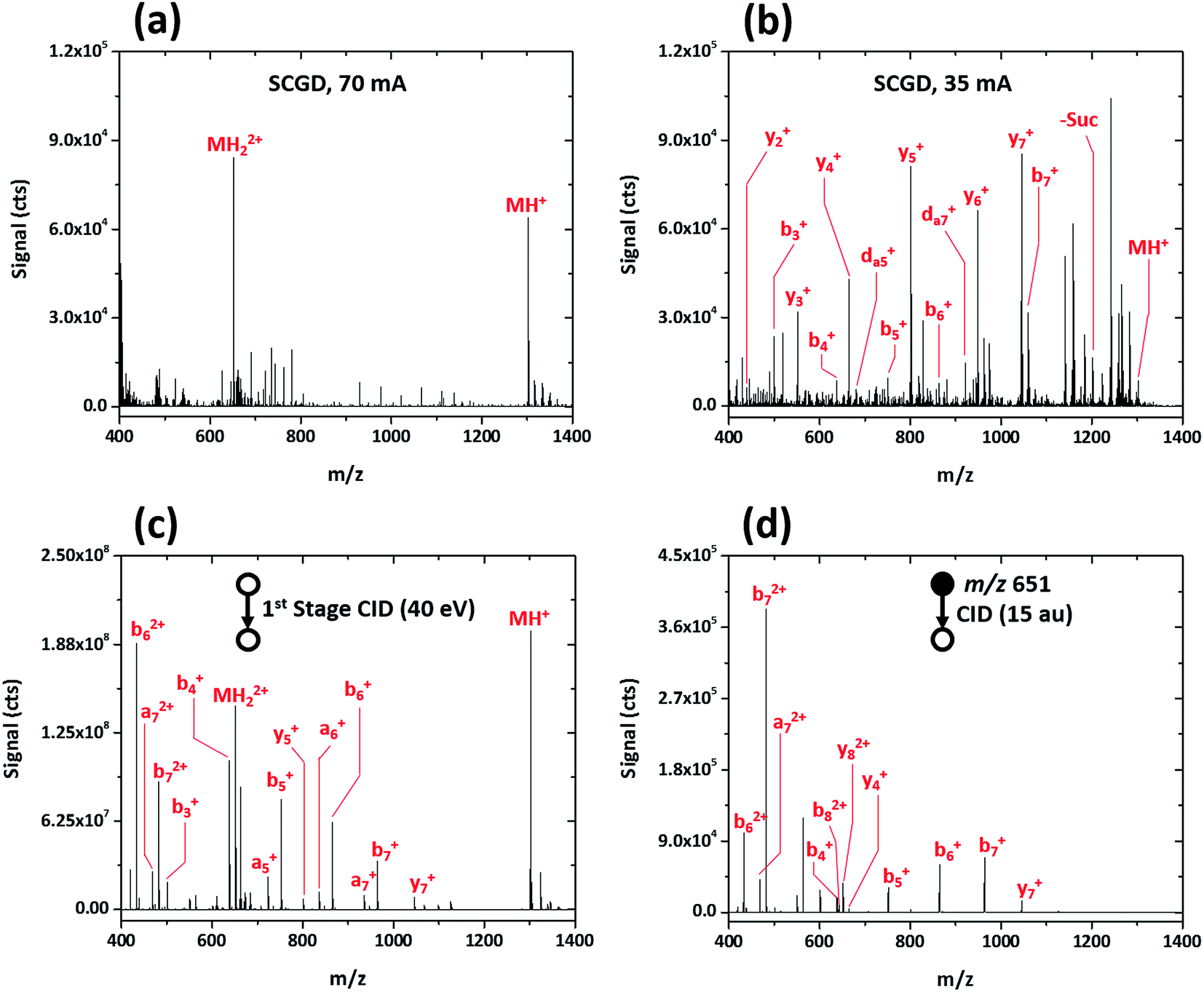 | ||
| Fig. 2 Mass spectra obtained for the peptide renin substrate I (Suc-RPFHLLVY-AMC) with SCGD (a, b) and traditional ESI with first-stage CID (c) and CID in a linear ion trap (d). Each spectrum represents the average signal produced by a 5 μL injection of 1.9 nmol renin substrate I. | ||
This seemingly electrospray-like mechanism supports earlier visual and photographic observations of the SCGD, where liquid jets, similar in structure to those seen with cone-jet electrospray, were observed on the solution surface in contact with the plasma.24 This earlier study also reported that liquid-jet nucleation sites were strongly influenced by current applied to the discharge—higher currents produced more liquid-jet nucleation sites than did low currents. To assess whether this effect might influence the SCGD mass spectrum of renin substrate I, the SCGD current was dropped to 35 mA. At this lower current, the spectrum is dramatically different (cf.Fig. 2b); the doubly protonated ion is no longer detected and the protonated molecular ion appears at rather low abundance. Instead, the spectrum is dominated by a number of ions of lower m/z. Examination of the mass differences among these lighter ions reveals successive losses of the C-terminal succinyl modification and amino-acid residues along the peptide chain. Surprisingly, despite the presence of a basic amino acid (arginine) at the N-terminus of renin substrate I, the most abundant fragment ions consist of singly charged, C-terminal, y-type ions, while the more thermodynamically favored N-terminal b-ions occur at significantly lower abundance. Finally, and most notably, da5+ and da7+ ions are also present in the SCGD spectrum of renin substrate I. The d-type fragments, resulting from partial side-chain fragmentation of an ions, are typically produced only with high-energy CID25,26 or vacuum-ultraviolet photodissociation (UV-PD).27 These secondary d-type fragments are particularly useful for distinguishing between the isomeric amino acids leucine and isoleucine. In the case of renin substrate I, if isoleucine were instead present at position five, the observed da5+ ion would appear at m/z 694.355 and be accompanied by a db5+ fragment at m/z 708.370. Since neither of those masses was detected, the SCGD appears capable of distinguishing between these two isomeric residues. To date, no other atmospheric-pressure ionization and fragmentation method has been reported to produce these high-energy fragment ions.
