
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFatty acids' double role in the prebiotic formation of a hydrophobic dipeptide†
Sara
Murillo-Sánchez
a,
Damien
Beaufils
b,
Juan Manuel
González Mañas
c,
Robert
Pascal
 *b and
Kepa
Ruiz-Mirazo
*ad
*b and
Kepa
Ruiz-Mirazo
*ad
aBiophysics Unit (CSIC, UPV/EHU), University of the Basque Country, Spain. E-mail: kepa.ruiz-mirazo@ehu.eus
bInstitut des Biomolécules Max Mousseron (IBMM, UMR 5247, CNRS/Université de Montpellier/ENSCM), Montpellier, France. E-mail: robert.pascal@umontpellier.fr
cDepartment of Biochemistry and Molecular Biology, University of the Basque Country, Spain
dDepartment of Logic and Philosophy of Science, University of the Basque Country, Spain
First published on 9th February 2016
Abstract
In search of a connection between prebiotic peptide chemistry and lipid compartments, the reaction of a 5(4H)-oxazolone with leucinamide was extensively explored under buffered aqueous conditions, where diverse amphiphiles and surfactants could form supramolecular assemblies. Significant increases in yield and changes in stereoselectivity were observed when fatty acids exceeded their critical aggregation concentration, self-assembling into vesicles in particular. This effect does not take place below the fatty acid solubility limit, or when other anionic amphiphiles/surfactants are used. Data from fluorimetric and Langmuir trough assays, complementary to the main HPLC results reported here, demonstrate that the dipeptide product co-localizes with fatty acid bilayers and monolayers. Additional experiments in organic solvents suggest that acid–base catalysis operates at the water–aggregate interface, linked to the continuous proton exchange dynamics that fatty acids undergo at pH values around their effective pKa. These simple amphiphiles could therefore play a dual role as enhancers of peptide chemistry under prebiotic conditions, providing soft and hydrophobic organic domains through self-assembly and actively inducing catalysis at their interface with the aqueous environment. Our results support a systems chemistry approach to life's origin.
Introduction
Chemistry today is up to the challenge of working with increasingly diverse combinations of molecules and supramolecular structures, both under quasi-equilibrium and non-equilibrium conditions, providing new means to explain their complex dynamic behaviour. The field of ‘systems chemistry’ has been launched precisely to address this challenge1,2 with strong implications for studies in connection with biology and, in particular, with research on the origins of life.3 In this context, the idea that the chemical roots of biological phenomena cannot be found in the properties of a single type of molecule (often assumed to be a biopolymer, like RNA), but must be approached in a more encompassing way that includes reaction networks made of different bio-molecular precursors, is gaining momentum. This novel view has recently received further support from evidence that there could be a common prebiotic chemistry underlying the synthesis of a rich variety of these precursors.4 The obvious next step would be to determine the way in which some of these initial biomonomers engage in interactions that contribute to producing and maintaining higher levels of molecular complexity and diversity, as we actually observe in biology. Here we report experimental results demonstrating that a primitive synergy of this kind could occur between two such types of molecules, namely those whose prebiotic relevance is better grounded: amino acids, as peptide and protein precursors,5,6 and fatty acids, as lipid precursors.7,8Previous studies have shown that the activation of amino acid monomers or peptides can lead to their oligomerization in aqueous solution.9,10 Peptide oligomers are formed by the stepwise polymerization of α-amino acid N-carboxyanhydrides (NCAs), as prebiotically plausible activated monomers. In addition to this pathway, the activation of the C-terminus in peptides or acylated amino acids has been shown to proceed through the formation of 5(4H)-oxazolones from reagents compatible with early Earth conditions.11 In turn, fatty acids, as single-chain amphiphilic precursors of the standard (but remarkably more complex) phospholipids found in bacterial and eukaryote biomembranes, were long ago shown to self-assemble into vesicles at intermediate pH values.12,13 This aggregation state is favoured due to the fact that both the neutral and anionic forms of the fatty acid are present in similar proportions (at pH ∼ pKa) and tend to establish H-bonds between them, leading to molecular pairs with cylindrical geometry (see Fig. S1, in the ESI†), which spontaneously assemble into flat bilayers. In recent years, an increasing number of research groups have explored the properties of these supramolecular structures, given their potential as protocellular model systems.14–22
However, relatively little effort has been devoted to putting together these two prebiotic systems. Although the idea of combining peptide chemistry with lipid aggregates is not new, it has been implemented mostly in making use of standard liposomes (i.e., vesicles made of phospholipids). For instance, mixing peptides with lipid aggregates was advocated to improve the stability and functionality of the latter.23 In addition, the polymerization of aminoacyl adenylates into peptides was claimed to yield increased chain lengths in the presence of vesicles,24 even though the mechanism does not correspond to a direct process, but actually involves a prior conversion into NCAs.25,26 Luisi’s group also performed a series of investigations demonstrating the role of phospholipid compartments in the efficiency and stereoselectivity of peptide polymerization.27–30 In an interesting step forward, liposomes made of cationic surfactants were later shown to favour the oligomerization of encapsulated anionic amino acid monomers.31 And, more recently, the formation of tryptophan based dipeptides in the aqueous cavity of phosphatidylcholine vesicles has been reported.32
Much less is found in the literature regarding the use of fatty acid vesicles to perform similar experiments. As an exception, Matsuno's lab performed some experiments in which they reported the oligomerization of encapsulated glycine, not only in phospholipids but also in fatty acid vesicle compartments.33 More directly relevant to our approach, a couple of years ago, Szostak's group managed to carry out, within oleic acid vesicles and with the catalytic assistance of a pre-encapsulated di-peptide (Ser–His), the synthesis of a strongly hydrophobic peptide, which spontaneously joined the membrane and, thereby, contributed to its growth.34 In this article we point towards a similar idea but approach it from a complementary angle. We show how a prototypical prebiotic synthesis of a hydrophobic dipeptide (starting from precursors expected to be protected by lipophilic environments)35 is significantly enhanced, without the aid of a soluble catalyst, just in the presence of vesicles made of fatty acids. This effect, which relies on the specific fatty acid nature of the compartments, is also partially detected when reactants are put in organic solvents (indicative of direct chemical catalysis by the fatty acids). Thus, our results give fatty acids wider significance as prebiotic compounds than has been commonly assumed in the field of the origins of life.
