
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of Au-, Co-η1PCO and Cu-η2PCO complexes, conversion of an Ir–η1PCO complex into a dimetalladiphosphene, and an interaction-free PCO anion†
Liu Leo Liuab,
David A. Ruiza,
Fatme Dahcheha,
Guy Bertrand*a,
Riccardo Suterc,
Aaron M. Tondreauc and
Hansjörg Grützmacher*c
aUCSD-CNRS Joint Research Chemistry Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093-0343, USA. E-mail: guybertrand@ucsd.edu
bKey Laboratory for Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Department of Chemistry, Xiamen University, Xiamen, 361005, China
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch
First published on 4th January 2016
Abstract
Sodium phosphaethynolate reacts with [MCl(PDI)] (M = Co, Ir; PDI = pyridinediimine) to give metallaphosphaketenes, which in the case of iridium rearranges into a dimetalladiphosphene, via CO migration from phosphorus to the metal. Two different bonding modes of the PCO anion to CAAC-coinage metal complexes [CAAC: cyclic (alkyl)(amino)(carbene)] are reported, one featuring a strong Au–P bond and the other an η2 coordination to copper. The gold complex appears to be mostly unreactive whereas the copper complex readily reacts with various organic substrates. A completely free PCO anion was structurally characterized as the [Cu(La)2]+ (OCP)− salt. It results from the simple displacement of the PCO unit of the cationic (CAAC)Cu(PCO) complex by a second equivalent of CAAC.
Introduction
Despite their early synthesis in 1992 by Becker, Westerhausen et al.,1 the reactivity of phosphaethynolate salts M+(PCO)− remained unexplored until the recent development of simple syntheses which allow for the preparation of large quantities of pure material.2 These salts have been shown to undergo a variety of chemical transformations with organic substrates,3 including cycloaddition reactions leading to phosphorus containing heterocycles.4 In contrast, the strong reducing ability of PCO− salts5 has hindered their exploration in transition metal chemistry. In most cases the reactions with metal complexes lead to decomposition products.6 The only exceptions are the formation of a P2(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2 ring A by reacting Li(OCP) with (η5-C5R5)(CO)2FeBr,7 the isolation of [Re(PCO)(CO)2(triphos)] B,6 and uranium and thorium M(OCP)(amid)3 complexes C,8 in which the phosphaethynolate binds the metal via the oxygen center (Fig. 1). Noteworthy is an extensive computational study, showing that metallaphosphaketene complexes such as D may rearrange via CO migration into molecular metal phosphides E.9 The limited number of studies concerning the coordination chemistry of PCO− is in marked contrast with the large number of reports dealing with the lighter analogue, namely the cyanate anion (NCO−).10
O)2 ring A by reacting Li(OCP) with (η5-C5R5)(CO)2FeBr,7 the isolation of [Re(PCO)(CO)2(triphos)] B,6 and uranium and thorium M(OCP)(amid)3 complexes C,8 in which the phosphaethynolate binds the metal via the oxygen center (Fig. 1). Noteworthy is an extensive computational study, showing that metallaphosphaketene complexes such as D may rearrange via CO migration into molecular metal phosphides E.9 The limited number of studies concerning the coordination chemistry of PCO− is in marked contrast with the large number of reports dealing with the lighter analogue, namely the cyanate anion (NCO−).10
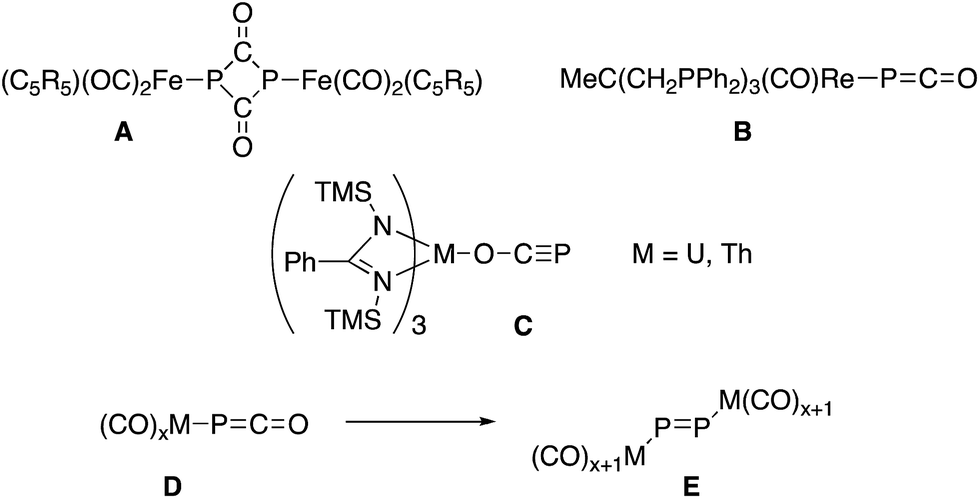 | ||
| Fig. 1 Previous experimental (A–C) and computational (D–E) works on phosphaethynolate with transition metals.6–9 | ||
Herein we describe salt metathesis reactions leading to both unstable and stable terminal PCO transition metal complexes, featuring different coordination modes, and reactivity. We also report the experimental demonstration of the predicted conversion of D to E, and the displacement of the PCO unit of the copper complex by a CAAC, which leads to a PCO anion with no coordinating solvents or binding agents.