The dramatic differences in spectra between the two studied SCGD current levels is evidence that ionization and fragmentation of peptides occur very near or within the plasma and not as a result of other factors (e.g. thermal decomposition in the heated capillary). It also demonstrates that peptide fragmentation within the SCGD is controllably tunable by simple adjustment of source parameters. Based on these early observations (cf.Fig. 2a and b), high-current operation provides a softer, more ESI-like spectrum, whereas the low-current mode produces extensive fragmentation and enables structural elucidation. Utility of SCGD-MS is further apparent when these spectra are compared to that from a conventional ESI source with either first-stage CID or CID performed within a linear ion trap, as shown in Fig. 2c and d, respectively. In contrast to either of the ESI-MS/MS spectra, which exhibit a combination of both singly and doubly charged b-, y-, and a-type ions, the SCGD mass spectrum is far simpler; primarily one ion type is dominant and there are no doubly charged fragments that complicate interpretation. Such an SCGD mass spectrum is simple to interpret, as the peptide sequence can be easily determined from the mass shifts between the abundant y-ion peaks.
Rather simple fragmentation patterns are observed also for the peptides angiotensin I (DRVYIHPFHL, MW 1297.486 Da) and angiotensin II (DRVYIHPF, MW 1047.189 Da), shown in Fig. 3a and b, respectively. Like renin substrate I, both angiotensin peptides exhibit a remarkably simple fragmentation spectrum with the C-terminal y-ions at highest abundance, though b-, x-, and a-type ions are also detected. Though not explicitly labeled in Fig. 3a due to their low relative abundance, da6+ and db6+ fragments, corresponding to the loss of either side chain from the doubly substituted beta carbon of isoleucine, are apparent in the spectrum of angiotensin I. This finding further demonstrates that SCGD-MS provides a means of differentiation between isomeric residues.
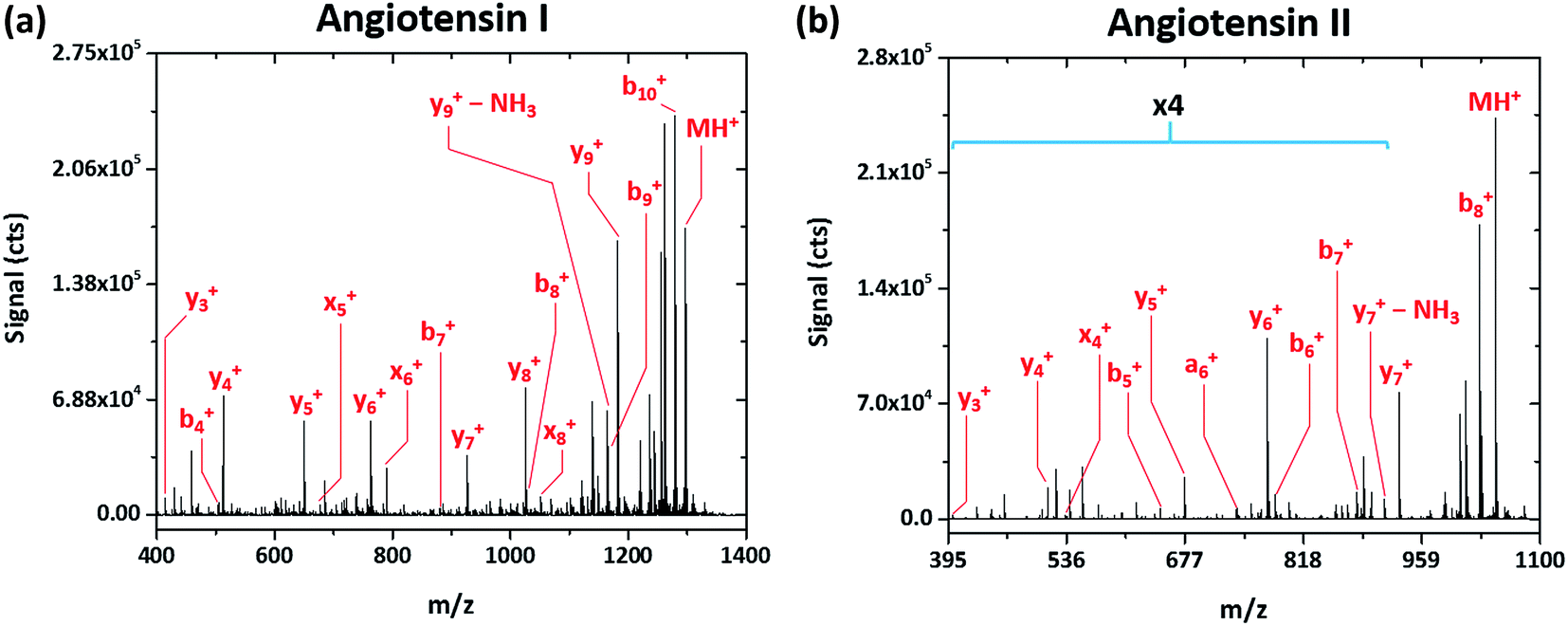 | ||
| Fig. 3 Fragmentation mass spectra recorded for the peptides angiotensin I (DRVYIHPFHL) and II (DRVYIHPF) with SCGD ionization-fragmentation at a SCGD current of 48 mA. Each spectrum represents the average signal from 5 μL injections of 1.9 and 2.5 nmol of angiotensin I and II, respectively. For easier viewing, the vertical axis of the angiotensin II spectrum is expanded by a factor of four in the m/z range of 395–900. | ||
Simple fragmentation patterns are not ubiquitous for all tested peptides—some produce significantly more information-rich spectra. For example, Fig. 4a shows the SCGD spectrum of the peptide bradykinin (RPPGFSPFR, MW 1061.218 Da). In contrast to the spectra presented earlier, SCGD fragmentation of bradykinin yields a-, b-, c-, y- and d-type ions, with no one ion type of dominant abundance. Substance P (RPKPQQFFGLM, MW 1349.624 Da), commonly used as a benchmark test for new fragmentation techniques,2,10–13 exhibits an even more complex fragmentation spectrum (cf.Fig. 4b). When fragmented by the SCGD, substance P generates an extensive series of a-, b-, c- and d-type ions. Complementary ion types, such as x-, y-, and z-type, are largely absent from the substance P spectrum with only y10+ and y8+ ions present.