Materials and methods
Chemicals and general information
N-Ethyl-N′-(3-dimethylaminopropyl)-carbodiimide·HCl (EDC), H-L-Leu-NH2, Ac-L-Tyr(Me)-OH and Boc-L-Tyr(Me)OH were purchased from Bachem. Piperazine-N,N′-bis(3-propanesulfonic acid) (PIPPS) was purchased from CalbioChem. 1,2-Dioleoyl-sn-glycero-phosphate (monosodium salt) was purchased from Avanti Polar Lipids. The rest of the reagents and solvents were purchased from Sigma Aldrich. All compounds were used without further purification. In all experiments pH was monitored using a Thermo Orion 3-STAR pH-meter with a VWR electrode. NMR spectra in DMSO-d6 solution were recorded on a Bruker Avance 300 spectrometer at 300 MHz for 1H. HPLC analysis (Method A) was carried out on a Waters Alliance system including a E2695 separation module and a 2998 photodiode array detector; column BDS Hypersil C18 (3 µm, 2.1 × 50 mm). The elution method used was: flow 0.2 mL min−1, solvent A 0.1% TFA in H2O, solvent B 0.1% TFA in CH3CN, gradient 90% B to 70% over 25 min; detection 273 nm. HPLC-ESI-MS analysis was carried out (Method B) using a Waters Synapt G2-S system connected to Waters Acquity UPLC H-Class apparatus equipped with an Acquity UPLC BEH C18, 1.7 µm 2.1 × 50 mm column; the gradient of solvent A was 0.01% formic acid in H2O and solvent B: 0.01% formic acid in acetonitrile; flow rate: 0.5 mL min−1; linear gradient 0–100% B over 3 min.Experimental procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v TFA/water reagent at r.t. for 30 min. The mixture was concentrated under reduced pressure and the product was precipitated by the addition of cold ethyl ether, filtered off, washed with ethyl ether and dried under vacuum. Acetylation was performed with acetic anhydride (1.5 eq.) and DIEA (2 eq.) in CH2Cl2 for 5 h at 0 °C. The mixture was diluted with water and extracted with ethyl acetate. The organic layers were washed with saturated Na2CO3 solution, then with 1 M KHSO4 and saturated NaCl and finally dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield the product as a white solid.
:10 v/v TFA/water reagent at r.t. for 30 min. The mixture was concentrated under reduced pressure and the product was precipitated by the addition of cold ethyl ether, filtered off, washed with ethyl ether and dried under vacuum. Acetylation was performed with acetic anhydride (1.5 eq.) and DIEA (2 eq.) in CH2Cl2 for 5 h at 0 °C. The mixture was diluted with water and extracted with ethyl acetate. The organic layers were washed with saturated Na2CO3 solution, then with 1 M KHSO4 and saturated NaCl and finally dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield the product as a white solid.
Peptide-forming reactions
Partition of dipeptide between solution and surfactant
The Langmuir–Blodgett monolayer technique (Kibron MicroTrough XS) was used to elucidate the interactions between the Ac-L-Tyr(Me)-L-Leu-NH2 dipeptide and the lipid. A monolayer of fatty acid was spread on a well with 1 mL of buffer at the corresponding pH registering an initial stabilized pressure (Πo). 10 µL of a 5 mM Ac-L-Tyr(Me)-L-Leu-NH2 dipeptide stock solution in MeOH was then added from the water face. After MeOH evaporation, any surface pressure increase resulted from a migration of the hydrophobic solute to the monolayer, stabilizing the signal at a new pressure (Πi). The difference between these two surface pressures (ΔΠ) was recorded and compared to the Πo in every individual experiment.Binding of Ac-L-Tyr(Me)-L-Leu-NH2 to the vesicles
