
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilising the lowest energy charge-separated state in a {metal chromophore – fullerene} assembly: a tuneable panchromatic absorbing donor–acceptor triad†
Maria A.
Lebedeva
*ab,
Thomas W.
Chamberlain
ac,
Paul A.
Scattergood
d,
Milan
Delor
d,
Igor V.
Sazanovich
de,
E. Stephen
Davies
a,
Mikhail
Suyetin
a,
Elena
Besley
a,
Martin
Schröder
af,
Julia A.
Weinstein
*d and
Andrei N.
Khlobystov
*ag
aSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: andrei.khlobystov@nottingham.ac.uk
bDepartment of Materials, University of Oxford, 16 Parks Road, Oxford, OX1 3PS, UK. E-mail: maria.lebedeva@materials.ox.ac.uk
cSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
dDepartment of Chemistry, University of Sheffield, S3 7HF, UK. E-mail: julia.weinstein@sheffield.ac.uk
eLaser for Science Facility, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Oxfordshire, OX11 0QX, UK
fSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
gNanoscale and Microscale Research Centre, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 19th May 2016
Abstract
Photoreduction of fullerene and the consequent stabilisation of a charge-separated state in a donor–acceptor assembly have been achieved, overcoming the common problem of a fullerene-based triplet state being an energy sink that prevents charge-separation. A route to incorporate a C60-fullerene electron acceptor moiety into a catecholate-Pt(II)-diimine photoactive dyad, which contains an unusually strong electron donor, 3,5-di-tert-butylcatecholate, has been developed. The synthetic methodology is based on the formation of the aldehyde functionalised bipyridine-Pt(II)-3,5-di-tert-butylcatechol dyad which is then added to the fullerene cage via a Prato cycloaddition reaction. The resultant product is the first example of a fullerene-diimine-Pt-catecholate donor–acceptor triad, C60bpy-Pt-cat. The triad exhibits an intense solvatochromic absorption band in the visible region due to catechol-to-diimine charge-transfer, which, together with fullerene-based transitions, provides efficient and tuneable light harvesting of the majority of the UV/visible spectral range. Cyclic voltammetry, EPR and UV/vis/IR spectroelectrochemistry reveal redox behaviour with a wealth of reversible reduction and oxidation processes forming multiply charged species and storing multiple redox equivalents. Ultrafast transient absorption and time resolved infrared spectroscopy, supported by molecular modelling, reveal the formation of a charge-separated state [C60˙−bpy-Pt-cat˙+] with a lifetime of ∼890 ps. The formation of cat˙+ in the excited state is evidenced directly by characteristic absorption bands in the 400–500 nm region, while the formation of C60˙− was confirmed directly by time-resolved infrared spectroscopy, TRIR. An IR-spectroelectrochemical study of the mono-reduced building block (C60-bpy)PtCl2, revealed a characteristic C60˙− vibrational feature at 1530 cm−1, which was also detected in the TRIR spectra. This combination of experiments offers the first direct IR-identification of C60˙− species in solution, and paves the way towards the application of transient infrared spectroscopy to the study of light-induced charge-separation in C60-containing assemblies, as well as fullerene films and fullerene/polymer blends in various OPV devices. Identification of the unique vibrational signature of a C60-anion provides a new way to follow photoinduced processes in fullerene-containing assemblies by means of time-resolved vibrational spectroscopy, as demonstrated for the fullerene-transition metal chromophore assembly with the lowest energy charge-separated excited state.
