
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-covalent S⋯O interactions control conformation in a scaffold that disrupts islet amyloid polypeptide fibrillation†
Hayden
Peacock
a,
Jinghui
Luo
a,
Tohru
Yamashita
a,
James
Luccarelli
a,
Sam
Thompson
*a and
Andrew D.
Hamilton
*ab
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: sam.thompson@chem.ox.ac.uk; Tel: +44 (0) 1865 275978
bDepartment of Chemistry, New York University, New York, New York 10003, USA. E-mail: andrew.hamilton@nyu.edu
First published on 1st July 2016
Abstract
Conformationally-constrained molecules that selectively recognise the surfaces of proteins have the potential to direct the path of protein folding. Such molecules are of therapeutic interest because the misfolding of proteins, especially that which results in fibrillation and aggregation, is strongly correlated with numerous diseases. Here we report the novel use of S⋯O interactions as a conformational control element in a new class of non-peptidic scaffold that mimics key elements of protein surfaces. These molecules disrupt the fibrillation of islet amyloid polypeptide (IAPP), a process that is implicated in the pathology of type II diabetes.
Introduction
Correctly folded proteins have myriad roles critical for life, including structural support, signal mediation and catalysis. Conversely, misfolded forms are associated with numerous diseases, including neurodegenerative disorders such as Alzheimer's and Parkinson's, and non-neuropathic conditions such as type II diabetes.1 The use of small molecules to mediate aberrant protein folding is a hugely appealing therapeutic strategy,2–4 however there are few rationally designed examples. A potential solution is the use of protein surface mimetics that modulate the energy landscape of protein folding via selective recognition of complementary protein surfaces, analogous to nature's chaperone proteins. Work by our group and others has established several families of synthetic protein surface mimetics that reproduce key recognition features of protein secondary structure5–9 and can thereby affect the pathways of protein folding.10,11In order to maximise target-binding affinity, protein surface mimetics are based on conformationally pre-organised scaffolds. This increases the population of the desired conformer and thus reduces the entropic cost of binding. Previous non-peptidic scaffolds have taken advantage of hydrogen bonding,12–16 the stereoelectronic preferences of secondary aryl amides,13,17,18 steric repulsion between vicinal cyclic systems,19,20 dipolar repulsion,21,22 cyclisation,23 and metal templation24 to provide conformational pre-organisation (Fig. 1).
