
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired supramolecular materials by orthogonal self-assembly of hydrogelators and phospholipids†
J.
Boekhoven‡
a,
A. M.
Brizard
a,
M. C. A.
Stuart
b,
L.
Florusse
a,
G.
Raffy
c,
A.
Del Guerzo
c and
J. H.
van Esch
 *a
*a
aDepartment of Chemical Engineering, Delft University of Technology, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: j.h.vanesch@tudelft.nl
bStratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747AG Groningen, The Netherlands
cUniv. Bordeaux, ISM, UMR 5255, 351 cours de la Libération, 33400 Talence, France
First published on 6th May 2016
Abstract
The orthogonal self-assembly of multiple components is a powerful strategy towards the formation of complex biomimetic architectures, but so far the rules for designing such systems are unclear. Here we show how to identify orthogonal self-assembly at the supramolecular level and describe guidelines to achieve self-sorting in self-assembled mixed systems. By investigating multicomponent self-assembled systems consisting of low molecular weight gelators and phospholipids, both at a molecular and a supramolecular level, we found that orthogonal self-assembly can only take place if the entities assemble via a strong and distinct set of interactions. The resulting supramolecular architectures consist of fibrillar networks that coexist with liposomes and thereby provide additional levels of compartmentalization and enhanced stability as compared to self-assembled systems of gelators or phospholipids alone.
Introduction
In the past decades, supramolecular chemistry has provided fascinating examples of objects based on the self-assembly of discrete elements,1–6 with potential applications in regenerative medicine,7,8 electronics, hydrogen production, catalysis and others.9,10 Although these advances are truly remarkable, the majority of man-made supramolecular materials suffer from a lack of complexity and a difficulty to be translated into higher ordered structures. To bring supramolecular materials to a new level of sophistication, much effort has gone into the synthesis of elaborate building blocks with high degrees of functionality.11,12 While this is a valid approach leading to elegant and functional designs, the preparation of new building blocks can be time-consuming and the corresponding assembly behavior remains poorly predictable.13An alternative approach towards the formation of more complex supramolecular architectures is based on multicomponent orthogonal self-assembly, i.e. the independent formation of distinct supramolecular structures within one system.14–17 Orthogonal self-assembly is inspired by self-assembly in biological cells, in which an astonishing variety of structures emerge from non-covalent and highly specific interactions between different self-assembling components like proteins, amphiphiles, biopolymers and others, leading to the formation of distinct coexisting architectures, such as membranes, cytoskeletal fibers and ribosomes.18,19 In supramolecular chemistry, highly specific complementary interactions have been exploited to achieve self-sorting into discrete sized objects such as cages and dimers,20,21 as well as to achieve orthogonal self-assembly by self-sorting of different molecular building blocks into distinct coexisting nanoarchitectures with high aggregate numbers.22 For instance, the orthogonal self-assembly of molecular gelators and liquid crystalline compounds has led to gelled liquid crystalline phases with enhanced anisotropic optical properties and ionic conductivity.14,23 Self-assembling interpenetrating networks have been prepared by orthogonal self-assembly of molecular gelators and surfactants and from different molecular gelators.15,16,24 Here, the coexistence of two different supramolecular networks leads to e.g. gels with new and adjustable rheological properties,25–28 and responsive super-structured networks.29 Interestingly, the orthogonal self-assembly of surfactants and supramolecular hydrogelators has also been used to fabricate the first examples of so-called “gellosomes”, liposomes encapsulating gel fibers, exclusively based on non-covalent interactions,15,30,31 and to construct a drug delivery gel with a post-production tunable release rate.32
These examples show that the orthogonal assembly approach can create supramolecular materials with a higher degree of functionality than their individual constituents, and can display new tunable properties and structures that arise from interactions between different self-assembled structures. Although the design rules for orthogonal self-assembling systems solely based on molecular gelators are beginning to evolve,24 it remains a challenge to identify and design new orthogonal self-assembling systems. The first reason for the limited availability of building blocks for orthogonal self-assembly should be sought in the set of requirements for such building blocks, as the candidates should not only assemble in the same environment (matching solvents, pH, and temperature), but also display a high degree of specificity in their non-covalent interactions to prevent co-assembly.15 Second, the lack of straightforward methods to clearly identify and understand self-sorting mechanisms also hampers its study and hence also the rational design of appropriate building blocks.24 Such knowledge is nonetheless essential to make bottom-up strategies towards the fabrication of new architectures and smart materials more powerful.
In this study, we propose a methodology to identify orthogonal self-assembling systems, and have applied this methodology to prototype orthogonal self-assembling systems consisting of molecular hydrogelators and phospholipids, for validation and to improve our understanding of self-sorting mechanisms. Similar to cell walls in conjunction with their extracellular matrix, we selected combinations of phospholipids with various low molecular weight gelators,33 molecules that assemble reversibly in gel-forming fiber matrices.34,35 In a previous study, intact liposomes were found to coexist with fibers of 1,3,5-triamide cis,cis-cyclohexane derivatives in water, suggesting interactions specific enough for orthogonal self-assembly into distinct supramolecular architectures.15 Here, we have selected a broader range of hydrogelators and investigated their possible interactions with phospholipid bilayers, both at a molecular and supramolecular level, to address the following questions: how can one evidence orthogonal self-assembly unambiguously? Why are some gelator–phospholipid configurations more prone to orthogonal self-assembly than others? Answering these questions will contribute to our understanding of orthogonal self-assembly and provide new insights towards the elaboration of more complex and functional self-assembled architectures, with a broader scope than hydrogels and amphiphiles.
Results and discussion
How to evaluate orthogonal self-assembly?