This experimental assay is based on the quenching action of acrylamide, which decreases the intrinsic fluorescence of Tyr residues as a function of its local concentration. Acrylamide was taken from a 6.25 M stock solution and added in 4 µL portions (5 additions, from 0 mM of acrylamide to 250 mM in the final volume) to the control and 1 mL samples containing vesicles (which were previously prepared, following the general procedure, in the presence of 1 µM Ac-L-Tyr(Me)-L-Leu-NH2 dipeptide). Fluorescence was monitored at a single wavelength (ex: 273 nm and em: 340 nm) after every addition. In parallel, a dilution control was carried out, adding the same volume of aqueous buffer instead of acrylamide solution. The fluorescence obtained as a result of the addition of only buffer (F0) was divided by the fluorescence obtained as a result of the addition of acrylamide (F) and compared against the concentration values of acrylamide added.Results
Experiments in aqueous media
We extensively studied the outcome of reactions taking place when a racemic mixture of hydrophobic 5(4H)-oxazolone enantiomers (L-2a and D-2a), derived from Ac-L-Tyr(Me)-OH L-1a (Scheme 1), was put in contact with H-L-Leu-NH2 in buffered media. Owing to its chiral instability, the 5(4H)-oxazolones undergo a kinetically stereoselective reaction with the nucleophiles, which have a fixed configuration,36 meaning that the timescale of the epimerization process is similar to or faster than the subsequent reaction and that the configuration of products is kinetically controlled. Reactions producing only peptide diastereomers and racemic Ac-Tyr(Me)-OH as a result of hydrolysis were carried out under different pH conditions and in the presence of different amounts of amphiphiles and surfactants. For each case, both the total dipeptide yield (relative to the amount of starting material) and the ratio of diastereomers were determined. Fig. 1 shows the characteristic results obtained when carboxylic acids are employed (in this case: decanoic acid (DA) at pH 7.2, but see Fig. 2 and Table S1 in the ESI† for further data). Typically, there is a 2 to 3-fold increase in the overall yield of dipeptide, together with a reversion in its diastereomeric ratio (from a preference for homochiral forms, in solution,36 toward more heterochiral ones).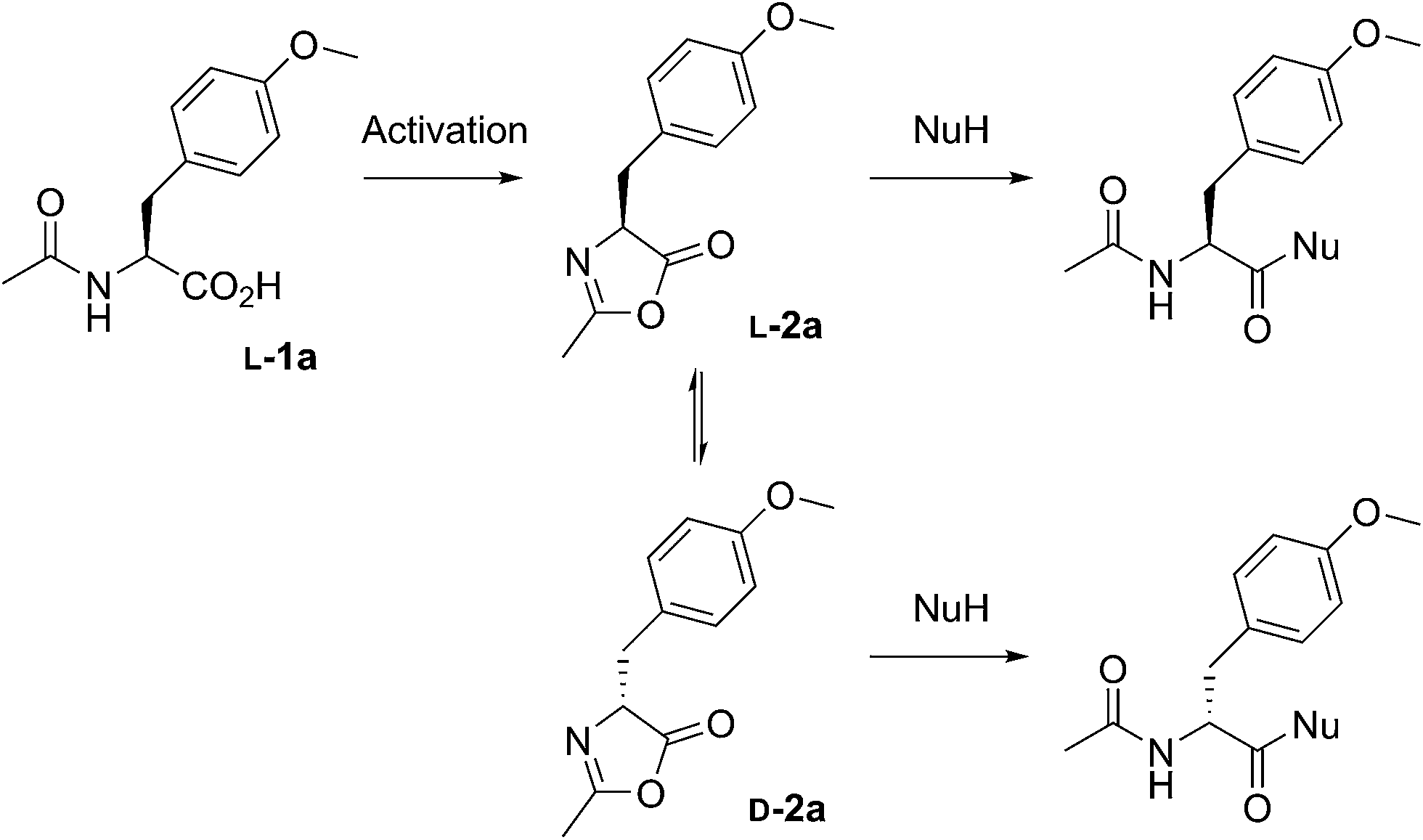 | ||
| Scheme 1 The reactions performed in this work from racemic mixtures of the 5(4H)-oxazolone enantiomers (L-2a and D-2a) with nucleophiles (NuH = H-L-Leu-NH2, H-L-Ala-NH2, H-Gly-NH2, or H-L-Leu-OH) in aqueous media, leading to the corresponding dipeptides as mixtures of enantiomers (Ac-L-Tyr(Me)-Gly-NH2 and Ac-D-Tyr(Me)-Gly-NH2) or diastereomers (Ac-L-Tyr(Me)-L-Leu-NH2 and Ac-D-Tyr(Me)-L-Leu-NH2, Ac-L-Tyr(Me)-L-Ala-NH2 and Ac-D-Tyr(Me)-L-Ala-NH2, or Ac-L-Tyr(Me)-L-Leu-OH and Ac-D-Tyr(Me)-L-Leu-OH). Although prepared from chiral Ac-L-Tyr(Me)-OH (L-1a), the oxazolone was present in a racemic state due to its chiral instability.36 | ||