Introduction
Creating artificial photosynthetic devices that can mimic natural light-driven processes is one of the major challenges for sustainable energy production.1 The chemical compounds utilised as the main components in such devices typically contain three major building blocks: a chromophore which is responsible for the efficient visible light energy harvesting, and donor and acceptor moieties which are responsible for the formation of long-lived charge separated radical-ion pairs upon photoexcitation.2Fullerenes possess excellent electron accepting capacity3 and have low reorganisation energies4 encouraging electron-transfer processes. They have therefore been extensively utilised as a component in artificial photosynthetic systems.5 Combining C60 fullerene with a suitable electron donor results in the formation of donor–acceptor (D–A) dyads potentially capable of generating the long-lived charge-separated states, the key intermediate in operation of photovoltaic devices, organic electronics and photocatalysis.6 A wide variety of organic and transition metal containing electron donor units have been linked to fullerene cages,7 including metalloporphyrins8 and phthalocyanines,9 cyanines, Ru and Re bipyridine complexes,10 boron dipyrrins,11 tetrathiafulvalenes12 and metallocenes.13 The photochemical properties of these compounds can be controlled by the nature of the chosen electron donor as well as the nature14 and the length15 of the linker between the donor and the acceptor moieties. Varying the method of addition of electron donor units via covalent functionalization of the fullerene cage with conjugated16 or non-conjugated17 linkers or via non-covalent supramolecular interactions such as axial coordination,18 hydrogen bonding, ammonia-crown ether coordination and mechanical interlocking into rotaxane and catenane type compounds19 has enabled the donor moiety to be positioned in close proximity to the fullerene cage or to spatially separate the D and A units. In this way the lifetimes of the resulting charge-separated states can be tuned. One of the major obstacles in stabilising charge separation in these systems is the formation of the fullerene triplet excited state (3C60*, 1.51 eV).20 This state is often lower in energy than the corresponding charge-separated state and therefore is formed as the final product instead of the desired long-lived radical-ion pair;21 in this case, energy transfer tends to outcompete the electron-transfer process. Attempts to address this issue have included shortening the linkers between the fullerene and donor moieties; for example, in RuII(bpy)-type MLCT chromophores where the extraordinarily short D–A distance should have a kinetically favoured electron-transfer process. Nonetheless this results in the formation of a fullerene triplet as the lowest excited state.10b Utilising Zn-porphyrin donor moieties brings the energy of the charge-separated state (CSS) slightly below the fullerene triplet (1.3–1.6 eV),22 and utilising a cascade Zn-porphyrin/ferrocene donor system lowers the energy of CSS state to as low as 1.05 eV.23 Only a small number of recent examples report systems with the CSS energy below 1 eV, which all utilise organic metal-free donors such as tetraalkylphenylenediamine.24
Clearly, in order to achieve the lowest energy charge-separated state involving a fullerene anion, a strong electron donor is required. Square-planar d8 Pt-diimine complexes are excellent chromophores due to ligand-to-ligand charge-transfer (LLCT) transitions in the visible region of the spectrum25 and, if suitable ligands are used, are also strong electron donors. Given an ultrafast intersystem crossing which is common in transition metal complexes,26 the lowest charge-separated state in Pt(II) complexes is expected to be triplet in nature, further lowering its energy. Phenolates/thiolates, catecholates and acetylides are commonly used as electron donor ligands.27 PtII-diimine chromophores28 have been exploited as building blocks for non-linear optical devices,29 light-emitting diodes,30 solar cells,31 singlet oxygen sensing devices,32 and photocatalytic applications.33,34 Surprisingly, only a few examples of combining C60 fullerene with Pt(II)-diimine chromophores have been reported, and none where the lowest excited state is a charge-separated one. Fullerene-modified Pt(II) diimine complexes containing phenothiazine35 and carbazole36 donor units have been reported. However, as both phenothiazine and carbazole groups are relatively weak electron donors, the energies of the corresponding charge separated states in these systems are higher than that of the 3C60*, which again acts as an energy sink.
This study details the design and a synthesis of a highly soluble and stable fullerene-Pt(II) assembly which incorporates the 3,5-di-tert-butylcatechol group as a strong electron donor D with the aim of significantly lowering the energy of the charge-separated state to below that of the fullerene triplet state. We have successfully prepared a cascade electron-transfer A2–A1–Pt–D system where a diimine ligand (a derivatized 2,2′-bipyridine) acts as the primary acceptor (A1) with C60 acting as a stronger, secondary acceptor (A2). In this charge-transfer triad, the (diimine)Pt(catecholate) moiety, A1–Pt–D, and the C60 can act as chromophores and initiate electron-transfer cascade events upon absorption of light. We investigated the ground state properties of this triad using absorption spectroscopy, cyclic voltammetry, UV/vis, infrared (IR) and electron paramagnetic resonance (EPR) spectroelectrochemical techniques, and DFT calculations. The photoinduced processes in the triad are investigated by transient absorption and time-resolved IR (TRIR) spectroscopy. In general, TRIR spectroscopy is a widely used tool to probe charge-transfer processes in the excited state as it is sensitive to the local electron density redistribution upon photoexcitation, and accordingly has been successfully used to study excited state dynamics in a diverse range of Pt(II) donor–acceptor assemblies.27,28,37,38 Yet application of TRIR to fullerene-containing assemblies has so far been reported for only a handful of systems, all of which involve photovoltaic polymer blends containing fullerene cages specially derivatized by an infrared-active group such as an ester.39 Here, we demonstrate how TRIR can be used to monitor charge-transfer and excited state dynamics in a fullerene-based donor–acceptor assembly based on the formation of the fullerene cage anion itself. The performance of this fullerene-bipyridine-Pt-catechol triad is compared with known fullerene-based D–A systems and bipyridine-Pt-catechol complexes. The first direct identification of a fullerene-anion by infrared and time-resolved infrared spectroscopies confirms the stabilisation of the lowest energy charge-separated state with an appreciable lifetime of 890 ps in the Pt(II)-fullerene donor–acceptor assembly.
