
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical control of neuronal firing via photoinduced electron transfer in donor–acceptor conjugates†
Yuta
Takano
a,
Tomohiro
Numata
bc,
Kazuto
Fujishima
a,
Kazuaki
Miyake
d,
Kazuya
Nakao
d,
Wesley David
Grove
a,
Ryuji
Inoue
b,
Mineko
Kengaku
*a,
Shigeyoshi
Sakaki
e,
Yasuo
Mori
*c,
Tatsuya
Murakami
a and
Hiroshi
Imahori
 *ad
*ad
aInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: kengaku@icems.kyoto-u.ac.jp
bDepartment of Physiology, Graduate School of Medical Sciences, Fukuoka University, Nanakuma 7-45-1, Johnan-ku, Fukuoka 814-0180, Japan
cDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: mori@sbchem.kyoto-u.ac.jp
dDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: imahori@scl.kyoto-u.ac.jp
eFukui Institute for Fundamental Chemistry, Kyoto University, Sakyo-ku, Kyoto 606-8103, Japan
First published on 17th February 2016
Abstract
A series of porphyrin–fullerene linked molecules has been synthesized to evaluate the effects of substituents and molecular structures on their charge-separation yield and the lifetime of a final charge-separated state in various hydrophilic environments. The selected high-performance molecule effectively achieved depolarization in a plasma cell membrane by visible light as well as two-photon excitation using a near-infrared light laser. Moreover, it was revealed that the depolarization can trigger neuronal firing in rat hippocampal neurons, demonstrating the potential and versatility for controlling cell functions using light.
Introduction
Exploring the use of excited-states of molecules has been an active area of research covering versatile fields of physics, chemistry, and biology. Molecular artificial photosynthesis and photovoltaics are two of the most representative, state-of-the-art examples in physics and chemistry.1–4 In biology, excited-states have been employed for imaging,5 sensing,6 and photothermal and photodynamic therapies.7,8 For instance, excited-states can react with oxygen via photoinduced energy-transfer (PEN) to generate singlet oxygen for cancer therapy.8 However, the inherent reactive nature of singlet oxygen often interrupts the other therapeutic effects caused by photoirradiation in biological systems.Photoinduced electron-transfer (PET) reactions between electron donors and acceptors involve generation of donor radical cations and acceptor radical anions. For instance, the corresponding holes and electrons are collected as photocurrent in photovoltaics, whereas further oxidation and reduction reactions by the ionic species lead to the formation of respective oxidized and reduced products in natural and artificial photosynthesis. This is in sharp contrast with PEN reactions, where PEN from energy donors to energy acceptors results in the formation of the excited-state of the energy acceptors. Since the energy level of the acceptor excited-state is typically higher than that of singlet oxygen,9 PEN reactions could be associated with cell damage. Nevertheless, biological applications with PET have been limited,10–12 and research efforts on the use of PET reactions have mainly focused on applications to chemical and electrical energy conversions.
To extract the full potential of PET reactions, it is essential to efficiently produce a long-lived, charge-separated state. However, molecular donor–acceptor (D–A) systems tend to form aggregates in highly polar solutions as well as in the solid state, which may reduce the net effect of the charge-separated states because of the cancellation and self-quenching of their excited-states by aggregation.13 Although a close correlation between the molecular structure and the net effect of charge-separated states in aggregates can be considered for energy and biological applications, such systematic studies have been limited.14
As a target for the control of cellular functions, the cell membrane potential, which is derived from the difference in charges between the outside and inside of the cell membrane, is one of the most attractive because it plays a key role in biological systems in the form of cell signaling.15 In particular, neural activity is precisely controlled by the membrane potential, which modulates neuronal firing to trigger the signaling and release of neurotransmitters.16,17
Here, we report the effects of substituents and molecular structures on the charge-separation (CS) yield and lifetime of a charge-separated state of D–A linked molecules in various hydrophilic environments to demonstrate a promising biological application for photocontrol of neuronal activity.