Due to the many possible interactions in multicomponent environments, recognizing true orthogonal self-assembling systems is a challenging task. Mixed systems comprise different assembly states for each component, leading to multiple equilibriums. For instance, phospholipids assemble into liposomes (Agg1) that are in equilibrium with their monomeric constituents (M1, Fig. 1).36 Similarly, in the case of hydrogelators, gel fibers (Agg2) emerge from the association of monomeric species (M2), due to strong intermolecular interactions. Therefore, in a self-sorting two-component situation, four possible states for the two self-assembling entities can occur: (1) both entities are in a monomeric state (M1M2); (2) the phospholipids are in a monomeric state, while the gelators mostly exist as fibers (M1Agg2); or conversely, (3) the liposomes are formed but the gelators are still in a monomeric state (Agg1M2); and finally, (4) the two entities are included into distinct self-assembled structures (Agg1Agg2), which corresponds to orthogonal self-assembly.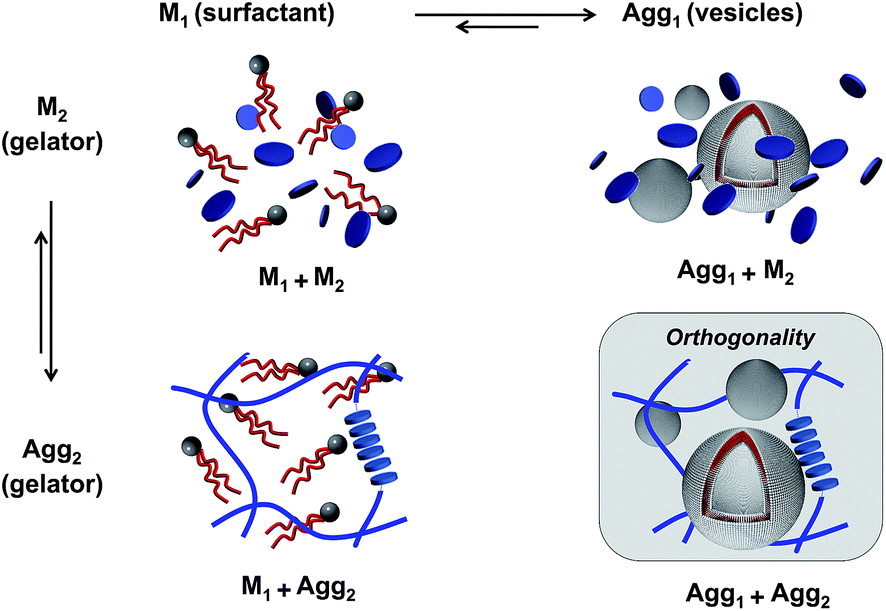 | ||
| Fig. 1 The different aggregation states in a two-component system of phospholipids and hydrogelators. Both gelators and phospholipids can exist in their monomeric state (Mi) or aggregated state (Aggi). This scheme represents only orthogonal self-assembly in which components do not interact with each other. | ||
In all cases above, the scheme represents ideal aggregation behavior where the components behave independently, and where the two self-assembly equilibriums are not affected by one another. Because gel fibers and vesicles can be considered as a separate phase, the assembly equilibriums of gelators and surfactants can, in a first approximation, be described by the pseudo-phase model.37 According to the pseudo-phase model, the Gibbs free energies for gel fiber or vesicle formation ΔGaggi are given by ΔGAggi = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(CACi), with CACi the critical aggregation concentration of component i. The CACi of each component i is equal to the concentration of the monomeric species CMi in equilibrium with its aggregated state Aggi. However, if monomers of different components have any affinity for each other, co-assembly into mixed aggregates can occur, which will alter the equilibrium concentrations associated with each component.38 To recognize orthogonal self-assembly processes, it is therefore adequate to demonstrate that the equilibrium concentrations of each component, and thus the individual equilibriums, remain unaffected in a multi-component environment.
ln(CACi), with CACi the critical aggregation concentration of component i. The CACi of each component i is equal to the concentration of the monomeric species CMi in equilibrium with its aggregated state Aggi. However, if monomers of different components have any affinity for each other, co-assembly into mixed aggregates can occur, which will alter the equilibrium concentrations associated with each component.38 To recognize orthogonal self-assembly processes, it is therefore adequate to demonstrate that the equilibrium concentrations of each component, and thus the individual equilibriums, remain unaffected in a multi-component environment.
Unfortunately, for the gelator component the determination of the CACs is not as straightforward as it seems, especially in mixed systems. For molecular hydrogelators, the CAC is equal to the solubility of the gelator (Csol) in equilibrium with the solid gel fibers. However, these solubility concentrations are cumbersome to measure accurately because of the presence of the gel state and other possibly mixed aggregates, and hence they are unsuited as a fast diagnostic parameter for orthogonal self-assembly. Therefore we turned our focus to the much easier to determine critical gelation concentration (CGC), which is widely used to characterize the gelation ability of supramolecular gelators. The CGC has been defined as the minimum concentration of a compound required to turn a liquid into a gel, and is the sum of the solubility of the gelator (Csol = CAC), and the minimum solid fraction of gelator required to form a self-supporting fiber network, (Cfib,min).42,43 Hence, CGC = Csol + Cfib,min = CAC + Cfib,min. Because Cfib,min cannot be neglected, the CGC can only be taken as an approximation of the CAC. The CGC can be evaluated by simple table-top rheology experiments like the inverted tube test or the dropping ball method, and usually reasonably accurate values of the CGC are obtained.34 Still, care needs to be taken because fiber and gel formation involve a nucleation step, which renders the measurement sensitive to hysteresis and assembly rates.44,45 Even if these experiments only allow estimations of the CGC, any significant alteration of this value in the presence of phospholipids can be considered as a relevant indicator for co-aggregation.
The determination of the CACs also turned out to be problematic for the surfactant component. Although the critical aggregation concentration (CAC) of surfactants and phospholipids are usually easy to determine using techniques like surface tension measurements, dynamic light scattering or NMR-spectroscopy, these latter methods cannot be applied to samples in the gel state, as the viscoelastic behavior interferes with the measurements. Moreover, liposome-forming phospholipids are usually characterized by CAC values in the nano-molar range,46 rendering their determination delicate. For these reasons, we measured the phase transition temperature (Tm) of the phospholipid physical state from the solid to the liquid phase. This specific property of phospholipid bilayers is sensitive to the incorporation of any molecules such as hydrophobic gelators, and it is therefore sensitive to co-aggregation.36,47 Finally, as a complement to CGC and phospholipid phase transition determinations, which reflect interactions at a molecular level, cryo-TEM was also used to examine the persistence of the original self-assembled structures.