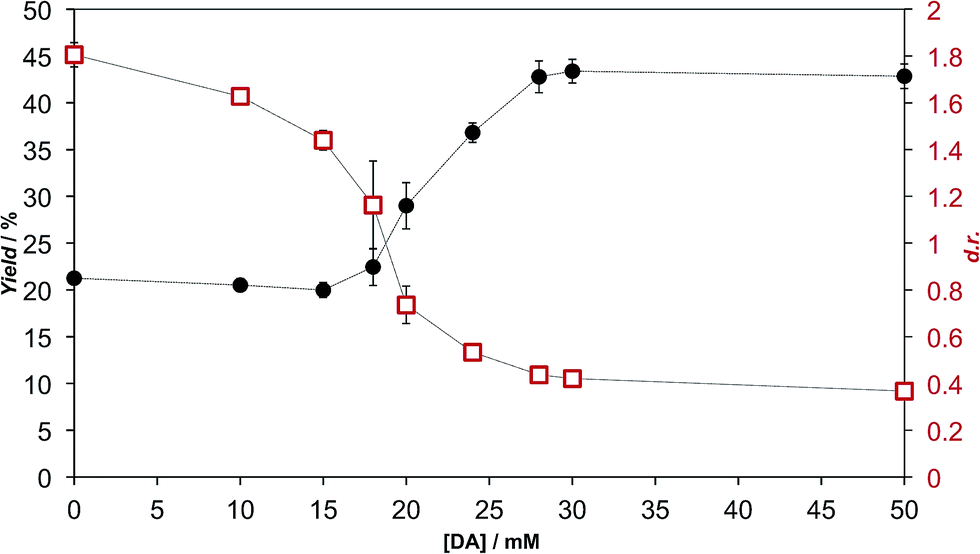 | ||
| Fig. 1 Formation of peptide diastereomers Ac-L-Tyr(Me)-L-Leu-NH2 and Ac-D-Tyr(Me)-L-Leu-NH2 by the reaction of 1 mM racemic 5(4H)-oxazolone 2a with 5 mM H-L-Leu-NH2 in 100 mM MOPS buffer (pH 7.2), at room temperature, in the presence of different concentrations of decanoic acid (DA). The effects are reported in terms of the relative total yield of the dipeptide (left axis, black filled circles) and of the diastereomeric ratio (right axis, red open squares). | ||
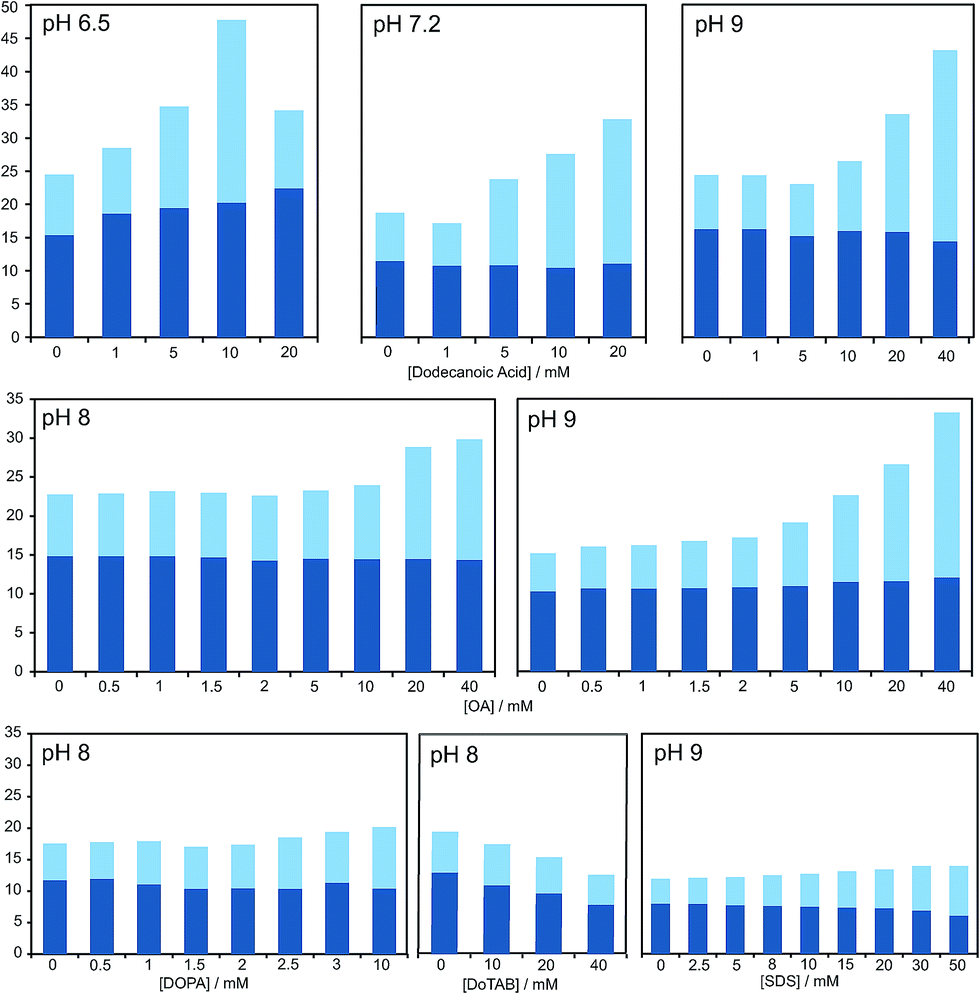 | ||
| Fig. 2 Bar graph representing the total yield of dipeptide (full bar size) and stereoselectivity (chiral conversion %: Ac-L-Tyr(Me)-L-Leu-NH2, in dark blue; and Ac-L-Tyr(Me)-L-Leu-NH2, in light blue) which are obtained in reactions carried out at room temperature, in aqueous buffer (100 mM) at various pH values, starting from Ac-Tyr(Me) oxazolone (1 mM), in the presence of H-L-Leu-NH2 (5 mM) and diverse amphiphile/surfactant concentrations. Each bar corresponds to a single experiment; error fluctuation was estimated with the control samples, without amphiphile (based on d.r. data and calculated for each pH value – see Table S1†) and remains within the range of 12–18% for all cases. OA stands for oleic acid. For a more exhaustive report of the results, see main text and Table S1 in the ESI.† | ||
As is apparent from Fig. 1, this double effect only happens provided that the concentration of fatty acid is above its critical aggregation concentration (CAC) value (the critical vesicular concentration, CVC, in this case), which was measured independently (Fig. S2 in the ESI†), and reaches a plateau soon after that critical value is crossed. Those two independent observations suggest that the reactions in which hydrophobic substrates are involved take place in a different environment, whenever fatty acid aggregates offer this possibility. The double effect may well depend on the fact that the nucleophilic reaction partners have a strong enough affinity for the hydrophobic environment (see data for alaninamide, glycinamide and leucine, instead of leucinamide, at the end of Table S1 in the ESI†). It is difficult to explain, otherwise, how the minimal variations in the chemical nature of the reactants (the reduced aliphatic side-chain in the first two cases, or the higher polarity in the third, zwitterionic one) make such a difference.
Additional assays were carried out to complement the previous results and check whether the product of the reaction co-localizes with the hydrophobic supramolecular domains. More specifically, a series of quenching experiments with acrylamide (Fig. S3 in ESI†) show that when DA vesicles are present in the medium, the fluorescent signal coming from the Tyr ring of the dipeptide could not be screened down, as occurs in free solution. Furthermore, data collected from Langmuir trough experiments (Fig. S4 in the ESI†) also show that the addition of the dipeptide to DA monolayers with different initial surface pressures (π0) induced an increase in the surface pressure (Δπ), which results from the adsorption of the dipeptide to the monolayer.