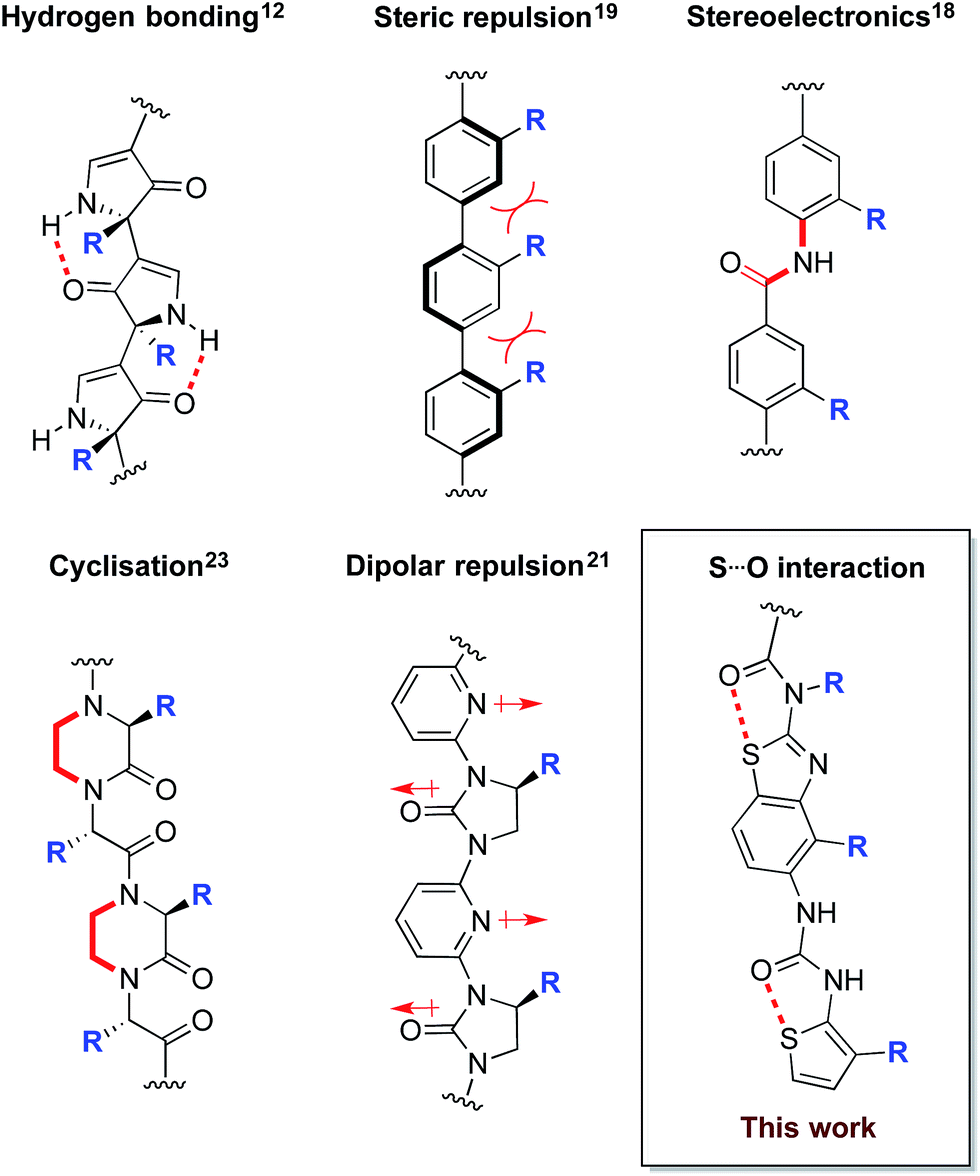 | ||
| Fig. 1 Conformational determinants of non-peptidic protein surface mimetics. | ||
With the aim of expanding the toolkit of scaffolds for protein surface mimicry we set out to explore additional conformational determinants that promote pre-organisation. The S⋯O interaction involves a low-lying C–S σ* molecular orbital that accepts electron density from an adjacent oxygen lone pair.25–27 Many observations of this effect have been made ex post facto – frequently upon inspection of X-ray diffraction data. However, there are few examples in which the interaction is incorporated as a conformational determinant of rationally designed compounds. The effect of the S⋯O interaction is highlighted by quantum mechanical simulation of a benzothiazole model system in which population distribution is evaluated as a function of two key dihedral angles (Φ1 and Φ2, Fig. 2A). A 1,5-S⋯O interaction heavily biases the conformation towards one in which the sulfur and oxygen atoms are co-planar and adjacent to each other (Φ1 and Φ2 of 0°, Fig. 2B), with a barrier to rotation about Φ1 of 7.4 kcal mol−1 when Φ2 is constant at 0°. In contrast, a model system containing an oxygen atom in place of sulfur is less biased towards a single conformation, and has an approximately equal preference for two out-of-plane geometries (Fig. 2B).28
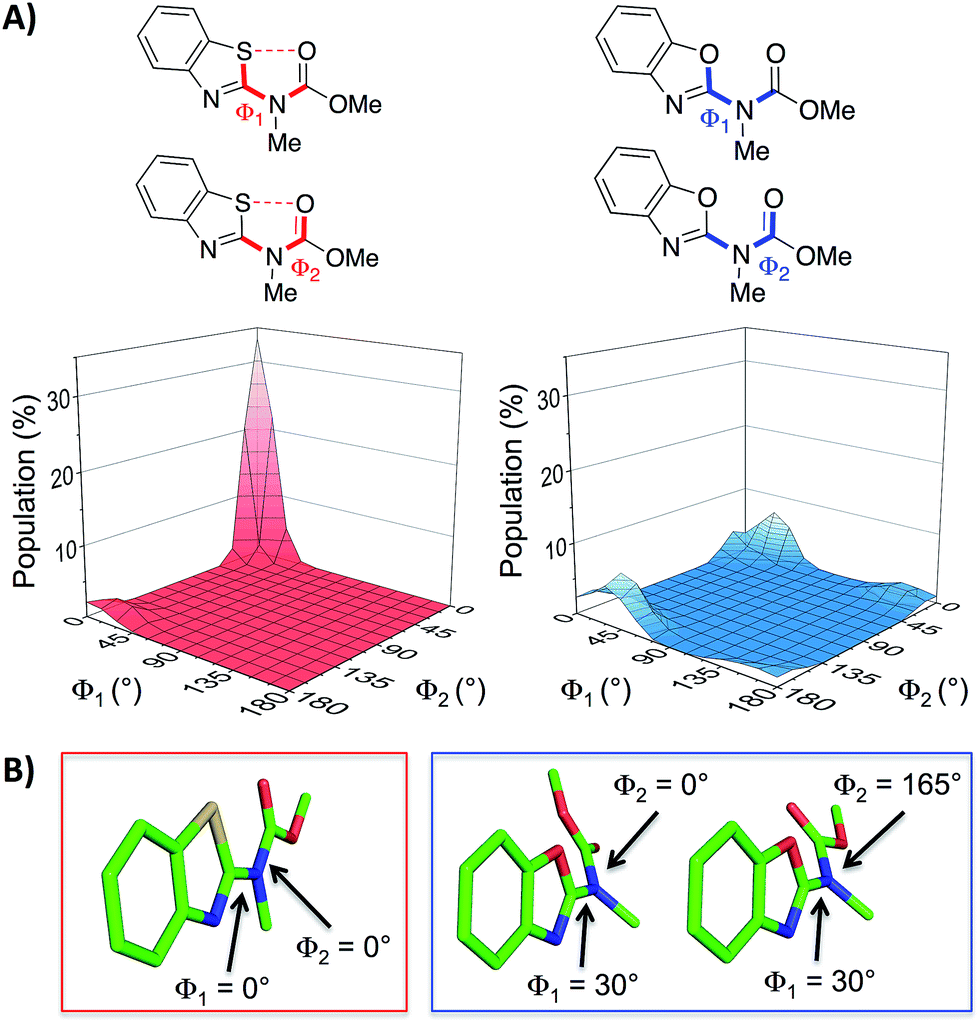 | ||
| Fig. 2 (A) Dihedral contour plots of a model system depicting the difference in conformational preference between benzothiazole and benzoxazole rings. The relevant dihedral angles are depicted in bold and the plots show the Boltzmann population of conformers at 298 K. (B) The model systems displayed in their most highly populated conformations (benzothiazole system in the red box and benzoxazole system in the blue box, see also the ESI†). The calculations were performed at the M06-2X/6-31G(d,p) level of theory with implicit aqueous solvent.29–34† | ||
Here we explore the 1,5-S⋯O interaction as a determinant of conformation in a benzothiazole–thiophene scaffold. This scaffold projects three substituents (R2, R3, R4) that mimic the i, i + 3/4 and i + 7 side-chains vectors of one face of an α-helix.
