
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the prevalence of bridged macrocyclic pyrroloindolines formed in regiodivergent alkylations of tryptophan†
Tristin E.
Rose‡
,
Brice H.
Curtin‡
,
Kenneth V.
Lawson‡§
,
Adam
Simon
,
K. N.
Houk
and
Patrick G.
Harran
*
Department of Chemistry and Biochemistry, University of California Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095, USA. E-mail: harran@chem.ucla.edu
First published on 9th March 2016
Abstract
A Friedel–Crafts alkylation is described that efficiently transforms tryptophan-containing peptides into macrocycles of varying ring connectivity. Factors are surveyed that influence the distribution of regioisomers, with a focus on indole C3-alkylations leading to bridged endo-pyrroloindolines. We probe the stability and stereochemistry of these pyrroloindolines, study their rearrangement to C2-linked indolic macrocycles, and demonstrate a scalable, stereoselective synthesis of this compound class. Placing the macrocyclization in sequence with further template-initiated annulation leads to extraordinary polycyclic products and further demonstrates the potential for this chemistry to drive novel peptidomimetic lead discovery programs.
Introduction
The pyrroloindoline (hexahydropyrrolo[2,3-b]indole) motif is present in numerous tryptophan- and tryptamine-derived natural products. Methods to synthesize this ring system typically parallel biosynthetic schemes, wherein an indolic precursor is activated by substitution at its C3 position by an external electrophile.1 The incipient indolenium ion is then captured internally by a proximal nitrogen nucleophile (Fig. 1A). Bimolecular reactions of tryptophan in this manner often proceed without diastereoselectivity.2 Where achievable, kinetically diastereoselective transformations typically favor an exo disposition of the C2-carboxyl group,3 the extent of which depends strongly on the nature of the electrophile and on Nα- and carboxyl substitution.4 Significant advances in enantioselective synthesis of pyrroloindolines have been recently reported,5 including catalyst systems that can override the inherent substrate bias of tryptophan.6 Here, we investigate a different modality wherein the indole activation step is itself a ring-forming reaction (Fig. 1B).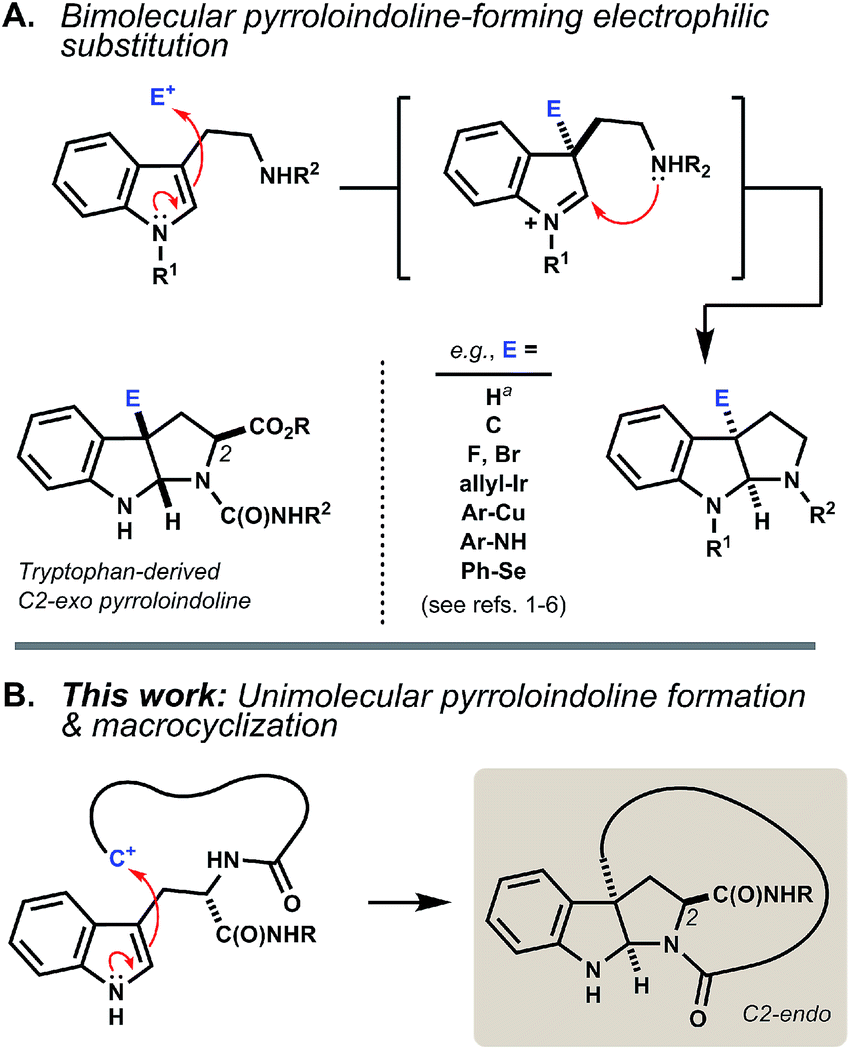 | ||
| Fig. 1 (A) Pyrroloindoline synthesis and biosynthesis typically proceed via bimolecular electrophilic substitution of indole at C3 and capture of the resulting indolenium ion by a proximal nitrogen nucleophile. The reaction of tryptophan is often exo-selective. aTautomerization of tryptophan by acid equilibrates to the C2-endo pyrroloindoline.4e,10 (B) Intramolecular C–C bond formation at C3 leads to ansa-bridged macrocyclic pyrroloindolines. | ||
Acid-promoted Friedel–Crafts alkylation enables procedurally straightforward, protecting group-free access to densely functionalized macrocycles.1 The indolic side chain of tryptophan is a powerful partner in this chemistry, because its multident nucleophilicity leads to diverse ring structures. Competing alkylation of indole C3 is particularly interesting as it embeds an endo-pyrroloindoline segment directly into an angularly linked, ansa-bridged peptidyl macrocycle.2 Pyrroloindolines bearing a C3a-linked macrocycle are rare, having been identified in only two classes of natural products, the chaetocochins,7 and nocardioazines.8 To our knowledge, synthesis of macrocyclic pyrroloindolines by intramolecular indole C3 substitution is limited to one example in an initial communication by us (vide infra).9 The present study accesses six new products bearing these motifs, as well as thirty-nine additional indolic macrocycle isomers, and characterizes their structures and reactivity in detail.
In a recent study of internal alkylations of tryptophan-containing peptides, we characterized the products derived from acidolysis of composite oligomer 2 (Scheme 1).9 When treated with Brønsted or Lewis acid, degradation of the cinnamyl carbonate in 2 led to competing internal Friedel–Crafts alkylations of proximal tryptophan and tyrosine side chains. The distribution of products was sensitive to acid promoter, solvent and temperature. Major products resulted from substitution at indole C5 of both tryptophan residues (4, 5, Scheme 1). The least polar isomer, a minor product of the reaction, was assigned as ansa-bridged polycycle 3, wherein the macrocyclization had formed an angular C–C linkage to a newly-formed endo-pyrroloindoline. The connectivity and relative stereochemistry of 3 was assigned by correlation NMR spectroscopy (HMBC, NOESY). Within limits of preparative isolation and analysis, a single diastereomer of 3 was identified in the product mixture.