Results and discussion
Because of the strongly reducing character of Na(OCP),6,7 we targeted complexes bearing pyridinediimine (PDI) ligands11 and cyclic (alkyl)(amino)carbenes (CAACs)12,13 which are known to efficiently stabilize metals in low oxidation states.14When the [CoCl(PDIiPr)] complex 1 was reacted with Na(OCP) in THF at −30 °C the color changed from pink to deep purple, and a single broad resonance in the 31P NMR spectrum at δ = −226 ppm [vs. δ = −392 ppm for Na(OCP)] indicated quantitative conversion. The metallaphosphaketene 3 was isolated in 61% yield and fully characterized. The IR spectrum showed the asymmetric stretching frequency of the phosphaketene unit at νasym = 1851 cm−1, intermediate between Na(OCP) (νasym = 1755 cm−1) and Ph3Sn–PCO (νasym = 1946 cm−1)2d indicating a cobalt phosphaketene structure, Co–PCO. This is confirmed by a single crystal X-ray diffraction analysis (Fig. 2). The P–C [1.633(4) Å] and C–O [1.179(6) Å] bond distances, the rather large Co–P–C angle [116.2(1)°] and long Co–C distance [3.325(4) Å] indicate a η1-coordination via the phosphorus atom of the OCP− anion.6 The structural parameters confirm that neither the cobalt center nor the PDI ligand are reduced by OCP−.
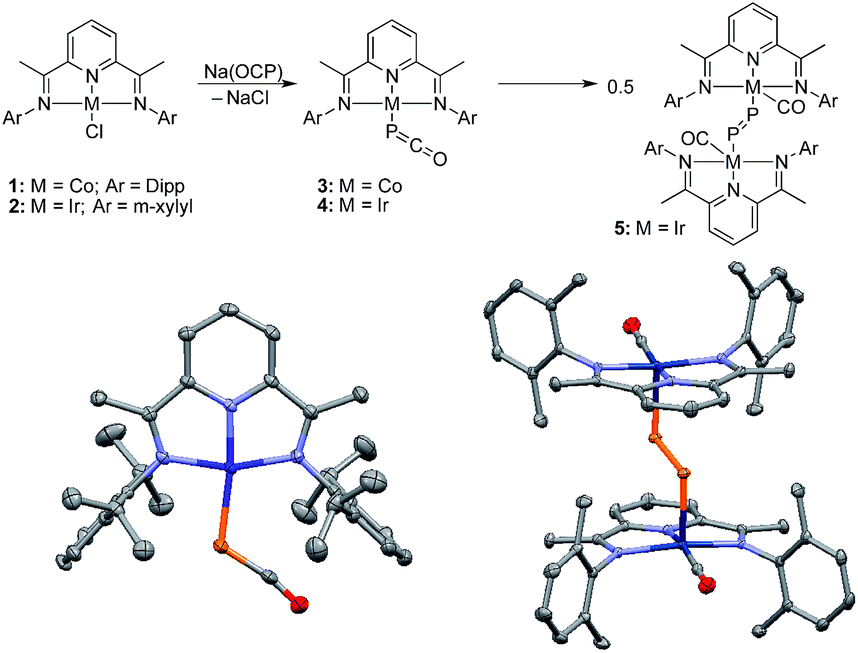 | ||
| Fig. 2 Synthesis of complexes 3 and 4, and conversion of the latter into dimetalladiphosphene 5, via CO migration. Solid-state structures of 3 (left) and 5 (right) (50% thermal ellipsoids are shown with hydrogen atoms and solvent molecules omitted for clarity); Dipp = 2,6-diisopropylphenyl, m-xylyl = 2,6-dimethylphenyl. Selected bond lengths [Å] and angles [°]: 3 P–Co 2.263(1), P–C 1.633(4), C–O 1.179(6); Co–P–C 116.2(1), P–C–O 169.9(4). 5 P–Ir 2.3886(6), P–P 2.021(1), C–Ir 1.862(2), C–O 1.153(3); Ir–P–P 103.76(3), C–Ir–P 92.787(6), Ir–C–O 177.8(2). | ||
The corresponding iridium complex [IrCl(PDIMe)] 2 with the less sterically encumbered PDIMe ligand reacts with Na(OCP) at low temperatures to cleanly give product 4. A 31P NMR resonance at δ = −316.7 ppm indicates a metallaphosphaketene (Ir–PCO) featuring a highly covalent metal phosphorus bond. Complex 4 could not be isolated. Keeping a THF solution at 20 °C for about 6 h leads cleanly to complex 5 (δ31P = +682 ppm; λmax = 524.1 nm, 732.5 nm), which was isolated as red crystals. A single crystal X-ray structure analysis shows compound 5 to be a dimetalladiphosphene (Fig. 2).15 The P–P distance [2.021(1) Å] is short and in the typical range of diphosphenes, R–PP–R. The iridium centers are bound to a redox-inactive PDI ligand with short CN bonds [N1–C2 1.340(2) Å; N3–C8 1.335(2) Å] and a carbonyl ligand. The rearrangement of 4 into 5 is the experimental confirmation of the computationally predicted transformation of D to E.9 This transformation is also comparable to the conversion of an iridium azido complex, Ir–N3, to a transient terminal nitrido complex, Ir![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, which could be spectroscopically characterized but also dimerizes to an Ir–NN–Ir complex.16
