
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heteronanowires of MoC–Mo2C as efficient electrocatalysts for hydrogen evolution reaction†
Huanlei
Lin
a,
Zhangping
Shi
b,
Sina
He
a,
Xiang
Yu
ac,
Sinong
Wang
b,
Qingsheng
Gao
*a and
Yi
Tang
*b
aDepartment of Chemistry, Jinan University, Guangzhou 510632, China. E-mail: tqsgao@jnu.edu.cn
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Laboratory of Advanced Materials and Collaborative Innovation Center of Chemistry for Energy Materials, Fudan University, Shanghai 200433, China. E-mail: yitang@fudan.edu.cn
cAnalytic and Testing Centre, Jinan University, Guangzhou 510632, China
First published on 12th February 2016
Abstract
Exploring efficient noble-metal free electrocatalysts for the hydrogen evolution reaction (HER) is one of the most promising pathways for facing the energy crisis. Herein, MoC–Mo2C heteronanowires composed of well-defined nanoparticles were accomplished via controlled carbonization, showing excellent HER activity, fast kinetic metrics and outstanding stability in both acid and basic electrolytes. In particular, the optimal one consisting of 31.4 wt% MoC displayed a low overpotential (η10 = 126 and 120 mV for reaching a current density of −10 mA cm−2), a small Tafel slope (43 and 42 mV dec−1) and a low onset overpotential (38 and 33 mV) in 0.5 M H2SO4 and 1.0 M KOH, respectively. Such prominent performance, outperforming most of the current noble-metal free electrocatalysts, was ascribed to the carbide surface with an optimized electron density, and the consequently facilitated HER kinetics. This work elucidates a feasible way toward efficient electrocatalysts via heteronanostructure engineering, shedding some light on the exploration and optimization of catalysts in energy chemistry.
Introduction
The rapid growth of global energy consumption and the associated environmental issues have triggered an urgent demand for renewable and clean energy sources.1,2 Hydrogen (H2) is a promising candidate as it stores energy from renewable sources (e.g., sunlight and wind) into the chemical bond via the electrolysis of water, which then can be released through the reverse reaction in fuel cells on demand.3 The hydrogen evolution reaction (HER) via water electrolysis essentially depends on the efficiency of electrocatalysts, which must be stable and capable of reducing water rapidly at potentials close to its thermodynamic value.4,5 Although noble metals, e.g., platinum, show high activity, they are severely limited by their high cost and low abundance.6,7 It is urgently demanded to develop noble-metal free catalysts with good activity, long-term stability, high element-abundance, and economical cost.5,8–10Remarkable advances have been recently made regarding the use of transition-metals and their carbides, nitrides, chalcogenides and phosphides.5,8,10,11 Presenting varied electronic features and catalytic properties related to tunable phases and composition,12–15 molybdenum carbides (MoCx) have received special attention as one of the promising noble-metal free catalysts. Among them, Mo2C demonstrates the best performance because of its electron configuration around the Fermi level (EF).13,16 Intense effort has been devoted to Mo2C nanostructures with enriched active-sites,17–26 and composites integrating a conducting matrix, e.g., carbon nanotubes (CNTs) and graphene (GR).27–31 However, the negative hydrogen-binding energy (ΔGH*) on Mo2C indicates a strong adsorption of H on the Mo2C surface, which benefits H+ reduction (i.e., Volmer step), but restricts Hads desorption (i.e., the Heyrovsky/Tafel step).16,32 Thus, an optimization of the electronic features are desired. The introduction of doping elements has even been employed,33,34via which the improvement is however limited due to inadequate modification and inevitable structure damage. It is notable that the electron density around the Mo active-sites mostly relies on the carbon in the lattice,35,36 which will be reduced with increasing C because of the electron-transfer from Mo to C.14 For example, with a high C content, MoC usually presents weaker hydrogen binding in comparison with that on Mo2C, and consequently a facilitated Heyrovsky/Tafel step, but a hindered Volmer reaction.13,29 Regarding the respectively promoted elementary reactions of HER on Mo2C and MoC, it's promising to achieve a synergistically-enhanced activity on MoC–Mo2C interfaces, which are rarely reported to the best of our knowledge.
Herein, we report novel MoC–Mo2C heteronanowires (HNWs) as efficient HER electrocatalysts, which are fabricated from MoOx–amine nanowires (NWs) via controlled carbonization. The HNWs denoted as MoC–Mo2C-n (where n refers to the MoC weight percentage) are one-dimensional (1D) heterostructures composed of defined nanoparticles (NPs), with rich nanoporosity, large surface area, and more importantly a tunable composition. This is remarkably improved from our previous work on nanoporous Mo2C,21 highlighted by the effective electron regulation and further improved activity via varying MoC/Mo2C in the HNWs. With an optimal composition, MoC–Mo2C-31.4 exhibits a low η10 (overpotential required to reach a current density of −10 mA cm−2) of 126 mV, a low Tafel slope of 43 mV dec−1, and a low ηonset (overpotential referring to the beginning of the linear regime in the Tafel plot) of 38 mV in 0.5 M H2SO4, outperforming most of the current noble-metal free electrocatalysts. The high HER activity should be ascribed to the moderated electron density on the carbide surface, which optimizes the hydrogen-binding and thus the HER kinetics. In addition, the good efficiency in basic electrolyte further verifies MoC–Mo2C HNWs as promising noble-metal free electrocatalysts.