Results and discussion
Synthesis of the target Pt(II) complexes
The synthesis of square-planar diimine-platinum(II) complexes are usually based on two key steps.40 First, a chelate dichloroplatinate complex is formed by reaction of diimine with Pt(DMSO)2Cl2, and this is followed by nucleophilic substitution of the two chloride ligands with the corresponding electron donor ligand(s) (Scheme 1a). Following this synthetic route we designed the fullerene containing bipyridine ligand 1 which was successfully prepared in a 40% yield via a Prato reaction41 of C60 fullerene with the corresponding 4′-methyl-2,2′-bipyridine-4-carbaldehyde 642 and N-substituted aminoacid 5. The 3,5-di-tert-butyl substituted phenyl ring was introduced at the nitrogen centre of the pyrrolidine ring in order to increase the solubility of the target compound,43 and this was achieved by targeting the corresponding N-substituted glycine 5. Compound 5 was synthesised in two steps44via reductive amination of 3,5-di-tert-butyl benzaldehyde with glycine methyl ester in the presence of Na[BH(OAc)3] to give the methyl ester 4 in 70% yield, followed by hydrolysis of 4 to give the carboxylic acid 5 in 80% yield (Scheme 1b). C60-bipyridine ligand 1 was reacted with Pt(DMSO)2Cl2, prepared according to the reported procedure45 from K2PtCl4 to give the fullerene containing chelate dichloroplatinum complex 2 in 90% yield.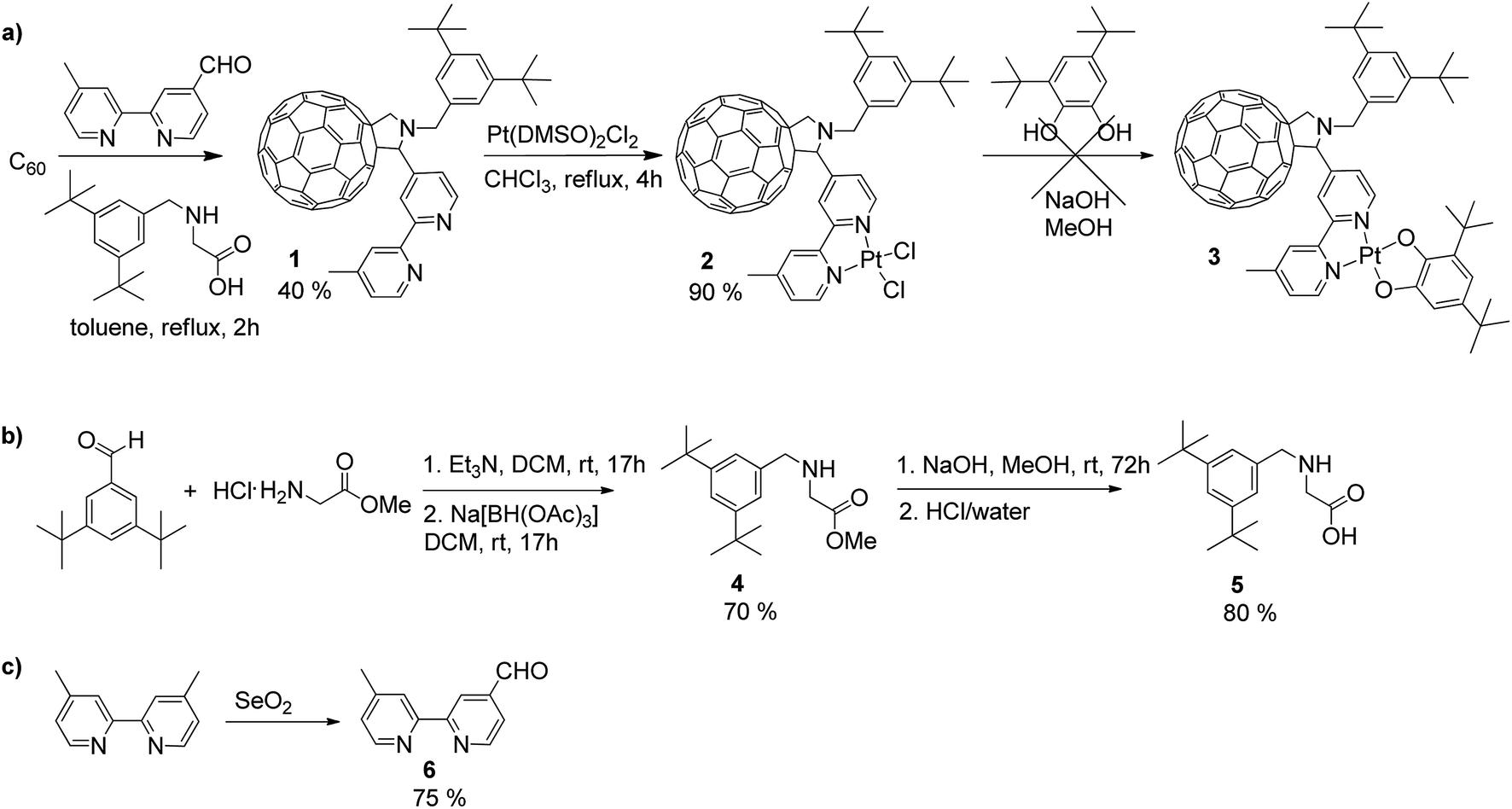 | ||
| Scheme 1 (a) Synthetic route towards fullerene-bipyridine-Pt-catechol complex 3via the formation of the fullerene-bipyridine-platinum dichloride complex 2, and synthesis of (b) N-functionalised glycine 5 and (c) the aldehyde group containing bipyridine derivative 6. | ||