Results and discussion
Molecular design and synthesis
With the application to biological systems under hydrophilic environments in mind, a cationic moiety was introduced into a series of electron D–A linked molecules (Fig. 1). The hydrophilic moiety is also beneficial for the formation of aggregates in these environments because of the introduction of amphiphilicity into the molecules. First, a ferrocene–porphyrin–fullerene triad 1 was chosen because its analogue 5 is known to exhibit an extremely high CS yield (0.99) as well as slow charge recombination (CR) (1.1 × 105 s−1) in benzonitrile (PhCN).18,19 A porphyrin–fullerene dyad 2 without the ferrocene was also selected to assess the effects of the lifetime of the charge-separated state, taking into account the high CS yield (0.99) but fast CR (1.3 × 106 s−1) of its analogue 6 in PhCN.13 Finally, a triad 3 possessing bulky long alkoxy chains around the porphyrin core was designed to modulate its amphiphilicity and determine the effects of intermolecular π–π interactions between the porphyrin–porphyrin and/or porphyrin–fullerene moieties. As a reference compound, cationic porphyrin 4 was prepared. A mesityl acridinium ion D–A linked molecule 7 is known to show a high CS yield (0.98) and slow CR (2.4 × 105 s−1) in acetonitrile.20 Hence, 7 was also used to assess the advantage of the porphyrin–fullerene-linked molecules (1–3). All the new compounds were synthesized and characterized by standard methods, including 1H NMR and FT-IR spectroscopies, and high-resolution mass spectrometry (see ESI, Fig. S1–S4†). | ||
| Fig. 1 Molecular structures of the compounds 1–7. | ||
Characterization of the aggregates
Dynamic light scattering (DLS) measurements were carried out to characterize the aggregation behavior in highly polar solvents (Table 1 and Fig. S5†). The mean diameters of 1–3 in DMSO are 5–11 nm. The values are larger than the single molecular lengths (4–5 nm) (Fig. S6†), indicating the formation of extremely small aggregates. 1 and 2 exhibit smaller aggregate sizes (≤7 nm) than 3, suggesting that they form in a different manner to that of 3. In aqueous DMSO solutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99 DMSO:H2O, v/v, hereafter denoted as DMSO/H2O unless specified), the aggregate sizes of 1–4 and 7 are much larger (46–89 nm) than those in DMSO, reflecting the higher degree of aggregation in DMSO/H2O due to the hydrophobic nature of the molecules in the more polar solvent. The relatively large size distributions in DMSO/H2O imply non-uniform growth of the aggregates. Atomic force microscopy (AFM) measurements visualize the aggregates on mica from DMSO solutions (Fig. S7†). They are uniform particles whose sizes are comparable to those obtained from the DLS measurements on 1–3 in DMSO. The larger mean sizes of the aggregates of 4 and 7 than those from DLS measurements would be caused by the spin-coating process that leads to intense aggregation of the compounds on mica.
:99 DMSO:H2O, v/v, hereafter denoted as DMSO/H2O unless specified), the aggregate sizes of 1–4 and 7 are much larger (46–89 nm) than those in DMSO, reflecting the higher degree of aggregation in DMSO/H2O due to the hydrophobic nature of the molecules in the more polar solvent. The relatively large size distributions in DMSO/H2O imply non-uniform growth of the aggregates. Atomic force microscopy (AFM) measurements visualize the aggregates on mica from DMSO solutions (Fig. S7†). They are uniform particles whose sizes are comparable to those obtained from the DLS measurements on 1–3 in DMSO. The larger mean sizes of the aggregates of 4 and 7 than those from DLS measurements would be caused by the spin-coating process that leads to intense aggregation of the compounds on mica.
| Compound | Mean size in DMSOb (nm) | Mean size in DMSO/H2Ob,c (nm) | Mean size on micad (nm) |
|---|---|---|---|
|
a Values ± S.D.
b Data were obtained from DLS measurements based on number distribution.
c DMSO/H2O, 1:99, v/v.
d Data were obtained from AFM measurements and image analyses of the particles. The samples were prepared from DMSO solutions.
e Value was less than the lower limitation (1 nm) of the apparatus.
|
|||
| 1 | 7 ± 2 | 60 ± 49 | 9 ± 4 |
| 2 | 5 ± 2 | 46 ± 41 | 8 ± 4 |
| 3 | 11 ± 2 | 80 ± 21 | 10 ± 5 |
| 4 | <1e | 89 ± 53 | 6 ± 2 |
| 7 | <1e | 48 ± 13 | 2 ± 1 |
The UV-visible absorption spectra of 1–3 in DMSO display similar spectral shapes and peak positions, arising mainly from the zinc-porphyrin (ZnP) moiety (Table 2 and Fig. S8†). The spectra of 1–3 in DMSO/H2O are considerably broader than those in DMSO due to the increase in aggregation in the more polar solvent. More importantly, the degree of decrease in the molar extinction coefficient of 3 in DMSO/H2O is smaller than those of 1 and 2. This can be rationalized by the weaker intermolecular interactions in 3 than in 1 and 2, owing to the steric hindrance around the porphyrin with the long bulky alkoxy chains. The Soret-bands of 1 and 2 in DMSO/H2O are red-shifted in comparison with those in DMSO, whereas 3 shows a blue-shift. This reflects the significant differences in the electronic environment and the intermolecular interactions of the porphyrin moieties.