Our HPLC results were consistent over a relatively wide range of concentrations, carboxylic acid species and pH values (see Fig. 2 and Table S1 in the ESI†). Nevertheless, stronger effects were typically obtained at pH values that do not largely exceed the pKa of the corresponding fatty acid in the charged aggregate environment, which suggests that the vesicles had the optimal supra-molecular configuration to achieve a more significant influence on the reaction (at lower pH less reliable results were collected, owing probably to the formation of droplets).
A series of additional control experiments was performed to check the specificity of our results with regard to the type of amphiphilic/surfactant molecule employed. Since fatty acid vesicles are, overall, negatively charged (due to the partial deprotonation of their monomeric units), we tried with negatively charged species containing aliphatic chains of a similar length, like the phosphatidic acid DOPA (1,2-dioleoyl-sn-glycero-3-phosphoric acid) or sodium dodecyl sulfate (SDS).
As reported in Fig. 2 and Table S1 (ESI†), no detectable effects on the yield of the reaction were found in those cases, even if we did observe changes in the diastereomeric ratios, which were altogether absent if a positively charged surfactant (like dodecyltrimethylammonium bromide, DoTAB) was used. Although an explanation for this inverted trend in the chiral nature of the products (preference for D-forms in the majority of cases when supramolecular structures are present) is not within the scope of this study, it is nevertheless useful as an indicator of the environment in which the reaction actually takes place. This view is also supported by the known effect of low polarity solvents favouring heterochiral adducts of 5(4H)-oxazolones.40 Finally, it should be mentioned that mixtures of decanoic acid with decanol (previously reported to confer higher stability to the resulting vesicles in aqueous solution)14 were tried, too, but the effects on the reaction were clearly diminished.
Experiments in organic solvents
All the results obtained in aqueous buffered solution indicate that the pathway through which fatty acids play a role does not simply correspond to the usual activity of hydrophobic assemblies on chemical reactivity. Indeed, a specific behaviour of carboxylic acids at pH values inducing the formation of bilayers might be related to the activity of their polar head groups, combined in both the neutral and anionic forms, at the lipid–water interface of the supramolecular structure (Fig. S1 in the ESI†). In order to determine what kind of chemical activity is effectively responsible for this behaviour, our reaction was also studied in organic solvents, in the presence of decanoic acid and acetic acid. Both acids induced a substantial increase in reaction rate in two different solvents, CH2Cl2 (Fig. 3) and CH3CN, and the hydrophobic chain of decanoic acid had no significant additional influence (see Table 1). The more acidic Ac-Leu-OH was studied in CH3CN to avoid solubility issues and had a slightly improved activity at low concentrations (2 mM), compared to acetic acid and decanoic acid under the same conditions (Table S2 in ESI†). Therefore, the activity of fatty acids does not depend on the presence of a hydrophobic aliphatic chain, which indicates that it is the result of the participation of the carboxylic acid moiety.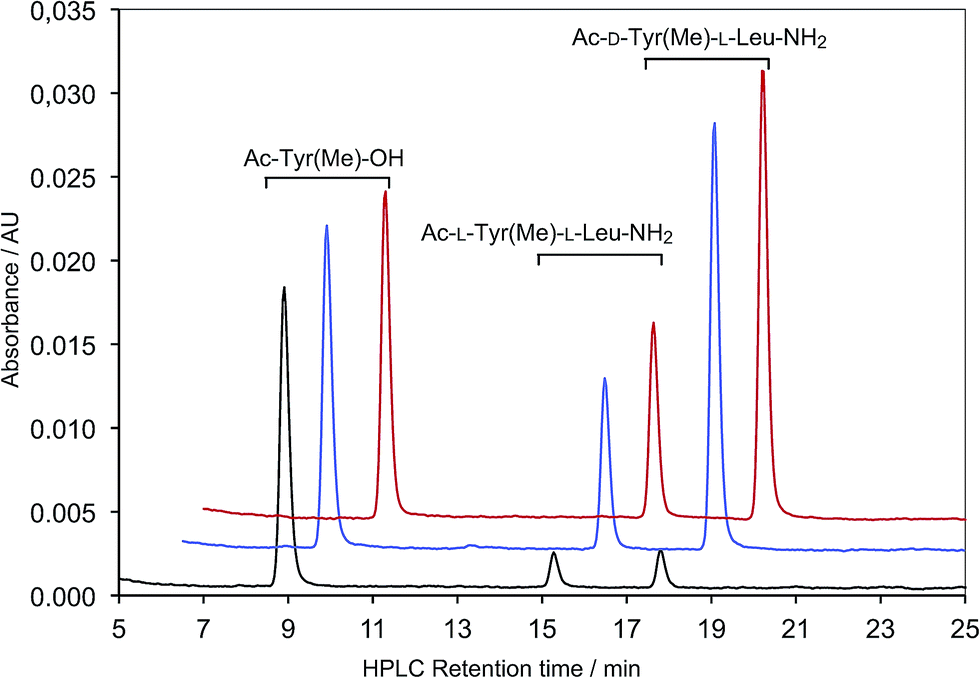 | ||
| Fig. 3 HPLC monitoring of the reaction (samples withdrawn after 1 h) of Ac-L-Tyr(Me)-5(4H)-oxazolone 2a (ca. 1 mM) with H-L-Leu-NH2 (5 mM) carried out in CH2Cl2 (control, black) or in the presence of added carboxylic acids (2 mM): decanoic acid (red) or acetic acid (blue). | ||
| Solvent | Carboxylic acid | Yield/% | d.r. |
|---|---|---|---|
| CH2Cl2 | Control without acid | 17.4 | 0.84 |
| CH2Cl2 | 10 mM acetic acid | 86.2 | 0.49 |
| CH2Cl2 | 10 mM DA | 86.9 | 0.54 |
| CH3CN | Control without acid | 2.7 | 0.62 |
| CH3CN | 10 mM acetic acid | 45.3 | 0.44 |
| CH3CN | 10 mM DA | 55.7 | 0.46 |
Discussion
Our experimental survey proves that fatty acid aggregates increase the yield of peptide formation from a hydrophobic 5(4H)-oxazolone and leucinamide. The product of the reaction was also demonstrated to have an affinity for the lipid phase. The favourable effect on the yield is specific of surfactants that bear a carboxylic acid moiety. This effect is accompanied by a reversal of stereoselectivity observed only above the CAC, which requires a more elaborate explanation but is beyond the scope of the present work.In organic solvents a similar preference for heterochiral products was observed, together with a rate enhancement promoted by carboxylic acids. In these media, carboxylic acids can either act as nucleophilic catalysts or acid–base catalysts (Scheme 2). Clues attesting to acid–base catalysis can neither be expected from the accumulation of intermediates nor from the detection of side-products. However, thanks to the presence of two electrophilic sites in the mixed anhydride intermediate, a transfer of activation from the 5(4H)-oxazolone 2a to the carboxylic acid R3–COOH leading to amide side-product 4 (rather than to the dipeptide Ac-Tyr(Me)-Leu-NH23) can, in contrast, be predicted during the nucleophilic pathway. This would come as a result of path (III), in addition to the aminolysis yielding product 3, as previously obtained from path (II). To assess the extent of nucleophilic catalysis, an experiment was carried out starting from 2 mM leucine 5(4H)-oxazolone 2b in acetonitrile and using 1 mM Ac-L-Tyr(Me)-OH 1a as a carboxylic acid component (R3COOH). The formation of Ac-L-Tyr(Me)-L-Leu-NH2 dipeptide was observed to be limited to 1% (with respect to 1a) after 1 h, and slowly increased up to 5% after 3 days (Fig. S5 in the ESI†). Importantly, Ac-L-Tyr(Me)-L-Leu-NH2 dipeptide was formed as a single stereoisomer, ruling out any reaction path occurring through the 5(4H)-oxazolone 2a, and supporting the mixed anhydride one (III). This experiment, in fact, suggests that the mixed anhydride pathway is a slowly proceeding process.