Selection of hydrogelators and amphiphiles
Various gelators in combination with phosphatidylcholine phospholipids were selected to screen for orthogonal self-assembly (Fig. 2). The first class consisted of 1,3,5-triamide cis,cis-cyclohexane derivatives,39,48 disk-like molecules that stack on top of each other via three hydrogen bonds per unit providing strong and unidirectional intermolecular interactions. The presence of hydrophobic amino acids (phenylalanine and methionine) coupled to this scaffold introduces additional aggregation forces. In this study, both symmetric (1, 2) and dissymmetric (3) derivatives were used. To understand why some gel–lipid configurations are more prone to self-sorting than others, the study was extended to hydrogelators differing by their melting points or molecular packing mechanism. The second class of supramolecular hydrogelators used in this study was based on amino acids known to produce supramolecular gels in water. For instance, dibenzoyl cysteine (4 or DBC) can form fibers based on the establishment of hydrogen bonds between the two cysteine moieties and π–π stacking originating from the benzoyl groups, creating a preferential one-dimensional organization.40,49 Bis(leucine) oxalyl-amide (5 or OxLeu2), another pseudo-dipeptide that assembles in fibrillar networks in water mostly due to an array of hydrogen bonds,41 was also chosen. Finally, two low molecular weight gelators based on amphiphilic molecules were selected for this study. The first was monourea gelator (6 or ureaG), containing a six-carbon alkyl tail substituted by ethyl serine.50 While assembly of this molecule is driven by hydrogen bonding together with hydrophobic effects, water solubility is promoted by the serine group. The second amphiphilic gelator was a tartrate gemini surfactant (7 or gemini tartrate), that assembled into fibrillar networks driven mostly by the hydrophobic effect.51 Overall, these gelators were selected because their association is driven by a combination of hydrogen bonding and hydrophobic interactions, i.e. a set of interactions similar to those that stabilize protein structures. Only for compound 7, the association is primarily driven by hydrophobic interactions, comparable to the self-assembly of phospholipids in bilayers.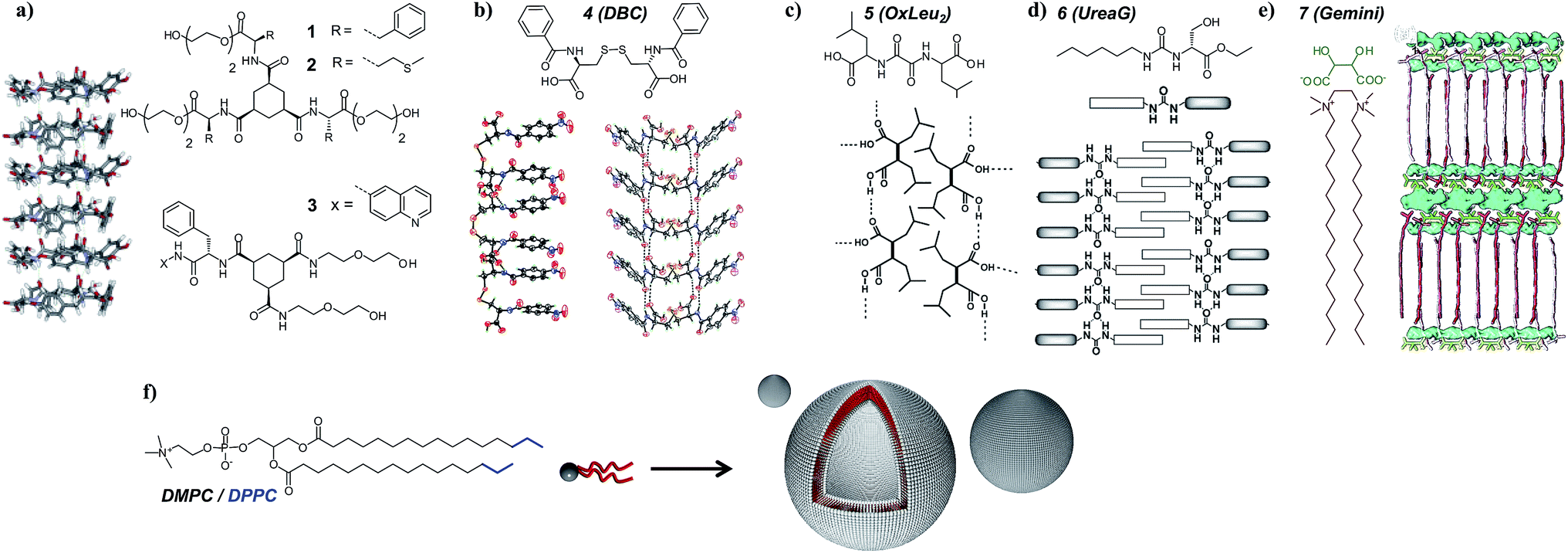 | ||
| Fig. 2 Various low molecular weight hydrogelators and their expected molecular arrangement in gel fibers: (a) 1,3,5-triamide cis,cis-cyclohexane derivatives 1, 2 and 3, (b) dibenzoyl cysteine (DBC), (c) bis(leucine) oxalyl-amide, (d) monourea ethyl serine gelator, and (e) gemini tartrate surfactant. The typical structure of the phosphatidylcholine phospholipids used in this study is also depicted (f). Crystal structures and schemes are reproduced with permission from ref. 39–41. | ||
Critical gelation concentrations and TGS
First we determined the gel–sol phase transition temperatures for each hydrogelator as a function of their concentration as an indication of the strength of the intermolecular interactions and molecular packing (Fig. 3). Of the chosen gelators, the 1,3,5-triamide cis,cis-cyclohexane derivatives 1–3 are likely to have the greatest intermolecular interactions that drive self-assembly, given the low gelation concentrations and gel-sol transition temperatures typically above 100 °C. Among those, gelators containing phenylalanine moieties appear to be driven by the highest non-covalent forces (1 and 3). The substitution of phenylalanine by methionine in 2 decreased the stability of the corresponding gel, but the gels remain stable up to 80–90 °C. For gelator 4, which contains only two hydrogen bonds per unit and benzoyl groups for π–π stacking, similar transition temperatures were found. When both the possibilities of π–π stacking and hydrogen bonding became limited with 5, 6 and the gemini tartrate 7, the gel–sol transition temperatures drastically dropped.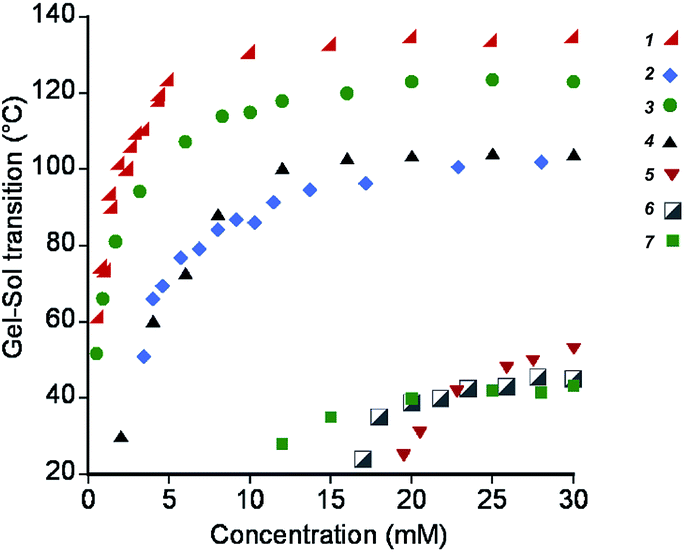 | ||
| Fig. 3 Gel–sol transition temperatures of hydrogels formed by compounds 1–7, as a function of their concentration. | ||
As a first indication of possible interactions between the various gelators and liposomes, the gelation ability of compounds 1–7 in the presence of 5 mM DMPC was investigated. If gelation was observed, the CGC in the presence of liposomes was determined using the inverted tube test. In line with previous observations,15 all the 1,3,5-triamide cis,cis-cyclohexane derivatives gelled water in the presence of liposomes, even if the phospholipid concentration was increased up to 100 mM in the case of 1 and 3, demonstrating the robustness of the gel network towards the phospholipids. This robustness of the system is supported by the almost unchanged CGC in the presence of liposomes (Table 1). This behavior was found to be general for other phenylalanine based cis,cis-cyclohexane derivatives.15 Compound 2, that lacks the presence of phenylalanine, was slightly more sensitive to the presence of phospholipids, with a minor increase of its CGC from ∼4 mM to 6.5 mM in the presence of 5 mM DMPC. Similarly, 10 mM gelator 4 (DBC) could still gel a 5 mM liposome solution. However, this gelator did show a CGC of ∼7 mM, which was twice the original CGC in water. Gelators 5, 6 and 7, which gelate pure water at 30 mM, 50 mM and 10 mM respectively, could not gel aqueous solutions at the same concentration containing 5 mM liposomes. Instead, turbid viscous solutions were obtained. Gelation of the latter three gelators only took place when the concentration of the gelator was at least 3 to 4 times as high as the original CGC in water.