| Compound | Medium | λ Soret (nm) | ε Soret (105 L mol−1 cm−1) | λ Q (nm) | ε Q (104 L mol−1 cm−1) |
|---|---|---|---|---|---|
|
a The spectroscopic data of the Soret band.
b The spectroscopic data of the most intense Q band.
c 1:99, v/v.
|
|||||
| 1 | DMSO | 431 | 3.7 | 562 | 1.5 |
| DMSO/H2Oc | 434 | 1.6 | 562 | 1.2 | |
| 2 | DMSO | 431 | 4.2 | 562 | 1.7 |
| DMSO/H2Oc | 435 | 1.8 | 563 | 1.3 | |
| 3 | DMSO | 432 | 3.9 | 562 | 1.9 |
| DMSO/H2Oc | 427 | 2.3 | 556 | 1.6 | |
Next, the photoinduced excited states of the molecules were studied to reveal the photophysical properties of the aggregates. The steady-state fluorescence spectra of the porphyrin–fullerene-based molecules 1–3 in DMSO and DMSO/H2O show strong quenching of the porphyrin fluorescence in comparison with 4 (Fig. S9†). This can be attributed to the occurrence of efficient PET from the porphyrin excited singlet state (1ZnP*) to the C60, as demonstrated previously.17,21 The degree of decrease in the fluorescence of 3 in DMSO/H2O relative to DMSO is smaller than those of 1 and 2, which is consistent with the trend in the absorption spectra. The formation of the final charge-separated states in 1–3 and their reference compounds was studied using nanosecond time-resolved transient absorption (TA) measurements (Fig. 2 and S10–S13†). The characteristic peak at 1000 nm arising from a C60 radical anion (C60˙−) appears in 1–3, whereas the fingerprint of a ZnP radical cation (ZnP˙+) is visible solely in 2.18 Note that the ferrocenium ion (Fc+) was not detected due to its small molar extinction coefficient at 800 nm.18 The time profiles of the C60˙− signal at 1000 nm in DMSO revealed the existence of two components showing fast (kCR1) and relatively slow CR (kCR2) in 1–3 (Table 3 and Fig. S10†). The fast minor components of 1 and 3 have kinetics similar to 5 with the single component and thus can be attributed to the monomer-like state. The existence of the slow major components is rationalized by the stabilization of C60˙− assisted by the other C60 in the aggregates, as the formation of fullerene dimers or oligomers has been previously proposed.22–24 DFT calculations predict that the two C60 moieties in a dimer of 1 can interact strongly in the aggregates (Fig. S14†).
 | ||
| Fig. 2 Nanosecond time-resolved TA spectra of (a) 1, (b) 2 and (c) 3 in deaerated DMSO taken at 1.5 μs, 0.19 μs and 2.0 μs, respectively, after nanosecond laser excitation. The spectra were obtained by excitation at 410 nm where the absorbances were adjusted to be identical (0.50). | ||
| Compound | Medium | ε r | ΔODmax at 1000 nm | k CR1 (s−1) | Amplitude (%) | k CR2 (s−1) | Amplitude (%) | Φ CS |
|---|---|---|---|---|---|---|---|---|
| a Standard error of the data shown here is less than 5%. b Data from ref. 25. c Value determined by using the Bruggeman equation as described in ref. 26. d Value obtained by comparison of the maximum intensity at 1000 nm with 5 in PhCN taking into account the different molar extinction coefficients caused by the solvent systems (Table S4†). e Not determined because of the low s/n ratio. f Value obtained from the maximum intensity at 490 nm, arising from the corresponding radical ion pair. g Data from ref. 20. h Value obtained from the maximum intensity at 490 nm in comparison with 7 in MeCN taking into account the different extinction coefficients caused by the solvent systems. | ||||||||
| 1 | DMSO | 46.7 | 8.3 × 10−3 | 1.4 × 105 | 85 | 2.2 × 104 | 15 | 0.38d |
| DMSO/H2O (1:99) |
79.7c | n.d.e | ||||||
| Liposome in H2O | n.d.e | |||||||
| 2 | DMSO | 46.7 | 2.8 × 10−3 | 1.5 × 106 | 57 | 2.9 × 105 | 43 | 0.12d |
| DMSO/H2O (1:99) |
79.7c | n.d.e | ||||||
| Liposome in H2O | n.d.e | |||||||
| 3 | DMSO | 46.7 | 1.4 × 10−2 | 7.7 × 104 | 17 | 2.5 × 104 | 83 | 0.62d |