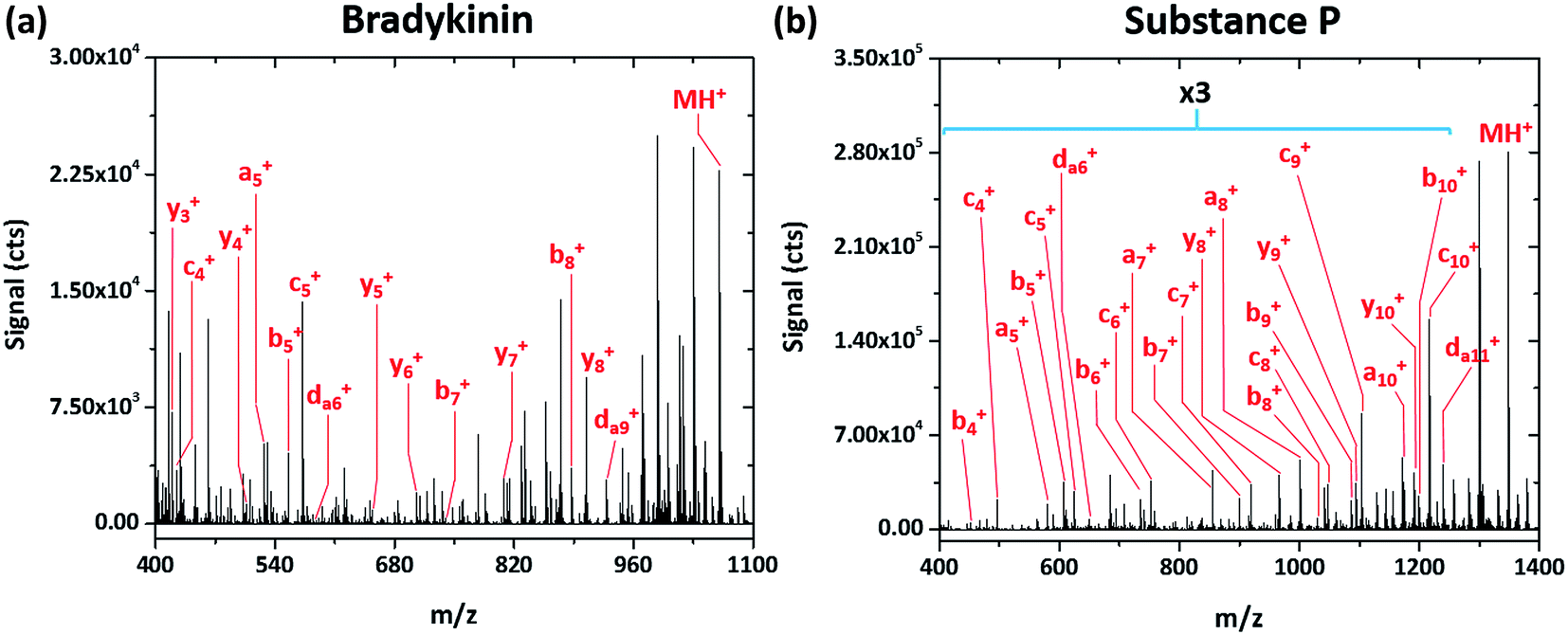 | ||
| Fig. 4 Mass spectra of the peptides bradykinin (RPPGFSPFR) and substance P (RPKPQQFFGLM) obtained with SCGD at a current of 55 mA. Each spectrum is the average produced by 5 μL injections of 2.4 and 1.9 nmol of bradykinin and substance P, respectively. For easier viewing, the vertical axis of the substance P spectrum is expanded by a factor of three in the m/z range of 400–1250. | ||
Interestingly, shorter peptides seem more resistant to fragmentation by the SCGD. Regardless of the SCGD current, the spectrum of Met5-enkephalin (YGGFM, MW 574.670 Da), shown in Fig. 5a, is dominated by the protonated molecular ion (MH+), oxygen adducts (MH+ + O and MH+ + O2), and a single fragment corresponding to the loss of a methanethiol group ([M − CH3SH]H+) from the methionine residue. In fact, no obvious fragment ions could be assigned for this peptide. The peptide [D-Ala2,D-Leu5]-enkephalin (YADGFLD, MW 570.658 Da), shown in Fig. 5b, is similarly resistant to fragmentation, with mainly the protonated molecular ion and a single oxygen adduct (MH+ and MH+ + O, respectively) apparent. In contrast to methionine enkephalin, the entire y-ion series as well as some a- and b-ions were detected for the substituted leucine enkephalin (not shown on the figure). However, these fragments were measured at less than 1% of the abundance of the molecular ion, which could make sequence identification difficult.
