DOI:
10.1039/C6SC00106H
(Edge Article)
Chem. Sci., 2016,
7, 3325-3330
Stapling of unprotected helical peptides via photo-induced intramolecular thiol–yne hydrothiolation†
Received
8th January 2016
, Accepted 3rd February 2016
First published on 5th February 2016
Abstract
Peptide stapling emerged as a versatile strategy to recapitulate the bioactive helical conformation of unstructured short peptides in water to improve their therapeutic properties in targeting intracellular “undruggable” targets. Here, we describe the development of photo-induced intramolecular thiol–yne macrocyclization for rapid access to short stapled peptides with enhanced biophysical properties. This new peptide stapling technique provides rapid access to conformationally constrained helices with satisfying functional group tolerance. Notably, the vinyl sulfide linkage shows distinct lipophilicity with reduced membrane toxicity compared to the corresponding all-hydrocarbon analogue. As a proof of principle, we constructed stabilized helices modulating intracellular estrogen receptor (ER)–coactivator interactions with a nanomolar binding affinity, enhanced serum stability, a diffuse cellular distribution and selective cytotoxicity towards ER-positive MCF-7 cells.
Introduction
Targeting aberrant intracellular protein–protein interactions (PPIs) is considered a promising therapeutic strategy.1–3 However, therapeutic intervention with small molecular entities has proven to be challenging due to generally flat protein–protein interfaces.4 Simplifying interacting proteins into structural elements and locking their functional conformation via synthetic manipulation is a promising approach to overcome this barrier and bridge the targeting space gap between small molecules and biologics.5–9 It is well-documented that over fifty percent of PPIs involve short α-helices.10,11 Short peptides are generally unstructured in aqueous solution as water molecules compete with the intramolecular hydrogen bonding of the peptide backbone. On the other hand, the unstructured conformation in solution is also entropically favorable. One of the most established approaches to restore and stabilize the bioactive helical conformation is peptide stapling, in which unstructured peptides are locked in their active conformation by introducing various sidechain-to-sidechain cross-link architectures at the same face of the helix via macrocyclization.12 The preorganization and structural reinforcement of peptides lead to an increased target binding affinity due to a reduced entropic penalty. In addition, shielding by this “staple” reduces exposure of the peptide backbone and enhances protease resistance.13 Current stapling cross-links include lactam,14–16 disulfide,17 “click” triazole,18–20 all-hydrocarbon,21–23m-xylene,24 perfluoroaryl25 and dithioether26 linkers. Different strengths and weaknesses of the cross-links exist according to the chemistry for their generation and the cross-link itself influences the properties of peptides.20,27 In particular, the polarity of the cross-link has an impact on the overall properties of peptides when penetrating the cell membrane as the cross-link directly interacts with membrane components and is associated with the solvation and desolvation process. The pioneering work of Verdine, Walensky and others made great impact on this field by cross-linking hydrocarbon α-methylated amino acids to afford potent modulators successfully targeting various intracellular PPIs.28–32 The incorporation of all-hydrocarbon cross-links via olefin metathesis provides highly lipophilic staples which increase the overall hydrophobicity of the peptides with a significant improvement in cellular entry and binding affinity.33,34 This approach usually requires high loading of a ruthenium catalyst to drive the reaction to completion. In addition, a ∼10-fold increase of hemolysis induced by helical antimicrobial peptides with an all-hydrocarbon staple was observed.35,36 Finally, some stapled peptides are more prone to aggregate as most key residues located at the interfaces of PPIs are hydrophobic.32,37 Taking this into consideration, a stapling architecture with tuned lipophilicity is needed to provide an alternative balance of cell permeability, off-target effects and toxicity.