Results and discussion
As shown in Fig. 1a, a series of MoCx HNWs can be achieved via the controlled carbonization of various MoOx–amine precursors (Table S1†), which were firstly fabricated through reacting molybdate with aniline (An) or p-methylaniline (MeAn). The wire-like precursors (Fig. S1†) obtained with An at pH 4.0 (MoAn-4.0) and 3.5 (MoAn-3.5) were respectively confirmed as Mo3O10(C6H8N)2·2H2O (JCPDS no. 50-2402) and its mixture with Mo8O25(C6H8N)2·2H2O (JCPDS no. 49-2068), using X-ray diffraction (XRD, Fig. S2†). For those obtained with MeAn (MoMeAn-4.0), a similar XRD pattern with an obvious shift to lower degree values suggests an analogous crystalline structure with an expanded lattice due to the large MeAn molecule. Their composition was further evidenced using Fourier transform infrared (FT-IR), thermogravimetric analysis coupled with differential scanning calorimetry (TGA/DSC), and CHN elemental analysis (Fig. S3†). Obviously, the different carbon content will benefit the controlled synthesis of MoCx.37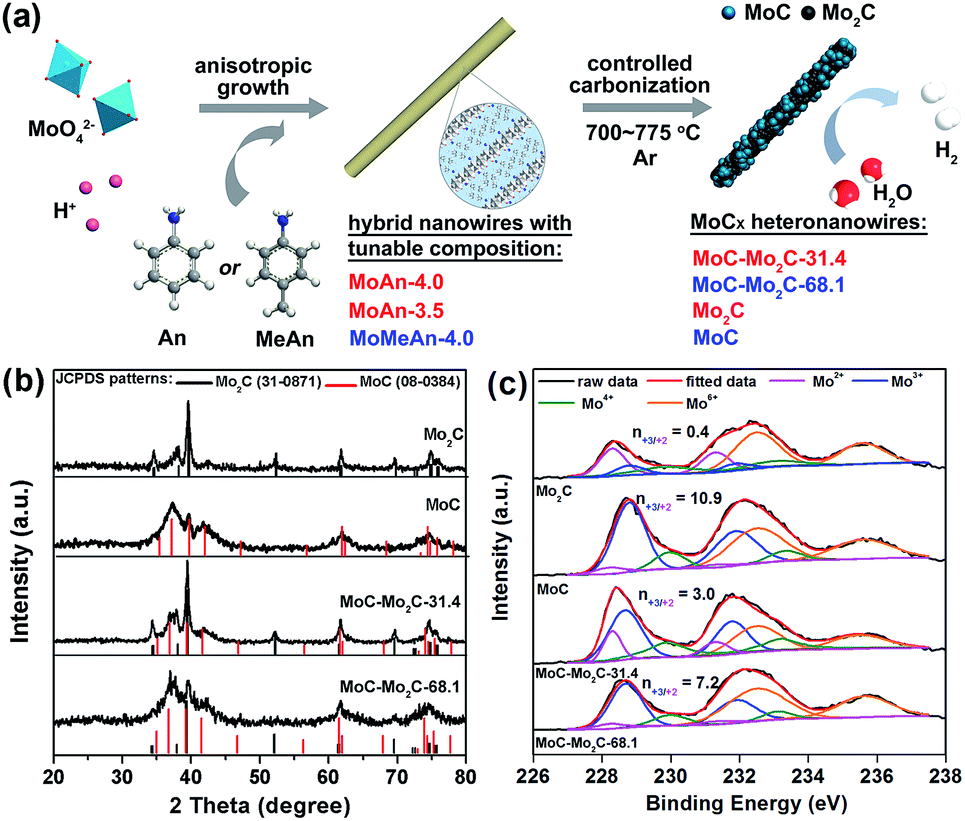 | ||
| Fig. 1 (a) Schematic illustration for the fabrication of MoCx HNWs from MoOx–amine NWs with tunable composition. (b) XRD patterns and (c) Mo 3d XPS profiles of the as-obtained MoCx NWs. | ||
The XRD investigation clearly confirms the achievement of various MoCx (Fig. 1b), whose composition was further determined through the combined measurements of XRD, CHN elemental analysis and inductively coupled plasma (ICP) (Table S2†). The product α-Mo2C (JCPDS no. 31-0871) was obtained from calcining MoAn-3.5 at 775 °C, and η-MoC (JCPDS no. 08-0384) was obtained from MoMeAn-4.0 at 700 °C. The heterostructures of MoC–Mo2C-31.4 and MoC–Mo2C-68.1 were harvested from MoAn-4.0 and MoMeAn-4.0, respectively, at 775 °C. As expected, the carbon source in the hybrid precursors contributes to the tailored generation of carbides. The higher carbon content of MoAn-4.0 (22.9%) compared to that of MoAn-3.5 (20.7%) benefits the formation of some MoC in Mo2C, and having sufficient carbon (25.3%) in MoMeAn-4.0 leads to the pure phase of MoC.