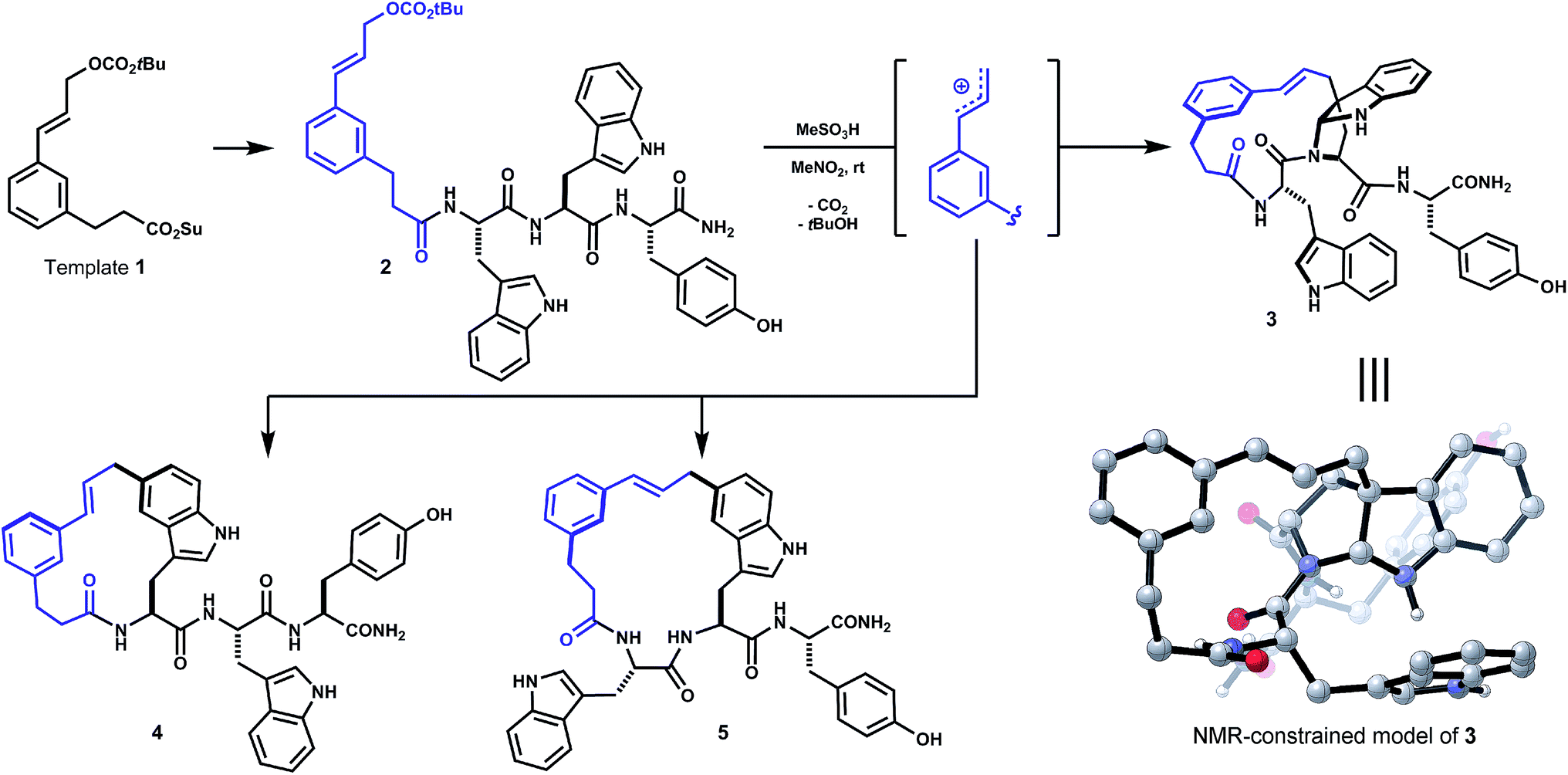 | ||
| Scheme 1 Initial unoptimized discovery (see ref. 9). Oligomer 2, derived from template 1 and Trp–Trp–Tyr, forms isomeric macrocycles by direct internal Friedel–Crafts alkylation under acidic conditions (e.g. MeSO3H, MeNO2). Major products 4 and 5 result from substitution at indole C5. Bridged endo-pyrroloindoline 3 was obtained as a minor product. | ||
Large ring-forming alkylations of this type were designed to generate topologically varied macrocycles with the intent of studying how ring connectivity and side chain rotational freedom influence pharmacological properties. Macrocyclization alone often improves performance relative to linear counterparts by restricting conformation and masking polar functional groups.11 Recent efforts have focused on confronting the pharmacokinetic limitations typically faced by macrocycles.11a,c,12 In particular, stereochemistry and backbone N-methylation patterns have been identified in cyclic penta- and hexapeptide lactams,11d,13 and of related analogs,11b,14 that enable passive diffusion through membranes and enhance oral bioavailability. More general means to affect these changes would be highly desirable. Along these lines, the increased rigidity and fewer main chain N–H bonds in 3 made this compound particularly interesting.
Here, we carefully examine the structure and stability of ansa-bridged pyrroloindolines related to 3. We survey their prevalence in mixtures of isomeric macrocycles generated by internal Friedel–Crafts alkylations of tryptophan. We evaluate structural features and reaction conditions that influence pyrroloindoline formation, and examine the origin of the observed diastereoselectivity. In addition, we have prepared a member of this new structural class using conventional target-based synthesis, thereby confirming the structure and establishing a scalable route to the group in general. Lastly, we show that this new unimolecular pyrroloindoline formation also proceeds in more complex settings.
Results and discussion
Analogs of linear oligomer 2 were designed to block major pathways of C5 alkylation in an attempt bias macrocyclization towards electrophilic substitution at indole C3. Substrates 6 and 7 bearing 5-methyl- and 5-fluoro-L-tryptophan, respectively, blocked C5 alkylation and removed competing side chain nucleophiles of Trp1 and Tyr3 (Scheme 2). Treating 6 or 7 with either methanesulfonic acid or triflimide in nitromethane¶ led to complete conversion to a mixture of isomeric products within minutes at room temperature. Abundant products were isolated and characterized as C–C and C–N linked macrocycles 8a–d and 9a–d. Bridged pyrroloindolines 8d and 9d were assigned as endo-22S,24R,25R on the basis of sequential NOE correlations about the newly formed pyrrolidine ring (see Scheme 2A). However, these materials were again obtained only as minor products. While using triflimide (4–6 eq.) as the acid promoter increased overall yield and remarkably shortened reaction times (1–2 min), it did not appreciably alter product distribution as had been observed for parent substrate 2.9 Blocking C5 with an electron-donating substituent (i.e. –CH3) had not suppressed reaction at the benzenoid ring or enhanced pyrroloindoline formation as intended, but an electron-withdrawing substituent (i.e. –F) was somewhat more effective in this regard. Both, however, shifted reactivity at the benzenoid ring from C5 to C4. Nonetheless, the formation of 15-membered macrocyclic endo-pyrroloindoline products in substrates bearing P2 tryptophan residues appeared to be generally diastereoselective.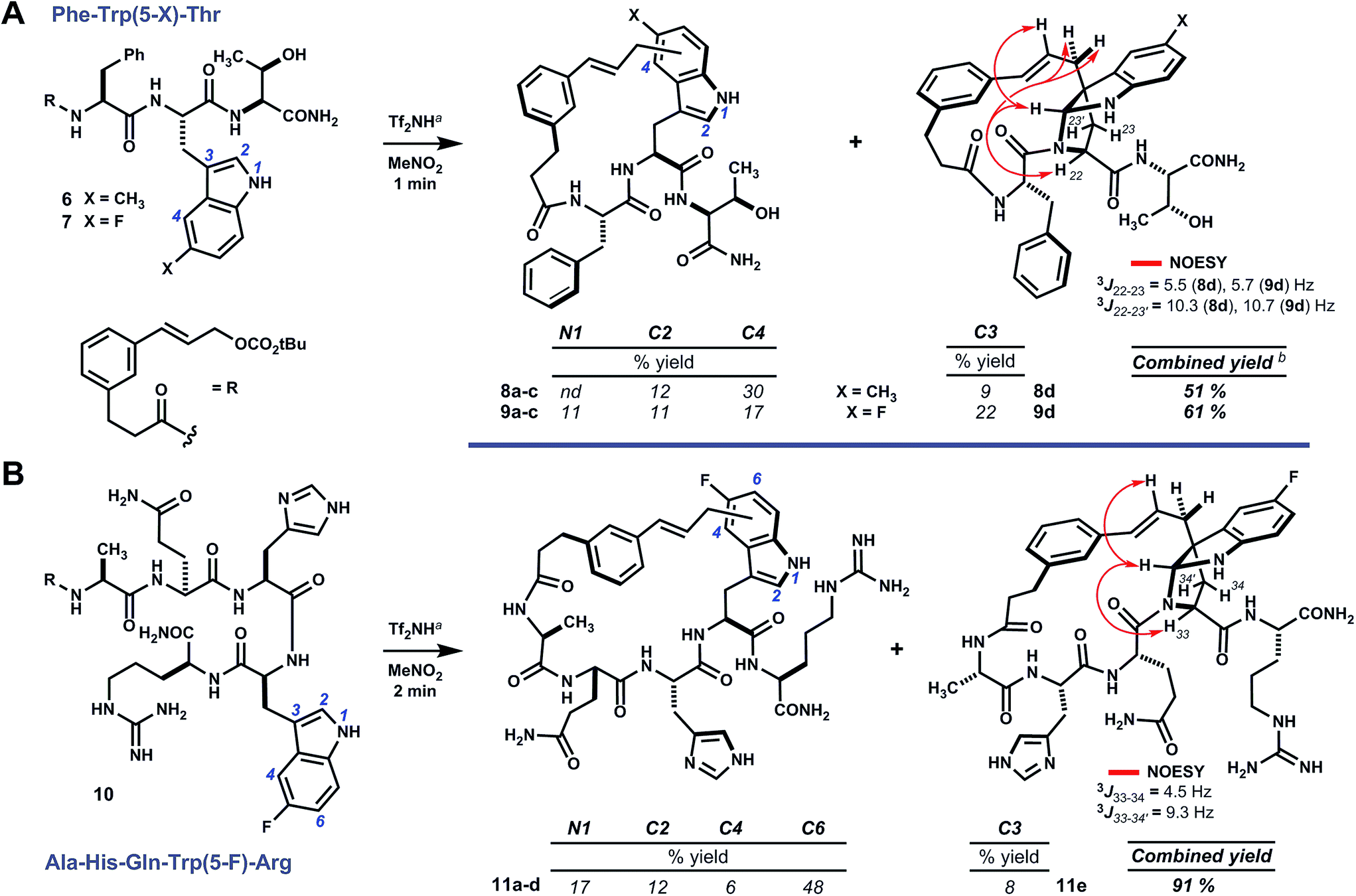 | ||