Our approach is to substitute one carbon atom in the all-hydrocarbon cross-link with a non-polar sulfur atom, which only minimally perturbs the cross-link, avoiding bulky aromatic rings that can engage in non-specific interactions.26 The previous work by Fairlie et al. showed that an alkyl thioether is not a helical constraint in water, which is likely due to the lack of rigidity and increased structural freedom upon rotation of the C–S bond.15 Inspired by alkenyl all-hydrocarbon cross-links, we envisioned a vinyl sulfide with enhanced rigidity and balanced lipophilicity. A vinyl sulfide was previously reported to mimic the redox-sensitive disulfide bond in the cyclic analogues of angiotensin II.38 The synthetic procedure of reacting a thiol with the formyl group of allysine is challenging. Thus, we turned to the more versatile thiol–yne click chemistry. The bio-orthogonal and “double” functionality nature of thiol–yne hydrothiolation is frequently exploited for constructing highly functionalized materials and bioconjugates.39–42 An intramolecular thiol–yne reaction has recently been reported to synthesize thioglycals, which generates 5-exo and 6-endo cyclized products.43 However, the potential use of this intramolecular thiol–yne chemistry for macrocyclization remain largely unexplored. The analogous intramolecular thiol–ene reaction has been previously used for peptide macrocyclization.44,45 More recently, an efficient two-component thiol–ene coupling has been reported to nucleate long helical peptides including Axin and p53 mimetics.26 This methodology circumvents the use of unnatural amino acids and could potentially be applied to recombinant proteins. In the reported i, i + 4 variant, whether the relatively flexible 11- or 12-membered dithioether cross-link could nucleate the helical conformation of even shorter peptides remains unknown.15,26 In this work, we report a one-component preparation of short stapled helical peptides as well as conformational mimetics of the disulfide bond via photo-induced intramolecular thiyl radical addition to an alkyne (Fig. 1).
 |
| | Fig. 1 Schematic presentation of photo-induced intramolecular thiol–yne hydrothiolation for peptide stapling. | |
Results and discussion
In the early work reported by Verdine et al., the stereochemistry and cross-link length were systematically investigated.22 In the i, i + 4 system, the most effective helix induction tether was an 8-membered hydrocarbon cross-link with a double bond in the center. According to this well-established stapling system, the nonapeptides 1a and 1b were synthesized through solid phase peptide synthesis. After cleavage from the resin, the peptide was subjected to UV irradiation in the presence of a photoinitiator to afford an 8-membered vinyl sulfide staple with a double bond in the center of the linkage, shown as entries 1 and 2 (Fig. 2A). The vinyl sulfide products showed distinct retention times (Fig. 3A). Further evidence for the intramolecular thiol–yne reaction was obtained using 1H-NMR spectroscopy. The double bond proton signals of the vinyl sulfide products appear as shown in Fig. 3B, which are assigned as the E/Z isomers. However, we found that this 8-membered cross-link failed to nucleate the helical conformation of model peptides 2a and 2b as indicated by the circular dichroism (CD) spectrum (Fig. 2B). Nevertheless, the Z-isomer with a longer retention time (2a-B) revealed a weak positive maximum molar ellipticity at 190 nm. We hypothesized that this linking pattern provides a preference for helicity induction but that the 8-membered tether was too flexible. Thus, peptides 2c and 2d were synthesized to investigate whether shortening the linker could achieve better helix induction. Fortunately, the 7-membered cross-link with cysteine located at the N-terminus successfully nucleated model peptide 2d into a helical conformation in water as Cotton effects and zero-crossing were observed in the CD spectrum at 190 nm and 206 nm, and 222 nm and 196 nm, respectively. The HPLC traces of the crude reaction mixture at different time intervals revealed the rapid kinetics of intramolecular thiol–yne reactions in peptides, which was consistent with previous kinetics studies (Fig. S2†).54 Interestingly, the Z-isomers were preferred in different 7-membered cross-linked peptides. A different peptide sequence led to comparable helicity (see Fig. S1†). Only a trace amount of peptide 2c was observed after UV irradiation. This is likely due to a conformational preference for trapping the thiyl radical at the N-terminus when contracting to a 20-membered macrocycle. We further optimized the conditions for improved thiol–yne efficiency and ease of purification based on the HPLC conversions as shown in Fig. 2A. To test the chemical stability of the vinyl sulfide cross-link, peptide 2d was incubated under acidic and basic conditions. We found that the peptide remained intact after 24 h of incubation suggesting the potential for further biological applications (Fig. 2C).