However, in our hands, substitution of chloride ligands in 2 with sodium 3,5-di-tert-butylcatecholate did not result in the formation of the desired complex 3, rather an insoluble material was isolated. This may be due to side reactions of one or more of the double bonds of the fullerene cage with the nucleophilic catecholate anion resulting in the formation of oligomers and polymers. To avoid this, we developed an alternative synthetic route in which chloride substitution was performed prior to the introduction of the fullerene cage to the platinum complex (Scheme 2). Thus, Pt(DMSO)2Cl27 was treated with sodium catecholate to give the Pt(II)-catecholate complex 8 in 70% yield. The DMSO ligands in 8 were then substituted with 4′-methyl-2,2′-bipyridine-4-carbaldehyde 6 to give the bipyridine-platinum catecholate complex 9. Complex 9 was then successfully utilised in a Prato reaction with C60 fullerene and the glycine derivative 5 to give the desired complex 3 in 20% yield. Complex 3 was purified by chromatography silica gel using a mixture of toluene/acetonitrile (97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 followed by 94:6) as eluent. Complex 3 is highly soluble and stable in common organic solvents such as toluene, THF, DMF, CH2Cl2, and CS2, but decomposes rapidly in CHCl3 possibly due to the reaction with trace amount of HCl in the solvent.
:3 followed by 94:6) as eluent. Complex 3 is highly soluble and stable in common organic solvents such as toluene, THF, DMF, CH2Cl2, and CS2, but decomposes rapidly in CHCl3 possibly due to the reaction with trace amount of HCl in the solvent.