These samples were further analyzed using X-ray photoelectron spectroscopy (XPS, Fig. 1c). The peak fitting of Mo 3d profiles suggests that there are four states (+2, +3, +4 and +6) for Mo on the surface.17,38 The Mo4+ and Mo6+ species result from the inactive MoO2 and MoO3, respectively, which are commonly observed as carbides are exposed to air.39 We focus on the Mo2+ and Mo3+ species with peaks at 228.0–229.0 eV (Mo 3d5/2) and 231.0–232.0 eV (Mo 3d3/2), because they are the active centres for electrocatalytic HER.13,17 The Mo3+/Mo2+ mole ratios (n3+/2+) on the MoCx surface can provide useful information to understand the nature of the active-sites (Table S3†). The n3+/2+ values for Mo2C and MoC are 0.4 and 10.9 (Fig. 1c), respectively, which suggests that Mo2+ is dominant to Mo3+ on Mo2C, while Mo3+ is prevailing on MoC. In the heterostructures, n3+/2+ visibly changed to 3.0 for MoC–Mo2C-31.4, and 7.2 for MoC–Mo2C-68.1. Such a variation of Mo3+/Mo2+ will influence the HER activity, related to the different electron density around Mo3+ and Mo2+.13
Meanwhile, the Raman spectra of the above MoCx samples display the D- and G-bands of carbon at 1350 and 1590 cm−1, respectively, confirming the presence of free carbon (Fig. S4†).25 In addition, N2 isothermal sorption reveals the large surface of the MoCx NWs (Fig. S5†). Particularly, MoC–Mo2C-31.4 HNWs present a specific surface area of 58.5 m2 g−1, larger than that of Mo2C (39.3 m2 g−1), MoC–Mo2C-68.1 (33.7 m2 g−1) and MoC (26.0 m2 g−1). A major pore distribution at around 5.5 nm is observed for Mo2C and MoC–Mo2C-31.4.
Taking MoC–Mo2C-31.4 as the model sample, the heteronanowires can be well confirmed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Wire-like products several micrometres in length and 80–150 nm in width are observed in Fig. 2a, maintaining the 1D morphology of the precursors. TEM further displays that such NWs are composed of NPs (∼10 nm), and the selected area electron diffraction (SAED) pattern corresponds to those of α-Mo2C and η-MoC (Fig. 2b). Accordingly, the (121) and (021) lattice fringes of α-Mo2C and the (101) and (006) fringes of η-MoC are identified in the high resolution TEM (HR-TEM, Fig. 2c). Noticeably, the interfaces between close-stacking MoC and Mo2C NPs are visible, which would benefit synergy of the surface activity. Analogously, the Mo2C and MoC NWs composed of the corresponding NPs are also verified through the TEM investigation, and in the MoC–Mo2C-68.1 HNWs, both the MoC and Mo2C NPs are identified (Fig. S6†). With nanosized crystallites, enriched nanoporosity, large surface areas, a conducting carbon matrix, and more importantly tunable Mo3+/Mo2+ centres, MoC–Mo2C HNWs are expected to efficiently catalyse the HER.
 | ||
| Fig. 2 (a) SEM, (b) TEM and (c) HR-TEM images of MoC–Mo2C-31.4 HNWs. Inset of (b) is the SAED pattern obtained on the marked area. | ||