N, which could be spectroscopically characterized but also dimerizes to an Ir–NN–Ir complex.16
Monitoring by 31P NMR spectroscopy the reaction of gold and copper complexes 6a,b and 7 with Na(OCP) in benzene showed in each case the formation of a single product giving a 31P NMR signal (8a: δ = −360; 8b: −364; 9: −387 ppm) slightly up-field shifted compared to that of Na(OCP) (δ = −392 ppm). Single crystals of 8a and 9 were grown and subjected to X-ray diffraction studies (Fig. 3). Only very subtle structural differences between both complexes were observed. The P–C [8a: 1.640(3); 9: 1.636(2) Å] and C–O bond lengths [8a: 1.176(4); 9: 1.184(2) Å] are similar, and the M–P–C angle is slightly more acute for the copper complex 9 [8a: 86.2(1), 9: 79.15(5)°].
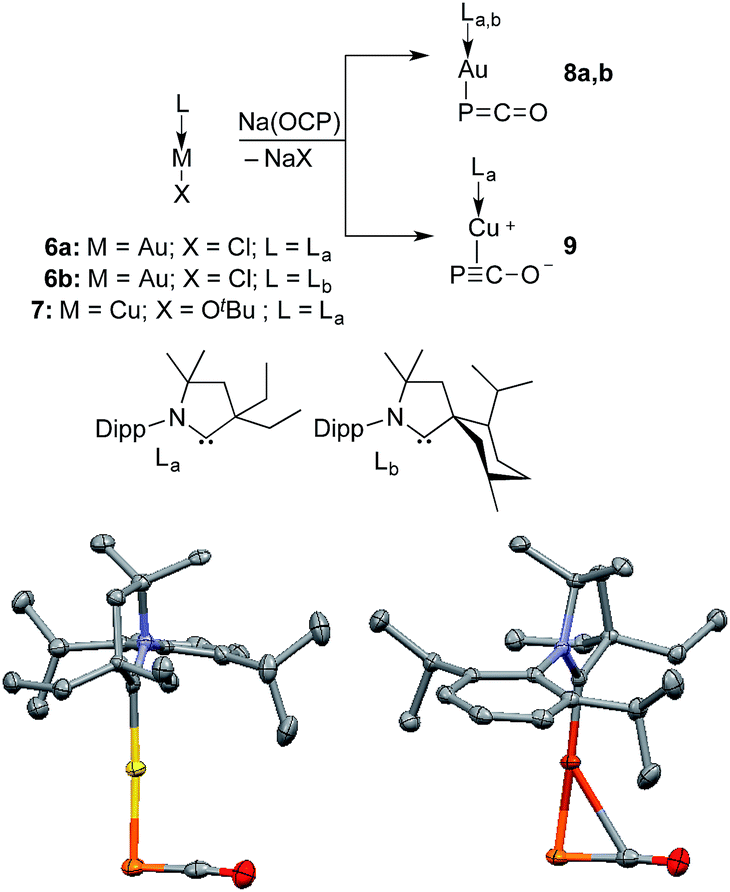 | ||
| Fig. 3 Synthesis and solid-state structures of 8a (left) and 9 (right) (50% thermal ellipsoids are shown with hydrogen atoms omitted for clarity); Dipp = 2,6-diisopropylphenyl. Selected bond lengths [Å] and angles [°]: 8a P–Au 2.354(1), P–C 1.640(3), C–O 1.176(4), Au–C 2.779(3); Au–P–C 86.2(1), P–C–O 176.5(3). 9 P–Cu 2.2244(5), P–C 1.636(2), C–O 1.184(2), Cu–C 2.501(2); Cu–P–C 79.15(5), P–C–O 176.4(1). | ||
Despite the similarities of the solid state structures, natural bond orbital (NBO) analysis at the M06/6-311++G(2d,p)+SDD//M06/6-31+G(d)+LANL2DZ(+f) level of theory shows significant differences between the electronic structures of 8a and 9. The NBO charges of Au and PCO in 8a are +0.39 and −0.56 a.u., respectively, whereas those of Cu and PCO in 9 are +0.58 and −0.68 a.u., suggesting that the PCO anion in 9 is more ionic and “free” than that in 8a. This is in agreement with the different 31P NMR chemical shifts of 8a (δ = −360 ppm) and 9 (δ = −387 ppm). The NBOs corresponding to the M(PCO) (M = Au or Cu) fragments are quite different as shown in Fig. 4. The phosphorus center of 8a forms three bonds (Au–P σ, P–C σ and P–C π) (Fig. 4a–c). In contrast, for 9 no Cu–P σ bond could be located. Instead, the phosphorus center of 9 forms one P–C σ bond and two P–C π bonds (Fig. 4f–h). Moreover, there are two bonds between the C and O atoms (C–O σ and C–O π) in 8a (Fig. 4d and e), while only one C–O σ bond in 9 (Fig. 4i). These computational results suggest that the coordination modes of PCO with gold and copper are η1 and η2, respectively.