 | ||
| Fig. 5 Solution-cathode glow discharge mass spectra of the peptides Met5-enkephalin (YGGFM) and [D-Ala2,D-Leu5]-enkephalin (TAGFL) obtained at a current of 55 mA. Spectra are the average produced from a 5 μL injection of 4.4 nmol peptide. | ||
Though fragmentation of enkephalin peptides is trivial by reduced-pressure dissociation methods,28 this is not the case with the SCGD. However, it is well known that enkephalin peptides often form multimeric aggregates in solution29 as well as in the gas-phase electrospray process,30,31 particularly under native pH conditions used here for the sample. Further, Bleiholder et al.32 showed that chiral substitutions of Leu-enkephalin greatly influence the propensity for peptide aggregation, with YADGFLD being the least likely to form dimers and aggregates. It is possible that the excess energy imparted by the SCGD to these smaller peptides is expended on breaking higher-order aggregates into monomeric protonated molecular ions. This hypothesis is supported by the fact that fragment ions were not detected for the chirally pure Met-enkephalin, which should be more prone to aggregation. Meanwhile, the chirally substituted YADGFLD did yield fragment ions when introduced into the SCGD. With fewer peptide clusters present, the excess energy from the SCGD would be able to fragment covalent bonds. This phenomenon will be investigated in greater detail in the near future.
To determine whether post-translational modifications are retained upon fragmentation with the SCGD, biotinylated (as 6-biotinylamino-hexanoic acid), phosphorylated SAMS peptide (biotin-HMRSAMpSGLHLVKRR, MW 2200.597) was analyzed. As shown in Fig. 6a, SAMS peptide exhibits extensive fragmentation in the SCGD. The protonated molecular ion is not observed at low currents. Instead, internal fragments, such as GLHLV+ and SGLHLVKR+ with a loss of NH3, and several a-, b-, and c-type ions (with a loss of a single m/z unit) appear. All the (a-1), (b-1), and (c-1) ions maintain the N-terminal biotin modification. Since no fragment ions are observed beyond (c6-1)+ in the SCGD spectrum, it is difficult to determine if phosphorylation is maintained. The mass of the internal fragment (SGLHLVR−NH3)+ suggests it is not, as only the dephosphorylated form of this ion is observed despite containing the phosphorylation site.
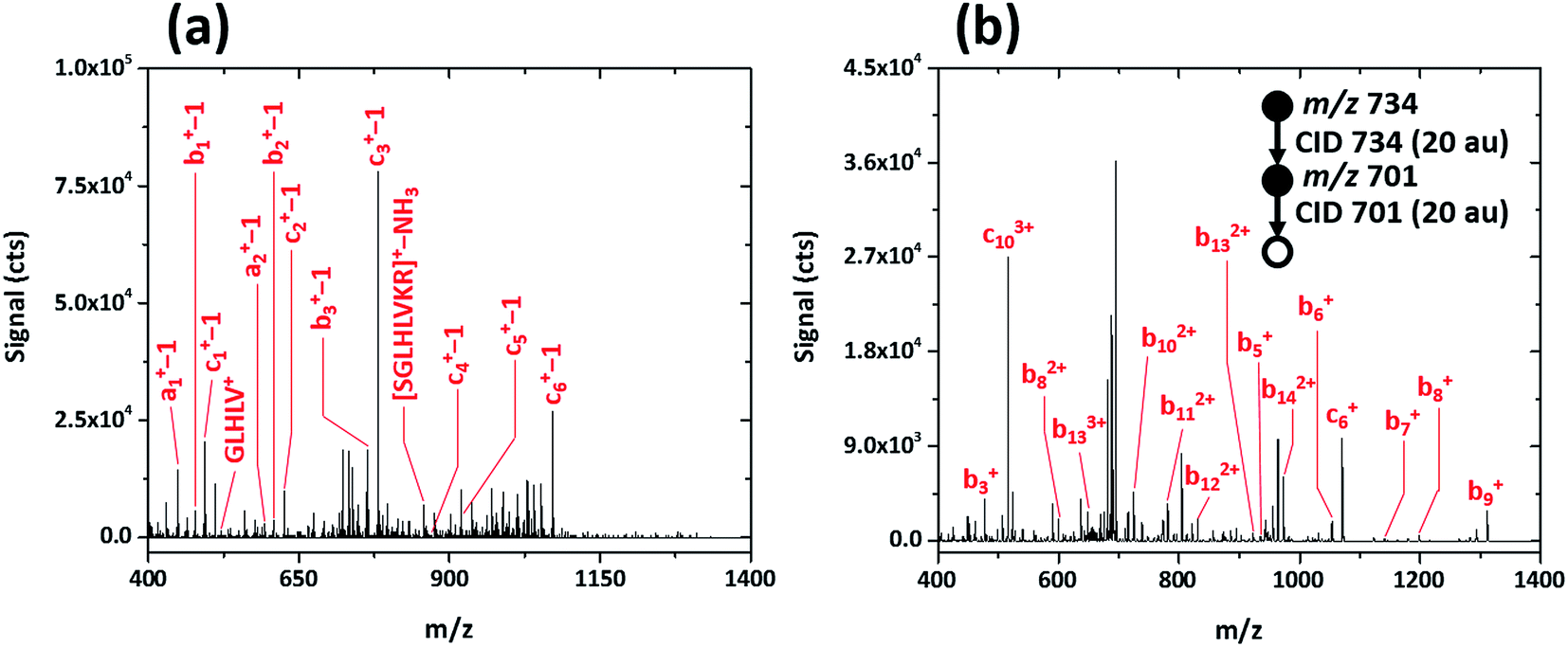 | ||
| Fig. 6 Comparison of the SCGD-MS (a) and ESI-MS3 (b) spectra of biotinylated, phosphorylated SAMS peptide (biotin-HMRSAMpSGLHLVKRR). Each spectrum represents the average ion signal from a 5 μL injection of 1.1 nmol of SAMS peptide. Note the significant differences between signals and signal-to-noise ratio between the two spectra. | ||
For comparison, an ESI-MSn spectrum of the same modified SAMS peptide was acquired on a linear ion-trap MS (cf.Fig. 6b). Notably, to obtain fragmentation comparable to the SCGD, two sequential CID activation steps were required. Activation of the triply charged molecular ion (MH33+) produced only the loss of phosphoric acid (H3PO4), common in CID of phosphorylated peptides.3 Subsequent activation resulted in incomplete b- and c-ion series, all retaining the N-terminal biotin, of multiple charge states (+1 to +3). In contrast, the SCGD provides fragmentation that is easier to interpret as all internal fragments and a-, b-, and c-type ions are singly charged, at signal levels higher than those afforded by multiple stages of CID, and without the requirement of a tandem-in-time instrument (i.e. an ion trap MS).
Discussion