| DMSO/H2O (1:99) |
79.7c | 5.5 × 10−3 | 2.6 × 106 | 66 | 1.6 × 105 | 34 | 0.57d | |
| Liposome in H2O | 2.3 × 10−3 | 5.9 × 105 | 68 | 2.2 × 104 | 32 | 0.27d | ||
| 7 | MeCN | 37.5 | 2.8 × 10−2 | 2.4 × 105 | 100 | 0.98g | ||
| DMSO/H2O (1:99) |
79.7 | 4.2 × 10−3 | 3.5 × 104 | 100 | 0.37h | |||
A notable feature of 3 is the largest intensity of ΔOD at 1000 nm (ΔODmax), corresponding to the CS yield (2 (0.12) < 1 (0.38) < 3 (0.62) in DMSO; 1 and 2 (<0.01) < 3 (0.57) in DMSO/H2O) on the measurement time scale. Since the 1ZnP* state is quenched efficiently by the C60 moiety via PET,18 the porphyrin-excited triplet state (3ZnP*) does not contribute to the CS yield. Note that we might miss a fast partial CR from the singlet ZnP˙+–C60˙− states of 1 and 2 occurring in the picosecond region,27 which would account for the low CS yields relative to 3. Nevertheless, the highest CS yield of 3 in the polar solvents can be attributed mainly to the suppression of cancellation of the charge-separated states (vide infra) as well as self-quenching in the aggregates due to the long bulky alkoxy chains around the porphyrin moiety. The decay of the porphyrin fluorescence of 1–3 in DMSO was fitted by two components (Table S1†). The slow minor component (520–1100 ps) may originate from an impurity or degradation during the measurements. The lifetimes (50–80 ps) of the fast major components are close to those of 5 (95 ps) and 6 (100 ps) in PhCN,18 but they are shorter. This suggests the involvement of aggregation quenching, even if the size of the aggregates in DMSO is small. The fluorescence lifetimes of the short components in DMSO are in the order of 3 (54 ps) < 1 (67 ps) < 2 (77 ps). Although the trend in the CS yields apparently agrees with that in the fluorescence lifetimes, conventionally corresponding to the CS yields, the difference in the CS yields may mainly result from the degree of self-quenching in the aggregates, cancellation of the charge-separated state in the aggregates, and plausible fast partial CR from the singlet ZnP˙+–C60˙− state occurring in the picosecond region.27
Solvent dependent properties
To clarify the solvent dependence of the aggregates, steady-state and TA spectra were recorded in various solvent systems with different dielectric constants (Table S2 and Fig. S15–20†). The addition of PhCN to the DMSO solutions of 1–3 reveals unique changes in their molar extinction coefficients of the characteristic peaks (Fig. S15, S16 and Table S3†). Both 2 and 3 show an increase in the extinction coefficients of the Soret bands with the addition of PhCN. In contrast, 1 does not exhibit a simple trend. Because 1–3 possess amphiphilic properties, their solvation patterns and structures in the solvents are affected by the balance of the hydrophilic and hydrophobic parts of the molecules. The observed trend suggests that 2 and 3 show a simple correlation with the dielectric constant, whereas 1 does not follow this trend in the less polar solvent systems. Meanwhile, the addition of water to the DMSO solution drastically decreases the molar extinction coefficients of 1 and 2 due to the susceptibility of the porphyrin unit with the 3,5-di-tert-butyl phenyl groups to water. By contrast, that of 3 decreased gradually owing to the protection of the porphyrin core from water by the surrounding long alkoxy chains.The superiority of 3 is evident from the TA spectra in the various solvents. The highest CS yields among 1–3 are attained for 3, irrespective of the dielectric constants (Table S2†). It is noteworthy that the CS yield of 3 in PhCN is comparable to that of 5 (0.99) in PhCN. Therefore, 3 is expected to become a basal molecule for utilizing CS in various environments.