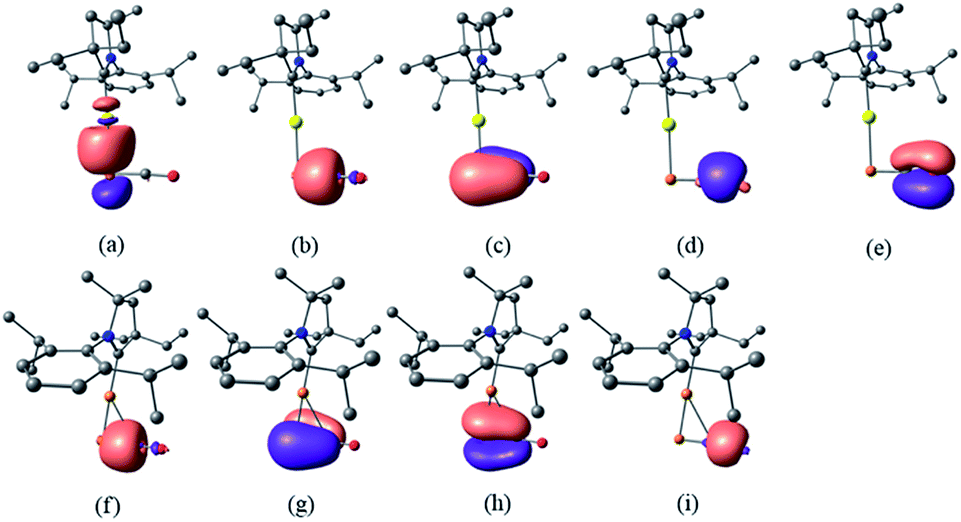 | ||
| Fig. 4 NBOs corresponding to M(PCO) fragments of 8a and 9 (isovalue = 0.05). NBOs of 8a: (a) Au–P σ; (b) P–C σ; (c) P–C π; (d) C–O σ; (e) C–O π. NBOs of 9: (f) P–C σ; (g) P–C π; (h) P–C π; (i) C–O σ. | ||
The different bonding modes in 8 and 9 lead to a difference in reactivity. In solution 9 decomposes after a few hours giving a complex mixture, whereas 8b rearranges into complex 10 over the course of a week when left standing in THF at room temperature (Fig. 5). The trinuclear nature of 10 [(LbAu)3P], as determined by an X-ray diffraction study, is reminiscent of the rearrangement product of Ph3Sn(PCO), namely (Ph3Sn)3P.2d,17 Three gold atoms surround a single P atom, leading to a phosphine supported solely by metals. The 31P NMR spectrum displays a signal at δ = −200 ppm, which is considerably high-field shifted compared to alkyl and aryl phosphines. The electron rich nature as well as the steric crowding around the P center could make 10 an interesting redox active ligand for transition metals.18
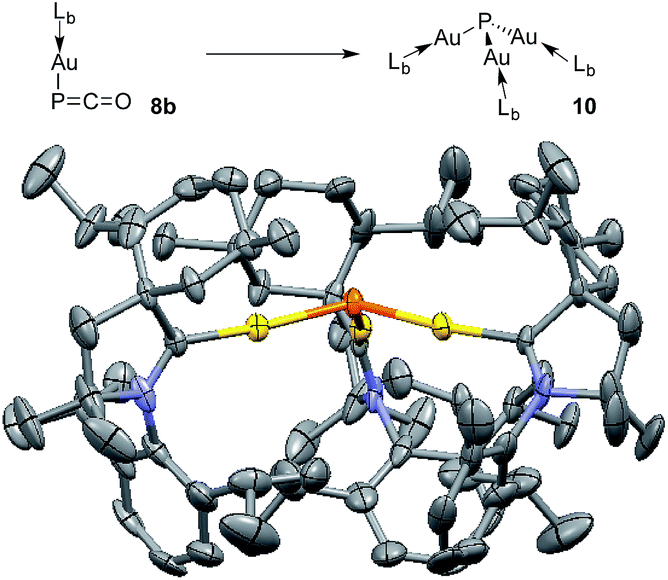 | ||
| Fig. 5 Conversion of 8b to (LbAu)3P 10, and solid state structure of 10 (50% thermal ellipsoids are shown with hydrogen atoms omitted for clarity). | ||
The gold complexes 8a,b are rather inert and do not react with heavier group 14 element halides to give R3E-PCO derivatives. Equally, no reaction with N,N′-dicyclohexylcarbodiimide or with carbene La are observed. On the contrary, the copper salt 9 does react with these reagents similarly to Na(OCP) (see the ESI† for details).2b,d Remarkably, 9 reacts with carbene La to afford the cationic bis(CAAC)Cu complex 11, in which the PCO fragment is the anionic counterpart. The 31P NMR signal appears at δ = −400 ppm, which is more downfield shifted than Na(OCP) (δ = −392 ppm), implying that PCO− is less coordinated. The IR spectrum showed the asymmetric stretching frequency of the PCO unit at νasym = 1791 cm−1, suggesting a more cumulenic nature than in the two crystalline forms of Na(OCP) (νasym = 1780 or 1755 cm−1).6,19a,b Lastly, although a disorder precludes accurate determination of the geometric parameters, the X-ray diffraction study revealed that the PCO anion has no close contacts with the cationic part of the complex (Fig. 6). This is the first time that the PCO anion has been structurally observed without any binding agents or coordinating solvents.
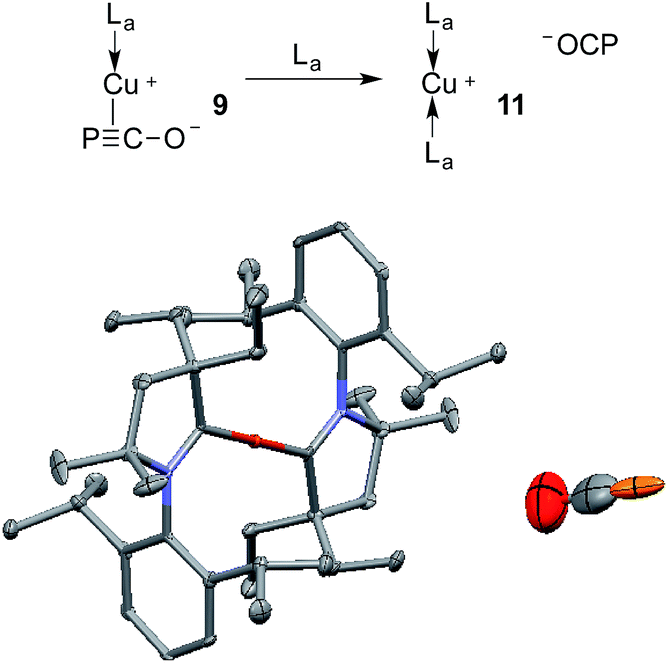 | ||
| Fig. 6 Solid-state structure of 11 showing the free PCO anion. 50% thermal ellipsoids are shown and hydrogen atoms are omitted for clarity. | ||