To investigate the HER performance in an acidic electrolyte, the as-prepared MoCx NWs were loaded onto glassy carbon electrodes (GCEs) with a mass loading of 0.14 mg cm−2. Fig. 3a displays their polarization curves with iR-drop corrections in 0.5 M H2SO4, along with that of the benchmark Pt/C catalyst (40 wt% Pt on carbon black from Johnson Matthey) for reference. Among the MoCx catalysts, MoC–Mo2C-31.4 exhibits the highest activity. To achieve a current density (j) of −10 mA cm−2, MoC–Mo2C-31.4 requires a η10 of 126 mV, which is obviously lower than those of α-Mo2C (182 mV) and η-MoC (232 mV). This suggests a synergic enhancement between Mo2C and MoC in the HNWs. Meanwhile, the mechanically-mixed MoC–Mo2C NWs with a similar MoC content of 30 wt% (denoted as MoC–Mo2C-30 (mixed)) displayed a lower activity (η10 = 222 mV), indicating that the MoC–Mo2C interfaces on the nanoscale in MoC–Mo2C-31.4 contribute to the efficient HER. Such a synergic effect is prohibited by the high percentage of MoC in the HNWs, and as this was increased to 68.1%, the activity obviously reduced. A summary of the HER activity of the above MoCx is listed in Table 1.
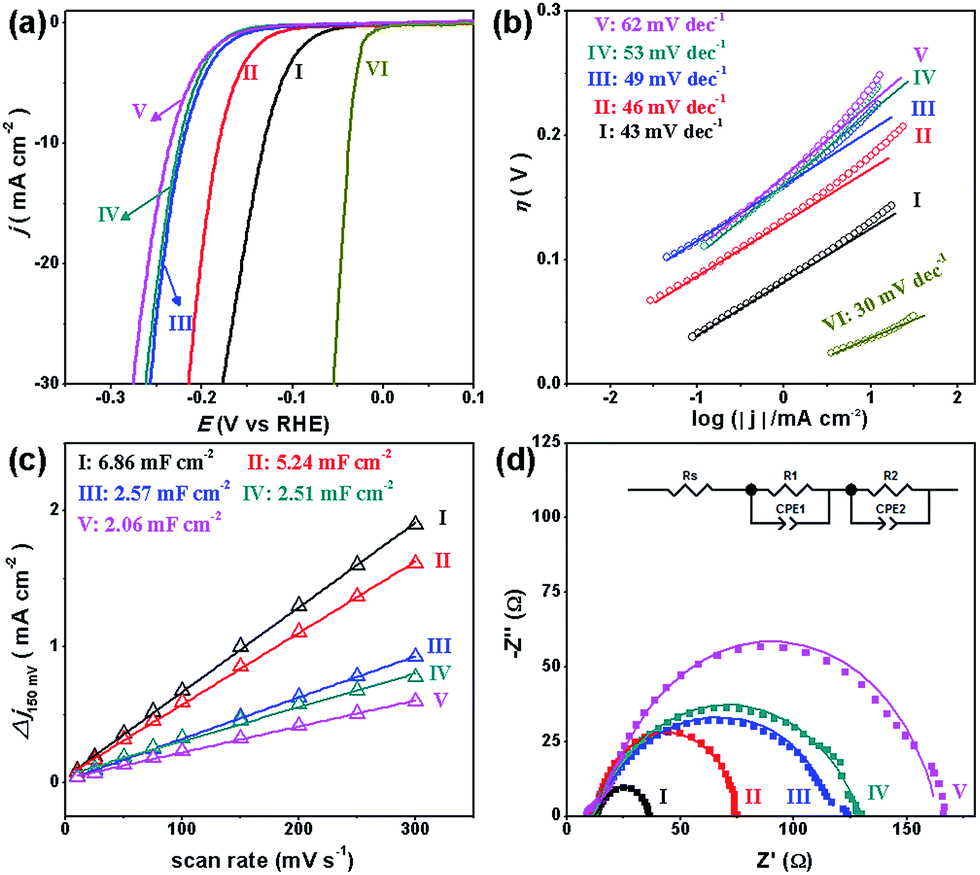 | ||
| Fig. 3 (a) Polarization curves and (b) Tafel plots for the HER on modified GCEs comprising (I) MoC–Mo2C-31.4, (II) Mo2C, (III) MoC–Mo2C-68.1, (IV) MoC–Mo2C-30 (mixed), (V) MoC, and (VI) commercial Pt/C in 0.5 M H2SO4. (c) Estimation of Cdl through plotting the current density variation (Δj = (ja − jc)/2, at 150 mV vs. RHE; data obtained from the CV in Fig. S7†) against scan rate to fit a linear regression, and (d) Nyquist plots (at η = 200 mV) of the above MoCx electrocatalysts. | ||
| Cat. | η 10 (mV) | η onset (mV) | Tafel slope (mV dec−1) | R ct (Ω) | C dl (mF cm−2) | j 0 (mA cm−2) |
|---|---|---|---|---|---|---|
| a Data was measured at η = 200 mV. b Data was calculated according to the CV results (Fig. S7). c Exchange current densities (j0) were obtained from Tafel curves by using extrapolation methods. | ||||||
| MoC–Mo2C-31.4 | 126 | 38 | 43 | 21.8 | 6.86 | 1.1 × 10−2 |
| Mo2C | 182 | 61 | 46 | 60.4 | 5.24 | 1.2 × 10−3 |
| MoC–Mo2C-68.1 | 218 | 65 | 52 | 101.7 | 2.57 | 7.2 × 10−4 |
| MoC–Mo2C-30 (mixed) | 222 | 100 | 53 | 116 | 2.51 | 3.2 × 10−4 |
| MoC | 232 | 105 | 62 | 145 | 2.06 | 5.0 × 10−4 |
Accordingly, the Tafel plots of the above carbides present the same order in HER kinetics (Fig. 3b and Table 1). Among them, MoC–Mo2C-31.4 shows a ηonset of 38 mV and a Tafel slope of 43 mV dec−1, which are obviously lower than those of Mo2C, MoC, MoC–Mo2C-68.1 and MoC–Mo2C-30.0 (mixed). The small Tafel slope of MoC–Mo2C-31.4 indicates a fast increase of the hydrogen generation rate with the applied overpotential, corresponding to the high activity presented in the polarization curves. According to the classic theory, the HER in acidic aqueous media proceeds in two steps (eqn (1)–(3)),40,41 where the * indicates the active site, and H* is a hydrogen atom bound to an active site. The first one is an electrochemical reduction step (H+ reduction, Volmer-reaction) with a Tafel slope of 118 mV dec−1 (eqn (1)), and the second one (Hads desorption) is either the ion and atom reaction (Heyrovsky-reaction) with a slope of 40 mV dec−1 (eqn (2)) or the atom combination reaction (Tafel-reaction) with a slope of 30 mV dec−1 (eqn (3)).8,40,41 Although the Tafel slope alone is insufficient to determine the specific mechanism, the evidently reduced slope for MoC–Mo2C-31.4, compared with MoC and MoC–Mo2C-68.1, still confirms the promoted Volmer-step in the HER kinetics.42,43 In addition, the exchange current density (j0) of the above electrocatalysts was further calculated by extrapolating the Tafel plots, which is the most inherent measure of HER activity. As expected, the j0 of 1.1 × 10−2 mA cm−2 for MoC–Mo2C-31.4 is higher than that of the other MoCx (Table 1).