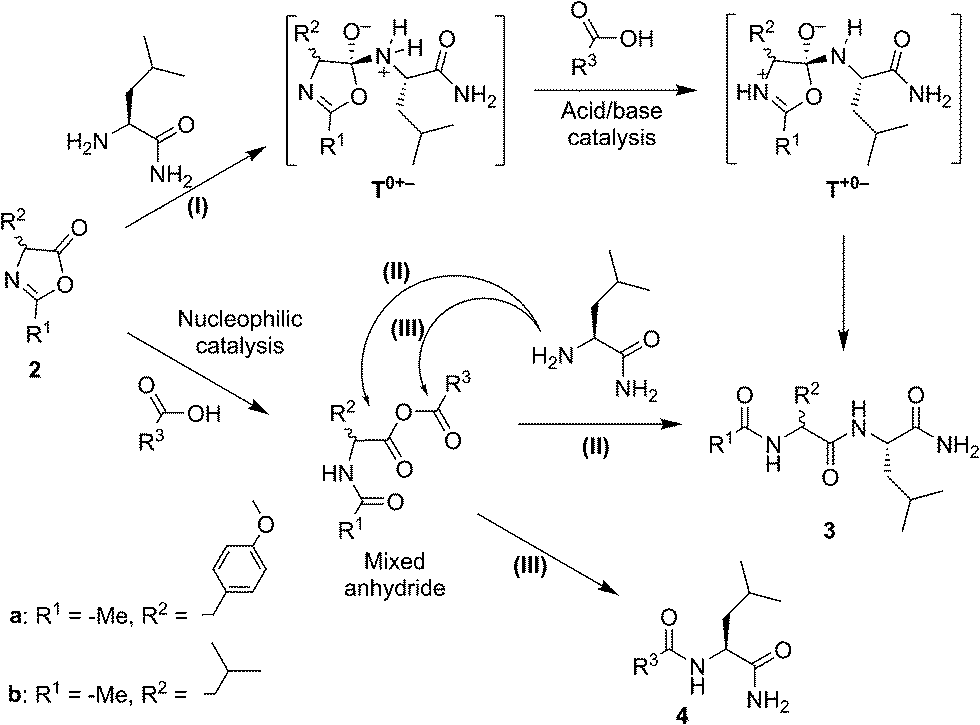 | ||
| Scheme 2 Potential chemical pathways of acid–base (I) and nucleophilic (II and III) catalysis (during the aminolysis of 5(4H)-oxazolones 2a and 2b by leucinamide in organic solvents) suggested as a way to account for the activity of carboxylic acids R3COOH (which include acetic acid, decanoic acid and the acylated α-amino acid Ac-D,L-Leu-OH and optically active Ac-L-Leu-OH and Ac-L-Tyr(Me)-OH). | ||
The fast and highly efficient increase in the rate (ca. 10-fold) observed by adding 2 mM Ac-Leu-OH (as R3COOH) to 1 mM Ac-Tyr(Me) 5(4H)-oxazolone 2a must therefore be considered to be the result of the acid–base-catalysed path (I). The faster reaction of 5(4H)-oxazolone 2a, compared to that of the mixed anhydride, is not surprising if one takes into account two key points: (i) the lesser importance of steric hindrance for the nucleophilic attack at the 5(4H)-oxazolone carbonyl group, relative to that occurring at the two reacting centres of the mixed anhydride; and (ii) the better leaving group ability of the amide oxygen (pKa ∼ −1–0 in water for the conjugate acid) relative to that of a carboxylate group (pKa ∼ 4–5 in water for carboxylic acids – although a significantly higher value is expected in the anionic environment of the aggregate).
The occurrence of acid–base catalysis of any kind is directly connected to the need for proton transfer during the course of the reaction, given the large change in pKa of the reacting groups involved.41,42 In path (I) two proton transfers are needed for conversion into the product. The tetrahedral intermediate T0+− is produced by the direct addition of the nucleophile to the 5(4H)-oxazolone. The need for reaching T+0− by proton transfer before any conversion into products takes place can account for the catalysis by carboxylic acid observed in organic solvents (Scheme 2, path (I)). The most straightforward process is a pathway starting from this intermediate by protonation of the nitrogen atom of the leaving group by the carboxylic acid, thus facilitating breakdown. The alternative possibility of the carboxylate anion acting as a base can be ruled out by the fact that the base, H-Leu-NH2, present at the same time in the medium (in 5 mM concentration) was not sufficient to promote the reaction (Fig. 3, control). An additional advantage of carboxylic acids could be their ability to act as both an acid and base in a bifunctional way,43 which could make the decomposition of the tetrahedral intermediate easier by the direct conversion of T0+− into T+0−.