On the basis of DFT calculations, the HOMOs of 8a and 9 are mainly localized on the PCO fragments (see ESI†) and the oxygen atoms carry the largest negative charge (−0.56 a.u. in 8a and −0.59 a.u. in 9). Thus, we were curious to see if the terminal oxygen atom could react with a Lewis acidic borane. Indeed, adding one equivalent of B(C6F5)3 to either 8b or 9 led to the same type of heterocycle 12 and 13, respectively (Fig. 7). In the case of gold, the corresponding product immediately crystallized out and became insoluble in the tested solvents. However, for copper the product was soluble and the 31P NMR spectrum showed two broad peaks at δ = 261 and δ = 137 ppm. Single crystal X-ray diffraction studies of 12 and 13 revealed a four-membered P2C2 heterocycle that arose from borane coordination to oxygen followed by a dimerization and lastly a migration of a LM fragment from one phosphorus center to the other. The four-membered P2C2 heterocycles have a planar geometry and, as expected, the bond lengths of the PCO fragments become elongated compared to 8b and 9 as a result of the delocalization over the ring. This small ring represents a novel bonding mode for the rapidly growing field of molecular polyphosphorus clusters and cages.20
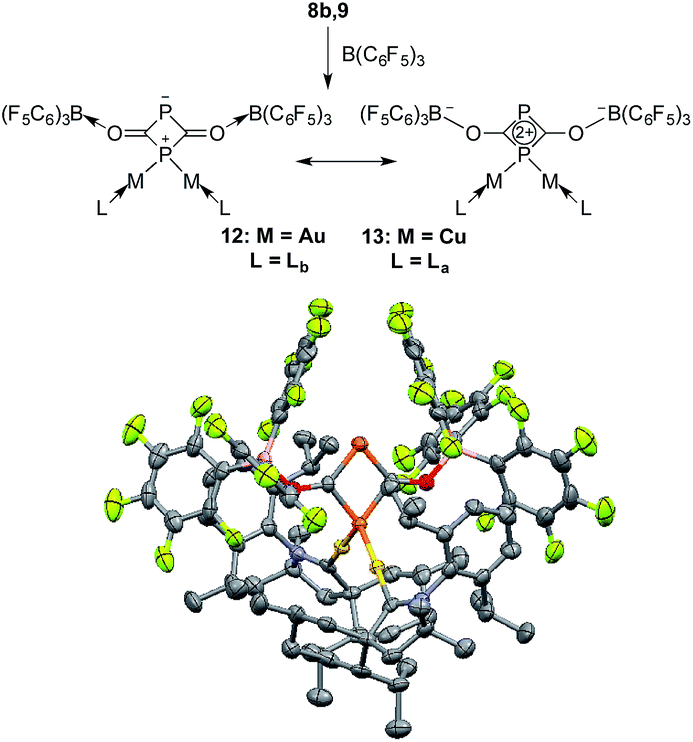 | ||
| Fig. 7 Reaction of 8b,9 with B(C6F5)3 and two resonance structures of 12 and 13; solid state-structure of the gold complex 12 (50% thermal ellipsoids are shown with hydrogen atoms omitted for clarity). | ||
Conclusions
PCO complexes with electron rich metal centers such as copper, gold and also cobalt can be prepared and are relatively stable. On the other hand, the iridium phosphaketene 4 rapidly rearranges via CO migration to give a genuine dimetalladiphosphene. The copper complex features an η2 coordination mode, which leads to an active PCO fragment that can undergo further reactions. A free PCO anion, resulting from simple displacement of the PCO unit with a carbene, was also isolated. For both the copper and gold complexes, borane coordination to the oxygen of the OCP unit induces a [2 + 2] cycloaddition into a P2C2 heterocycle. These results demonstrate that the uncharted chemistry of transition metal PCO complexes is rich and the formation of new metal phosphides and the mechanisms leading to them merit further exploration.Experimental section
General considerations
All air- and moisture-sensitive manipulations were carried out using standard vacuum line Schlenk techniques or an MBraun dry-box under argon. THF was distilled over sodium benzophenone-ketyl before use. THF-d8, CD2Cl2, and C6D6 were purchased from Cambridge Isotope Laboratories and dried over 4 Å molecular sieves. (iPrPDI)CoCl 1,21 (MePDI)IrCl 2,11d La,bAuCl 6a,b22 and LaCuOtBu 723 were synthesized according to literature procedures. 