| H3O+(a.q.) + e− + * → H* + H2O(l) | (1) |
| H3O+(a.q.) + e− + H* → * + H2(g) + H2O(l) | (2) |
| H* + H* → 2* + H2(g) | (3) |
The electrochemical surface area (ECSA) and resistant charge-transfer (Rct) were further evaluated to provide insight into the MoCx electrocatalysts (Table 1, Fig. 3c and d). Although the accurate measurement of ECSA is difficult owing to the unclear capacitive behaviour, it can be visualized through calculating the double-layer capacitances (Cdl) which are proportional to the ECSA values.44 An estimation of Cdl using the cyclic voltammograms (CV, Fig. S7†) in 0.5 M H2SO4 were alternatively utilized to provide a relative comparison.25,45 As shown in Fig. 3c, the Cdl of 6.86 mF cm−2 presented by MoC–Mo2C-31.4 is higher than those on α-Mo2C (5.24 mF cm−2), η-MoC (2.06 mF cm−2), MoC–Mo2C-68.1 (2.57 mF cm−2), and MoC–Mo2C-30 (mixed) (2.51 mF cm−2). Regarding the Cdl associated with the active surface area, the current density divided by Cdl can further reflect the intrinsic activity,46,47 from which the remarkably high one for MoC–Mo2C-31.4 indicates intrinsic optimization of the active-sites (Fig. S8†). Meanwhile, their electrochemical impedance spectroscopy (EIS) measurements show the consistent order in Rct, and a Rct as low as 21.8 Ω delivered by MoC–Mo2C-31.4 confirms the rapid electron transport for hydrogen evolution (Fig. 3d).28
It has been reported that the HER activity of MoCx depends on the active Mo2+ and Mo3+ centres exposed on the catalyst surface,17,38 which present various Mo–H resulting from the different electron densities of Mo.16 As displayed in Fig. 4a, the HER activities of Mo2C, MoC–Mo2C-31.4, MoC–Mo2C-68.1 and MoC are dependent on the variation of the ratio of active Mo3+/Mo2+ (n3+/2+) on the surface, featured by the both of the current densities at η = 0 (j0) and 150 mV (j150). With a higher n3+/2+ of 3.0 in comparison with Mo2C (n3+/2+ = 0.4), MoC–Mo2C-31.4 shows an obviously improved activity, which suggests that the enriched Mo3+ species with fewer electrons benefits HER. Furthermore, the narrowed valance-band (VB) distribution of MoC–Mo2C-31.4 (Fig. S9†) indicates the lower electron density around the Fermi level (EF) than that of Mo2C. Regarding the strong hydrogen binding on Mo2C and the consequently restricted Hads desorption, the decreased electron density in the MoC–Mo2C-31.4 HNWs would reduce the strength of Mo–H towards the promoted Hads desorption and thus remarkably improved the HER activity (Fig. 4b). Moreover, with n3+/2+ increased to 7.2 and 10.9, respectively, MoC–Mo2C-68.1 and MoC display further reduced electron density around EF (Fig. S9†) and decreased HER activity in comparison with MoC–Mo2C-31.4 (Fig. 4a). Their higher Tafel slopes (53 mV dec−1 for MoC–Mo2C-68.1, and 62 mV dec−1 for MoC) suggest that the limitation of the Volmer step becomes more obvious, because of weak hydrogen-binding involving less electron donated by Mo (Fig. 4b). It's reasonable that the high activity of the MoC–Mo2C-31.4 HNWs is ascribed to the optimized electronic properties of the MoC–Mo2C interfaces with a well-defined composition.
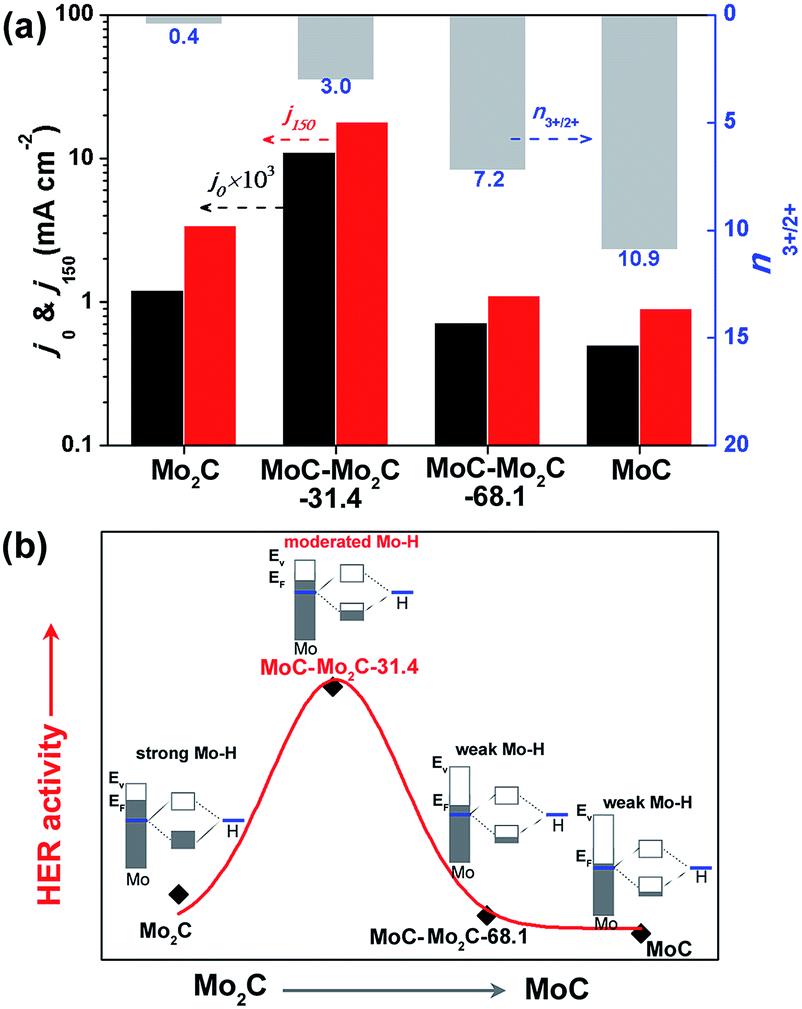 | ||
| Fig. 4 (a) j0 (obtained by extrapolating the Tafel curves to η = 0 mV) and j150 (current density at η = 150 mV) of the above MoCx, which are associated with the ratio of surface Mo3+/Mo2+ determined through XPS analysis. (b) Schematic illustration of the HER activity relying on the electron density of Mo in a series of MoCx electrocatalysts. | ||
The HER activity of MoC–Mo2C-31.4 is superior to most of the carbide-based HER electrocatalysts that have ever been reported in acidic electrolytes (Table S4†). The η10 of 126 mV delivered by MoC–Mo2C-31.4 is obviously lower than that of the reported nanoporous Mo2C NWs (130 mV),21 MoCN NPs (140 mV),17 MoCx nano-octahedrons (142 mV),48 Mo2C nanotubes (172 mV),26 and even that of supported MoCx (Mo2C/CNT-GR: 130 mV;27 Mo2C/N-doped CNT: 147 mV;49 Mo2C/RGO: 150 mV;29 Mo2C/CNT: 152 mV 28). To the best of our knowledge, the lower η10 than our MoC–Mo2C-31.4 has been only achieved on GR or N-doped carbon encapsulated Mo2C NPs, which require precise control over the N-doping and thickness of the carbon shells.38,45 In regard of the high mass loading of the previously reported electrocatalysts (0.21–2.0 mg cm−2), the remarkably low one in this work (0.14 mg cm−2) strongly supports the superior activity of the MoC–Mo2C-31.4 HNWs. Meanwhile, the fast HER kinetics of MoC–Mo2C-31.4 are also confirmed by its low ηonset (38 mV) and Tafel slope (43 mV dec−1), which outperform most of the reported MoCx (Table S4†). Furthermore, the HER performance of the MoC–Mo2C-31.4 HNWs is among the best reported when compared with many representative noble-metal free electrocatalysts, e.g., transition-metals and their carbides, nitrides, chalcogenides and phosphides (Table S4†).