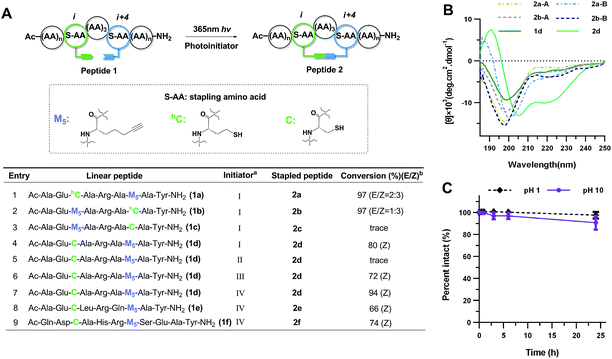 |
| | Fig. 2 Cross-link optimization and chemical stability of vinyl sulfide stapled peptides. (A) Screening for reaction conditions. aInitiator: (I) 0.5 eq. DMPA, 1 h; (II) no initiator, 1 h; (III) 0.5 eq. DMPA, 0.5 eq. MAP, 1 h; (IV) 0.5 eq. IHT-PI 659, 0.5 h. Abbreviations: MAP, 4-methoxyacetophenone; DMPA, 2,2-dimethoxy-2-phenylacetophenone; IHT-PI 659, 2-hydroxy-1-[4-(2-hydroxyethoxy)-phenyl]-2-methyl-1-propanone. bConversions [desired product/(desired product + starting material)] were determined by integration of reverse-phase HPLC. For peptides 2d–f, the Z-isomer is dominant. (B) CD spectra of vinyl sulfide stapled peptides in 10 mM phosphate buffer, pH 7.4, 20 °C; (A) is the E-isomer and (B) is the Z-isomer. (C) Chemical stability of peptide 2d in 3% TFA or 0.1 mM NaOH aqueous buffer. Percentage intact, mean ± s. d. and n = 3. | |
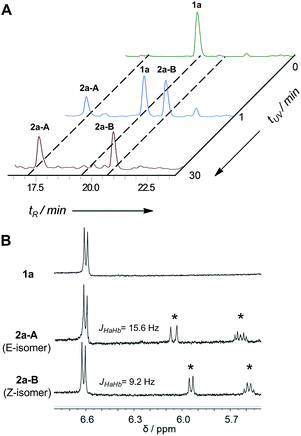 |
| | Fig. 3 Monitoring the reaction for intramolecular thiol–yne hydrothiolation in model peptide 1a. (A) HPLC traces of the reaction mixture at different UV irradiation times, monitored at 220 nm; (B) 1H-NMR spectra of 1a, 2a-A and 2a-B (measured in DMSO-d6 at 400 MHz). Asterisks indicate the formation of a vinyl sulfide double bond after UV irradiation. | |
To further understand the effect of vinyl sulfide constraint on peptide conformational preference in aqueous solution, a detailed 1D and 2D 1H-NMR study was performed in 10% D2O in PBS (pH 5.0) at 25 °C. As expected, the Rotating-frame Overhauser Effect (ROE) signals of vinyl sulfide stapled peptide 2d were substantially different from its linear counterpart (Fig. 4A). Although the assignment of a few key cross-peaks was obscured due to peak overlap, 2d revealed a series of low amide coupling constants (3JNHCHα ≤ 6), which is indicative of a helical folded structure. However, residues 2 and 7 have 3JNHCHα > 6, suggesting substantial fraying of the helical conformation at either termini of the peptide. The ROESY spectrum of 2d also displayed several dαN(i, i + 3), dαN(i, i + 4) and dαβ(i, i + 3) ROEs, which further provide evidence for the helical propensity. In addition, the chemical shift Δδ (Hα) of the residues in 2d (Ala1 not determined due to peak overlap) displayed upfield shifts compared to its linear precursor, which is in good agreement with the helical folding state observed by others (Fig. 4B).16 Moreover, sequential low temperature dependence for amide NH chemical shifts (Δδ/T ≤ 4.5 ppb K−1) was also observed, which is evidence for hydrogen bonding. These results confirm the potential of the vinyl sulfide cross-link as a helical constraint in water. To understand the conformational features of nonapeptides 1d and 2d, a total of 500 ns molecular dynamics (MD) simulation was run in a NPT ensemble (details in the ESI†). We characterized the secondary structure distributions for the ensemble structures of each analog (Fig. 4C). The major residues of 1d were in random coil conformations. Meanwhile, residues 3–6 of 2d displayed a notable extent of the α-helix (45%) and 310-helix (25%) conformations. The remaining residues beyond the stapled region were major random coil, which is highly consistent with the 1H-NMR spectra. The Ramachandran plots of each residue for 1d and 2d are shown in Fig. S4.† Both the ROEs and MD simulation results suggested that residues positioned within the stapled region showed a notable helical propensity, while residues beyond this region possessed more conformational fluctuations in this short nonapeptide in aqueous solution.