| Gelator | CGCa/mM | [Gelator]b/mM | In waterc | +DMPCd (5 mM) | CGC*e/mM |
|---|---|---|---|---|---|
| a CGC value of pure gelator in water. b Gelator concentration used to test gelation in 5 mM DMPC liposome solutions. c Appearance of pure gelator solution at the given concentration in water. d Appearance of gelator solution at the given concentration in a 5 mM DMPC liposome solution in water. e Critical gelation concentration of the gelator in a 5 mM DMPC liposome solution. | |||||
| 1 | ∼0.6 | 2 | Hydrogel | Hydrogel | ∼0.8 |
| 2 | ∼4 | 10 | Hydrogel | Hydrogel | ∼6.5 |
| 3 | ∼0.6 | 2 | Hydrogel | Hydrogel | ∼0.9 |
| 4 | ∼3 | 10 | Hydrogel | Weak hydrogel | ∼7 |
| 5 | ∼16 | 30 | Hydrogel | Weak gel/viscous solution | ∼45 |
| 6 | ∼22 | 50 | Hydrogel | Viscous solution | ∼75 |
| 7 | ∼5 | 10 | Hydrogel | Viscous solution | ∼30 |
Ultrasensitive DSC
To investigate the interactions between vesicles and gel networks in more detail, specific properties of liposomes were also examined. Phospholipid bilayers are characterized by a phase transition corresponding to a chain packing transition from solid to liquid-like (Tm). Phosphatidylcholine bilayer phase transitions are usually accompanied by a pre-transition phase transition (typically 5–10 °C below the main transition) with smaller enthalpy. Studies have shown that both transitions are strongly affected by any alterations in bilayer packing caused by the presence of entities interfering with the phospholipid chains.52 This property can be measured using microcalorimetry making it a powerful technique to investigate orthogonal self-assembly.The DMPC and DPPC phase transitions were examined in the absence and presence of gelators 1 to 7 (Fig. 4). In all cases, these mixed systems were obtained by addition of liposomes to a preformed hydrogel, and subsequent annealing above the melting point of the gel to ensure homogeneity. In other words, the vesicle bilayers had the opportunity to interact with monomeric gelators before being cooled down. The same preparative conditions were applied to control solutions of DMPC and DPPC alone. In the absence of gelators, Tm of 23 °C and 41.5 °C were found for DMPC and DPPC respectively, together with pre-transition temperatures of 16 °C and 34 °C, in agreement with literature values.47,53 In the presence of gelators 1–3, the Tm and the pre-transition temperature were unchanged compared to pure water. However, in the presence of 4, a significant broadening and a slight shift of the Tm appeared for both DPPC and DMPC. The presence of 5 also decreased the Tm of the phosphatidylcholine phospholipids, to around 37 °C and 17 °C for DPPC and DMPC respectively. Similar behavior was observed for urea gelator 6. Finally, the mixing of gemini tartrate 7 with DMPC and DPPC liposomes led to relatively broad peaks. For gelators 4–7, no pre-transitions were observed for DMPC or DPPC liposomes.
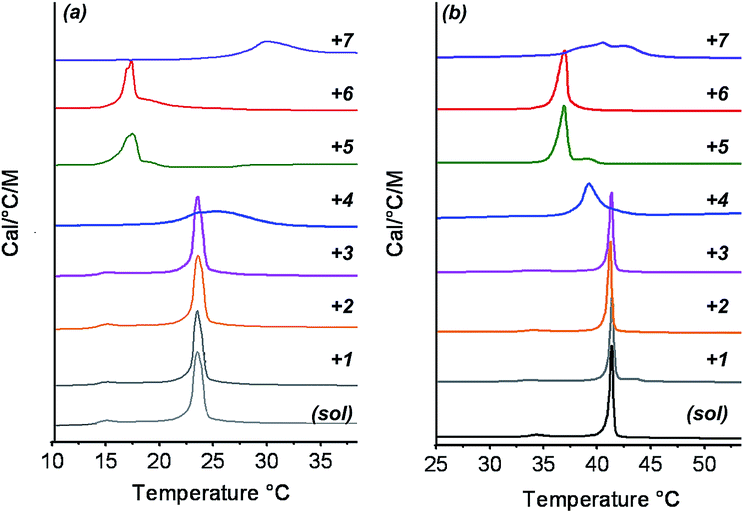 | ||
| Fig. 4 DSC traces of DMPC (a) and DPPC (b): liposomes in pure water or in the presence of 1,3,5-triamide cis,cis-cyclohexane derivatives (1, 2 and 3), dibenzoyl cysteine 4, bis(leucine) oxalyl-amide 5, monourea ethyl serine gelator 6 and gemini tartrate surfactant 7. | ||
Cryo-TEM
To investigate if the previous results translated to the supramolecular level, the mixed systems were also examined using cryogenic-transmission electron microscopy (cryo-TEM, Fig. 5). In order to ensure complete gelator dissolution in the mixed systems of liposomes and gel fibers, all systems were heated to 100 °C and cooled prior to analysis. Cryo-TEM of the control solutions of liposomes showed mostly unilamellar liposomes 200 nm in diameter. However, some degree of polydispersity as well as multilamellar vesicles was found, likely a result of the annealing process. When gelators 1–3 were assembled in the presence of liposomes, cryo-TEM revealed the presence of similar liposomes coexisting with gel fibers (Fig. 5a–c). These results strongly suggest that the molecular packing of the DMPC phospholipids is not altered by the self-assembly of gelators 1–3. Some ripple-phase could even be observed with DMPC vesicles (Fig. 5d), which relates to the persistence of pre-transitions observed using DSC.54,55 Interestingly, we did observe supramolecular interactions between the architectures’ evidenced non-spherical vesicles (Fig. 5, indicated by arrows). As a result of the confined space due to the porous gel structure, the gel network forced the vesicles to accommodate their shape by stress exerted on their walls. Conversely, the fibers of gels 1–3 remained unaffected by the presence of the liposomes. Gelator 1 and 2 give polydisperse fibers, rendering the study of their alteration by liposomes using TEM alone quite delicate (see ESI†). Fortunately, compound 3 displays characteristic features in water, i.e. a high persistence length and a well-defined diameter of 5 nm. Identical fiber morphologies were observed in the presence of liposomes, suggesting no influence on the assembly process of the gelator molecules. | ||
| Fig. 5 Cryo-TEM micrographs of DMPC liposomes with 1,3,5-triamide cis,cis-cyclohexane derivatives 1 (a), 2 (b) and 3 (c). (d) Magnification of c, showing DMPC ripple phase. Micrographs of dibenzoyl cysteine 4 bis(leucine) (e), oxalyl-amide 5 (f), monourea ethyl serine gelator 6 (g) and gemini tartrate 7 (h) in the presence of DMPC liposomes. The inset in f shows a vesicle that is disrupted by the presence of 5. Arrows indicate non-spherical vesicles that are squeezed by the gel network. | ||
In the presence of gelator 4, mostly unilamellar DMPC liposomes were attached to fibers (Fig. 5e). For the mixed system of DMPC liposomes with gelator 5, broken vesicles were observed within the gel network. Some micrographs revealed partly dissolved liposomes as shown by missing fragments of bilayers (Fig. 5f). The fibers of 5 also appeared thinner compared to their original morphology. In the presence of oxyl leucine derivative 6, the vesicle disruption was stronger as compared to samples with fibers of 5, and some liposomes had lost their original structure, while gel fibers were hardly observed (Fig. 5g). Finally, mixed systems of gemini tartrate 7 with DMPC did not reveal any liposomes. Instead, assemblies somewhat reminiscent of bicelles seemed to prevail (Fig. 5h). The twisted fibers that evolve in helical ribbons, normally observed for gemini tartrate in pure water (see ESI†), were not found.