1H, 13C, 11B, 19F, and 31P NMR spectra were recorded on a Varian VX 500, Bruker 300, Bruker 500 and Jeol 500 spectrometer at 25 °C. All 1H and 13C NMR chemical shifts are reported relative to SiMe4 using the 1H (residual) and 13C chemical shifts of the solvent as a secondary standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, sept = septet, m = multiplet, br = broad signal. Chemical shifts are given in ppm and coupling constants J are given in Hz. Peak widths at half heights (in Hz) are given for broad signals. Infrared spectra were collected on a Perkin-Elmer-Spectrum 2000 FT-IR-Raman and Bruker ALPHA FT-IR spectrometer. Elemental analyses were performed at the Mikrolabor of ETH Zürich. Single crystals suitable for X-ray diffraction were coated with polyisobutylene oil in a dry-box, transferred to a nylon loop and then transferred to the goniometer of a Bruker X8 APEX2 diffractometer equipped with a molybdenum X-ray tube (λ = 0.71073 Å) or on a Bruker Apex II-CCD detector using Mo-Kα radiation (λ = 0.71073 Å) or Cu-Kα radiation (λ = 1.54178 Å). The data were processed using the Bruker SAINT+ program and corrected for absorption using SADABS. The structures were solved using direct methods (SHELXS) completed by Fourier synthesis and refined by full-matrix least-squares procedures. Mass spectra were performed at the UC San Diego Mass Spectrometry Laboratory. Melting points were measured with an electrothermal MEL-TEMP apparatus.O d, JPC = 100.4 Hz), 146.1, 135.3, 130.9, 126.0, 81.0, 63.0, 43.0, 32.4, 20.1, 29.5, 27.7, 23.7, 10.3; 31P {1H} NMR (C6D6, 121 MHz) δ = −359.5. HRMS was attempted but a peak corresponding to M+ could not be located, probably due to the weak P metal bond.O d, JPC = 101.1 Hz), 146.2, 145.8, 136.1, 130.6, 129.2, 125.8, 77.6, 65.4, 53.4, 52.0, 50.0, 36.4, 31.3, 30.1, 29.8, 28.8, 28.0, 27.3, 25.6, 23.8, 23.7, 23.6, 20.7; 31P {1H} NMR (C6D6, 121 MHz) δ = −364.2. HRMS was attempted but a peak corresponding to M+ could not be located, probably due to the weak P metal bond.O d, JPC = 97.8 Hz), 145.9, 135.5, 130.7, 125.6, 81.2, 63.1, 43.1, 31.9, 29.9, 29.4, 27.9, 23.1, 10.4; 31P {1H} NMR (C6D6, 121 MHz) δ = −387.4. HRMS was attempted but a peak corresponding to M+ could not be located, probably due to the weak P metal bond.Acknowledgements
Thanks are due to the NSF (CHE-1359809) for financial support, the China Scholarship Council for a graduate fellowship (L. L.) and the U.S. Department of Education for a GAANN fellowship (D. A. R.). Further support by the ETH Zürich and the Swiss National Science Foundation (SNF) is acknowledged.Notes and references
- (a) G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS; (b) M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643, 189–193 CrossRef.