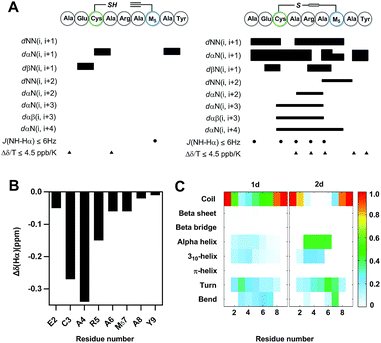 |
| | Fig. 4 NMR characterization and molecular dynamics (MD) simulation. (A) ROE summary diagram of 1d and 2d (measured in 10% D2O in PBS, pH 5.0, 25 °C). Bar thickness reveals the intensity of the ROE signals. (B) Plot of the chemical shift of Hα (Δδ = δ2d − δ1d). (C) Comparison of the secondary structure profile for each residue derived from the MD simulation. | |
To demonstrate the biological potential of this new stapling technique, we synthesized the vinyl sulfide stapled analogues modulating intracellular estrogen receptor (ER)–coactivator interactions.17 ERs belong to the nuclear receptors (NR) subfamily, which are DNA-binding transcription factors activated by the hormone estrogen. The dysfunction of ERs is implicated in the pathogenesis of many diseases including cancer, osteoporosis, and others.46 Peptides that mimic the short helical leucine rich NR box motif (LXXLL, where X represents any amino acid) could function as ER antagonists.47 Several groups have reported peptides targeting ERs identified through phage display or ribosomal display.47–49 The peptides studied in this context are shown in Fig. 5A. Peptide 2h conserved the NR box motif necessary for ER binding. To test the biological activity of our vinyl sulfide stapled peptide, we evaluated the binding affinity between fluorescein-labeled derivatives and ER-α/ER-β using a fluorescence polarization (FP) assay. 5-Carboxyfluorescein (FAM) was site-specifically incorporated into the ε-NH2 group of a lysine residue. Vinyl sulfide stapled 2h displayed a similar affinity compared to the linear analog 1h as shown in Fig. 5A and S5.† Flow cytometry analysis was performed to quantify the cellular uptake of peptides in HEK293T cells, and 2h-FAM revealed a 5-fold higher mean fluorescence than its linear 1h-FAM or unstapled 1g-FAM analogues (see Table S1 and Fig. S6†). An enhanced helicity and serum stability of 2h were also observed compared to the linear analog (Fig. 5B and S1†). Interestingly, we found that the serum half-life of 2h was significantly prolonged (18-fold longer) while the helicity enhancement was moderate, which suggested that macrocyclization was the key to improving protease resistance. The location of the staple also influences the helicity as peptide 2j showed a better helical content than 2h (see Fig. S1† for the CD spectra and structure of 2j). Despite the fact that numerous cyclic peptides were shown to target the ER–coactivator binding region, the effect of these agents on ER-positive cell viability is not well explored. We therefore tested whether our vinyl sulfide stapled peptides, which bind to ER-α at around 100 nM, could inhibit the viability of MCF-7 cells using an MTT assay. Peptide 2h showed selective cytotoxicity towards ER-positive MCF-7 cells but not the ER-negative MDA-MD-231 cell line (Fig. 5C). The confocal microscopy images of the HEK293T cells treated with FAM-labeled peptide 2h-FAM displayed a diffuse cellular