Theoretical calculations
DFT calculations were also performed to shed light onto the intermolecular interactions in the aggregates. For a systematic study, the C60 (8), porphyrin (9 possessing tert-butyl groups and 10 possessing alkoxy groups), and ferrocene (11) units were chosen for the calculations (Fig. S21–S24 and Tables S5–S7†). The binding energies of the dimers of the porphyrins (hereafter denoted as 9/9 and 10/10) were found to be larger than those of the other combinations (8/8, 8/9, 8/10, 8/11 and 11/11) (Table S5†), contributing to the formation of the aggregates in the polar solvents. In particular, the binding energy of the dimer of the porphyrins with tert-butyl groups (9/9) is larger than that with alkoxy groups (10/10), rationalizing the more intense aggregation tendencies of 1 and 2 than that of 3 (Table S5†). The solvent effect does not have an influence on the order of the binding energies, which parallels with the similar trend in DMSO and DMSO/H2O. The intermolecular interactions in the charge-separated states were also evaluated. The stabilization of the C60˙− unit and the porphyrin˙+ with tert-butyl groups (8•−/9•+) is the largest among the formable combinations in the charge-separated states (8•−/8•−, 8•−/9•+, 8•−/11•+, 9•+/9•+ and 11•+/11•+) (Tables S6 and S7†). This is in accordance with the more likely occurring cancellation of the charge-separated states of 2 in the aggregates by the change in the molecular orientation in the aggregates (vide supra). Taking everything into consideration, the calculation results support the observed highest CS yield of 3.Application for biological membrane systems
Next, we investigated the effects on membrane potential to explore the potential biological application of the charge-separated states of 1–3 and 7. We envisioned that the membrane potential would be affected by the charge-separated states of 1–3 and 7 that are equivalent to the large electric field on the nanometer scale.19,28 The compounds were delivered to PC12 cells by adding their DMSO/H2O solution, followed by incubation for 3 min. The incorporation of 1–3 into the surface membrane was successfully visualized by confocal laser scanning microscopy (Fig. 3a and S26†). The inhomogeneous distribution suggests that the aggregates of 1–3 are retained in the cell membrane. In contrast, 7 showed localization inside of the cells instead of in the surface membrane (Fig. 3b). Although 7 showed comparable CR and CS yield compared to those of 3 in polar environments (Table 3 and Fig. S13†), the observed localization of 7 persuaded us to abandon the investigation of its direct effects on the membrane potential in the present study. Nevertheless, further elaboration of the design of 7 would fulfill the requirements for future consideration. | ||
| Fig. 3 Confocal microscopy images of PC12 cells that were stained using wheat germ agglutinin (WGA)–Alexa Fluor®488 conjugate and treated with (a) 3 and (b) 7, where the red bars denote 5 μm. Merged image (left), compounds (magenta, center), and the Alexa Fluor488 conjugate (green, right). | ||
Photophysical measurements in the lipid bilayer were conducted to shed light on the final charge-separated states of the aggregates in the cell membrane. First, we attempted to measure the TA spectra of the aggregates in PC12 cells. Unfortunately, intense aggregation of the PC12 cells hampered the measurements. Then, we applied an artificial lipid bilayer system (COATSOME® EL-11-A) to evaluate the D–A properties in a hydrophobic membrane environment. The incorporation of 1–3 into the liposome was complete in 1 h. The UV-vis absorption spectra show the broadening and red-shift of the absorptions as a result of the incorporation (Fig. S27†). The TA spectrum of the 3-incorporated liposome exhibits a distinct contrast from those with 1 and 2. The characteristic peak derived from C60˙− is evident solely for 3, confirming the more efficient formation of the charge-separated state of 3 than of 1 and 2 even in the hydrophobic membrane environment (Fig. S12b and d,†Table 3). This behavior is rather close to that observed in DMSO/H2O (1:99, v/v). Moreover, the CR of 3 (2.2 × 104 s−1) in the liposome is the slowest among the present solvent systems. The dielectric constant of the lipid bilayer is generally estimated to be ca. 2.29 In low polarity environments the energy levels of the charge-separated states of 1–3 may be higher than the excited-states of the porphyrin and the C60 moieties, resulting in faster CR than in PhCN due to the rapid decay to the excited-states.30 The observed slow CR would reflect the dynamic motion of the bilayer at room temperature causing communication between the surface water molecules and inner compounds.31 Overall, it can be concluded that the CS property in aqueous solutions would provide useful insight to predict the CS property in the lipid bilayer.