Results and discussion
The synthesis began with the preparation of 5-nitrobenzo[d]thiazol-2-amine 1 on a multi-gram scale from 2-fluoro-5-nitroaniline in two steps.35† A range of R1 and R2 groups were introduced either by N-acylation and N-alkylation (Scheme 1, Route 1), or an alternative sequence in which the amine was diazotized and substituted with chlorine, allowing introduction of substituted amines by nucleophilic substitution followed by N-acylation (Scheme 1, Route 2).† Reduction of the aromatic nitro group of compounds 5–7 with hydrogen under palladium on carbon catalysis gave anilines that were regioselectively iodinated to give 8–10. Negishi or Suzuki cross-coupling allowed introduction of various R3 groups followed in the latter case by hydrogenation of the resultant alkene. A range of thiophene ureas, and thus R4 groups, were introduced via nucleophilic attack of anilines 11–15 on isocyanates formed in situ from 2-amino thiophenes 16–18 with phosgene.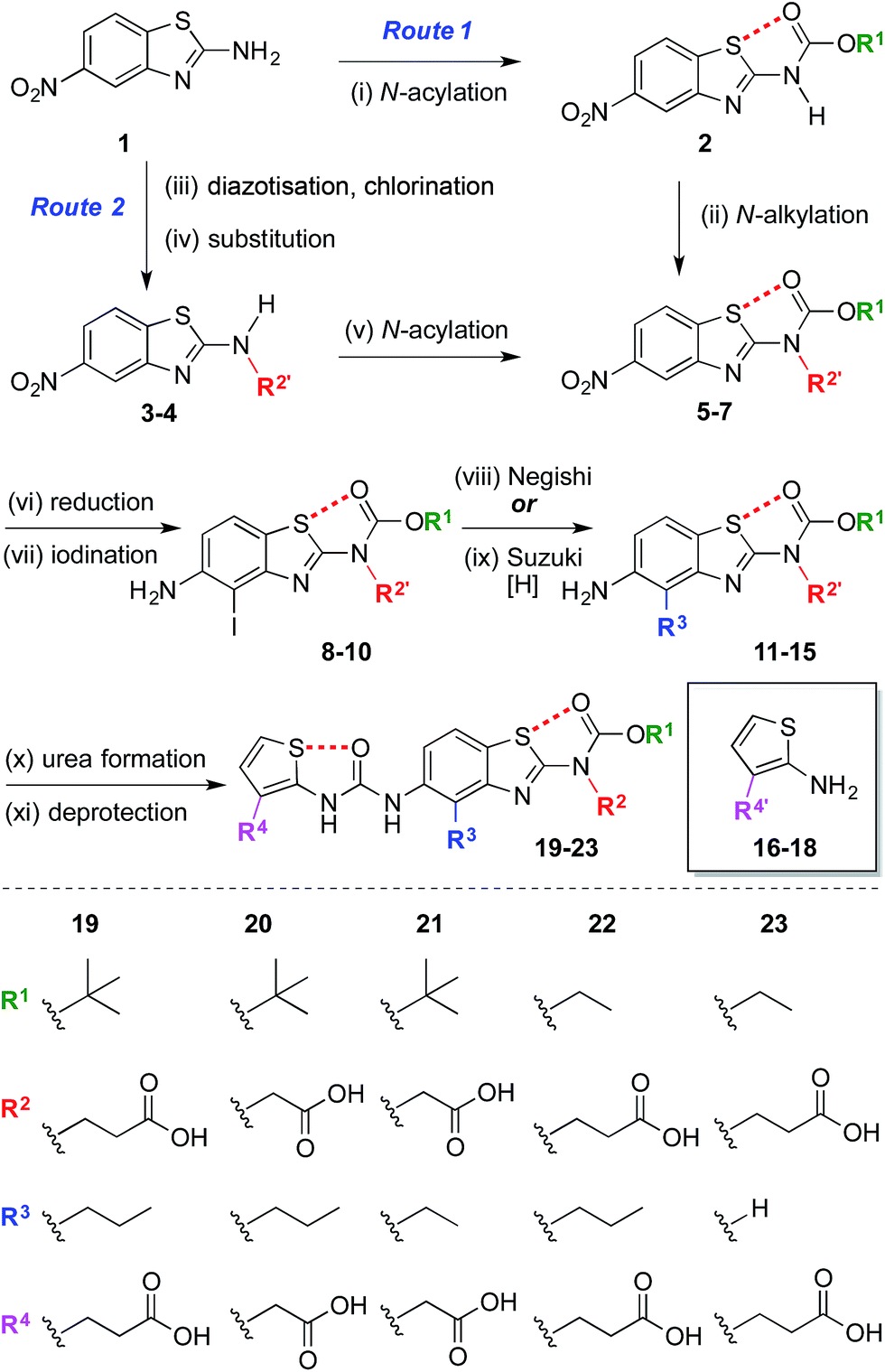 | ||
| Scheme 1 Modular synthesis of protein surface mimetics stabilised by 1,5-S⋯O interactions. Reagents and conditions: (i) Boc2O, DMAP, DMF, 60 °C, 4 h, 47%; (ii) NaH, BrCH2CO2Me, DMF, 0 °C – r.t., 1 h, 69%; (iii) i-BuONO, CuCl2·H2O, MeCN, r.t. – 60 °C, 1 h; (iv) NH2R2′, i-Pr2NEt, DMF, r.t., 16 h, 41–51% over two steps; (v) (CO2Et)2O, DMAP, MeCN, 1 h, r.t., 92%; (vi) H2, Pd/C, THF–MeOH, r.t., 1.5–4 h, 74–85%; (vii) NIS, MeCN, 0 °C – r.t., 10–30 min, 84–99%; (viii) n-PrMgCl, ZnCl2, PdCl2(dppf), THF, 65 °C, 2 h, 14–35%; (ix) H2CCHBF3K, PdCl2(dppf), Et3N, EtOH, 70 °C, 3 h, then H2, Pd/C, THF–MeOH, r.t., 30 min, 42% over two steps; (x) 16–18, COCl2, Et3N, CH2Cl2, 0 °C, 30 min, then Et3N, CH2Cl2, aniline 11–15, r.t., 1 h, 17–78%; (xi) Me/Et ester: NaOH, THF–MeOH, r.t. – 60 °C, 1 h, 33–59%; allyl ester: piperidine, Pd(PPh3)4, THF, r.t., 30 min, 44%; t-Bu ester: TFA, CH2Cl2, r.t., 30 min, 96%. n = normal, Boc = CO2t-Bu, DMAP = 4-dimethylaminopyridine, DMF = N,N-dimethylformamide, Me = methyl, Bu = butyl, Pr = propyl, THF = tetrahydrofuran, NIS = N-iodosuccinimide, dppf = bis(diphenylphosphino)ferrocene, Et = ethyl, Ph = C6H5, TFA = trifluoroacetic acid. R′ denotes a carboxylic acid side-chain protected as an ester [Me, Et, allyl, or t-Bu].† | ||