distribution. The majority of the peptide was localized in the cytoplasm and a small fraction was also detected in the nucleus (Fig. 5D). The moderate cellular activity observed in peptide 2h is likely to be attributed to the insufficient nuclear envelope penetration which is consistent with the immuno-staining of ER-α in MCF-7 cells where ER-α was mainly localized at the nucleus (Fig. S7†). Erythrocytes lysis generally precludes the direct administration of these agents intravenously and often enhances the toxicity when delivered via other routes.50,51 We therefore tested the influence of the all-hydrocarbon cross-link and the vinyl sulfide cross-link on the hemolytic activity of the peptides. Monosubstituted olefinic amino acids were used for constructing the all-hydrocarbon stapled peptides as the α-methyl group in the olefinic amino acid may not be essential.52 This platform allows us to solely compare how the cross-link itself influences the properties of peptides. Notably, in this case we observed substantially stronger hemolysis induced by the all-hydrocarbon stapled peptide compared to the vinyl sulfide analog. A lactate dehydrogenase (LDH) release assay was performed to measure the membrane integrity of Hela cells upon treatment with these agents. As expected, we observed notable LDH release at low concentrations of the all-hydrocarbon stapled peptide, which indicates the membrane disruptive effect of the highly lipophilic amphiphile compared to 2h (Fig. S8†).50 The membrane toxicity is associated with the overall lipophilicity of these agents. This data suggested that the vinyl sulfide cross-link provides alternative lipophilicity with a balance between cell permeability and membrane toxicity.
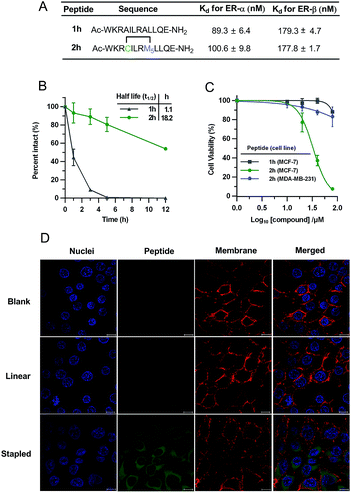 |
| | Fig. 5 Biological characterization of peptides targeting estrogen receptors. (A) FP assay of FAM labeled peptide 2h-FAM binding to ER-α and ER-β. mP, mean ± s. d. and n = 2. (B) Serum stability. Percentage intact, mean ± s. d. and n = 2. (C) Cell viability assay in ER-positive MCF-7 cells and ER-negative MDA-MB-231 cells. Percentage viability, mean ± s. d. and n = 3. (D) Confocal microscopy images of HEK293T cells treated with 5 μM FAM-labelled peptides (blank: DMSO; linear: 1h-FAM; stapled: 2h-FAM) at 37 °C for 3 h. Scale bar, 10 μm. | |
Conclusions
In conclusion, we demonstrated a facile stapling technique based on a one-component intramolecular thiol–yne hydrothiolation upon UV irradiation to constrain unprotected helical peptides. No metal catalyst is needed in this approach and the bio-orthogonal nature provides satisfying functional group tolerance for constraining peptides in their native form. As a proof of principle, we were able to stabilize helical peptides modulating intracellular estrogen receptor (ER)–coactivator interactions via this strategy. Furthermore, in contrast to the all-hydrocarbon stapled peptides constructed through olefin metathesis, the vinyl sulfide linkages show distinct lipophilicity with reduced membrane toxicity which is beneficial for clinical translation. In addition, the vinyl sulfide bond provides the opportunity to further functionalize the staple and we are now exploring a sequential thiol–yne/thiol–ene coupling for labelling and modulating peptide activity.20,40,53 