Discussion on orthogonal self-assembly
Gelation tests, calorimetry experiments and cryo-TEM investigations suggest that all the studied 1,3,5-triamide cis,cis-cyclohexane derivatives lead to orthogonal self-assembly with phosphatidylcholine (PC) liposomes. The unmodified pre-transition and main phase transition of the phospholipid bilayers revealed their intact organization in the presence of fibers of 1–3. Conversely, gelation properties of these derivatives were also retained with comparable CGC measured in pure water or in the presence of PC vesicles. At a supramolecular level, cryo-TEM also evidenced unmodified liposomes and gel morphologies. Together, these results demonstrate the orthogonal self-assembly of phospholipids and 1,3,5-triamide cis,cis-cyclohexane-based low molecular weight gelators, leading to the formation of distinct supramolecular structures within the same system. We hypothesize that strong intermolecular interactions in this class of gelators, evidenced by the high melting points and low CGC, explains why these gelators do not form mixed assemblies with phospholipids. This stability originates from the strong intermolecular hydrogen bonds and the hydrophobic interactions due to the amino acids that presumably do not only act as an additional intermolecular force, but also shield the amides from water.The formation of liposomes in conjunction with self-assembled 4 fibers was also revealed by cryo-TEM. However, the small shift and the broadening of the main phase transition of the phospholipid bilayers, the disappearance of the associated pre-transition, as well as a tendency of the vesicles to stick to the fibers, indicate that liposomes and gel fibers interact. Moreover, the CGC of 4 also increased in the presence of liposomes. Similar to gelator 2, gels formed by 4 are characterized by relatively high melting temperatures and gelator 4 has a CGC comparable to compound 2. The presence of interactions between the gels and the vesicles can therefore not be explained by a lower stability of gels formed by 4. We hypothesize that in this case the molecular packing of the 4 molecules (Fig. 2) facilitates access of the phospholipid molecules to stacks of 4. In the case of the cyclohexane derivatives 1–3, the hydrophobic domains of the stacks are held together by three hydrogen bonds located at the core of the fibers that are protected by a shell of ethylene glycol moieties. In contrast, stacks formed by 4 only present two hydrogen bonds per unit that are not protected by ethylene glycol groups, thereby exposing the hydrophobic groups to the phospholipids. Similar to PEGylated liposomes that do not fuse because of the resulting steric hindrance, the absence of ethylene glycol at the surface of the gel fibers could well explain the interactions between 4 and the phospholipids.
For gelator 5 (OxLeu2), the interactions between the hydrogelators and vesicles were clearly noticeable as both the liposomes and gel properties were affected at a molecular and supramolecular level. Derivative 5 is a relatively weak gelator (Fig. 3) and the model proposed by Žinić et al. suggests that hydrogen bonds primarily drive its assembly.41 Such intermolecular interactions between the gelator molecules do not appear to be sufficiently strong and specific to counteract some intermixing between phospholipids and gelator molecules. Gelators based on amphiphilic molecules did not show orthogonal self-assembly with liposomes. Cryo-TEM could not evidence the presence of gel fibers and the liposomes were either partially dissolved (6) or absent (7). These microscopic observations correlated with the DSC experiments since the phase transitions of the liposomes were strongly shifted to lower temperatures in the case of the urea gelator 6. The effect of the gemini tartrate 7 on the phase transitions of the DMPC liposomes was even more pronounced, since an intermediate transition between the respective phase transitions of the phospholipids (23 °C) and the gel (melting point at 43 °C) was found. These results suggest that both gelators 6 and 7 co-assemble with DMPC. Cryo-TEM indeed revealed the presence of bilayer fragments rather than liposomes in conjunction with gel fibers.
The methods used in this study allow the rapid identification of orthogonal self-assembling systems. Together, these results suggest that orthogonal self-assembly between phospholipids and gelators can only take place when: (1) the gelator and the phospholipid assemblies are characterized by high melting points, or in other words, strong intermolecular interactions; (2) both self-assembled structures are based on a distinct set of interactions; (3) the hydrophobic domains of the gelator stacks are shielded from the environment by hydrophilic groups. With these guidelines, the molecular design rules that underpin orthogonal self-assembly in water emerge. Such rules could be applied to other self-assembling systems and possibly lead to even more complex self-assembled architectures. Similar to the systems described in this study, we believe for instance that micelles, hydrogels, polymersomes, viruses, or any other protein structures could well coexist with each other as long as a different set of strong interactions characterizes each assembly.
Stability of liposomes in a gel network
In biological systems, the extracellular matrix surrounds cells and offers them physical protection. Therefore, we wondered if the presence of a network in conjunction with a phospholipid membrane could contribute to a higher robustness of liposomes, with respect to their fusion and their permeability to small molecules. To study the first aspect, the fusion rate between DOPS vesicles embedded in self-assembled hydrogel networks was investigated and compared to DOPS in solution. DOPS liposomes were chosen because they fuse within minutes in the presence of calcium ions, contrary to phosphocholine phospholipids. Resonance energy transfer between phospholipid analogues N-NBD-PE (λex = 450 nm; λem = 530 nm) and N-Rh-PE (λex = 540 nm; λem = 590 nm) was used to monitor the progress of fusion. In our experiments, one population of DOPS vesicles was labeled with both fluorescently-tagged phospholipid analogues at such a concentration that their density resulted in efficient energy transfer (DOPS/N-NBD-PE/N-Rh-PE = 98:1:1). Upon fusion with unlabeled DOPS liposomes, the distance between tagged phospholipids increases, effectively decreasing the energy transfer efficiency, resulting in an increase in fluorescence intensity of donor N-NBD-PE. In order to quantify the degree of liposome fusion, the measured intensities at 530 nm were divided by the intensity of liposomes in the presence of triton X-100 corresponding to the maximal phospholipid intermixing.