2D NOESY data acquired on mimetic 20 are consistent with population of the desired conformation in the solution phase (Fig. 3A). Substituents R2 and R3 of 20 are separated by approximately 5 Å in the desired conformation. This separation is greater in all other conformations and is a maximum of approximately 6.5 Å if an anti-arrangement of these groups is adopted. Since 5 Å is the accepted cut-off distance for an observable nOe in a non-flexible system,36 the presence of an nOe correlation between the hydrogens of these substituents indicates that the desired conformation is likely to be a significant contributor to the solution phase conformational ensemble.
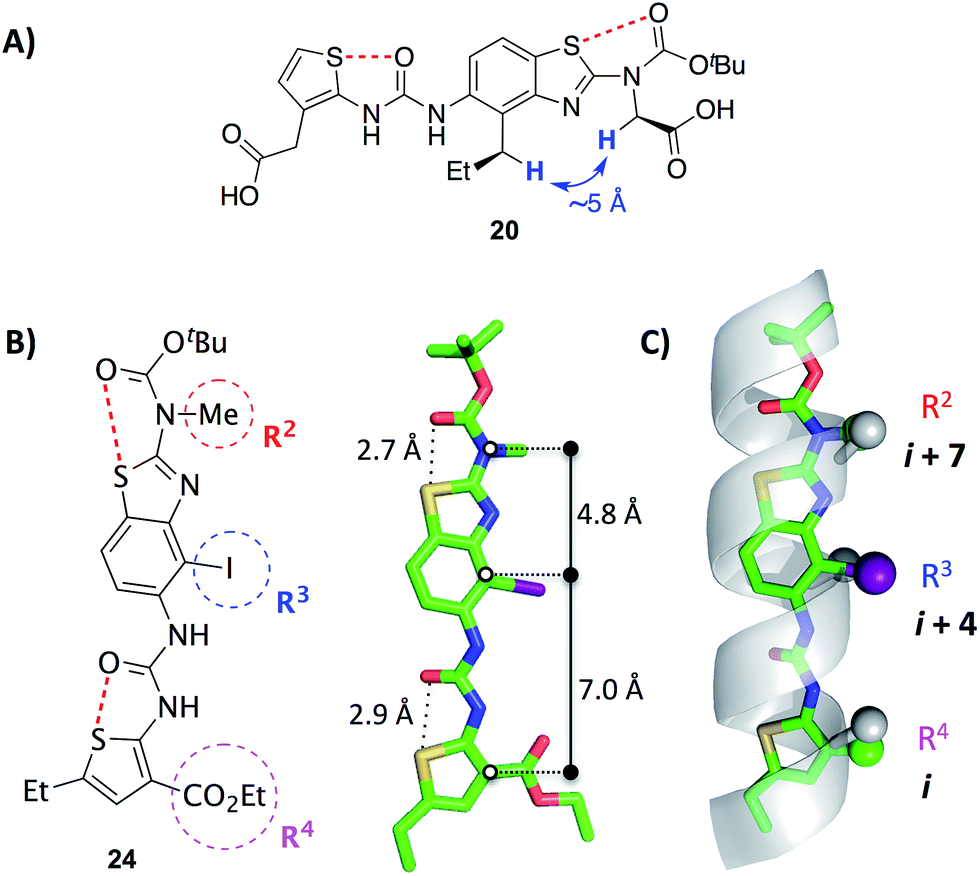 | ||
| Fig. 3 (A) A key 1H homonuclear nOe correlation observed in 20 (solution in d6-DMSO). The distance between the highlighted hydrogens is approximately 5 Å in the desired conformation.† (B) X-ray crystal structure of the benzothiazole–thiophene scaffold 24.† The solid lines indicate the distances between the atoms on the scaffold that superimpose with the α-carbons at positions i, i + 4 and i + 7 of a canonical α-helix. The red dashed lines indicate the 1,5-S⋯O interactions. (C) Superposition of the scaffold with a canonical α-helix (residues 19–31 of pdb 1HLO). The β-carbons of the helix and corresponding atoms of the scaffold are shown as spheres. The ethylcarboxy substituent of the scaffold (R4) is truncated for clarity. | ||
The X-ray crystal structure of iodide 24, an analogue of 19–23, confirms the presence of two 1,5-S⋯O interactions (Fig. 3B).† There is planarity about both the carbamate–benzothiazole and urea–thiophene units, and the S⋯O distances are 2.7 Å and 2.9 Å, respectively, smaller than the sum of the van der Waals radii of sulfur and oxygen (3.3 Å). The projection of three substituents (R2, R3 and R4) is in good agreement with side-chains i, i + 4 and i + 7 of a canonical α-helix (Fig. 3C). This positioning is analogous to the previously reported oligopyridylamide and oligobenzamide scaffolds.10,13 A six-point RMSD of 1.4 Å was calculated from the superposition of three α-carbons and three β-carbons of the α-helix with the corresponding atoms of the scaffold.37
In order to assess the capacity of the benzothiazole–thiophene scaffold to act as a protein surface mimetic, we studied the ability of appropriately functionalised variants to disrupt the fibrillation of islet amyloid polypeptide (IAPP). The self-association of intrinsically-disordered 37-residue IAPP (Fig. 4A) ultimately leads to amyloid fibrils (highly-ordered β-rich aggregates) which are the main component of the protein deposits in the pancreatic islets of type II