| Scheme 2 Pyrroloindoline-forming macrocyclizations of 5-substituted tryptophans. (A, B) Acidolysis of oligomers 6, 7 or 10 promotes internal substitution at indole N1, C2, C3, C4 or C6 (blue). The connectivity and relative stereochemistry of bridged pyrroloindolines 8d, 9d and 11e was assigned by 1H–13C-HMBC and 1H–1H-NOESY (red arrows), respectively. aTf2NH 4–6 eq., MeNO2, 5 mM in substrate, rt. bAdditional products were detected by HPLC; combined yield underestimates actual yield due to characterization of only major products, shown. nd = not determined. | ||
Accordingly, we next surveyed substrates varying in the position of tryptophan and in composition of the surrounding peptide. Substrate 10, bearing 5-fluoro-L-tryptophan at P4, underwent rapid cyclization unimpeded by the basic guanidine or imidazole side chains of arginine and histidine, respectively (Scheme 2B). Triflimide improved reaction yield considerably in this case, and afforded 21-membered ring bridged endo-pyrroloindoline 11e and isomeric macrocycles 11a–d in 91% combined yield. Notably, the major product resulted from alkylation at indole C6 (i.e.11d), rather than C4 as had been observed in reactions of 6 and 7. Taken together, these data suggest that regioselectivity tracks the inherent indole reactivity, which can be influenced by substitution of the indole nucleus. However, unlike bimolecular variants, the course of these cyclizations also depends on the geometry attainable by a given substrate and therefore the sequence of the embedded peptide. Thus, blocking the highly reactive C5 position of native tryptophan offers a useful means to finely tune the topology of macrocyclic products by shifting ring connectivity to adjacent positions C4 and C6.
To further test the effect of chain length on reaction regioselectivity and pyrroloindoline formation, we examined cyclizations of isomeric substrates 12–15 bearing 5-bromo-L-tryptophan in positions 1 through 4 (Scheme 3). Acidolysis of P1 variant 12 resulted primarily in cyclization at indole C6 (i.e.16a) and, to a lesser extent, at N1 and C7. No pyrroloindoline was observed in this case, presumably due to unfavorable strain associated with formation of a 13-memered ring by C3 substitution. As expected, P2 isomeric sequence 13 led to the pyrroloindoline 17e bearing the core 15-membered ring shared by products 3, 8d and 9d. Surprisingly, however, 17e was formed as the major product in 28% yield. It is not yet known whether this improved yield reflects the inherent reactivity of 5-bromoindole. Regioselectivity for C3 alkylation may also benefit from the smaller steric bulk of the serine side chain in 17e relative to tryptophan in 3 or to phenylalanine in 8d and 9d. Intriguingly, acidolysis of isomeric P3 sequence (14) also led to C3 alkylation and cyclization giving endo-pyrroloindoline 18c as the major product in 27% yield. The final P4 variant (15), however, did not give an analogous pyrroloindoline but instead afforded exo-pyridoindoline 19b by cyclization of the terminal carboxamide subsequent to C3 cinnamylation. Interestingly, the observed exo configuration had resulted from initial pro-R substitution, a facial bias identical to that observed for the pyrroloindoline outcomes. In another case, however, a substrate bearing 5-bromotryptophan at P4 yielded both exo- and endo-pyridoindolines (1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, see ESI Fig. S8†). This suggests that the mechanism for diastereoselection leading to exo-19b is distinct from pyrroloindoline outcomes (vide infra). The substrates surveyed here indicate that regioselectivity in tryptophan-based macrocyclizations is sensitive to oligomer composition, but bridged endo-pyrroloindoline and pyridoindoline products appear to form frequently.
:1 dr, see ESI Fig. S8†). This suggests that the mechanism for diastereoselection leading to exo-19b is distinct from pyrroloindoline outcomes (vide infra). The substrates surveyed here indicate that regioselectivity in tryptophan-based macrocyclizations is sensitive to oligomer composition, but bridged endo-pyrroloindoline and pyridoindoline products appear to form frequently.