From the preceding, it is clear that the SCGD is capable of atmospheric-pressure ionization and fragmentation of peptides directly from solution, though fragmentation patterns vary significantly from one peptide to another. Met5-enkephalin and [D-Ala2,D-Leu5]-enkephalin were resistant to fragmentation, renin substrate I, angiotensin I and II exhibit primarily an abundant y-ion series, while bradykinin, substance-P and SAMS peptides fragment with several ion series at similar abundance. Table 1 summarizes the identified fragments formed in the SCGD, including those not labeled in Fig. 2–6, and also shows the sequence coverage for each peptide as determined from the fragment ions. Of the peptides that dissociated within the SCGD, fragmentation is extensive (100% sequence coverage is obtained) for renin substrate I, bradykinin, substance P, angiotensin I and II, and [D-Ala2,D-Leu5]-enkephalin. By comparison, the SCGD provides lower sequence coverage, 40% and 0%, for biotinylated, phosphorylated SAMS peptide and Met-enkephalin, respectively. Compared to the coverage offered by ESI-MS with CID, also shown in Table 1 (spectra used to determine CID sequence coverage are available in Fig. S3†), the SCGD provides superior coverage for all peptides except biotinylated, phosphorylated SAMS and the enkephalin peptides, though the former peptide necessitated multiple isolation and CID-activation steps. If only a single stage of CID is used, SAMS peptide exhibits only the loss of phosphate and no sequence information is obtained for CID.| Name, # residues | Peptide sequence | an | bn | cn | xn | yn | dn | Internal fragments | Identified residues | |
|---|---|---|---|---|---|---|---|---|---|---|
| SCGD | ESI-MS/MS | |||||||||
| a Sequence coverage achieved only through MS3 as MS/MS produced exclusively [M − H3PO4 + H3]3+. b Detected fragment ions were all less than 1% abundance relative to the protonated molecular ion of the peptide. | ||||||||||
| Renin substrate I, 8 | Suc-RPFHLLVY-AMC | — | 2–8 | — | — | 1–8 | a5, a7 | — | 8/8, 100% | 4/8, 50% |
| Angiotensin I, 10 | DRVYIHPFHL | — | 4, 7–10 | — | 5, 8 | 2–10 | a6, b6 | — | 10/10, 100% | 3/10, 30% |
| Angiotensin II, 8 | DRVYIHPF | 6 | 5–8 | — | 4 | 3–8 | — | — | 8/8, 100% | 2/8, 25% |
| Bradykinin, 9 | RPPGFSPFR | 5 | 5, 7–9 | 4, 5 | — | 2–9 | a6, a9 | — | 9/9, 100% | 3/9, 33% |
| Substance P, 11 | RPKPQQFFGLM | 5, 7, 8, 10 | 3–11 | 4–10 | — | 8–11 | a6, a11 | — | 11/11, 100% | 6/11, 55% |
| SAMS peptide, 15 | Biotin-HMRSAMpSGLHLVKRR | 1, 2 | 1–3 | 1–6 | — | — | — | GLHLV+, [SGLHLVKR]+–NH3 | 6/15, 40% | 9/15, 60%a |
| [D-Ala2,D-Leu5]-enkephalin, 5 | YADGFLD | 4–5 | 3–5 | — | — | 2–5 | — | — | 5/5, 100%b | 5/5, 100% |
| Met-enkephalin, 5 | YGGFL | — | — | — | — | — | — | [M − CH3SH]H+ | 0/5, 0% | 5/5, 100% |
At present, we have too little information to predict, a priori, peptide fragment ions formed within the SCGD. As Table 1 demonstrates, there is no obvious correlation between the amino-acid sequence of a peptide and the observed fragment ions. Ultimately, this finding suggests that the mechanism of dissociation is complex or that fragmentation results from a combination of several mechanisms. Due to the intricate chemical and physical properties of energetic plasmas, there are many potential mechanisms that might lead to peptide dissociation within the SCGD; these include CID, ECD/ETD, photodissociation, thermal dissociation, chemical fragmentation or a combination thereof. At the present stage of investigation, proposal of an exact mechanism for the SCGD-induced fragmentation of peptides is premature, though general insights can be gained from a comparison of these early SCGD-MS results with those in the literature.