Photoinduced changes in the membrane potential were recorded using patch clamp techniques (Fig. 4a–c). When PC12 cells were treated with 1–3, depolarizations occurred under illumination with light (400–450 nm) over time, reaching a constant membrane potential. Meanwhile, the illumination of PC12 cells in the absence of 1–3 did not result in a change in the membrane potential (Fig. S28†). It is noteworthy that the amplitude of the depolarizations is in the order of 2 < 1 < 3 (Fig. 4d). Moreover, it was found that 3 can induce a one and a half times larger depolarization than 1. This order parallels that of the CS yields in DMSO, DMSO/H2O and even in the liposome, rather than the lifetime of the charge-separated state, suggesting the importance of the CS yield for the depolarization. In terms of the nanoscale electric fields that are induced by the CS, one can expect a comparable magnitude of the electric fields of 1 and 3 based on the same edge-to-edge distance (3.9 nm) between the radical ion pairs (Fig. S6†). Thus, the experimental result also supports the importance of the CS yield for the depolarization. Although the shorter edge-to-edge distance (2.5 nm) of the charge-separated state of 2 can generate a 1.56 times larger electric field than those of 1 and 3, the smallest depolarization is observed in 2. These comparisons indicate that the CS yield and the lifetime of the final charge-separated state rather than the magnitude of the electric field have a large impact on the depolarization. We also confirmed that the depolarization does not correlate with the order of the reactive oxygen species (ROS) generation ability, cell death inducing ability (Fig. S29†) and number of incorporated molecules per single cell (Table S8†). This ensures that the depolarization is not caused by membrane damage.32 Therefore, it can be concluded that the generation of charge-separated states with high CS yields is a key to achieving large photoinduced depolarization.
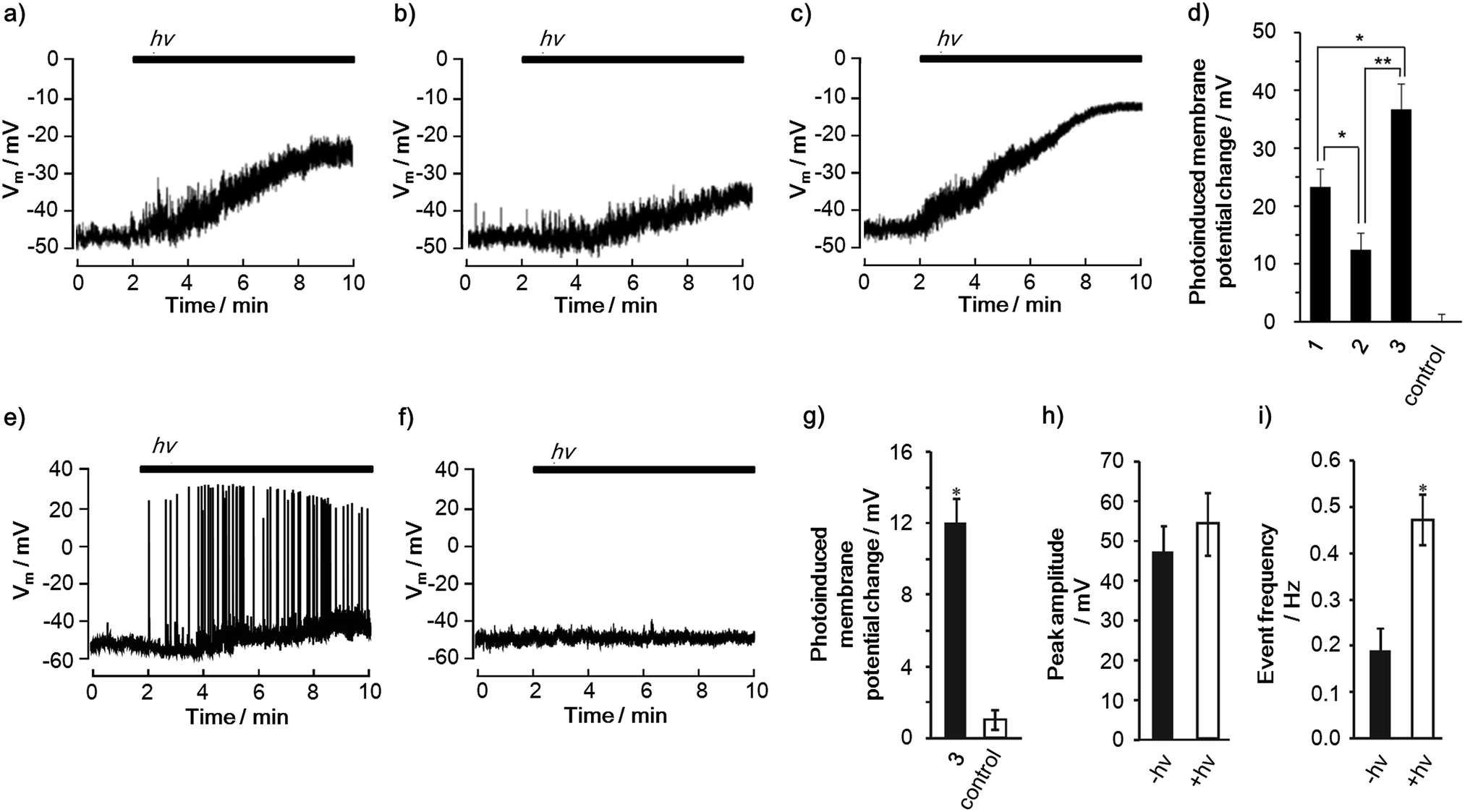 | ||
| Fig. 4 Incorporation of the CS molecules into mammalian cells and photoinduced change of cell membrane potentials. Representative traces of photoinduced depolarization in a PC12 cell incorporated with (a) 1 (n = 10), (b) 2 (n = 10) and (c) 3 (n = 7), and (d) degrees of change in the membrane potential of PC12 cells after 8 min of illumination (240 mJ). Photoinduced depolarization and neuronal firing in a rat hippocampal neuron (e) incorporated with 3 (n = 10), (f) negative control without any compounds (n = 8), (g) degrees of change in the membrane potentials of the neurons after 8 min of illumination (240 mJ cm−2), (h) peak amplitude and (i) event frequency of the action potentials before (−hv) and during (+hv) the illumination, obtained using patch clamp techniques (400–450 nm, 2.0 mW cm−2). Error bars indicate S.D. Statistically significant differences between the negative control and each compound are indicated with asterisks (**P < 0.01, *P < 0.05). | ||