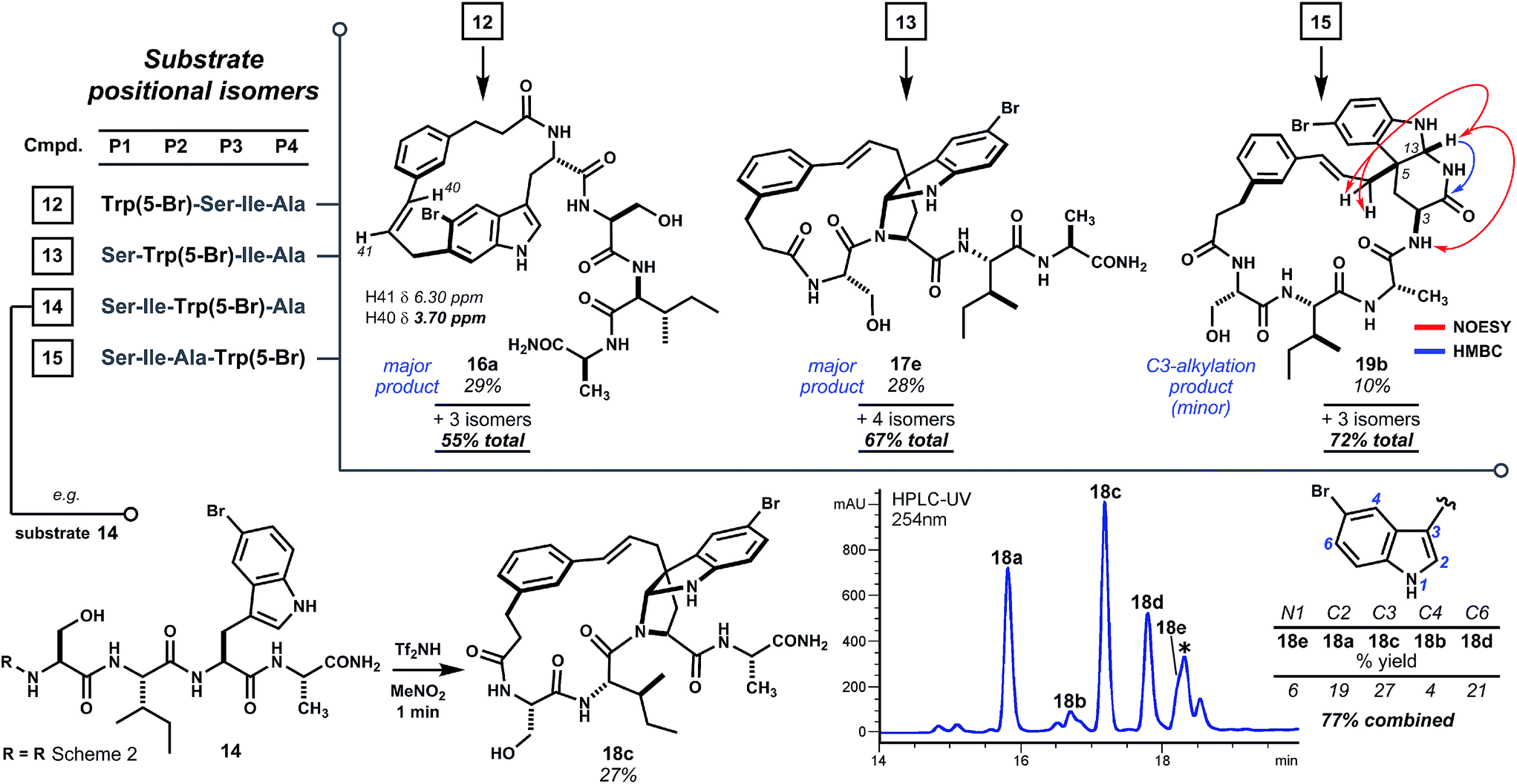 | ||
| Scheme 3 Cyclization scan of oligomers having Trp(5-Br) shifted along the chain (P1 → P4). Pyrroloindoline formation is sensitive to sequence composition and ring size, but favored by 5-bromotryptophan. No pyrroloindoline is formed from P1 isomer 12, whereas P2 and P3 variants 13 and 14 lead to pyrroloindolines 17e and 18c, respectively, as major products. Internal C3 alkylation of the P4 variant leads instead to cyclization of the terminal carboxamide to exo-pyridoindoline 19b. Reaction conditions as in Scheme 2. * Denotes non-isomeric impurity. For detailed product isomer distribution see ESI Fig. S4–S7.† | ||
While facial bias offers one potential rationale for the observed diastereoselectivity, we remained cognizant of the possibility for rearrangement of C3-linked macrocycles to other isomers under the reaction conditions.15 Indeed, bimolecular electrophilic substitution at indole C2 often proceeds by initial C3 addition and 1,2-migration.16 When isolated pyrroloindoline 18c was re-subjected to the reaction conditions with Tf2NH (4 eq.) in MeNO2, partial 1,2-rearrangement to the corresponding C2-linked isomer 18a was observed over a period of several hours. Consistent with previous observations, this slow equilibration of pyrroloindoline products suggests that large ring-forming cinnamylations proceed under kinetic control.17 Under forcing conditions with 20 vol% TFA in MeNO2, complete rearrangement of 18c to 18a was observed within 3 hours (t1/2 = 44 min, Fig. 2). Trace formation of other macrocycle isomers indicates that 1,2-rearrangement competes with reversion to a cinnamyl carbocation. The formation of C2-linked 18a in brief acidolysis reactions of linear substrate 14 may result from direct substitution at C2.16b,18 Alternatively, 18a may result from initially unselective C3 alkylation and rapid 1,2-rearrangement of one diastereomer, that corresponding to the exo-pyrroloindoline (i.e. pro-S addition at C3).19 Though a discrete exo diastereomer has not been observed, the latter possibility is supported by the near equal ratio of C3- to C2-linked products in the aforementioned seven examples. In two additional acid-promoted cyclizations, however, indole C2-linked macrocycles were obtained in the absence of pyrroloindoline products (see ESI Fig. S8 and S9†). Yet, in the case of 2 → 3 (Scheme 1), the product of C3 alkylation was obtained without concurrent C2 alkylation.9
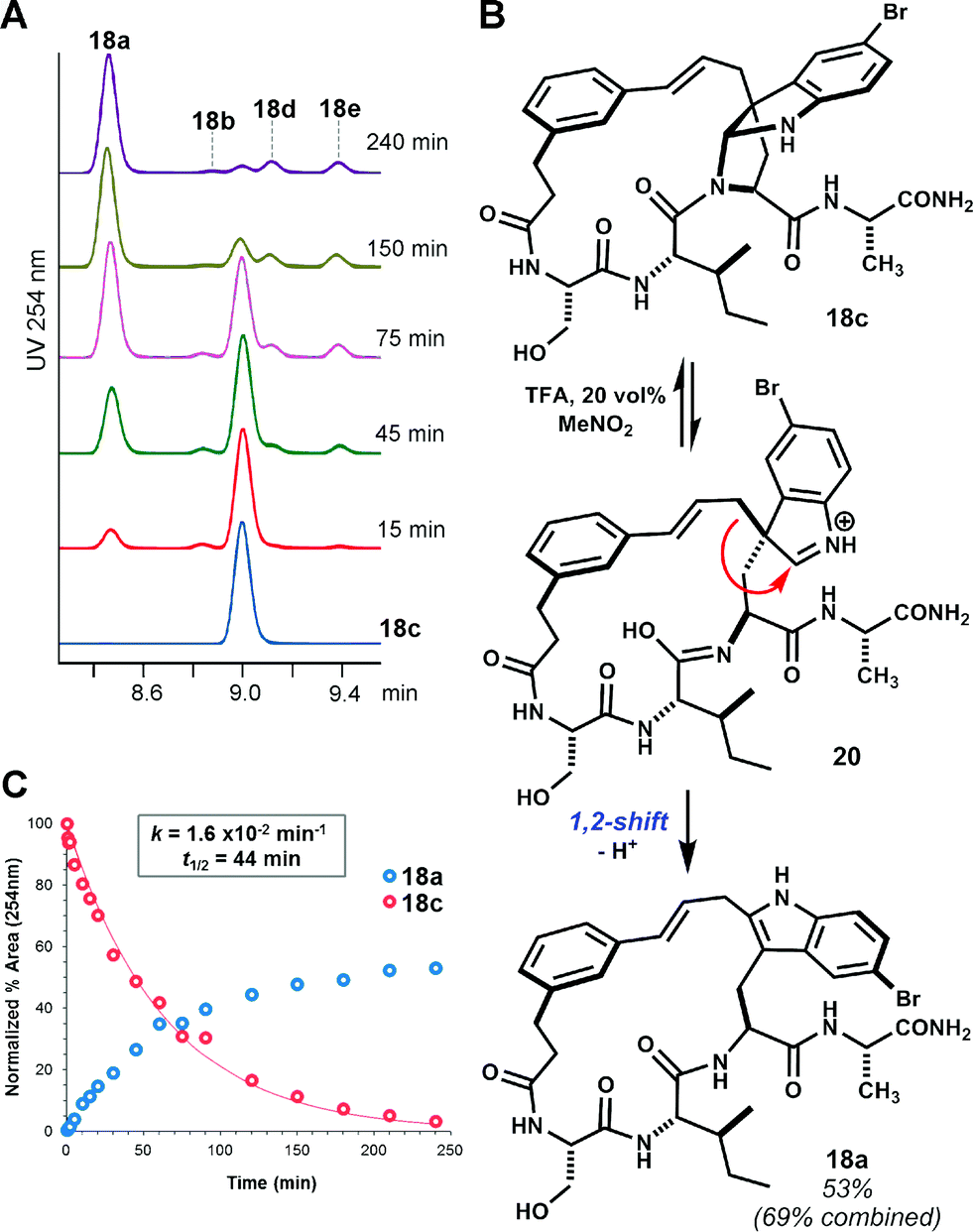 | ||
| Fig. 2 Rearrangement of pyrroloindoline 18c. (A) Time-course HPLC-UV (254 nm) analysis showing rearrangement of isolated 18c to isomer 18a in TFA/MeNO2 solution. Trace regioisomers (labelled) also form in this reaction. (B) Proposed mechanism for 1,2-rearrangement.18 (C) Kinetic plot showing pseudo-first-order reaction of 18c and accumulation of major product 18a. | ||
To further probe the origin of diastereoselectivity in pyrroloindoline-forming macrocyclizations, we examined the 1,2-rearrangement of model exo- and endo-pyrroloindolines 21a and 21b (Fig. 3A). Under pseudo-first-order conditions using 20 vol% TFA in MeNO2, rearrangement of exo-21b to indole C2-linked product 22 proceeded at a rate nearly 30-times faster than that of endo-21a (Fig. 3B). An Eyring plot was constructed from rate data at five different temperatures, which revealed activation energies ΔG‡exo = 20.5 kcal mol−1 and ΔG‡endo = 22.4 kcal mol−1 for these processes at 22 °C. We next explored this reaction computationally in order to better understand this kinetic difference. Using density functional theory (DFT), endo-pyrroloindoline 21a (R = Me) was calculated to be 1.0 kcal mol−1 more