At t0, CaCl2 was added to DOPS liposomes in buffer or added on top of the liposome-gel system. In solution, the fusion between pure DOPS liposomes and labeled liposomes occurred rapidly. After 10 minutes, the relative liposome fusion stabilized at around 0.6, representing the maximal dilution of the fluorescent phospholipids with the non-labeled liposomes (Fig. 6a–c). In order to test the capacity of gel networks to prevent fusion, we incorporated the liposomes in a gel of 2. We chose 2 because it forms thixotropic gels, i.e. it becomes a liquid under mechanical stress and returns to a gel after release of the stress, and we used this feature to mix the liposomes into the gel by vortexing. In the gel environment, stabilization of the relative fusion was much slower than in water and leveled off after about 50 hours (Fig. 6c). The difference in fusion rates is likely due to slow calcium diffusion through the gel network, rather than slow fusion events themselves.§ Nevertheless, the final value of the relative fusion after stabilization was three times lower than in solution. This experiment was repeated with different liposome sizes (30, 200 and 1000 nm diameter) and similar curves were obtained, indicating no significant influence of the liposomes’ size on the fusion events. Interestingly, within seconds after macroscopically breaking the gel by vortexing, the relative fluorescence intensity reached a value similar to those obtained by fusing liposomes in solution (Fig. 6c).
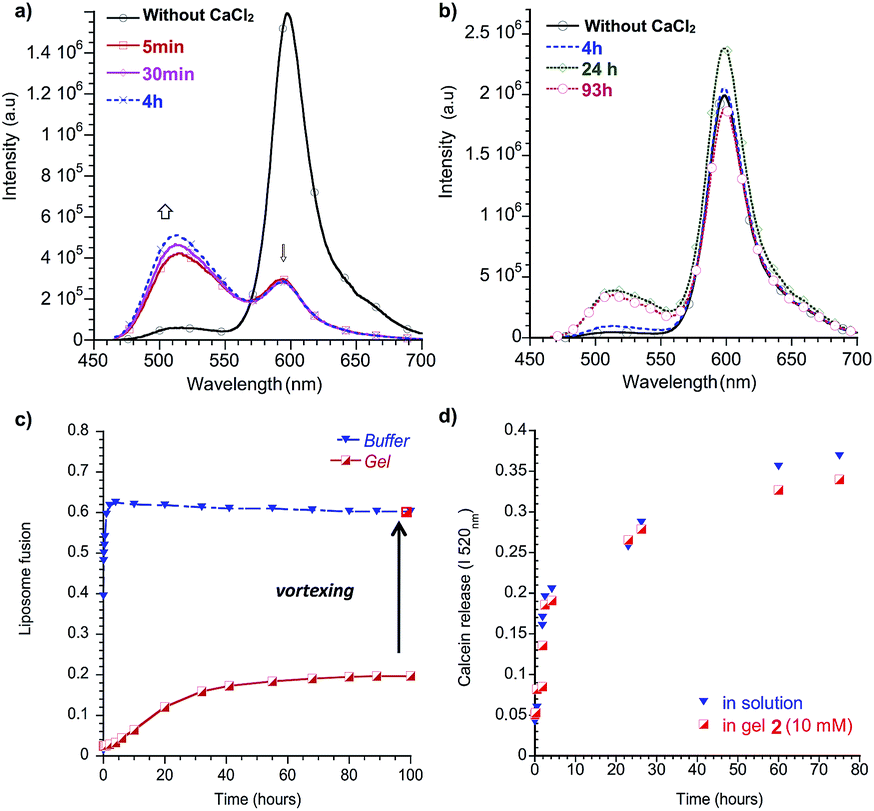 | ||
| Fig. 6 Emission spectra of DOPS vesicles containing 1 mol% fluorescent phospholipids N-NBD-PE and N-Rh-PE before and after the addition of CaCl2 (a) in buffer and (b) in a gel network of 2. (c) DOPS fusion as a function of time in buffer or in a gel of 2. (d) Calcein release from DOPC vesicles in buffer or embedded in a gel network of 2, as followed by the progressive increase of the fluorescence at 520 nm. | ||
Encouraged by the results above, 200 nm DOPC liposomes were entrapped in a gel network of 3, by using the pH-responsiveness of the gel. An acidic 100 μL solution of 50 mM 3 could be gelated rapidly by the addition of 1 mL of vesicles in neutral buffer. When the liposomes were released three weeks later by the addition of acid, the original size as measured by dynamic light scattering (DLS) was preserved while the same liposomes in buffer only had evolved to larger aggregates, with higher polydispersity (see ESI†).
The release of the internal contents of the liposomes was also monitored in the presence or absence of an orthogonally self-assembled matrix. In order to measure leakage, calcein was encapsulated in the interior of the liposomes at a self-quenching concentration. Upon release of the calcein from the interior, the fluorophore dilutes to below its self-quenching concentration thus recovering its fluorescence. We therefore measured the fluorescence recovery over time (Fig. 6d).56 In buffer solutions, the leakage was initially fast and stabilized at around 80% within a few days. Interestingly, the same rates were obtained for calcein-loaded liposomes in a gel of 2. These results suggest no improved stability of the liposomes with respect to their leakage.
The result above show that orthogonal self-assembly allows the mimicking of some aspects of natural cells. In biological systems, the extracellular matrix surrounds cells and offers them physical protection. Even if the configurations of synthetic self-assembled networks and intact phospholipid bilayers developed in this study constitute only very primitive and even naive model systems for cells with an extracellular matrix, they do allow the study of the effect of a self-assembled network on the liposomes’ stability. The results also showed that liposomes are stabilized towards fusion by confinement in a gel network, most likely by preventing the occurrence of fusogenic contact between neighboring liposomes. Using this approach, liposomes could be stored for at least three weeks without any change in their diameter. On the other hand, while limited fusion could be shown, no increased stability towards the leakage of calcein was observed.