The use of porphyrins as a sensitizer enables us to utilize a non-linear optical response, i.e. two-photon excitation,33 using a laser of near-infrared (NIR) light, which possesses less photocytotoxicity and higher penetrability in tissues than blue light. 3 can be excited by two-photon absorption as evidenced by its excitation wavelength-dependent fluorescence (Fig. S30a†). Consequently, it was found that the depolarization of the cell membrane can be induced by a NIR pumped pulsed laser at 860 nm in the presence of 3 (Fig. S30c†).
Furthermore, the triad 3 exemplified its ability to change the cell membrane potential of a primary cell. In rat hippocampal neurons, 3 successfully led to the photoinduced depolarization of the cell membrane (Fig. 4e–g). Remarkably, the depolarization was accompanied by an increase in the frequency of the action potentials because of the change in the basal membrane potential toward neuronal firing (Fig. 4h and i).34 Since action potentials are one of the most important neural activities, i.e., cell signaling and gene expression,16,173 is a promising molecule for controlling neuronal cellular functions using light. These results corroborate the potential and versatility of the porphyrin-based CS molecules for controlling cell functions using light.
Conclusions
To extract the full potential of PET toward practical biological applications, the effects of the substituents and molecular structures of electron D–A linked molecules on their final CS yield and lifetime of the charge-separated state have been examined in biological hydrophilic environments. After exploration, the selected high-performance CS molecule successfully achieved induction of neuronal firing by illumination. The results are beneficial not only for understanding the effects on the CS parameters closely associated with rational molecular design, but also for realizing photocontrol of cellular functions using D–A linked molecules, which could become a new indispensable tool for phototherapy.Acknowledgements
This work is supported by Grants-in-Aid for Scientific Research (S) (25220801), (C) (15K05563), and Grants-in-Aid for Young Scientists (B) (25810098), MEXT, Japan. The iCeMS is supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan. The CeMI is acknowledged for the microscopic measurements. We thank Prof. Nikolai V. Tkachenko and Ms Kati Stranius (Tampere University of Technology, Finland) for conducting the fluorescence lifetimes measurements and the discussion of the photophysical measurements.Notes and references
- T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- J. S. Wu, W. M. Liu, J. C. Ge, H. Y. Zhang and P. F. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- M. Ethirajan, Y. H. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- T. Gunnlaugsson, H. D. P. Ali, M. Glynn, P. E. Kruger, G. M. Hussey, F. M. Pfeffer, C. M. G. dos Santos and J. Tierney, J. Fluoresc., 2005, 15, 287–299 CrossRef CAS PubMed.