The results reported here for the reactions carried out in aqueous media could be explained by the possibility that fatty acid supramolecular aggregates have a catalytic effect similar to the micellar catalysis already demonstrated for bimolecular reactions between hydrophobic substrates (i.e., selective concentration effect in a reduced volume).44,45 The need for a concentration of surfactant exceeding the CAC (in order to have an observable effect) indeed supports the involvement of hydrophobic aggregates. However, this explanation is not sufficient to account for the highly specific behaviour that fatty acid aggregates (as compared to other lipid or surfactant aggregates) were observed to have, inducing significant increases in the yield of peptide formation reactions from 5(4H)-oxazolones. A more comprehensive explanation of all the data collected in this work, coherent with the results obtained in organic solvents, would include an acid–base type of catalysis taking place at the lipid–water interface of the supramolecular structures (Scheme 3), where the polar heads of the carboxylic acids undergo continuous protonation–deprotonation dynamics. Precisely the same ‘chemical trick’ that enables the stability of fatty acid vesicles as prebiotic compartments or microreactors (a balanced combination of neutral and anionic forms of the monomer) could be the critical feature to account for their catalytic power.
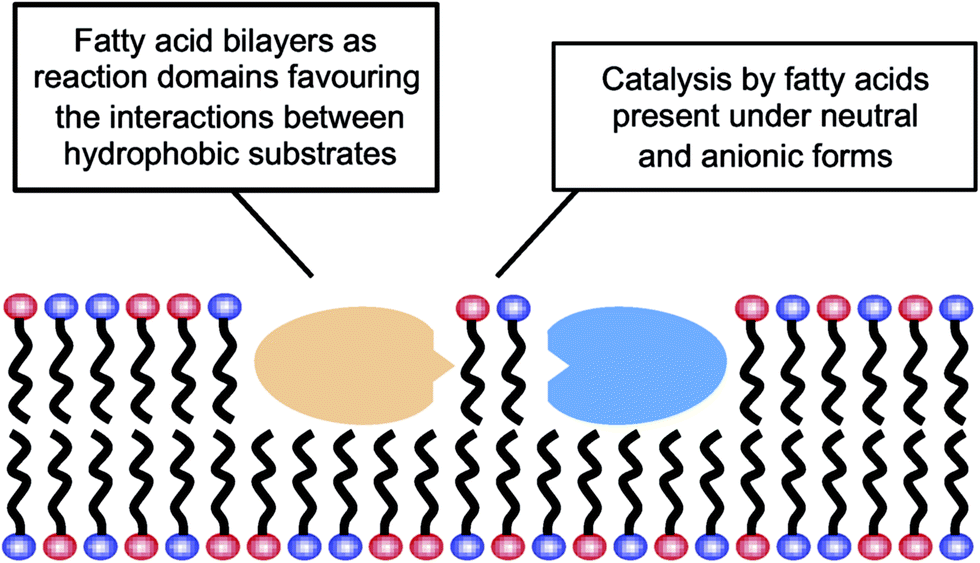 | ||
| Scheme 3 Fatty acids’ dual role in peptide bond formation. | ||
In this context, it is noteworthy that most aminolysis processes required for peptide syntheses involve large changes in the pKa of the reacting centres and, therefore, are susceptible to acid–base catalysis in a way similar to the reactions of 5(4H)-oxazolones. Bilayers made of fatty acids demonstrate that unique ability at neutral pH, which is not realised for other types of lipid compartments, like the phospholipid-based boundaries of modern cells. So fatty acids, in addition to providing a different local environment for the reaction (through their spontaneous assembly into supramolecular structures), could also act as acid–base catalysts for the formation of hydrophobic peptides, and potentially for any other chemical processes in which rate-determining proton transfer is required. These peptides, in turn, would likely contribute to membrane stability and would foster growth and potential reproduction of the vesicles. Therefore, one can envision a scenario in which lipid and protein precursors (i.e. fatty acids and hydrophobic peptides) would come together and mutually reinforce each other, in an integrated approach to the origin of the first functional protocells. This incipient stage is fully congruent with the modern structure of biological membranes, which consist of phospholipids and also of other hydrophobic components, most prominently membrane-associated or trans-membrane proteins. Accordingly, heterogeneous compartments, made of much simpler but already diverse kinds of hydrophobic compounds, would be present from the earliest phases of evolution, preceding the selection of current biomembranes.
More broadly speaking, this scenario is in line with recent perspectives on the problem of how chemistry could open the way towards biological phenomena, within a systems framework.3,4 The main idea underlying this new conception is that life, from its first steps, cannot be reduced to the chemistry of a single type of compound (e.g., nucleotide chemistry). Instead, prebiotic systems (taken as integrated entities, not just ‘populations of molecules’) ought to encompass several types of biomolecules – or rather, biomolecule precursors – in continuous interaction. In other words, the whole process of the origins of life would be understood as the evolutionary development of heterogeneous chemical mixtures with diverse emergent functionalities. Complex biopolymers (like the ones making up extant living cells) would be the final outcome of this long evolutionary process, not the starting point. Our results, from experiments carried out precisely with two main kinds of biomolecular precursor, fatty acids and amino acids, support this alternative view of the problem, in line with other recent experimental reports32,34 that highlight the importance of connecting peptide chemistry and compartments.
Acknowledgements
SMS and KRM received support from the Basque Government (IT 590-13 Grant) and from the Spanish Ministry of Economía y Competitividad (FFI2014-52173-P Grant). DB and RP thank the Agence Nationale de la Recherche for supporting the PeptiSystems project (grant ANR-14-CE33-0020). The authors also thank COST Action CM1304 (Emergence and Evolution of Complex Chemical Systems) for eliciting the collaboration between our groups and for supporting this project through a Short-Term Scientific Mission for SMS, who holds a PhD Grant from the University of the Basque Country (UPV/EHU).References
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- G. von Kiedrowski, S. Otto and P. J. Herdewijn, J. Syst. Chem., 2010, 1, 1–6 CrossRef.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef PubMed.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- S. Pizzarello and E. Shock, Cold Spring Harbor Perspect. Biol., 2010, 2, a002105 Search PubMed.