stable than the corresponding exo diastereomer 21b. This finding is consistent with reported thermodynamic preferences of related pyrroloindolines.3b,4e,19 Preference for the endo-pyrroloindoline in this case is primarily due to 1,3-allylic strain resulting from the tertiary amide (ESI Fig. S15†). DFT was also used to calculate the free energy profiles for the reactions of 21a and 21b (ESI Fig. S11†). In each case, 1,2-shift of the cinnamyl group from an intermediate indolium ion (i.e.23a,b) was found to be rate limiting (see ESI Fig. S14 and S16†). Diastereomeric transition structures TS-2a and TS-2b bear nearly enantiomeric geometries with respect to the indole ring and migrating cinnamyl group, but differ in orientation of the S-alanyl moiety relative to the indolic nucleus. This leads to greater stabilization of transition state TS-2b relative to TS-2a (ΔΔG‡calc = 2.9 kcal mol−1), in agreement with the experimentally observed difference in activation energy (ΔΔG‡exp = 1.9 kcal mol−1). Thus, the more rapid rearrangement of exo-pyrroloindoline 21b results both from the higher energy of 21b relative to 21a, and from the lower kinetic barrier for the reaction 21b → 22 relative to 21a → 22. These findings suggest that the diastereoselectivity observed in pyrroloindoline-forming macrocyclizations arises, at least in part, from the facility with which exo-pyrroloindolines rearrange to the corresponding indole C2-linked isomers.
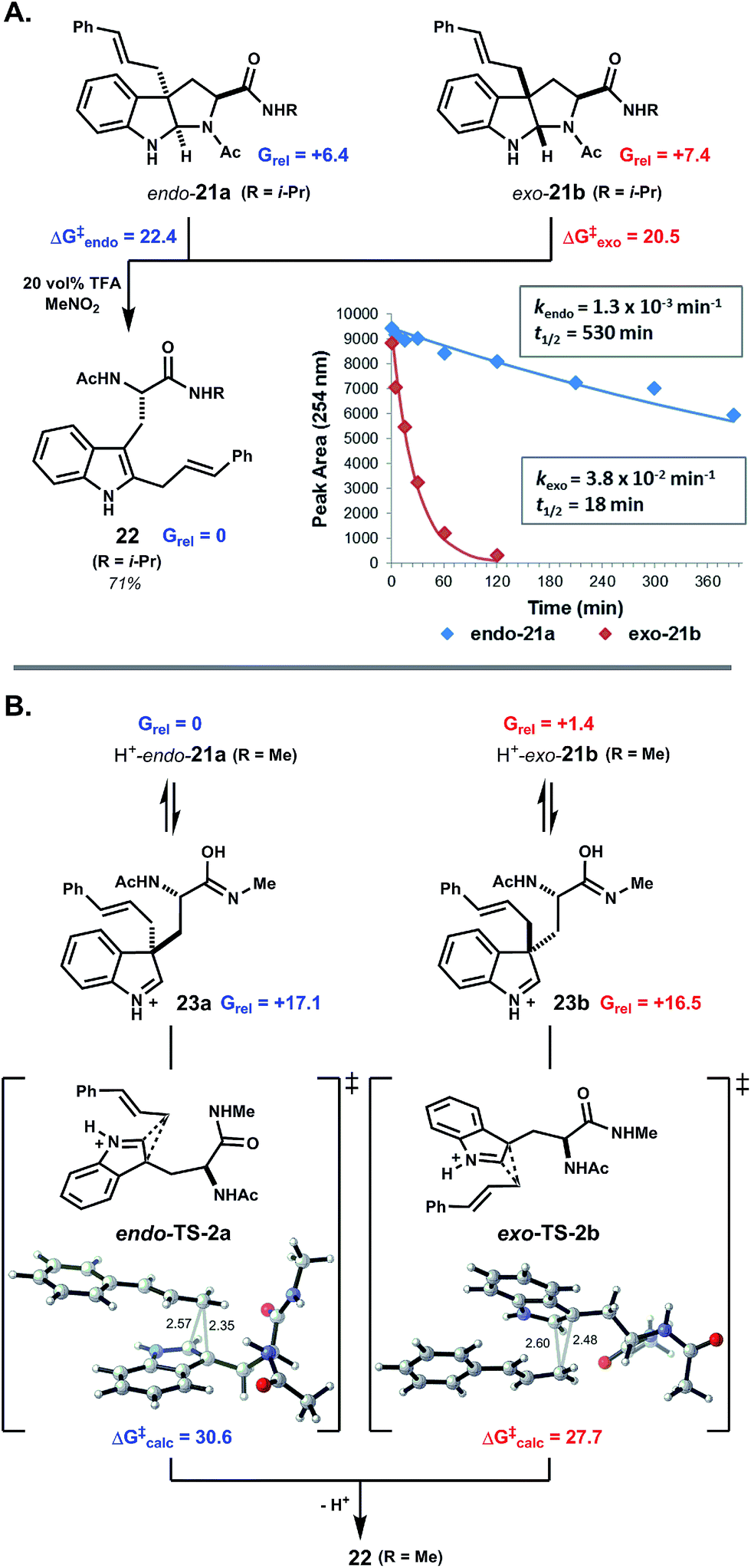 | ||
| Fig. 3 exo-Pyrroloindolines rearrange more readily than endo-pyrroloindolines. (A) Under acidic conditions, C3a-cinnamyl pyrroloindolines undergo ring-chain tautomerism and 1,2-rearrangement to indole C2-linked isomers. The kinetic plot for rearrangement of 21a and 21b in 20 vol% TFA at 5 °C shows the faster rate of reaction for exo-pyrroloindoline 21b. From Eyring analysis, the free energy of activation was 1.9 kcal mol−1 higher for exo-21b relative to endo-21a. (B) DFT calculations indicate that endo-pyrroloindoline 21a is the thermodynamically more stable than exo-21b. Cinnamyl 1,2-shift is rate limiting in both cases, and the reaction of exo-21b proceeds via a lower kinetic barrier (see text and further discussion in ESI†). Note: all free energies are in kcal mol−1. | ||
The Friedel–Crafts macrocyclizations under study generate collections of macrocycle isomers that would be time-consuming to prepare individually. Exploring ring diversity in this manner is our approach to refining biological activity. Importantly, isomers of particular interest may also be synthesized by convergent means. This has been demonstrated by a selective synthesis of fluorinated pyrroloindoline 9d (Scheme 4). Starting from 5-fluoro-L-tryptophan methyl ester and cinnamyl alcohol 24, a derivative of template 1, intermolecular Pd0-catalyzed allylation promoted by Et3B led to selective C3 cinnamylation and afforded endo-pyrroloindoline 26 in 80% yield as a single diastereomer.20 The remaining two amino acids were then introduced by first amidation of 26 with N-Boc-L-phenylalanine, then saponification of the methyl ester and coupling of the resulting carboxylate to L-threonine amide to give 27. Deprotection of the N-Boc and tert-butyl groups with TFA:DCM (1:1) at 0 °C minimized C3–C2 rearrangement, and lactamization of this seco acid with HBTU completed bridged pyrroloindoline 9d. This material was spectroscopically identical to that obtained by the acidolysis of 7 (see annotations Scheme 4). Convergent routes such as this are useful for preparing larger quantities of material, whereas Friedel–Crafts cyclization forms pyrroloindolines and additional macrocycle isomers rapidly and directly.