diabetes patients.38,39 In the presence of phospholipid membranes_– such as those found within biological systems_– the N-terminus of IAPP (residues 5–22) populates α-helical states before undergoing fibrillisation (Fig. 4B).40,41 The stabilisation of these membrane-bound α-helical states attenuates the formation of IAPP fibrils41 and, as recent evidence suggests, decreases the cellular toxicity of IAPP.41,42 This indicates therapeutic applications of IAPP helix stabilisers. In our previous work, scaffolds functionalised with acidic, hydrophobic and acidic groups that overlay with positions i, i + 3/4 and i + 7 of an α-helix, respectively, were highly effective disruptors of membrane-catalysed IAPP fibrillation.10,11,42 These surface mimetics were complementary to side-chains of the α-helical N-terminus of IAPP (postulated to be Arg11, Asn14/Phe15 and His18) and stabilised its helix formation (Fig. 4C).42
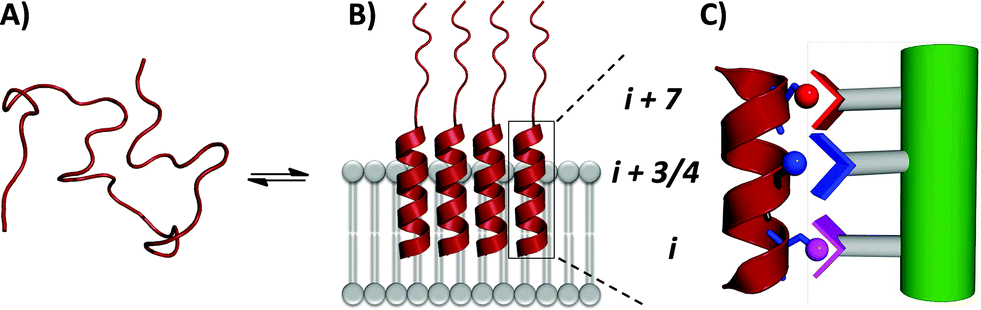 | ||
| Fig. 4 (A) IAPP is intrinsically disordered in solution. (B) IAPP populates α-helical states in the presence of a lipid membrane. (C) α-Helical states are targets for surface-mimetics that form complementary interactions with side-chains of the protein. | ||
We measured the fibrillation of IAPP using a thioflavin T (ThT) assay43 in the presence of a model membrane system.44 The fluorescence intensity of ThT correlates with the extent of protein fibril formation. The assay confirmed that ability of the benzothiazole–thiophenes to disrupt the fibrillation of IAPP. This can be seen by the lower end-point fluorescence intensity of the samples containing 19, 22 and 23 relative to the IAPP control (Fig. 5A). Additionally, the IAPP fibrillation process is delayed in the presence of these three compounds in comparison to control, seen by the extended times to half-maximal fibrillation (t1/2) of these samples (Fig. 5B). The results of the ThT assay were confirmed by electron microscopy, which showed that treatment of IAPP with 19 and 22 gives rise to amorphous aggregates whereas 21 gives rise to a combination of amorphous aggregates and ordered fibrils (ESI Fig. 3†). Furthermore, circular dichroism (CD) confirmed that compound 19 disrupts the transition of IAPP from its initial α-helical conformation (a spectrum characterised by two minima at 208 and 222 nm) to a β-sheet conformation (a spectrum characterised by a single minimum at approximately 215 nm) in the presence of a model membrane system (Fig. 6).