Our MoC–Mo2C-31.4 HNWs are also active for the HER in basic solution (1.0 M KOH), showing the best activity and kinetics in comparison with Mo2C, MoC, MoC–Mo2C-68.1 and MoC–Mo2C-30 (mixed) (Fig. 5a and b). This shows good consistency with its high j0, high Cdl and low Rct (Table S5, Fig. S10 and S11†). Obviously, the synergy between MoC and Mo2C also promotes the HER performance in a basic electrolyte due to the optimized electronic properties of the Mo species. The η10 of 120 mV, ηonset of 33 mV and Tafel slope of 42 mV dec−1, observed for MoC–Mo2C-31.4, verify the outstanding activity performing among the best of the current MoCx materials,18,20,23,25,26,34,45,49 and other noble-metal free electrocatalysts (Table S6†).
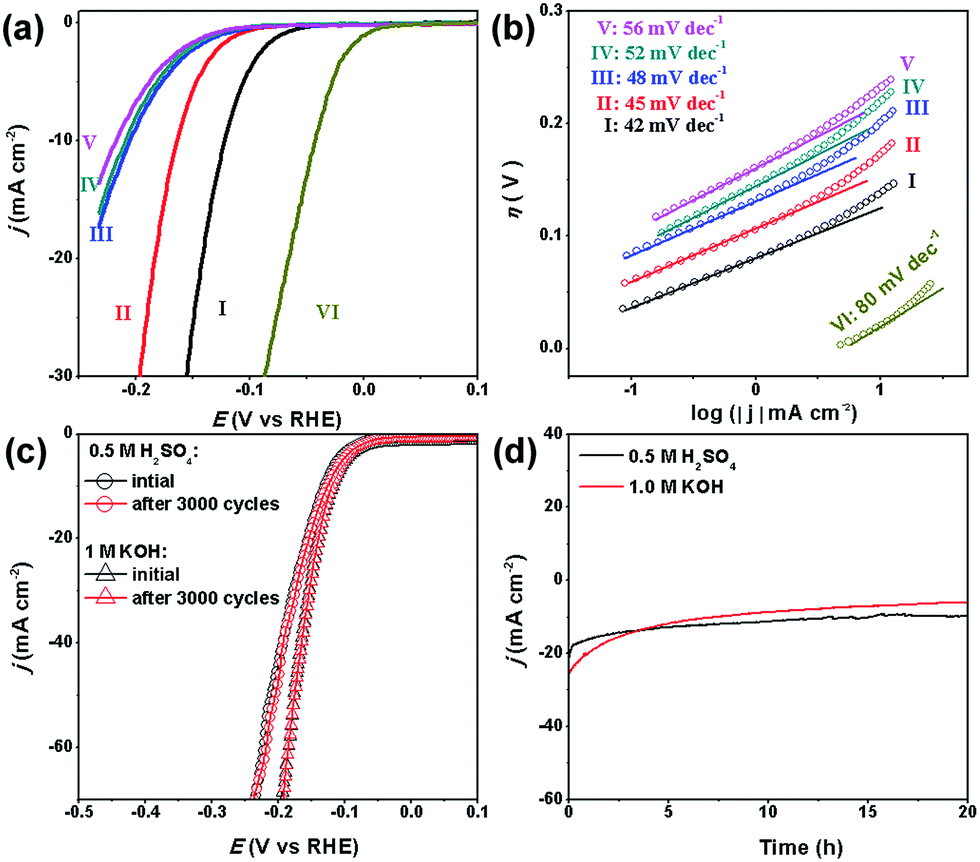 | ||
| Fig. 5 (a) Polarization curves and (b) Tafel plots for the HER on modified GCEs comprising (I) MoC–Mo2C-31.4, (II) Mo2C, (III) MoC–Mo2C-68.1, (IV) MoC–Mo2C-30 (mixed), (V) MoC, and (VI) commercial Pt/C in 1.0 M KOH. (c) Stability of the MoC/Mo2C-31.4 modified electrodes with an initial polarization curve and after 3000 cycles in 0.5 M H2SO4 and 1.0 M KOH, and (d) the long-term durability tests at η = 190 mV. | ||
Interestingly, the activity of our MoCx NWs in basic electrolyte is slightly higher than that in acidic solution. Similar situations have been observed with Mo2C@N-doped carbon,30 MoP,50 Mo2C NPs.18 This can be explained by the fact that the surface oxidized species on MoC–Mo2C can be dissolved by KOH, exposing rich active-sites for the HER (Fig. S12†).