Compartmentalization in orthogonally self-assembled systems
Compartmentalization is another essential feature of biological cells, to organize incompatible functions in the confined intracellular space. Despite many efforts, most synthetic compartmentalized systems comprise at best only a single hydrophobic compartment and a single aqueous compartment. Here, the orthogonal self-assembly of phospholipids with low-molecular weight gelators provides four distinct compartments: the inner aqueous compartment (i) of the liposomes is separated by a hydrophobic bilayer (ii) from the aqueous phase (iii) filling the pores of the gel network, which provides another hydrophobic compartment (iv). While the two aqueous compartments are not trivial to discriminate, the hydrophobic compartments of liposomes and gelators can easily be addressed separately, because they are assembled by distinct sets of interactions. For that purpose, we have synthesized 3a, a fluorescent analog of 3 (Fig. 7a, see ESI†). When 3a was incorporated into gel fibers of 3 (0.1 mol%) and analyzed by fluorescent lifetime imaging microscopy, an average lifetime of 6 ns was measured when excited at 385 nm (see ESI†). In contrast, NBD phospholipids (1 mol%) incorporated in liposomes showed a higher lifetime of typically around 9 ns (see ESI†). Upon orthogonal self-assembly of the two architectures, two distinct supramolecular aggregates were clearly evidenced based on their fluorescence lifetimes (Fig. 7b), i.e. bundles of fibers of 3 selectively incorporated 3a, as evidenced by the shortest lifetimes measured on these aggregates (green, below 7.0 ns), while the liposomes mostly contained fluorescent species of higher lifetimes, typical for NBD-PE (red, above 7.0 ns). This result demonstrates the self-sorting of species in two distinct self-assembled architectures.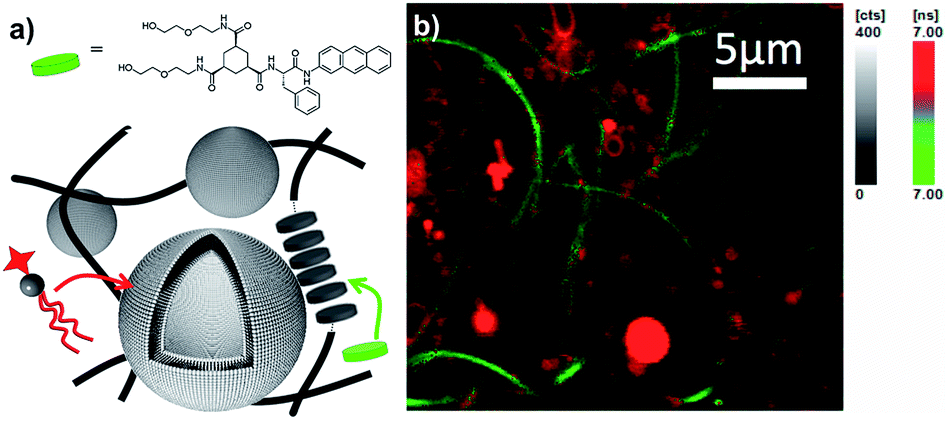 | ||
| Fig. 7 (a) Preferential incorporation of fluorescent phospholipids (NBD-PE) in liposomes and 3a in the self-assembled fibers, (b) as evidenced by fluorescent lifetime imaging microscopy based on lifetime discrepancies (λex = 385 nm, λem > 405 nm, green reflects fluorescence lifetimes below 7 ns, red above 7 ns). | ||
Conclusions
This study provides guidelines to achieve orthogonal self-assembly. By investigating specific features of low molecular weight gelators and phospholipids, both at a molecular and a supramolecular level, we showed that orthogonal self-assembly can take place if the entities assemble via strong but also distinct sets of interactions. Further study is needed to provide quantitative insight into e.g. the minimal differences required for orthogonal self-assembly, shielding of interactions and spatial mismatch.26,57 The resulting supramolecular architectures consisting of fibrillar networks coexisting with liposomes already provide additional levels of compartmentalization and enhanced stability as compared to self-assembled systems of gelators or phospholipids alone. Therefore, and even if this study is restricted to hydrogelators and phospholipids, we foresee that the application of orthogonal self-assembly approaches to self-assembled systems from other molecular components, polymersomes, protein filaments, viral capsids, and nanoparticle assemblies, will offer new avenues to much more complex self-assembled architectures with multiple levels of compartmentalization and functionality, that are unattainable by single component self-assembly.Experimental
Materials
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine] (DOPS) were obtained from Avanti Polar phospholipids. Dioctadecyldimethylammonium bromide (DODAB), ditetradecyl phospate (DTP) and gelator 4 (N,N′-dibenzoyl-L-cysteine, DBC) were purchased from Sigma Aldrich. N-(7-Nitro-2,1,3-benzoxadiazol-4-yl)-dipalmitoyl-L-α-phosphatidylethanolamine (N-NBD-PE) and N-(lissamine rhodamine B sulfonyl) diacyl-phosphatidylethanolamine (N-Rh-PE) were obtained from Invitrogen, Molecular Probes.The synthesis and full characterization of compounds 1, 2 and 3 have been reported before in the literature.39 The bis(leucine) oxalyl amide (5, Ox(Leu)2) was prepared according to ref. 41. NMR spectra and mass were consistent with the described procedures (see ESI†). The synthesis of compound 6 (UreaG) was slightly modified from ref. 50 (see ESI†). Compound 7 (gemini tartrate) was prepared according to the literature procedure.51 NMR spectra and mass were consistent with the described procedures. For the FLIM experiments, an anthracene derivative of gelator 3 was synthesized (see ESI†).
General procedures
:1. After evaporation of the solvent, these films were hydrated at a typical concentration of 20 mM in double distilled water or PBS buffer (pH 7.4, 10 mM phosphate, 27 mM KCl, 137 mM NaCl). These multilamellar vesicles were extruded 21 times above their main transition temperature, through polycarbonate filters (Nuclepore Whatman), typically with a pore size of 200 nm, using a small-volume extruder (Avanti polar phospholipids), to give unilamellar vesicles with an average diameter of 200 nm, as confirmed by Dynamic Light Scattering and Cryo-TEM.
:1:1, were prepared in Tris buffer (10 mM), 100 mM NaCl, pH = 7.4, and extruded with polycarbonate filters of 30 nm, 200 nm or 1 μm porosity. Equal volumes of each solution (0.5 mL) were diluted with 1 mL of buffer (control experiment) or vortexed with 1 mL of hydrogel, to ensure incorporation of the vesicles in the gel matrix (2 = 10 mM). Gelation reversibly took place after 2 min (thixotropic gel). This method was preferred over the heating/cooling cycle across the sol–gel transition to prevent extensive fusion of the liposomes prior to the experiment. In order to induce fusion, calcium ions were required. 0.5 mL of CaCl2 solution at 20 mM were therefore added in both samples (in the case of the gel, the calcium solution was deposited on top of the gel), so that the final concentrations of calcium, labeled and unlabeled liposomes were respectively 5 mM, 0.25 mM and 1 mM. Fluorescence measurements were performed at 25 °C with a Perkin Elmer LS-50B spectrophotofluorimeter using an excitation wavelength of 450 nm. The fusion events were followed as a function of time, by monitoring the quenching of the fluorescence intensity of N-NBD-PE (I520 nm/I595 nm). The maximal level of fusion was determined by adding 10 μL of Triton (10%) to the samples and subsequent vortexing, to ensure complete lipid mixing (the same values were obtained in solution or the gel state). The fluorescence level of this suspension was set to 100%.