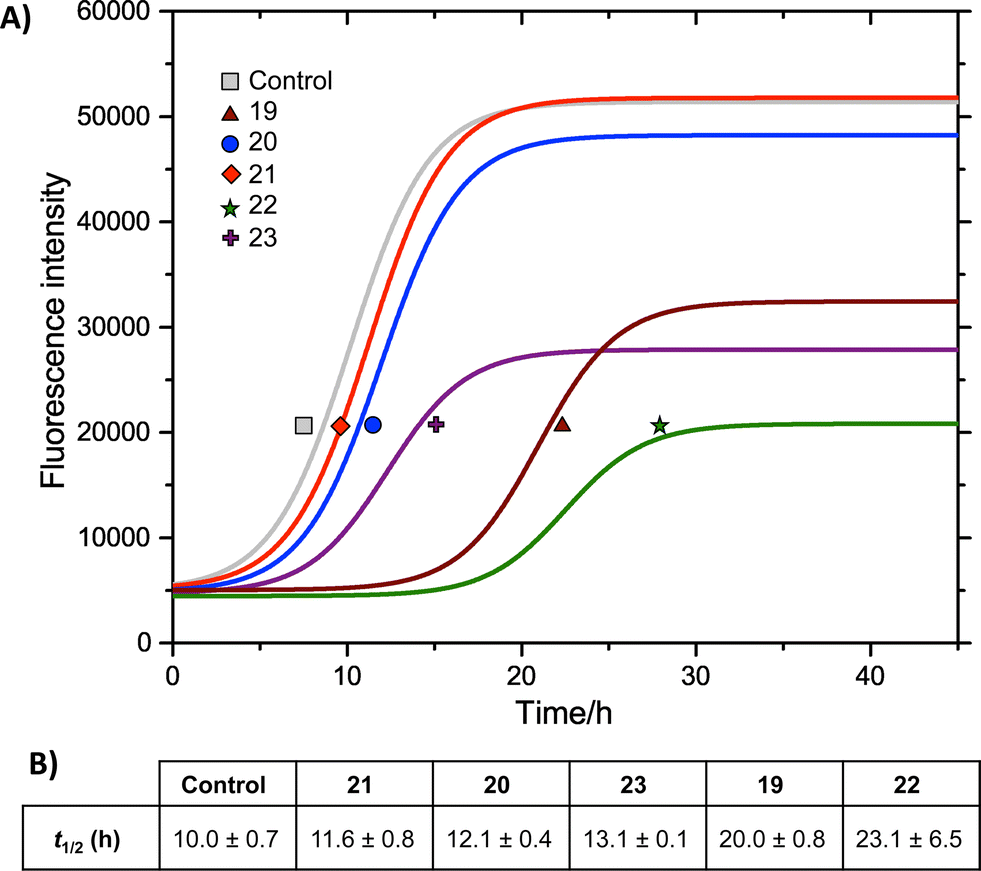 | ||
Fig. 5 (A) ThT fibrillation kinetics of 10 μM IAPP in the absence (grey curve) or presence of 50 μM test compounds. Each curve represents the average of six replicates. (B) Times to half-maximal fibrillation (t1/2). Conditions: 500 μg mL−1 DOPG/DOPC liposomes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 20 mM potassium phosphate buffer, 100 mM sodium chloride, pH 7.2, 25 °C.† :1), 20 mM potassium phosphate buffer, 100 mM sodium chloride, pH 7.2, 25 °C.† | ||
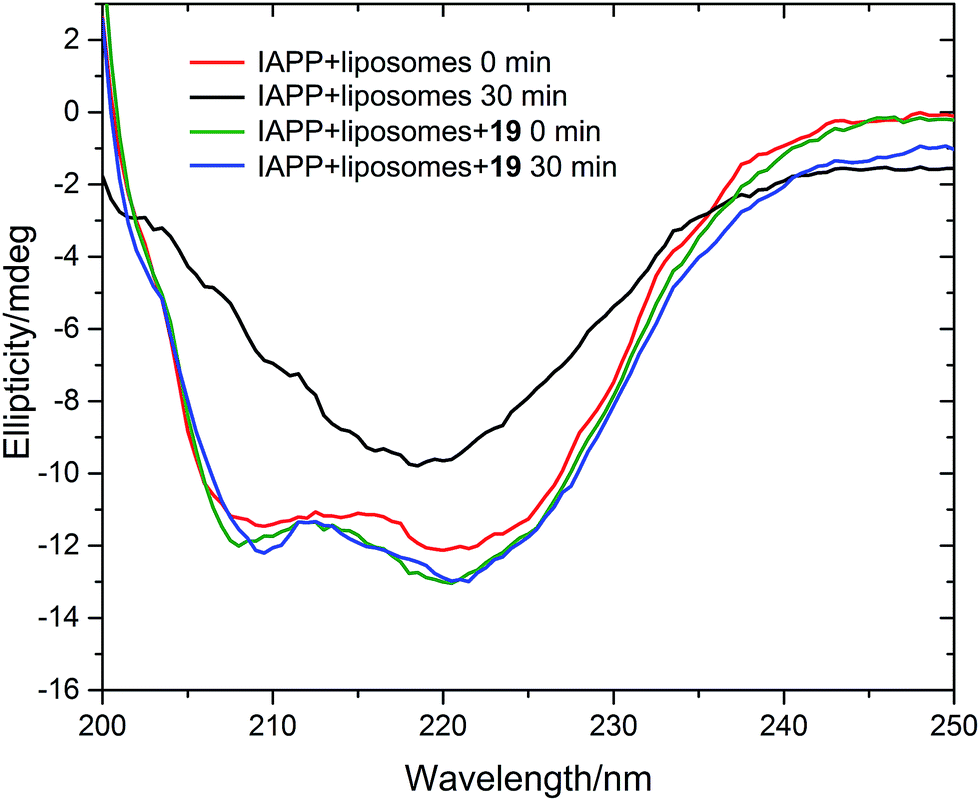 | ||
| Fig. 6 Circular dichroism spectra of IAPP in the absence (red and black curves) and presence (green and blue curves) of 19. Conditions: 20 μM IAPP, 20 μM 19, 1 m g mL−1 DOPG/DOPC liposomes (1:1), 20 mM potassium phosphate buffer, 100 mM sodium chloride, pH 7.2, 25 °C. | ||
The most striking feature of the structure–activity relationship is that the active compounds bear three-carbon carboxylic acid substituents at positions R2 and R4, however the inactive compounds, 20 and 21, both bear two-carbon carboxylic acids substituents at these positions. This indicates that the length of the acidic substituents at positions R2 and R4 is critical to the interaction between the protein and the small molecule, consistent with the formation of specific contacts. The central hydrophobic substituent (R3) was also sensitive to modification. Compound 23, which differs from 22 only by the absence of this central substituent, was a less effective inhibitor of fibrillation. This is consistent with a previous observation that the central hydrophobic substituent is sensitive to modification within an oligopyridylamide scaffold displaying an analogous set of functional groups.43
Conclusions
We have synthesised a new non-peptidic protein surface mimetic based on a benzothiazole–thiophene scaffold that is conformationally pre-organised by two 1,5-S⋯O interactions. The scaffold directs three side-chains to positions i, i + 3/4 and i + 7 of an α-helix. The S⋯O interaction was evaluated as a determinant of conformation by calculation, X-ray crystallography and solution-phase NMR spectroscopy. A focussed library of conformationally constrained scaffolds displaying a range of functional groups act as protein surface mimetics that disrupt membrane-catalysed islet amyloid polypeptide (IAPP) fibrillation, a pathway strongly associated with the pathology of type-II diabetes.Acknowledgements