Among the potential pathways to dissociation in the SCGD, only those that occur on short timescales (i.e. vertical electronic transitions) are likely to contribute to fragmentation. Since fragmentation within the SCGD occurs at atmospheric pressure, collisional cooling would preclude effective fragmentation by thermal and/or vibrational mechanisms. As a result, both CID and thermal fragmentation seem unlikely to contribute to peptide dissociation in the SCGD, though collisions with energetic ions and neutral species could induce fragmentation by a mechanism similar to high-energy CID or surface-induced dissociation (SID). A comparison of the SCGD spectra of renin substrate I and angiotensin I (Fig. 2b and 3) to those obtained with SID33 shows major differences. Specifically, the SCGD produces predominantly y-ion series with b-, x-, and d-ions at lower levels for both of these peptides, whereas SID exhibits strong series of a- and b-type ions. Differences are observed also between SCGD fragmentation and high-energy CID,34 which produces mainly w- and v-type fragments (z- and y-ions with side-chain losses) that are not observed in the SCGD spectra. Combined, these comparisons suggest that other mechanisms that induce vertical excitation (ECD, ETD, UV-PD and chemical fragmentation) are more likely to contribute to fragmentation in the SCGD than those of collisions with energetic species.
Past studies have revealed that the free-electron number density in the SCGD is on the order of 1014 cm−3.35 Electron temperature in the SCGD has also been determined to be, on average, 5000 K,36 which corresponds to a mean electron energy of approximately 0.4 eV. Although the SCGD electron energy is higher than typically utilized in ECD (<0.2 eV), it is possible that some peptide fragmentation occurs within the SCGD as a result of ECD. Also conceivable is ETD arising from anions (NO3−, O3−, etc.) formed within the SCGD. However, SCGD fragmentation patterns differ substantially from those in atmospheric-pressure ECD,10 or reduced-pressure ECD5 and ETD.3 Both atmospheric-pressure ECD and reduced-pressure ECD/ETD produce primarily series of c- and z-ions and comparatively minor amounts of a- and y-ions.3,5,10 In contrast, the SCGD produces series of a-, b-, c-, x-, y- and d-type ions, depending on the peptide. If either ECD or ETD were a significant pathway to dissociation in the SCGD, the absent z-type ions should also be prominent in the spectra. In addition to these observations, the SCGD mass spectra reveal that fragmentation occurs at proline residues, which is not found with either ECD or ETD. Together, these observations hint that both ECD and ETD are unlikely to contribute significantly to peptide fragmentation in the SCGD.
From earlier optical-emission studies, several reactive radical species form within the SCGD (OH˙, NO˙, etc.)15,37 that are known to result in backbone and side-chain cleavage of peptides in both the solution and gas phase.38,39 Presence of oxygen adducts in the SCGD spectra of Met5 enkephalin and [D-Ala2,D-Leu5]-enkephalin (Fig. 5a and b) suggests that peptides interact with reactive species within the plasma, and hints that radical-induced dissociation (RID) might occur. Further, earlier work by optical emission has revealed that radical, molecular species are more prevalent in the SCGD at reduced currents.40 Accordingly, the concentration of molecular radicals goes up as SCGD current is lowered, and might explain the seemingly counter-intuitive enhanced fragmentation under low-power conditions. Vilkov et al.2 presented RID spectra for the peptides substance P and bradykinin. Though the SCGD spectra differ greatly from those presented in that earlier study, heating of the peptides by the plasma would influence the rates of reaction of any radical species with the amino-acid side chains and backbone.2,41,42 Earlier RID experiments were performed at temperatures of 20–400 °C, much lower than in the SCGD, which exhibits gas-kinetic temperatures as high as 3500 K, (approximately 0.3 eV).43 These thermal energies are insufficient to lead to peptide-bond fragmentation alone, which requires at least a few electron volts of energy. However, once an initial peptide fragment is formed, the potential barrier for subsequent cleavages and rearrangements is lowered. Jue et al.44 have shown that fragmentation spectra in a reduced-pressure ion trap can change significantly based on a comparatively mild increase in bath-gas temperature to 160 °C. Thus, differences between SCGD fragmentation spectra and those obtained with RID would be expected due to the higher gas temperatures and larger number of collisions leading to subsequent cleavages or interconversion of fragment ions. From these observations, RID might contribute to fragmentation in the SCGD.