- H. J. Cleaves, J. H. Chalmers, A. Lazcano, S. L. Miller and J. L. Bada, Origins Life Evol. Biosphere, 2008, 38, 105–115 CrossRef CAS PubMed.
- L. P. Dworkin, D. W. Deamer, S. A. Sandford and L. Allamandola, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 815–819 CrossRef PubMed.
- T. M. McCollom, G. Ritter and B. R. Simoneit, Origins Life Evol. Biospheres, 1999, 29, 153–166 CrossRef CAS.
- G. Danger, R. Plasson and R. Pascal, Chem. Soc. Rev., 2012, 41, 5416–5429 RSC.
- R. Pascal, L. Boiteau and A. Commeyras, Top. Curr. Chem., 2005, 259, 69–122 CrossRef CAS.
- G. Danger, A. Michaut, M. Bucchi, L. Boiteau, J. Canal, R. Plasson and R. Pascal, Angew. Chem., Int. Ed., 2013, 52, 611–614 CrossRef CAS PubMed.
- W. R. Hargreaves and D. W. Deamer, Biochemistry, 1978, 17, 3759–3768 CrossRef CAS PubMed.
- J. M. Gebicki and M. Hicks, Nature, 1973, 243, 232–234 CrossRef CAS PubMed.
- C. L. Apel, D. W. Deamer and M. N. Mautner, Biochim. Biophys. Acta, Biomembr., 2002, 1559, 1–9 CrossRef CAS.
- M. M. Hanczyc and J. W. Szostak, Curr. Opin. Chem. Biol., 2004, 8, 660–664 CrossRef CAS PubMed.
- T. Namani and P. Walde, Langmuir, 2005, 21, 6210–6219 CrossRef CAS PubMed.
- K. Morigaki and P. Walde, Curr. Opin. Colloid Interface Sci., 2007, 12, 75–80 CrossRef CAS.
- S. E. Maurer, D. W. Deamer, J. M. Boncella and P.-A. Monnard, Astrobiology, 2009, 9, 979–987 CrossRef CAS PubMed.
- S. E. Maurer and P.-A. Monnard, Entropy, 2011, 13, 466–484 CrossRef CAS.
- I. A. Chen and P. Walde, Cold Spring Harbor Perspect. Biol., 2010, 2, a002170 CrossRef PubMed.
- P. Walde, H. Umakoshi, P. Stano and F. Mavelli, Chem. Commun., 2014, 50, 10177–10197 RSC.
- J. Blain and J. W. Szostak, Annu. Rev. Biochem., 2014, 83, 615–640 CrossRef CAS PubMed.
- H. Yanagawa, Y. Ogawa, K. Kojima and M. Ito, Origins Life Evol. Biosphere, 1988, 18, 179–207 CrossRef CAS PubMed.
- D. W. Armstrong, R. Seguin, C. J. McNeal, R. D. Macfarlane and J. H. Fendler, J. Am. Chem. Soc., 1978, 100, 4605–4606 CrossRef CAS.
- A. Brack, Origins Life Evol. Biosphere, 1987, 17, 367–379 CrossRef CAS PubMed.
- Z. Liu, D. Beaufils, J.-C. Rossi and R. Pascal, Sci. Rep., 2014, 4, 7440 CrossRef CAS PubMed.
- M. Blocher, D. Liu, P. Walde and P. L. Luisi, Macromolecules, 1999, 32, 7332–7334 CrossRef.
- M. Blocher, D. Liu and P. L. Luisi, Macromolecules, 2000, 33, 5787–5796 CrossRef.
- M. Blocher, T. Hitz and P. L. Luisi, Helv. Chim. Acta, 2001, 84, 842–848 CrossRef CAS.
- T. Hitz, M. Blocher, P. Walde and P. L. Luisi, Macromolecules, 2001, 34, 2443–2449 CrossRef CAS.
- H. H. Zepik, S. Rajamani, M.-C. Maurel and D. Deamer, Origins Life Evol. Biosphere, 2007, 37, 495–505 CrossRef CAS PubMed.
- A. Grochmal, L. Prout, R. Makin-Taylor, R. Prohens and S. Tomas, J. Am. Chem. Soc., 2015, 137, 12269–12275 CrossRef CAS PubMed.
- R. Furuuchi, E. I. Imai, H. Honda, K. Hatori and K. Matsuno, Origins Life Evol. Biosphere, 2005, 35, 333–343 CrossRef CAS PubMed.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495–501 CrossRef CAS PubMed.
- Z. R. Todd and C. H. House, Astrobiology, 2014, 14, 859–865 CrossRef CAS PubMed.
- D. Beaufils, G. Danger and L. Boiteau, et al. , Chem. Commun., 2014, 50, 3100–3102 RSC.
- F. M. F. Chen, K. Kuroda and N. L. Benoiton, Synthesis, 1979, 230–232 CrossRef CAS.
- F. M. F. Chen and N. L. Benoiton, Int. J. Pept. Protein Res., 2009, 30, 683–688 CrossRef.
- J. L. Cape, P.-A. Monnard and J. M. Boncella, Chem. Sci., 2011, 2, 661–671 RSC.
- N. L. Benoiton, K. Kuroda and F. M. F. Chen, Tetrahedron Lett., 1981, 22, 3361–3364 CrossRef CAS.
- W. P. Jencks, Acc. Chem. Res., 1976, 9, 425–432 CrossRef CAS.
- W. P. Jencks, Acc. Chem. Res., 1980, 13, 161–169 CrossRef CAS.
- A. C. Satterthwait and W. P. Jencks, J. Am. Chem. Soc., 1974, 96, 7031–7044 CrossRef CAS.
- C. A. Bunton, F. Nome, F. H. Quina and L. S. Romsted, Acc. Chem. Res., 1991, 24, 357–364 CrossRef CAS.
- S. Otto and J. B. F. N. Engberts, Org. Biomol. Chem., 2003, 1, 2809–2820 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Exhaustive data on the outcome of reactions, the aggregation state of the surfactants and the affinity of the dipeptide product for the aggregates. See DOI: 10.1039/c5sc04796j |
| ‡ HPLC peaks of all the other dipeptide diastereomers used in this work were identified by comparison with authentic samples prepared earlier.36 |
| This journal is © The Royal Society of Chemistry 2016 |