We thank The University of Oxford for funding, The Netherlands Organisation for Scientific Research (NWO) for a Rubicon Fellowship (J. L.), Dr Peter C. Knipe for assistance with X-ray crystallography, and Takeda Pharmaceutical Company Limited for their generosity in providing financial and logistical support during a sabbatical position for T. Y. as a Visiting Scientist in The University of Oxford (2014). We thank Diamond Light Source for an award of beam time (MT9981).Notes and references
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- L. K. Gavrin, R. A. Denny and E. Saiah, J. Med. Chem., 2012, 55, 10823–10843 CrossRef CAS PubMed.
- L. M. Young, J. C. Saunders, R. A. Mahood, C. H. Revill, R. J. Foster, L.-H. Tu, D. P. Raleigh, S. E. Radford and A. E. Ashcroft, Nat. Chem., 2015, 7, 73–81 CrossRef CAS PubMed.
- G. Tóth, S. J. Gardai, W. Zago, C. W. Bertoncini, N. Cremades, S. L. Roy, M. A. Tambe, J.-C. Rochet, C. Galvagnion, G. Skibinski, S. Finkbeiner, M. Bova, K. Regnstrom, S.-S. Chiou, J. Johnston, K. Callaway, J. P. Anderson, M. F. Jobling, A. K. Buell, T. A. Yednock, T. P. J. Knowles, M. Vendruscolo, J. Christodoulou, C. M. Dobson, D. Schenk and L. McConlogue, PLoS One, 2014, 9, e87133 Search PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- M. K. P. Jayatunga, S. Thompson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2014, 24, 717–724 CrossRef CAS PubMed.
- A. M. Watkins and P. S. Arora, Eur. J. Med. Chem., 2015, 94, 480–488 CrossRef CAS PubMed.
- D. Xin, A. Raghuraman and K. Burgess, J. Org. Chem., 2015, 80, 4450–4458 CrossRef CAS PubMed.
- M. Oh, J. H. Lee, W. Wang, H. S. Lee, W. S. Lee, C. Burlak, W. Im, Q. Q. Hoang and H.-S. Lim, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11007–11012 CrossRef CAS PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736–739 CrossRef CAS PubMed.