Photodissociation is another possible mechanism of peptide fragmentation. The SCGD emits strongly in the ultraviolet region as a result of molecular band emission from NO˙ (A2E+–χ2Π system, 204.7–345.9 nm), OH˙ (A2E–χ2Π system, 244.4–402.2 nm), and N2 (C3Πu–B3Πg system, 280.3–497.6 nm).45 Further, since the SCGD was operated with a support solution containing 6% methanol in these experiments, additional UV emission would result from CO molecular emission (a3Π–χ1E system, 198.9–257.5 nm).37 Ultimately, this emission results in a photon flux from the SCGD with energies as high as 6.2 eV, lower than is employed for atmospheric-pressure photodissociation (AP-PD)11,46 or reduced-pressure UV-PD27 (6.4–10 eV). Regardless of differences in photon energies, the fragmentation patterns of the SCGD are similar to those of AP-PD and UV-PD. With substance P as a basis for comparison, AP-PD produces a-, b-, c-, and y-type ions, similar to the SCGD.11 Unlike the SCGD, no d-type ions were reported with AP-PD fragmentation, but are highly abundant in the reduced-pressure UV-PD spectrum of substance P.27 Despite similarities in the observed ion series, the SCGD spectrum differs in the abundances and sequence coverage for the respective ion types produced by both AP-PD and UV-PD. This comparison suggests that photodissociation could play a significant role in fragmentation of peptides with the SCGD, though it is likely accompanied by other mechanisms.
Though a definitive mechanism for peptide fragmentation cannot be offered at this stage of study, the potential impact of the SCGD on the field of proteomics is clear. Inexpensive, compact, and capable of ionizing and controllably fragmenting peptides at atmospheric pressure, the SCGD opens up the possibility of peptide sequencing on MS instruments that could not otherwise provide structural information. This capability could enable future instrumentation to be greatly simplified through elimination of the need for reduced-pressure fragmentation methods. Furthermore, the SCGD offers potential for more complete peptide analyses from a single ionization source and without the complex MS instrumentation on the back end. Many peptides were found to fragment extensively, exhibiting high sequence coverage through a variety of potential ion series (a-, b-, c-, x- and y-types). In addition, the presence of d-type fragment ions, resulting from side-chain cleavages, shows that the SCGD would be useful for accurate assignment of isomeric residues without the need for additional tagging experiments. Combined, these early results highlight the utility of the SCGD for peptide characterization and the potential impact of the source on proteomic instrumentation, and also suggest that the source might be useful for analysis of other large polymers (RNA, DNA, lipids, sugars, etc.), which will be investigated in future studies. Furthermore, beyond its potential for biological mass spectrometry, in a separate study47 we have also demonstrated that the SCGD is useful for ionization of non-biological species (atomic, inorganic and organic molecules, etc.), further demonstrating the utility and potential impact of the source in the area of mass spectrometry.
Acknowledgements
Supported in part by the U.S. Department of Energy through grant DOE DE-FG02-98ER 14890. The authors also thank Indiana University Mechanical Instrument Services for assistance in fabrication of the SCGD cell and capillary sampling inlet used in this work. Andrew Schwartz was supported, in part, by an E.M. Kratz Fellowship from Indiana University.References
- J. A. Loo, C. G. Edmonds and R. D. Smith, Anal. Chem., 1991, 63, 2488–2499 CrossRef CAS PubMed
.
- A. N. Vilkov, V. V. Laiko and V. M. Doroshenko, J. Mass Spectrom., 2009, 44, 477–484 CrossRef CAS PubMed
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed
- K. F. Haselmann, B. A. Budnik, F. Kjeldsen, N. C. Polfer and R. A. Zubarev, Eur. J. Mass Spectrom., 2002, 8, 461–469 CrossRef CAS
- N. A. Kruger, R. A. Zubarev, D. M. Horn and F. W. McLafferty, Int. J. Mass Spectrom., 1999, 185, 787–793 CrossRef
- R. J. Simpson, L. M. Connolly, J. S. Eddes, J. J. Pereira, R. L. Moritz and G. E. Reid, Electrophoresis, 2000, 21, 1707–1732 CrossRef CAS PubMed
- F. W. McLafferty, D. M. Horn, K. Breuker, Y. Ge, M. A. Lewis, B. Cerda, R. A. Zubarev and B. K. Carpenter, J. Am. Soc. Mass Spectrom., 2001, 12, 245–249 CrossRef CAS PubMed