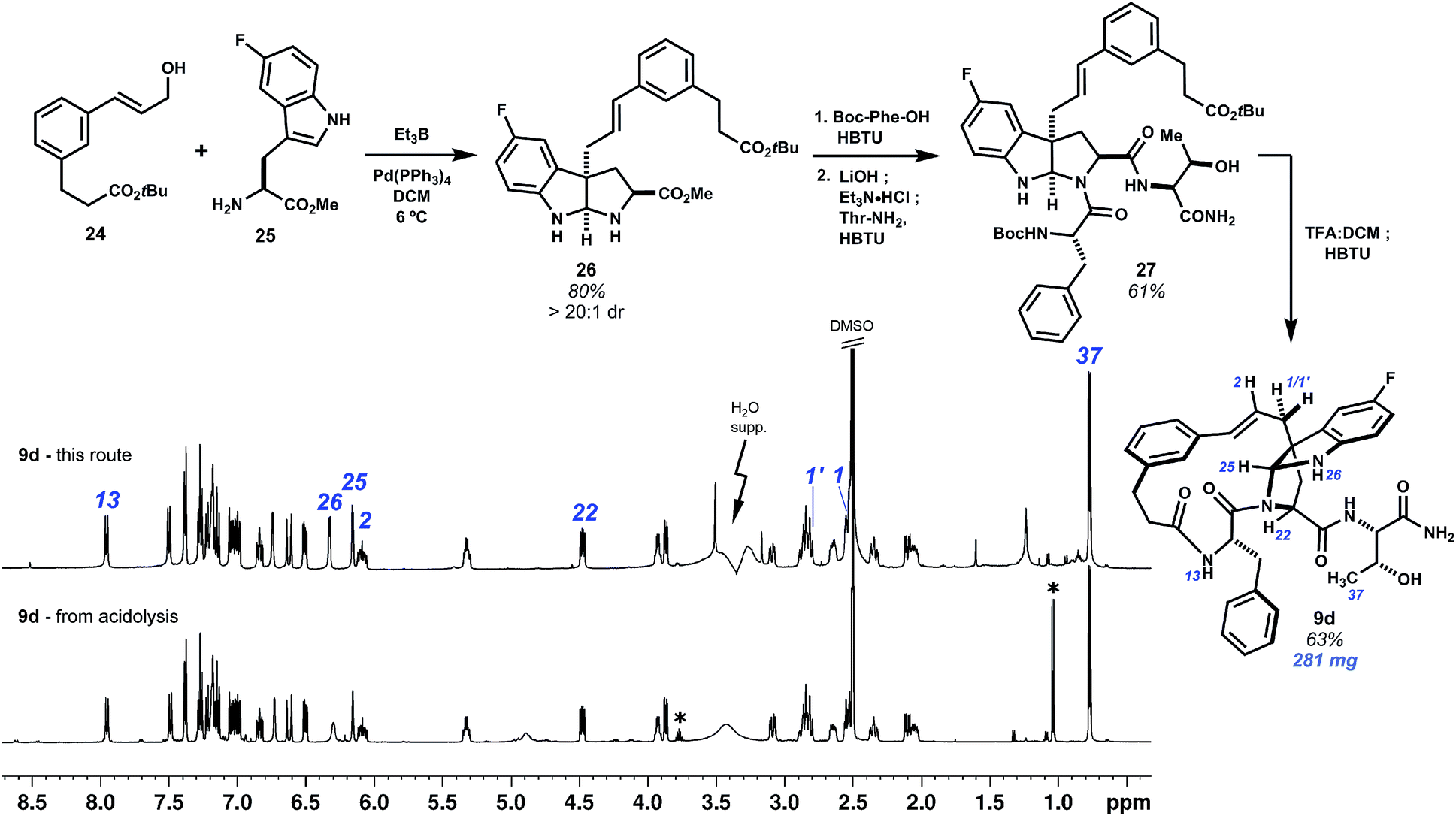 | ||
| Scheme 4 Selective synthesis of pyrroloindoline 9d. Bimolecular Pd0-catalyzed C3-selective cinnamylation of 5-fluoro-L-tryptophan promoted by Et3B sets up to form the bridging 15-membered ring by lactamization. 1H NMR spectra (500 MHz, DMSO-d6) of 9d obtained by this route match that of material isolated from the acid-promoted cyclization. Key resonance annotations in blue. * Denotes contaminant signals. | ||
Internal alkylations using simple template 1 form large rings with broad functional group tolerance and procedural ease. Creative opportunity exists to combine macrocyclizations of this type with additional template reactivity to further stabilize the peptide domain by rigidifying product structures. For example, variant 28 additionally bears a latent aldehyde designed to initiate N-acyliminium ion cyclizations (e.g.29, Scheme 5), the results of which depend on the nature of the P1 side chain.21 Extensive investigations of this reaction by the Meldal laboratory demonstrate flexibility and generally high diastereoselectivity.21 In direct comparison to template 1, we examined the performance of trifunctional variant 28 using prototypical tryptophan-containing oligomer Trp–Trp–Tyr.9,22 Acylation of the peptide N-terminus and subsequent treatment with aqueous acetic acid promoted diastereoselective Pictet–Spengler cyclization of Trp1 to give intermediate fused tryptoline 30 in 88% yield. When treated with Tf2NH (3 eq.) in MeNO2, this material was cleanly transformed to isomeric products 31a–f resulting exclusively from alkylation of Trp2 with the major product arising from substitution at indole C5, as expected. Alkylation ortho to the phenol of tyrosine, anticipated from previous studies of analogous substrate 2, was not observed.23 Alkylation of the P1 tryptoline moiety was also not observed, presumably due to strain associated with annulation of this rigid ring system. These results anticipate that templates 1 and 28 will exhibit subtly different regioselectivity in large ring-forming reactions. That said, we were pleased to find polycyclic pyrroloindoline 31e, despite these differences. This product possesses nine fused rings and a mere five rotatable bonds, whereas Trp–Trp–Tyr itself possesses eleven such bonds. Additionally, 31e bears less polar surface area (161 Å2) than the starting peptide (181 Å2), with only non-polar surface area introduced by the template. These alterations tend towards molecular properties advocated for the design of orally bioavailable drugs.11c,24 Though 31e has not yet been evaluated for biological activity, this outcome exemplifies marked structural alterations that can be quickly achieved. The pharmacological properties of such structures will almost assuredly improve relative to the starting peptide.