- O. V. Kulikov, S. Kumar, M. Magzoub, P. C. Knipe, I. Saraogi, S. Thompson, A. D. Miranker and A. D. Hamilton, Tetrahedron Lett., 2015, 56, 3670–3673 CrossRef CAS.
- A. Smith, M. Guzman, P. Sprengeler, T. Keenan, R. Holcomb, J. Wood, P. Carroll and R. Hirschmann, J. Am. Chem. Soc., 1994, 116, 9947–9962 CrossRef CAS.
- J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535–539 CrossRef CAS PubMed.
- M. J. Adler, R. T. W. Scott and A. D. Hamilton, Chem.–Eur. J., 2012, 18, 12974–12977 CrossRef CAS PubMed.
- S. Thompson and A. D. Hamilton, Org. Biomol. Chem., 2012, 10, 5780–5782 CAS.
- S. Thompson, R. Vallinayagam, M. J. Adler, R. T. W. Scott and A. D. Hamilton, Tetrahedron, 2012, 68, 4501–4505 CrossRef CAS.
- J.-M. Ahn and S.-Y. Han, Tetrahedron Lett., 2007, 48, 3543–3547 CrossRef CAS.
- F. Lu, S.-W. Chi, D.-H. Kim, K.-H. Han, I. D. Kuntz and R. K. Guy, J. Comb. Chem., 2006, 8, 315–325 CrossRef CAS PubMed.
- H. Yin, G. Lee, K. A. Sedey, O. Kutzki, H. S. Park, B. P. Orner, J. T. Ernst, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 10191–10196 CrossRef CAS PubMed.
- A. Raghuraman, E. Ko, L. M. Perez, T. R. Ioerger and K. Burgess, J. Am. Chem. Soc., 2011, 133, 12350–12353 CrossRef CAS PubMed.
- E. A. German, J. E. Ross, P. C. Knipe, M. F. Don, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2015, 54, 2649–2652 CrossRef CAS PubMed.
- T. Yamashita, P. C. Knipe, N. Busschaert, S. Thompson and A. D. Hamilton, Chem.–Eur. J., 2015, 21, 14699–14702 CrossRef CAS PubMed.
- B. B. Lao, I. Grishagin, H. Mesallati, T. F. Brewer, B. Z. Olenyuk and P. S. Arora, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7531–7536 Search PubMed.
- L. Nevola, J. M. Rodrigeuz, S. Thompson and A. D. Hamilton, Supramol. Chem., 2013, 25, 586–590 CrossRef CAS PubMed.
- F. Bernardi, I. G. Csizmadia and A. Mangini, Organic sulfur chemistry, Elsevier Scientific Publishing Co., Amsterdam, 1985 Search PubMed.
- Y. Nagao, T. Hirata, S. Goto, S. Sano, A. Kakehi, K. Iizuka and M. Shiro, J. Am. Chem. Soc., 1998, 120, 3104–3110 CrossRef CAS.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed.
- The conformational control of the 1,5-S⋯O interaction was also observed in an alternative model system based on a thiophenylurea (ESI Fig. 2†).
- Maestro, version 10.2, Schrödinger, LLC, New York, NY, 2015 Search PubMed.
- Jaguar, version 8.8, Schrödinger, LLC, New York, NY, 2015 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- R. J. Woods, M. Khalil, W. Pell, S. H. Moffat and V. H. Smith, J. Comput. Chem., 1990, 11, 297–310 CrossRef CAS.
- T. Herz, R. Krauss, M. Kubbutat, M. Lang, C. Schaechtele, S. Tasler and F. Totzke, US Pat., 7514460B2, 2009.
- D. Neuhaus and M. P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, Wiley, 2nd edn, 2000 Search PubMed.
- For a discussion of the interpretation of peptidomimetic RMSD values see: D. Xin, L. M. Perez, T. R. Ioerger and K. Burgess, Angew. Chem., Int. Ed., 2014, 53, 3594–3598 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- P. Cao, A. Abedini and D. P. Raleigh, Curr. Opin. Struct. Biol., 2013, 23, 82–89 CrossRef CAS PubMed.
- J. A. Williamson, J. P. Loria and A. D. Miranker, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- C. A. De Carufel, N. Quittot, P. T. Nguyen and S. Bourgault, Angew. Chem., Int. Ed., 2015, 54, 14383–14387 CrossRef CAS PubMed.
- S. Kumar, D. E. Schlamadinger, M. A. Brown, J. M. Dunn, B. Mercado, J. A. Hebda, I. Saraogi, E. Rhoades, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2015, 22, 369–378 CrossRef CAS PubMed.
- L. S. Wolfe, M. F. Calabrese, A. Nath, D. V. Blaho, A. D. Miranker and Y. Xiong, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16863–16868 CrossRef CAS PubMed.
- An equimolar mixture of dioleoylphosphatidylglycerol (DOPG) and dioleoylphosphatidylcholine (DOPC) were extruded in to 100 nm vesicles.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; 1H, 13C and NOESY spectra; HRMS, and IR values. CCDC 1453550 (24), ThT assay data, computation, electron microscopy. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00756b |
| This journal is © The Royal Society of Chemistry 2016 |