- H. Takakura, R. Kojirna, M. Kamiya, E. Kobayashi, T. Komatsu, T. Ueno, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4010–4013 CrossRef CAS PubMed.
- J. G. Rohan, Y. R. Citron, A. C. Durrell, L. E. Cheruzel, H. B. Gray, R. H. Grubbs, M. Humayun, K. L. Engisch, V. Pikov and R. H. Chow, ACS Chem. Neurosci., 2013, 4, 585–593 CrossRef CAS PubMed.
- S. Fukuzumi, R. Hanazaki, H. Kotani and K. Ohkubo, J. Am. Chem. Soc., 2010, 132, 11002–11003 CrossRef CAS PubMed.
- E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. J. Chen, F. Würthner, P. Marsal, D. Beljonne, J. Cornil and R. A. J. Janssen, J. Am. Chem. Soc., 2006, 128, 649–657 CrossRef CAS PubMed.
- J. Haag and A. Borst, J. Neurosci., 1998, 18, 7972–7986 CAS.
- R. D. Fields, F. Eshete, B. Stevens and K. Itoh, J. Neurosci., 1997, 17, 7252–7266 CAS.
- M. S. Santos, A. J. Moreno and A. P. Carvalho, Stroke, 1996, 27, 941–950 CrossRef CAS PubMed.
- H. Imahori, K. Tamaki, D. M. Guldi, C. P. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS PubMed.
- T. Numata, T. Murakami, F. Kawashima, N. Morone, J. E. Heuser, Y. Takano, K. Ohkubo, S. Fukuzumi, Y. Mori and H. Imahori, J. Am. Chem. Soc., 2012, 134, 6092–6095 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- H. Imahori, H. Yamada, Y. Nishimura, I. Yamazaki and Y. Sakata, J. Phys. Chem. B, 2000, 104, 2099–2108 CrossRef CAS.
- D. P. Voronin, A. S. Buchelnikov, V. V. Kostjukov, S. V. Khrapatiy, D. Wyrzykowski, J. Piosik, Y. I. Prylutskyy, U. Ritter and M. P. Evstigneev, J. Chem. Phys., 2014, 140, 104909 CrossRef PubMed.
- K. G. Thomas, V. Biju, D. M. Guldi, P. V. Kamat and M. V. George, J. Phys. Chem. B, 1999, 103, 8864–8869 CrossRef CAS.
- T. E. Shubina, D. I. Sharapa, C. Schubert, D. Zahn, M. Halik, P. A. Keller, S. G. Pyne, S. Jennepalli, D. M. Guldi and T. Clark, J. Am. Chem. Soc., 2014, 136, 10890–10893 CrossRef CAS PubMed.
- Recommended Reference Materials for the Realization of Physicochemical Properties, ed. K. N. Marsh, Blackwell Scientific Publications, Oxford, 1987 Search PubMed.
- J. F. Lou, T. A. Hatton and P. E. Laibinis, J. Phys. Chem. A, 1997, 101, 5262–5268 CrossRef CAS.
- T. Higashino, T. Yamada, M. Yamamoto, A. Furube, N. V. Tkachenko, T. Miura, Y. Kobori, R. Jono, K. Yamashita and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 629–633 CrossRef CAS PubMed.
- D. Gosztola, H. Yamada and M. R. Wasielewski, J. Am. Chem. Soc., 1995, 117, 2041–2048 CrossRef CAS.
- W. Huang and D. G. Levitt, Biophys. J., 1977, 17, 111–128 CrossRef CAS PubMed.
- M. Fujitsuka, O. Ito, H. Imahori, K. Yamada, H. Yamada and Y. Sakata, Chem. Lett., 1999, 28, 721–722 CrossRef.
- F. Zhou and K. Schulten, J. Phys. Chem., 1995, 99, 2194–2207 CrossRef CAS.
- G. Stark, J. Membr. Biol., 2005, 205, 1–16 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- D. Fricker, J. A. H. Verheugen and P. Miles, J. Physiol., 1999, 517, 791–804 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all new compounds, supplementary characterizations, and optimized structures by theoretical calculations. See DOI: 10.1039/c5sc04135j |
| This journal is © The Royal Society of Chemistry 2016 |