Acknowledgements
The authors thank the Netherlands Organization for Scientific Research (NWO) for financial support through a VICI grant and a Rubicon grant, and NanoFM (Groningen, The Netherlands) for providing the molecular gelators. JB acknowledges the European Cooperation in Science and Technology (COST) action D31 for a Short Term Scientific Mission (STSM) grant.Notes and references
- M. C. T. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393 CrossRef CAS.
- J. M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295, 2403 CrossRef CAS PubMed.
- F. Diederich, Angew. Chem., Int. Ed., 2007, 46, 68 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642 CrossRef CAS PubMed.
- J. Boekhoven, R. H. Zha, F. Tantakitti, E. Zhuang, R. Zandi, C. J. Newcomb and S. I. Stupp, RSC Adv., 2015, 5, 8753 RSC.
- H. Nakanishi, K. J. M. Bishop, B. Kowalczyk, A. Nitzan, E. A. Weiss, K. V. Tretiakov, M. M. Apodaca, R. Klajn, J. F. Stoddart and B. A. Grzybowski, Nature, 2009, 460, 371 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014 Search PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS PubMed.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009 CrossRef CAS PubMed.
- J. H. van Esch, Langmuir, 2009, 25, 8392 CrossRef CAS PubMed.
- T. Kato, T. Kutsuna, K. Hanabusa and M. Ukon, Adv. Mater., 1998, 10, 606 CrossRef CAS.
- A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063 CrossRef CAS PubMed.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316 RSC.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 4th edn, 2002 Search PubMed.
- G. Vereb, J. Szöllsi, J. Matko, P. Nagy, T. Farkas, L. M. L. W. T. A. Vigh, L. Matyus, T. A. Waldmann and S. Damjanovich, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8053 CrossRef CAS PubMed.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831 CrossRef CAS PubMed.
- P. Mukhopadhyay, A. Wu and L. Isaacs, J. Org. Chem., 2004, 69, 6157 CrossRef CAS PubMed.
- A. Heeres, C. van der Pol, M. C. A. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252 CrossRef CAS PubMed.
- T. Kato, Science, 2002, 295, 2414 CrossRef CAS PubMed.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170 RSC.
- A. M. Brizard, M. C. A. Stuart and J. H. van Esch, Faraday Discuss., 2009, 143, 345 RSC.
- A. Pal, S. Karthikeyan and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 7842 CrossRef CAS PubMed.
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719 RSC.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171 CrossRef CAS PubMed.
- E. R. Draper, E. G. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848 CrossRef CAS PubMed.
- H. K. Lee, S. Soukasene, H. Jiang, S. Zhang, W. Feng and S. I. Stupp, Soft Matter, 2008, 4, 962 RSC.
- S. Himmelein, V. Lewe, M. C. A. Stuart and B. J. Ravoo, Chem. Sci., 2014, 5, 1054 RSC.
- J. Boekhoven, M. Koot, T. A. Wezendonk, R. Eelkema and J. H. van Esch, J. Am. Chem. Soc., 2012, 134, 12908 CrossRef CAS PubMed.
- A. M. Brizard and J. H. Van Esch, Soft Matter, 2009, 5, 1320 RSC.
- R. G. Weiss and P. Terech, Molecular gels: materials with self-assembled fibrillar networks, Springer, Dordrecht, 2006 Search PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- G. Cevc, Phospholipids handbook, CRC Press, 1993 Search PubMed.
- D. F. Evans and H. Wennerstrom, Colloidal domain, Wiley-Vch, 1999 Search PubMed.
- J. H. Clint, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 1327 RSC.
- K. J. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed. Engl., 2004, 43, 1663 CrossRef CAS PubMed.
- F. Menger and K. Caran, J. Am. Chem. Soc., 2000, 122, 11679 CrossRef CAS.
- J. Makarevic, M. Jokic, B. Peric, V. Tomisic, B. Kojic-Prodic and M. Žinić, Chem.–Eur. J., 2001, 7, 3328 CrossRef CAS.
- P. Terech, C. Rossat and F. Volino, J. Colloid Interface Sci., 2000, 227, 363 CrossRef CAS PubMed.
- B. Escuder, M. Llusar and J. Miravet, J. Org. Chem., 2006, 71, 7747 CrossRef CAS PubMed.
- J. M. Poolman, J. Boekhoven, A. Besselink, A. G. L. Olive, J. H. van Esch and R. Eelkema, Nat. Protoc., 2014, 9, 977 CrossRef CAS PubMed.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and surface forces: revised third edition, Academic Press, St Louis, Missouri, U.S.A., 2010 Search PubMed.
- H. Heerklotz, J. Phys.: Condens. Matter, 2004, 16, R441 CrossRef CAS.
- K. Hanabusa, A. Kawakami, M. Kimura and H. Shirai, Chem. Lett., 1997, 191 CrossRef CAS.
- C. G. L. Wolf and E. K. Rideal, Biochem. J., 1922, 16, 548 CrossRef CAS PubMed.
- G. Wang and A. Hamilton, Chem. Commun., 2003, 310 RSC.
- A. Brizard, C. Aime, T. Labrot, I. Huc, D. Berthier, F. Artzner, B. Desbat and R. Oda, J. Am. Chem. Soc., 2007, 129, 3754 CrossRef CAS PubMed.
- F. D. Gunstone, J. L. Harwood and A. J. Dijkstra, The lipid handbook, CRC Press, Boca Raton, 3th edn, 2007 Search PubMed.
- C. Huang and S. Li, Biochim. Biophys. Acta, 1999, 1422, 273 CrossRef CAS.
- M. J. Janiak, D. M. Small and G. G. Shipley, Biochemistry, 1976, 15, 4575 CrossRef CAS PubMed.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159 CrossRef CAS.
- T. M. Allen and L. G. Cleland, Biochim. Biophys. Acta, 1980, 597, 418 CrossRef CAS.
- A. Pal, P. Besenius and R. P. Sijbesma, J. Am. Chem. Soc., 2011, 133, 12987 CrossRef CAS PubMed.
- A. Takahashi, M. Sakai and T. Kato, Polym. J., 1980, 12, 335 CrossRef CAS.
- D. K. Struck, D. Hoekstra and R. E. Pagano, Biochemistry, 1981, 20, 4093 CrossRef CAS PubMed.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M. T. Kao, A. G. Olive and A. Del Guerzo, J. Am. Chem. Soc., 2011, 133, 316 CrossRef CAS PubMed.
- F. Szoka and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, cryo-TEM micrographs, fluorescence lifetime imaging micrographs. See DOI: 10.1039/c6sc01021k |
| ‡ Current address: Department of Chemistry, Technische Universität München, Lichtenbergstr, 485748 Garching b. München, Germany. |
| § Conductivity experiments indeed showed that the concentration of CaCl2 initially deposited in a gel network reached its equilibrium in the same time scale. |
| This journal is © The Royal Society of Chemistry 2016 |