- (a) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed; (b) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed; (c) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC; (d) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC.
- Z.-J. Quan and X.-C. Wang, Org. Chem. Front., 2014, 1, 1128–1131 RSC.
- (a) X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed; (b) D. Heift, Z. Benkő and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757–6761 CrossRef CAS PubMed; (c) L. Liu, J. Zhu and Y. Zhao, Chem. Commun., 2014, 50, 11347–11349 RSC; (d) D. Heift, Z. Benkő, H. Grützmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC; (e) A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed; (f) M. B. Geeson, A. R. Jupp, J. E. McGrady and J. M. Goicoechea, Chem. Commun., 2014, 50, 12281–12284 RSC; (g) T. P. Robinson and J. M. Goicoechea, Chem.–Eur. J., 2015, 21, 5727–5731 CrossRef CAS PubMed; (h) A. R. Jupp, G. Trott, É. Payendella Garanderie, J. D. G. Holl, D. Carmichael and J. M. Goicoechea, Chem.–Eur. J., 2015, 21, 8015–8018 CrossRef CAS PubMed.
- G. Becker, G. Heckmann, K. Hübler and W. Schwarz, Z. Anorg. Allg. Chem., 1995, 621, 34–46 CrossRef CAS.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem.–Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- L. Weber, B. Torwiehe, G. Bassmann, H.-G. Stammler and B. Neumann, Organometallics, 1996, 15, 128–132 CrossRef CAS.
- C. Camp, N. Settineri, J. Lefevre, A. R. Jupp, J. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- W. Lü, C. Wang, Q. Luo, Q. Li, Y. Xie, R. B. King and H. F. Schaefer III, New J. Chem., 2015, 39, 1390–1403 RSC.
- (a) U. Ray, D. Banerjee, B. Gopal Chand, J. Cheng, T.-H. Lu and C. Sinha, J. Coord. Chem., 2005, 58, 1105–1113 CrossRef CAS; (b) J. Carranza, M. Julve and J. Sletten, Inorg. Chim. Acta, 2008, 361, 2499–2507 CrossRef CAS; (c) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, Nat. Chem., 2010, 2, 30–35 CrossRef CAS PubMed; (d) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed; (e) M. Anselmi, V. Bonuccelli, T. Funaioli, P. Leoni, F. Marchetti, L. Marchetti, S. K. Mohapatra and M. Pasquali, Dalton Trans., 2013, 42, 10855–10866 RSC.
- (a) V. A. Schmidt, J. M. Hoyt, G. W. Margulieux and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 7903–7914 CrossRef CAS PubMed; (b) K. M. Lewis, C. C. H. Atienza, J. L. Boyer, P. J. Chirik, J. G. P. Delis and A. K. Roy, Saturated and unsaturated silahydrocarbons via iron and cobalt pyridine diamine catalyzed olefin silylation, US 2014/0330036 A1, 2014 Search PubMed; (c) C. C. Hojilla Atienza, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 16343–16345 CrossRef CAS PubMed; (d) A. C. Bowman, C. Milsmann, C. C. H. Atienza, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 1676–1684 CrossRef CAS PubMed; (e) A. C. Bowman, C. Milsmann, E. Bill, E. Lobkovsky, T. Weyhermueller, K. Wieghardt and P. J. Chirik, Inorg. Chem., 2010, 49, 6110–6123 CrossRef CAS PubMed; (f) J. Scott, I. Vidyaratne, I. Korobkov, S. Gambarotta and P. H. M. Budzelaar, Inorg. Chem., 2008, 47, 896–911 CrossRef CAS PubMed; (g) I. Vidyaratne, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, Inorg. Chem., 2005, 44, 1187–1189 CrossRef CAS PubMed.
- For reviews on CAACs, see: (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (b) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- For synthesis of CAACs, see: (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (b) R. Jazzar, J.-B. Bourg, R. D. Dewhurst, B. Donnadieu and G. Bertrand, J. Org. Chem., 2007, 72, 3492–3499 CrossRef CAS PubMed; (c) R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899–2902 CrossRef CAS PubMed.