In this regard, the thiol–yne stapling technique provides an alternative approach to access bioactive stabilized helical peptides. This thiol–yne chemistry could also provide rapid access to vinyl sulfide bonds mimicking the redox-sensitive disulfide bond with similar conformational properties.38 We envision that this peptide stapling technique will enrich the chemical toolbox of current stapling methodologies with unique properties and will revitalize the century-old chemistry of thiyl radical addition to alkynes towards up-to-date applications in the field of chemical biology.
Acknowledgements
We acknowledge financial support from National Natural Science Foundation of China (Grant 21102007 and 21372023), MOST 2015DFA31590, the Shenzhen Science and Technology Innovation Committee (SW201110018, SGLH20120928095602764, ZDSY20130331145112855 and JSGG20140519105550503) and the Shenzhen Peacock Program (KQTD201103). We thank Beijing NMR Center and the NMR facility of the National Center for Protein Sciences at Peking University. We thank Prof. Olaf Wiest for discussions on the manuscript.
Notes and references
- A. L. Hopkins and C. R. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS PubMed.
- G. Zinzalla and D. E. Thurston, Future Med. Chem., 2009, 1, 65–93 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 4130–4163 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- A. L. Jochim and P. S. Arora, Mol. BioSyst., 2009, 5, 924–926 RSC.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- Y. H. Lau, P. De Andrade, Y. T. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- G. H. Bird, W. C. Crannell and L. D. Walensky, Curr. Protoc. Chem. Biol., 2011, 3, 99–117 Search PubMed.
- J. W. Taylor, Biopolymers, 2002, 66, 49–75 CrossRef CAS PubMed.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
- N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS PubMed.
- A. M. Leduc, J. O. Trent, J. L. Wittliff, K. S. Bramlett, S. L. Briggs, N. Y. Chirgadze, Y. Wang, T. P. Burris and A. F. Spatola, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11273–11278 CrossRef CAS PubMed.
- S. Cantel, A. L. C. Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D'Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- S. A. Kawamoto, A. Coleska, X. Ran, H. Yi, C.-Y. Yang and S. Wang, J. Med. Chem., 2012, 55, 1137–1146 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, S. T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- Y. W. Kim, P. S. Kutchukian and G. L. Verdine, Org. Lett., 2010, 12, 3046–3049 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Drug Discovery Today: Technol., 2012, 9, e41–e47 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. K. Zou, J. J. Ling, H. T. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- Y. Wang and D. H. C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- R. E. Moellering, M. Cornejo, T. N. Davis, C. Del Bianco, J. C. Aster, S. C. Blacklow, A. L. Kung, D. G. Gilliland, G. L. Verdine and J. E. Bradner, Nature, 2009, 462, 182–188 CrossRef CAS PubMed.
- C. Phillips, L. R. Roberts, M. Schade, R. Bazin, A. Bent, N. L. Davies, R. Moore, A. D. Pannifer, A. R. Pickford, S. H. Prior, C. M. Read, A. Scott, D. G. Brown, B. Xu and S. L. Irving, J. Am. Chem. Soc., 2011, 133, 9696–9699 CrossRef CAS PubMed.