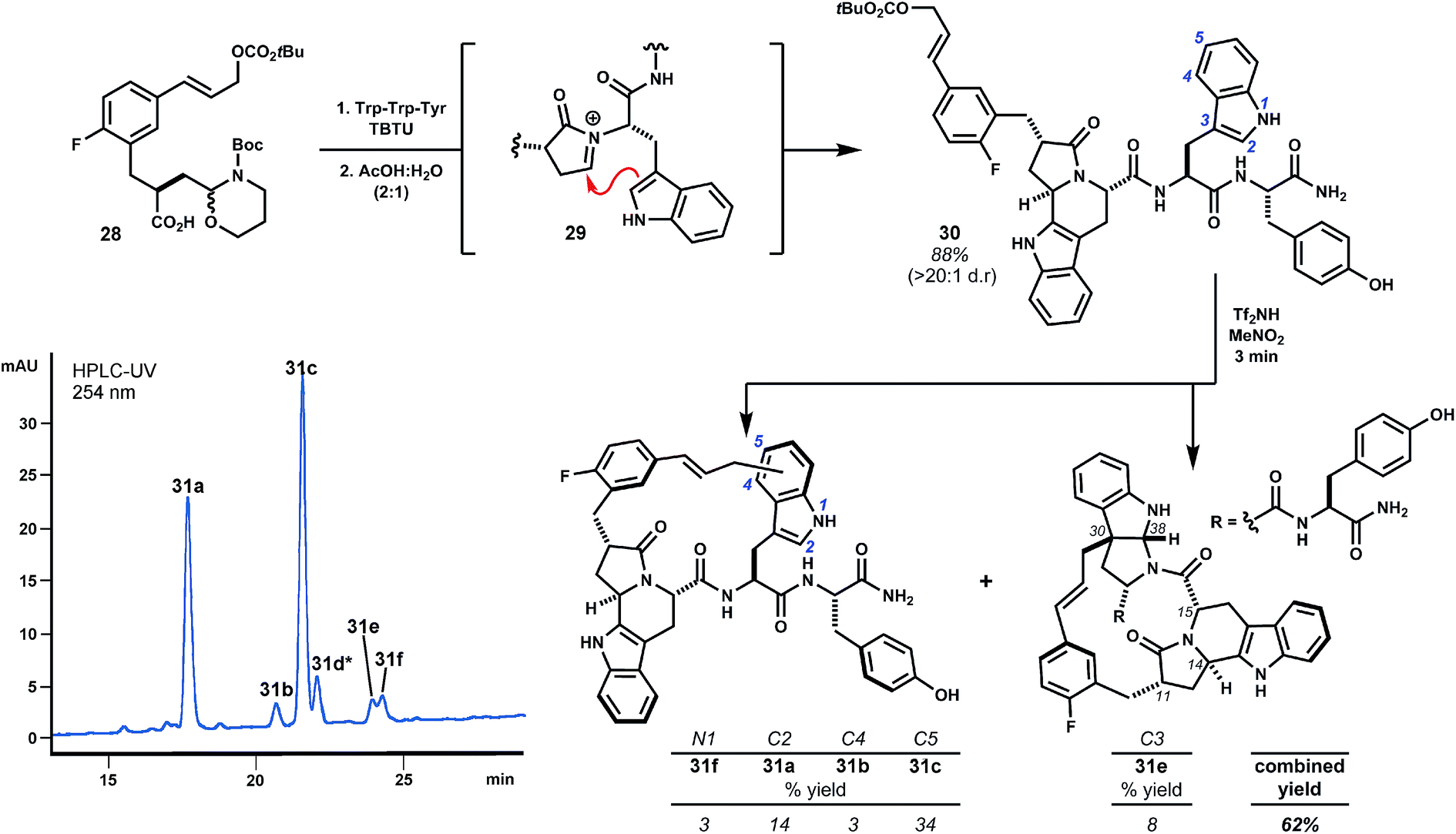 | ||
| Scheme 5 Combining trifunctional template 28 with Trp–Trp–Tyr rapidly forms complex polycycles. Initial amidation and Pictet–Spengler cyclization of Trp1 (i.e.29) followed by acid-promoted cyclization leads to macrocycle isomers 31a–f substituting the periphery of Trp2. Polycycle 31e results from indole C3 alkylation and cyclization to the endo-pyrroloindoline, analogously to related products obtained from template 1. *Note: peak 31d contained two product isomers that were not identified. | ||
Conclusions
Regiodivergent internal alkylations of tryptophan create macrocycles of varying connectivity in two or three steps from linear peptides. Macrocyclic products bearing embedded endo-pyrroloindolines are a valuable facet of this chemistry. The simplicity of the acidolysis method permits even minor constituents to be isolated, screened for function, and characterized with relative ease. In this regard, the general presence of pyrroloindolines and predictability of regiochemical outcomes are more important than the abundance of any one product. For structures of particular interest, convergent target-oriented synthesis is always an option, as we demonstrate. These more step-intensive routes offer scalable access when refined medicinal chemistry is appropriate. Finally, methods combining large ring-formations with additional template-initiated annulation, such as in reactions of 28, hold unique potential to quickly build peptidomimetics of unprecedented structural complexity. Further experiments along these lines are ongoing.Acknowledgements
Funding was provided by the NIH (CA184772 to P. G. H.), the NSF (CHE-1361104 to K. N. H.), a grant from the University of California Cancer Research Coordinating Committee, the Donald J. & Jane M. Cram Endowment, and NSF Equipment Grant CHE-1048804. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE). T. E. R. was supported by an NSF Graduate Research Fellowship (DGE-0707424), and a UCLA Dissertation Year Fellowship. T. E. R. and A. S. were supported by the UCLA Chemistry-Biology Interface Training Program (T32GM008496).Notes and references
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem.–Eur. J., 2011, 17, 1388 CrossRef CAS PubMed.
- (a) T. F. Spande, M. Wilchek and B. Witkop, J. Am. Chem. Soc., 1968, 90, 3256 CrossRef CAS PubMed; (b) B. G. Mcfarland, Y. Inoue and K. Nakanishi, Tetrahedron Lett., 1969, 10, 857 CrossRef; (c) G. M. Loudon, D. Portsmouth, A. Lukton and D. E. J. Koshland, J. Am. Chem. Soc., 1969, 91, 2792 CrossRef CAS PubMed; (d) P. S. Baran, C. A. Guerrero and E. J. Corey, Org. Lett., 2003, 5, 1999 CrossRef CAS PubMed.
- (a) C. S. López, C. Pérez-Balado, P. Rodríguez-Graña and Á. R. De Lera, Org. Lett., 2008, 10, 77 CrossRef PubMed; (b) D. Crich and X. Huang, J. Org. Chem., 1999, 64, 7218 CrossRef CAS.
- (a) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151 CrossRef CAS PubMed; (b) T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119 CrossRef CAS PubMed; (c) K. M. Depew, S. P. Marsden, D. Zatorska, A. Zatorski, W. G. Bornmann and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11953 CrossRef CAS; (d) M. Nakagawa, H. Watanabe, S. Kodato, H. Okajima, T. Hino, J. L. Flippen and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4730 CrossRef CAS PubMed; (e) M. Taniguchi and T. Hino, Tetrahedron, 1981, 37, 1487 CrossRef CAS.
- (a) H. M. Nelson, S. H. Reisberg, H. P. Shunatona, J. S. Patel and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 5600 CrossRef CAS PubMed; (b) L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418 CrossRef CAS PubMed; (c) W. Shao, H. Li, C. Liu, C.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 7684 Search PubMed; (d) O. Lozano, G. Blessley, T. Martinez Del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS PubMed; (e) L. M. Repka and S. E. Reisman, J. Org. Chem., 2013, 78, 12314 CrossRef CAS PubMed; (f) J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS PubMed; (g) S. Zhu and D. W. C. MacMillan, J. Am. Chem. Soc., 2012, 134, 10815 CrossRef CAS PubMed; (h) J. R. Wolstenhulme, A. Cavell, M. Gredičak, R. W. Driver and M. D. Smith, Chem. Commun., 2014, 50, 13585 RSC; (i) Y. Huang, C. Zheng, L. Pan, Q. Jin and G. Zhao, J. Org. Chem., 2015, 80, 10710 CrossRef CAS PubMed.
- X. Zhang, W.-B. Liu, H.-F. Tu and S.-L. You, Chem. Sci., 2015, 6, 4525 RSC.