- (a) P. P. Samuel, K. C. Mondal, H. W. Roesky, M. Hermann, G. Frenking, S. Demeshko, F. Meyer, A. C. Stückl, J. H. Christian, N. S. Dalal, L. Ungur, L. F. Chibotaru, K. Pröpper, A. Meents and B. Dittrich, Angew. Chem., Int. Ed., 2013, 52, 11817–11821 CrossRef CAS PubMed; (b) A. P. Singh, P. P. Samuel, H. W. Roesky, M. C. Schwarzer, G. Frenking, N. S. Sidhu and B. Dittrich, J. Am. Chem. Soc., 2013, 135, 7324–7329 CrossRef CAS PubMed; (c) D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964–8967 CrossRef CAS PubMed; (d) P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123–17135 CrossRef CAS PubMed; (e) K. C. Mondal, P. P. Samuel, Y. Li, H. W. Roesky, S. Roy, L. Ackermann, N. S. Sidhu, G. M. Sheldrick, E. Carl, S. Demeshko, S. de, P. Parameswaran, L. Ungur, L. F. Chibotaru and D. M. Andrada, Eur. J. Inorg. Chem., 2014, 818–823 CrossRef CAS; (f) G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed; (g) D. S. Weinberger, N. Amin Sk, K. C. Mondal, M. Melaimi, G. Bertrand, A. C. Stückl, H. W. Roesky, B. Dittrich, S. Demeshko, B. Schwederski, W. Kaim, P. Jerabek and G. Frenking, J. Am. Chem. Soc., 2014, 136, 6235–6238 CrossRef CAS PubMed.
- To the best of our knowledge this is the first example of a dimetalladiphosphene. The closest structure analogue is [(silox)3M]2P2 which, however, contains a MP–PM (M = Ab, Ta) unit: E. B. Hulley, P. T. Wolczanski and E. B. Lobkovsky, Chem. Comm., 2009, 6412–6414 RSC Numerous complexes of organic diphosphenes, R–PP–R, have been reported, for examples see:
(a) L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS;
(b) L. Weber, K. Reizig, D. Bungardt and R. Boese, Organometallics, 1987, 6, 110–114 CrossRef CAS;
(c) J. Grobe, D. L. van, T. Pohlmeyer, B. Krebs, O. Conrad, E. Dobbert and L. Weber, Organometallics, 1998, 17, 3383–3386 CrossRef CAS;
(d) L. Weber, H. Bastian, A. Mueller and H. Boegge, Z. Naturforsch., B: Chem. Sci., 1992, 47, 231–237 CAS;
(e) L. Weber, M. Frebel and R. Boese, Organometallics, 1989, 8, 1718–1722 CrossRef CAS;
(f) W. Domańska-Babul, J. Chojnacki, E. Matern and J. Pikies, J. Organomet. Chem., 2007, 692, 3640–3648 CrossRef.
- B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef PubMed.
- C. C. Cummins, C. Huang, T. J. Miller, M. W. Reintinger, J. M. Stauber, I. Tannou, D. Tofan, A. Toubaei, A. Velian and G. Wu, Inorg. Chem., 2014, 53, 3678–3687 CrossRef CAS PubMed.
- (a) R. T. Boeré and Y. Zhang, J. Organomet. Chem., 2005, 690, 2651–2657 CrossRef; (b) S. Sasaki and M. Yoshifuji, Curr. Org. Chem., 2007, 11, 17–31 CrossRef CAS; (c) R. T. Boere, A. M. Bond, S. Cronin, N. W. Duffy, P. Hazendonk, J. D. Masuda, K. Pollard, T. L. Roemmele, P. Tran and Y. Zhang, New J. Chem., 2008, 32, 214–231 RSC; (d) X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed.
- The asymmetric stretching frequency of the naked PCO anion has been calculated several times to be 1789 cm−1 in ref. 6, 1804 cm−1 in: (a) C. Léonard, H. Gritli and G. Chambaud, J. Chem. Phys., 2010, 133, 124318 CrossRef PubMed; and 1838 cm−1 in: (b) Y. Lu, H. Wang, Y. Xie, H. Liu and H. F. Schaefer, Inorg. Chem., 2014, 53, 6252–6256 CrossRef CAS PubMed.
- (a) M. H. Holthausen and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639–6657 RSC; (b) R. S. P. Turbervill and J. M. Goicoechea, Chem. Rev., 2014, 114, 10807–10828 CrossRef CAS PubMed.
- S. Nueckel and P. Burger, Organometallics, 2001, 20, 4345–4359 CrossRef CAS.
- G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2008, 693, 1674–1682 CrossRef CAS PubMed.
- G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Chem.–Asian J., 2011, 6, 402–405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and crystallographic data. CCDC 1404128–1404135, 1418457 and 1418458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04504e |
| This journal is © The Royal Society of Chemistry 2016 |