Another important criterion for a good electrocatalyst is its high durability. Herein, the long-term stability of MoC–Mo2C-31.4 HNWs and the ability to continuously catalyse the generation of H2 were examined through continuous cycling for 3000 cycles and chronoamperometry in both 0.5 M H2SO4 and 1.0 M KOH. At the end of the cycling procedure, the catalyst affords similar j–V curves to the initial cycle with negligible loss of the cathodic current (Fig. 5c), confirming the satisfactory durability in both acidic and basic electrolytes. When further evaluated through prolonged electrolysis at a fixed potential (Fig. 5d), MoC–Mo2C-31.4 exhibited a catalytic current which remained at around 20 mA cm−2 for over 20 hours in 0.5 M H2SO4. However, the current in 1.0 M KOH slightly decreased.
Conclusions
In summary, we have reported the facile fabrication of MoC–Mo2C HNWs via the controlled carbonization of MoOx–amine. This strategy presents significance in regulating the crystalline structure, composition and electronic properties toward efficient HER. Showing an optimized electron density on the carbide surface, the MoC–Mo2C-31.4 HNWs exhibit high activity and good stability in both acidic and basic solutions. This work will open up new opportunities to develop high-performance electrocatalysts via rational engineering of nanostructures and interfaces.Experimental section
Catalyst preparation
Improved from our previous reports,51,52 MoAn-4.0 and MoAn-3.5 NWs were typically synthesized as follows: 2.48 g of ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O) was dissolved in 40 mL of water consisting of 3.28 mL of Aniline. Then, 1 M HCl aqueous solution was added to adjust the pH level to 4.0 for generating MoAn-4.0, and 3.5 for achieving MoAn-3.5, respectively. After reaction at 50 °C for 4 hour in an oil bath, the products were filtered and thoroughly washed with ethanol, and then dried at 50 °C overnight. MoMeAn-4.0 NWs were prepared through a similar process to that of MoAn, replacing the aniline with 3.83 g of p-methylaniline.The as-obtained MoOx-based hybrids (MoAn-4.0, MoAn-3.5 and MoMeAn-4.0) were transferred into a tube furnace and kept under an Ar flow for 4.0 h in order to remove air before heating. Then, the sample was heated to a target temperature and held for 5 h. The details for carbonization are listed in Table S1.†
Physical measurements
SEM and TEM investigations were undertaken on a ZEISS ULTRA55 and a JEOL JEM 2100F, respectively. XRD analysis was performed on a Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54056 Å). XPS was processed on a Perkin-Elmer PHI X-tool, using C 1s (B. E. = 284.6 eV) as a reference. TGA/DSC was tested on a NETZSCH STA449F3 under an air flow. FT-IR spectra were collected with a Nicolet 6700 FTIR spectrometer. The composition of the NWs was determined using ICP (for Mo), CHN elemental analysis using a Vario EL Elementar (for C, H and N) and an internal standard quantification in XRD (for the ratio of MoC/Mo2C). N2 adsorption–desorption isotherms were recorded on an automatic gas adsorption analyzer (Quantachrome Autosorb-iQ-MP). Raman spectra were recorded on a Raman spectrometer (Horiba), with an excitation laser wavelength of 632.81 nm.Electrochemical measurements
The MoCx electrocatalysts were loaded onto GCEs for testing in 0.5 M H2SO4 and 1.0 M KOH solutions using a typical three-electrode setup. Typically, 4 mg of catalyst and 40.0 μL of 5 wt% Nafion solution were dispersed in 1 mL of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v water/ethanol through at least 30 min of sonication to form a homogeneous ink. For the test in 1.0 M KOH, 10 μL of polyvinylidene fluoride (5 wt%) was further added into the above ink. Then 2.5 μL of catalyst ink was loaded onto a GCE of 3 mm in diameter. Linear sweep voltammetry (LSV) was conducted with the scan rate of 2 mV s−1 in 0.5 mol L−1 H2SO4 or 1.0 M KOH on a potentiostat of CHI760 (CH Instruments), using a saturated calomel electrode as the reference electrode, and a graphite electrode as the counter electrode. All of the potentials reported in our manuscript were referenced to a reversible hydrogen electrode (RHE) by adding a value of (0.241 + 0.059 pH) V. AC impedance measurements were carried out in the same configuration at η = 200 mV from 0.01 to 1000000 Hz and an amplitude of 5 mV.
:1 v/v water/ethanol through at least 30 min of sonication to form a homogeneous ink. For the test in 1.0 M KOH, 10 μL of polyvinylidene fluoride (5 wt%) was further added into the above ink. Then 2.5 μL of catalyst ink was loaded onto a GCE of 3 mm in diameter. Linear sweep voltammetry (LSV) was conducted with the scan rate of 2 mV s−1 in 0.5 mol L−1 H2SO4 or 1.0 M KOH on a potentiostat of CHI760 (CH Instruments), using a saturated calomel electrode as the reference electrode, and a graphite electrode as the counter electrode. All of the potentials reported in our manuscript were referenced to a reversible hydrogen electrode (RHE) by adding a value of (0.241 + 0.059 pH) V. AC impedance measurements were carried out in the same configuration at η = 200 mV from 0.01 to 1000000 Hz and an amplitude of 5 mV.