- G. Li, B. Li, T. Yang, J. Yan, G. Liu and G. Zhang, J. Nat. Prod., 2006, 69, 1374 CrossRef CAS PubMed.
- R. Raju, A. M. Piggott, X. C. Huang and R. J. Capon, Org. Lett., 2011, 13, 2770 CrossRef CAS PubMed.
- K. V. Lawson, T. E. Rose and P. G. Harran, Tetrahedron, 2013, 69, 7683 CrossRef CAS PubMed.
- T. Hino and M. Taniguchi, J. Am. Chem. Soc., 1978, 100, 5564 CrossRef CAS.
- (a) G. H. Goetz, L. Philippe and M. J. Shapiro, ACS Med. Chem. Lett., 2014, 5, 1167 CrossRef CAS PubMed; (b) A. T. Bockus, K. W. Lexa, C. R. Pye, A. S. Kalgutkar, J. W. Gardner, K. C. R. Hund, W. M. Hewitt, J. A. Schwochert, E. Glassey, D. A. Price, A. M. Mathiowetz, S. Liras, M. P. Jacobson and R. S. Lokey, J. Med. Chem., 2015, 58, 4581 CrossRef CAS PubMed; (c) A. T. Bockus, C. M. McEwen and R. S. Lokey, Curr. Top. Med. Chem., 2013, 13, 821 CrossRef CAS PubMed; (d) T. R. White, C. M. Renzelman, A. C. Rand, T. Rezai, C. M. McEwen, V. M. Gelev, R. A. Turner, R. G. Linington, S. S. F. Leung, A. S. Kalgutkar, J. N. Bauman, Y. Zhang, S. Liras, D. A. Price, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, Nat. Chem. Biol., 2011, 7, 810 CrossRef CAS PubMed; (e) Y. S. Tsantrizos, G. Bolger, P. Bonneau, D. R. Cameron, N. Goudreau, G. Kukolj, S. R. LaPlante, M. Llinàs-Brunet, H. Nar and D. Lamarre, Angew. Chem., Int. Ed., 2003, 42, 1356 CrossRef CAS PubMed; (f) A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC; (g) J. P. Maianti, A. McFedries, Z. H. Foda, R. E. Kleiner, X. Q. Du, M. A. Leissring, W.-J. Tang, M. J. Charron, M. A. Seeliger, A. Saghatelian and D. R. Liu, Nature, 2014, 511, 94 CrossRef CAS PubMed; (h) C. J. Dinsmore, M. J. Bogusky, J. C. Culberson, J. M. Bergman, C. F. Homnick, C. B. Zartman, S. D. Mosser, M. D. Schaber, R. G. Robinson, K. S. Koblan, H. E. Huber, S. L. Graham, G. D. Hartman, J. R. Huff and T. M. Williams, J. Am. Chem. Soc., 2001, 123, 2107 CrossRef CAS PubMed; (i) R. J. Cherney, L. Wang, D. T. Meyer, C.-B. Xue, Z. R. Wasserman, K. D. Hardman, P. K. Welch, M. B. Covington, R. A. Copeland, E. C. Arner, W. F. DeGrado and C. P. Decicco, J. Med. Chem., 1998, 41, 1749 CrossRef CAS PubMed.
- (a) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136 CrossRef CAS PubMed; (b) J. Kotz, Sci. Exch., 2012, 5, 1 Search PubMed.
- (a) T. A. Hill, R. Lohman, H. N. Hoang, D. S. Nielsen, C. C. G. Scully, W. M. Kok, L. Liu, A. J. Lucke, M. J. Stoermer, C. I. Schroeder, S. Chaousis, B. Colless, P. V. Bernhardt, D. J. Edmonds, D. A. Gri, C. J. Rotter, R. B. Ruggeri, D. A. Price, S. Liras, D. J. Craik and D. P. Fairlie, ACS Med. Chem. Lett., 2014, 5, 1148 CrossRef CAS PubMed; (b) C. K. Wang, S. E. Northfield, B. Colless, S. Chaousis, I. Hamernig, R. J. Lohman, D. S. Nielsen, C. I. Schroeder, S. Liras, D. A. Price, D. P. Fairlie and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17504 CrossRef CAS PubMed; (c) T. Rezai, J. E. Bock, M. V. Zhou, C. Kalyanaraman, R. S. Lokey and M. P. Jacobson, J. Am. Chem. Soc., 2006, 128, 14073 CrossRef CAS PubMed.
- D. S. Nielsen, H. N. Hoang, R.-J. Lohman, T. A. Hill, A. J. Lucke, D. J. Craik, D. J. Edmonds, D. A. Griffith, C. J. Rotter, R. B. Ruggeri, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 12059 CrossRef CAS PubMed.
- M. Mari, S. Lucarini, F. Bartoccini, G. Piersanti and G. Spadoni, Beilstein J. Org. Chem., 2014, 10, 1991 CrossRef PubMed.
- (a) A. H. Jackson, B. Naidoo and P. Smith, Tetrahedron, 1968, 24, 6119 CrossRef CAS; (b) A. H. Jackson and P. Smith, Chem. Commun., 1967, 264 RSC.
- T. E. Rose, K. V. Lawson and P. G. Harran, Chem. Sci., 2015, 6, 2219 RSC.
- G. Casnati, A. Dossena and A. Pochini, Tetrahedron Lett., 1972, 13, 5277 CrossRef.
- This possibility parallels reports by Crich and co-workers wherein related tryptophan-derived C2-exo-pyrroloindolines were found to ring open more readily, allowing ready isolation of the C2-endo diastereomer. See: D. Crich, M. Bruncko, S. Natarajan, B. Teo and D. Tocher, Tetrahedron, 1995, 51, 2215 CrossRef CAS , and ref. 4a.
- (a) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314 CrossRef CAS PubMed; (b) M. Kimura, M. Futamata, R. Mukai and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 4592 CrossRef CAS PubMed.
- (a) T. Groth and M. J. Meldal, J. Comb. Chem., 2001, 3, 34 CrossRef CAS PubMed; (b) T. E. Nielsen and M. Meldal, J. Org. Chem., 2004, 69, 3765 CrossRef CAS PubMed.
- H. Zhao, L. Negash, Q. Wei, T. G. LaCour, S. J. Estill, E. Capota, A. A. Pieper and P. G. Harran, J. Am. Chem. Soc., 2008, 130, 13864 CrossRef CAS PubMed.
- Tyrosine C-alkylation could likely be achieved by internal Pd0-catalyzed O-cinnamylation and acid-promoted O → Cortho rearrangement analogously to our previous report. See ref. 9 and 17.
- (a) D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS PubMed; (b) J. J. Lu, K. Crimin, J. T. Goodwin, P. Crivori, C. Orrenius, L. Xing, P. J. Tandler, T. J. Vidmar, B. M. Amore, A. G. E. Wilson, P. F. W. Stouten and P. S. Burton, J. Med. Chem., 2004, 47, 6104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S17: comparison of Tf2NH versus MeSO3H, structures of additional macrocycle isomers obtained from reactions of 12–15, three additional examples. Experimental details, characterization and NMR data for all new compounds. See DOI: 10.1039/c5sc04612b |
| ‡ T. E. R., B. H. C. and K. V. L. contributed equally to this work. |
| § Current address: Harvard University, Department of Chemistry and Chemical Biology, 12 Oxford Street, Cambridge, Massachusetts 02138, USA. |
| ¶ Pre-treatment of commercial grade nitromethane with either 3 Å molecular sieves (7 days) or activated neutral alumina (12 h) is essential for optimal results in Friedel–Crafts cyclizations. Interestingly, adding H2O (up to 1000 ppm) to the resultant dry nitromethane has no deleterious effects. For further discussions see: T. E. Rose, Ph.D. Dissertation [Online], University of California, Los Angeles, 2015, pp. 158–160, http://escholarship.org/uc/item/0mx7x1st (accessed Oct 2, 2015), UMI: 3706064. |
| This journal is © The Royal Society of Chemistry 2016 |