- S. G. Jennings, J. Aerosol Sci., 1988, 19, 159–166 CrossRef CAS
- S. A. McLuckey, G. J. Van Berkel, D. E. Goeringer and G. L. Glish, Anal. Chem., 1994, 66, 737A–743A CrossRef CAS PubMed
- D. B. Robb, J. C. Rogalski, J. Kast and M. W. Blades, Anal. Chem., 2012, 84, 4221–4226 CrossRef CAS PubMed
- D. Debois, A. Giuliani and O. Laprevote, J. Mass Spectrom., 2006, 41, 1554–1560 CrossRef CAS PubMed
- V. D. Berkout and V. M. Doroshenko, Int. J. Mass Spectrom., 2012, 325–327, 113–120 CrossRef CAS PubMed
- J. R. Lloyd and S. Hess, J. Am. Soc. Mass Spectrom., 2010, 21, 2051–2061 CrossRef CAS PubMed
- M. R. Webb, F. J. Andrade and G. M. Hieftje, Anal. Chem., 2007, 79, 7807–7812 CrossRef CAS PubMed
- M. R. Webb, F. J. Andrade and G. M. Hieftje, Anal. Chem., 2007, 79, 7899–7905 CrossRef CAS PubMed
- T. Cserfalvi, P. Mezei and P. Apai, J. Phys. D: Appl. Phys., 1993, 26, 2184 CrossRef CAS
- T. A. Doroski, A. M. King, M. P. Fritz and M. R. Webb, J. Anal. At. Spectrom., 2013, 28, 1090–1095 RSC
- R. Shekhar, D. Karunasagar, M. Ranjit and J. Arunachalam, Anal. Chem., 2009, 81, 8157–8166 CrossRef CAS PubMed
- A. J. Schwartz, S. J. Ray and G. M. Hieftje, Spectrochim. Acta, Part B, 2015, 105, 77–83 CrossRef CAS
- S. J. Gaskell, M. S. Bolgar, I. Riba and S. G. Summerfield, NATO Adv. Sci. Inst. Ser., Ser. C, 1997, 504, 3–16 CAS
- S.-C. Cheng, S.-S. Jhang, M.-Z. Huang and J. Shiea, Anal. Chem., 2015, 87, 1743–1748 CrossRef CAS PubMed
- Y. Shen, C. Han, J. Chen and X. Wang, Chromatographia, 2007, 66, 319–323 CrossRef CAS
- J. J. Coon and W. W. Harrison, Anal. Chem., 2002, 74, 5600–5605 CrossRef CAS PubMed
- A. J. Schwartz, S. J. Ray, E. Elish, A. P. Storey, A. A. Rubinshtein, G. C. Y. Chan, K. P. Pfeuffer and G. M. Hieftje, Talanta, 2012, 102, 26–33 CrossRef CAS PubMed
- R. S. Johnson, S. A. Martin and K. Biemann, Int. J. Mass Spectrom. Ion Processes, 1988, 86, 137–154 CrossRef CAS
- R. S. Johnson, S. A. Martin, K. Biemann, J. T. Stults and J. T. Watson, Anal. Chem., 1987, 59, 2621–2625 CrossRef CAS PubMed
- W. D. Cui, M. S. Thompson and J. P. Reilly, J. Am. Soc. Mass Spectrom., 2005, 16, 1384–1398 CrossRef CAS PubMed
- J. Sztáray, A. Memboeuf, L. Drahos and K. Vékey, Mass Spectrom. Rev., 2011, 30, 298–320 CrossRef PubMed
- C. Bignardi, A. Cavazza, M. Marini and L. G. Roda, Int. J. Mass Spectrom., 2014, 357, 22–28 CrossRef CAS
- R. Guevremont and R. W. Purves, J. Am. Soc. Mass Spectrom., 1999, 10, 492–501 CrossRef CAS PubMed
- J. C. Jurchen, D. E. Garcia and E. R. Williams, J. Am. Soc. Mass Spectrom., 2003, 14, 1373–1386 CrossRef CAS PubMed
- C. Bleiholder, N. F. Dupuis and M. T. Bowers, J. Phys. Chem. B, 2013, 117, 1770–1779 CrossRef CAS PubMed
- A. L. McCormack, J. L. Jones and V. H. Wysocki, J. Am. Soc. Mass Spectrom., 1992, 3, 859–862 CrossRef CAS PubMed
- L. Sleno and D. A. Volmer, J. Mass Spectrom., 2004, 39, 1091–1112 CrossRef CAS PubMed
- M. R. Webb, G. C. Y. Chan, F. J. Andrade, G. Gamez and G. M. Hieftje, J. Anal. At. Spectrom., 2006, 21, 525–530 RSC
- T. Cserfalvi and P. Mezei, J. Anal. At. Spectrom., 1994, 9, 345–349 RSC
- C. G. Decker and M. R. Webb, J. Anal. At. Spectrom., 2016, 31, 311–318 RSC
- J. Q. Guan and M. R. Chance, Trends Biochem. Sci., 2005, 30, 583–592 CrossRef CAS PubMed
- S. D. Maleknia, J. W. H. Wong and K. M. Downard, Photochem. Photobiol. Sci., 2004, 3, 741–748 Search PubMed
- M. R. Webb, F. J. Andrade and G. M. Hieftje, J. Anal. At. Spectrom., 2007, 22, 766–774 RSC
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196–219 CrossRef CAS
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS
- M. R. Webb, F. J. Andrade, G. Gamez, R. McCrindle and G. M. Hieftje, J. Anal. At. Spectrom., 2005, 20, 1218–1225 RSC
- A. L. Jue, A. H. Racine and G. L. Glish, Int. J. Mass Spectrom., 2011, 301, 74–83 CrossRef CAS
- P. Jamroz, P. Pohl and W. Zyrnicki, J. Anal. At. Spectrom., 2012, 27, 1032–1037 RSC
- A. Delobel, F. Halgand, B. Laffranchise-Gosse, H. Snijders and O. Laprevote, Anal. Chem., 2003, 75, 5961–5968 CrossRef CAS PubMed
- A. J. Schwartz, K. L. Williams, J. T. Shelley and G. M. Hieftje, presented in part at the SciX 2015, Providence, RI, September 29, 2015, 2015.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02032a |
| ‡ Current address: Department of Chemistry and Chemical Biology, Rensselaer Polytechnic Institute, Troy, NY 12180, USA. |
| This journal is © The Royal Society of Chemistry 2016 |