Acknowledgements
This work is financially supported by the National Basic Research Program of China (2013CB934101), the National Natural Science Foundation of China (21373102 and 21433002) and the Fundamental Research Funds for the Central Universities (21615402). Q. S. Gao also thanks the support from the Natural Science Foundation of Guangdong Province (2015A030306014 and 2014TQ01N036) and the Guangdong Higher Education Institute (YQ2013022).Notes and references
- H. B. Gray, Nat. Chem., 2009, 1, 7–8 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39–44 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 CAS.
- M. Zeng and Y. G. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 CAS.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- Y. N. Regmi, G. R. Waetzig, K. D. Duffee, S. M. Schmuecker, J. M. Thode and B. M. Leonard, J. Mater. Chem. A, 2015, 3, 10085–10091 CAS.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS PubMed.
- Q. S. Gao, N. Liu, S. N. Wang and Y. Tang, Nanoscale, 2014, 6, 14106–14120 RSC.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185 CrossRef CAS PubMed.
- R. Michalsky, Y. J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Am. Chem. Soc., 2015, 137, 110–113 CrossRef CAS PubMed.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 CAS.
- C. Tang, A. Sun, Y. Xu, Z. Wu and D. Wang, J. Power Sources, 2015, 296, 18–22 CrossRef CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 CAS.
- C. Ge, P. Jiang, W. Cui, Z. Pu, Z. Xing, A. M. Asiri, A. Y. Obaid, X. Sun and J. Tian, Electrochim. Acta, 2014, 134, 182–186 CrossRef.
- P. Xiao, Y. Yan, X. Ge, Z. Liu, J.-Y. Wang and X. Wang, Appl. Catal., B, 2014, 154, 232–237 CrossRef.
- K. Zhang, C. Li, Y. Zhao, X. Yu and Y. Chen, Phys. Chem. Chem. Phys., 2015, 17, 16609–16614 RSC.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef PubMed.
- F. X. Ma, H. B. Wu, C. Y. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 15395–15399 CrossRef CAS PubMed.
- D. H. Youn, S. Han, J. Y. Kim, J. Y. Kim, H. Park, S. H. Choi and J. S. Lee, ACS Nano, 2014, 8, 5164–5173 CrossRef CAS PubMed.
- W. F. Chen, C. H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943 CAS.
- C. He and J. Tao, Chem. Commun., 2015, 51, 8323–8325 RSC.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS PubMed.
- J. Zhu, K. Sakaushi, G. Clavel, M. Shalom, M. Antonietti and T.-P. Fellinger, J. Am. Chem. Soc., 2015, 137, 5480–5485 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- C. Wan and B. M. Leonard, Chem. Mater., 2015, 27, 4281–4288 CrossRef CAS.
- K. Xiong, L. Li, L. Zhang, W. Ding, L. S. Peng, Y. Wang, S. G. Chen, S. Y. Tan and Z. D. Wei, J. Mater. Chem. A, 2015, 3, 1863–1867 CAS.
- V. Heine, Phys. Rev. A, 1967, 153, 673 CrossRef CAS.
- L. Ramqvist, Appl. Phys., 1971, 42, 2113–2127 CAS.
- C. Wan, N. A. Knight and B. M. Leonard, Chem. Commun., 2013, 49, 10409–10411 RSC.
- R. G. Ma, Y. Zhou, Y. F. Chen, P. X. Li, Q. Liu and J. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS PubMed.
- M. Xiang, D. Li, W. Li, B. Zhong and Y. Sun, Catal. Commun., 2007, 8, 513–518 CrossRef CAS.
- S. A. Vilekar, I. Fishtik and R. Datta, J. Electrochem. Soc., 2010, 157, B1040–B1050 CrossRef CAS.
- C. G. Morales-Guio, L. A. Stern and X. L. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- N. Liu, Y. L. Guo, X. Y. Yang, H. L. Lin, L. C. Yang, Z. P. Shi, Z. W. Zhong, S. N. Wang, Y. Tang and Q. S. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 23741–23749 CAS.
- N. Liu, L. C. Yang, S. N. Wang, Z. W. Zhong, S. N. He, X. Y. Yang, Q. S. Gao and Y. Tang, J. Power Sources, 2015, 275, 588–594 CrossRef CAS.
- M. R. Gao, M. K. Y. Chan and Y. G. Sun, Nat. Commun., 2015, 6, 7493 CrossRef PubMed.
- Y. Liu, G. Yu, G.-D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. L. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- N. Liu, L. Yang, S. Wang, Z. Zhong, S. He, X. Yang, Q. Gao and Y. Tang, J. Power Sources, 2015, 275, 588–594 CrossRef.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef PubMed.
- K. Zhang, Y. Zhao, D. Fu and Y. Chen, J. Mater. Chem. A, 2015, 3, 5783–5788 CAS.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 CAS.
- Q. S. Gao, C. X. Zhang, S. H. Xie, W. M. Hua, Y. H. Zhang, N. Ren, H. L. Xu and Y. Tang, Chem. Mater., 2009, 21, 5560–5562 CrossRef CAS.
- Q. Gao, S. Wang, H. Fang, J. Weng, Y. Zhang, J. Mao and Y. Tang, J. Mater. Chem., 2012, 22, 4709–4715 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, XRD, FT-IR, TG/DSC and CHN elemental analysis of hybrid precursors, additional XPS results, SEM, TEM, N2-sorption isothermals, Raman spectra, CV and EIS of a series of MoCx and their comparison with previously reported noble-metal free catalysts. See DOI: 10.1039/c6sc00077k |
| This journal is © The Royal Society of Chemistry 2016 |