- C. J. Brown, S. T. Quah, J. Jong, A. M. Goh, P. C. Chiam, K. H. Khoo, M. L. Choong, M. A. Lee, L. Yurlova, K. Zolghadr, T. L. Joseph, C. S. Verma and D. P. Lane, ACS Chem. Biol., 2013, 8, 506–512 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. L. Cai, P. Berry, E. Feyfant, X. G. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- Y. W. Kim and G. L. Verdine, Bioorg. Med. Chem. Lett., 2009, 19, 2533–2536 CrossRef CAS PubMed.
- A. Y. L. Sim and C. Verma, J. Comput. Chem., 2015, 36, 773–784 CrossRef CAS PubMed.
- H. Chapuis, J. Slaninová, L. Bednárová, L. Monincová, M. Buděšínský and V. Čeřovský, Amino Acids, 2012, 43, 2047–2058 CrossRef CAS PubMed.
- T. T. T. Dinh, D. H. Kim, H. X. Luong, B. J. Lee and Y. W. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 4016–4019 CrossRef CAS PubMed.
- S. Bhattacharya, H. Zhang, D. Cowburn and A. K. Debnath, Biopolymers, 2012, 97, 253–264 CrossRef CAS PubMed.
- P. Johannesson, G. Lindeberg, A. Johansson, G. V. Nikiforovich, A. Gogoll, B. Synnergren, M. Le Greves, F. Nyberg, A. Karlen and A. Hallberg, J. Med. Chem., 2002, 45, 1767–1777 CrossRef CAS PubMed.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- A. Massi and D. Nanni, Org. Biomol. Chem., 2012, 10, 3791–3807 CAS.
- Y. M. Li, M. Pan, Y. T. Li, Y. C. Huang and Q. X. Guo, Org. Biomol. Chem., 2013, 11, 2624–2629 CAS.
- M. Minozzi, A. Monesi, D. Nanni, P. Spagnolo, N. Marchetti and A. Massi, J. Org. Chem., 2011, 76, 450–459 CrossRef CAS PubMed.
- V. Corce, L. McSweeney, A. Malone and E. M. Scanlan, Chem. Commun., 2015, 51, 8672–8674 RSC.
- A. A. Aimetti, R. K. Shoemaker, C. C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- C. Hoppmann, P. Schmieder, N. Heinrich and M. Beyermann, ChemBioChem, 2011, 12, 2555–2559 CrossRef CAS PubMed.
- B. J. Deroo and K. S. Korach, J. Clin. Invest., 2006, 116, 561–570 CAS.
- J. D. Norris, L. A. Paige, D. J. Christensen, C. Y. Chang, M. R. Huacani, D. J. Fan, P. T. Hamilton, D. M. Fowlkes and D. P. McDonnell, Science, 1999, 285, 744–746 CrossRef CAS PubMed.
- S. Fuchs, H. D. Nguyen, T. T. P. Phan, M. F. Burton, L. Nieto, I. J. de Vries-van Leeuwen, A. Schmidt, M. Goodarzifard, S. M. Agten, R. Rose, C. Ottmann, L. G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2013, 135, 4364–4371 CrossRef CAS PubMed.
- T. Phan, H. D. Nguyen, H. Goksel, S. Mocklinghoff and L. Brunsveld, Chem. Commun., 2010, 46, 8207–8209 RSC.
- K. Saar, M. Lindgren, M. Hansen, E. Eiriksdottir, Y. Jiang, K. Rosenthal-Aizman, M. Sassian and U. Langel, Anal. Biochem., 2005, 345, 55–65 CrossRef CAS PubMed.
- H. T. Chen, M. F. Neerman, A. R. Parrish and E. E. Simanek, J. Am. Chem. Soc., 2004, 126, 10044–10048 CrossRef CAS PubMed.
- D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131–9133 RSC.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supporting tables and figures. See DOI: 10.1039/c6sc00106h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence