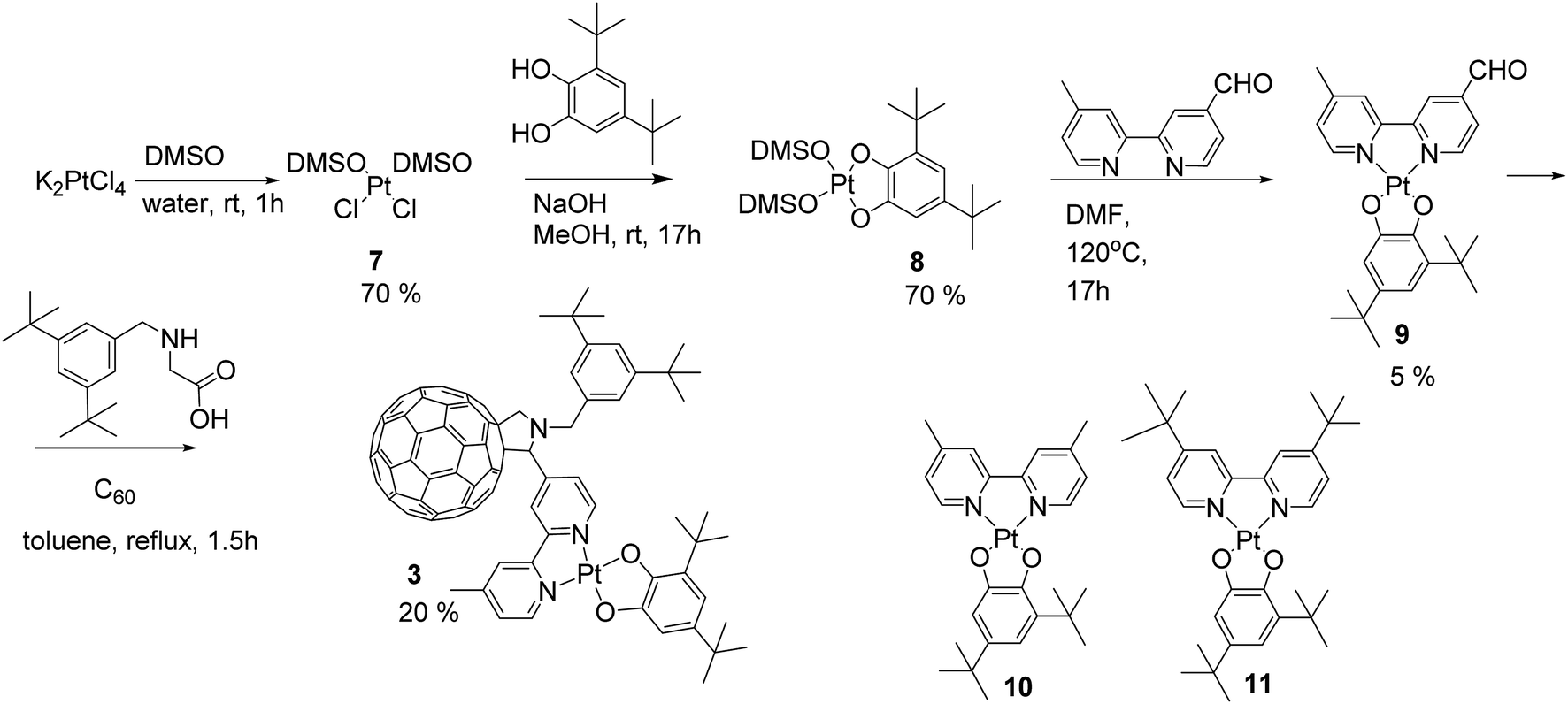 | ||
| Scheme 2 Alternative synthetic route towards the donor–acceptor complex 3via the Prato reaction of the aldehyde group tagged bipyridine-Pt-catecholate unit and C60 fullerene. Molecular structures of the model dyads (4,4′-diMe-bpy)Pt(cat) (10) and (4,4′-ditBu-bpy)Pt(cat) (11) are also shown. | ||
Spectroscopic characterisation
Complex 3 is a strong light-absorber and possesses rich electrochemical properties, with multiple redox states being accessible. The UV/vis spectrum of 3 can be considered a superposition of the UV/vis spectra of the functionalised C60 fullerene and the platinum diimine catecholate complex with no evidence of intramolecular charge-transfer from the bipyridine-Pt-catechol moiety to the fullerene cage observed in the ground state (Fig. 1a). This is a common phenomenon in fullerene containing dyads with organic46 and transition-metal binding47 groups where the donor and acceptor moiety are separated by non-conjugated linkers with one or more sp3 carbon atoms. The band corresponding to absorption of the pyrrolidine functionalised fullerene cage is observed at 436 nm; it is identical to the bands observed in the spectra of the (C60-bpy)PtCl22 and the free ligand 1 (see ESI file†), and is consistent with previously reported [6,6]-adducts of C60.48 A strong, broad and featureless absorption band at 500–700 nm is observed for 3 and in its fullerene-free analogue,38 but not observed for 1 or 2, and is assigned to ligand-to-ligand charge-transfer (LLCT) from the catecholate group to the bipyridine moiety of 3, with an additional contribution from the Pt centre d-orbitals, as confirmed by the spectroelectrochemical experiments discussed later.49 The LLCT band shows negative solvatochromic behaviour, where the energy of the lowest absorption band decreases linearly with a decrease in solvent polarity50 (Fig. 1),25 with the slope of 0.49 eV similar to that reported for other Pt(bpy)(cat) complexes.28 As the fullerene-based absorption band is not sensitive to the nature of the solvent, the solvatochromism of the LLCT transition of the (bpy)Pt(cat) moiety provides a useful mechanism by which to control the light absorption properties of fullerene-Pt(diimine) assemblies, enabling absorption of energy across the entire visible spectrum.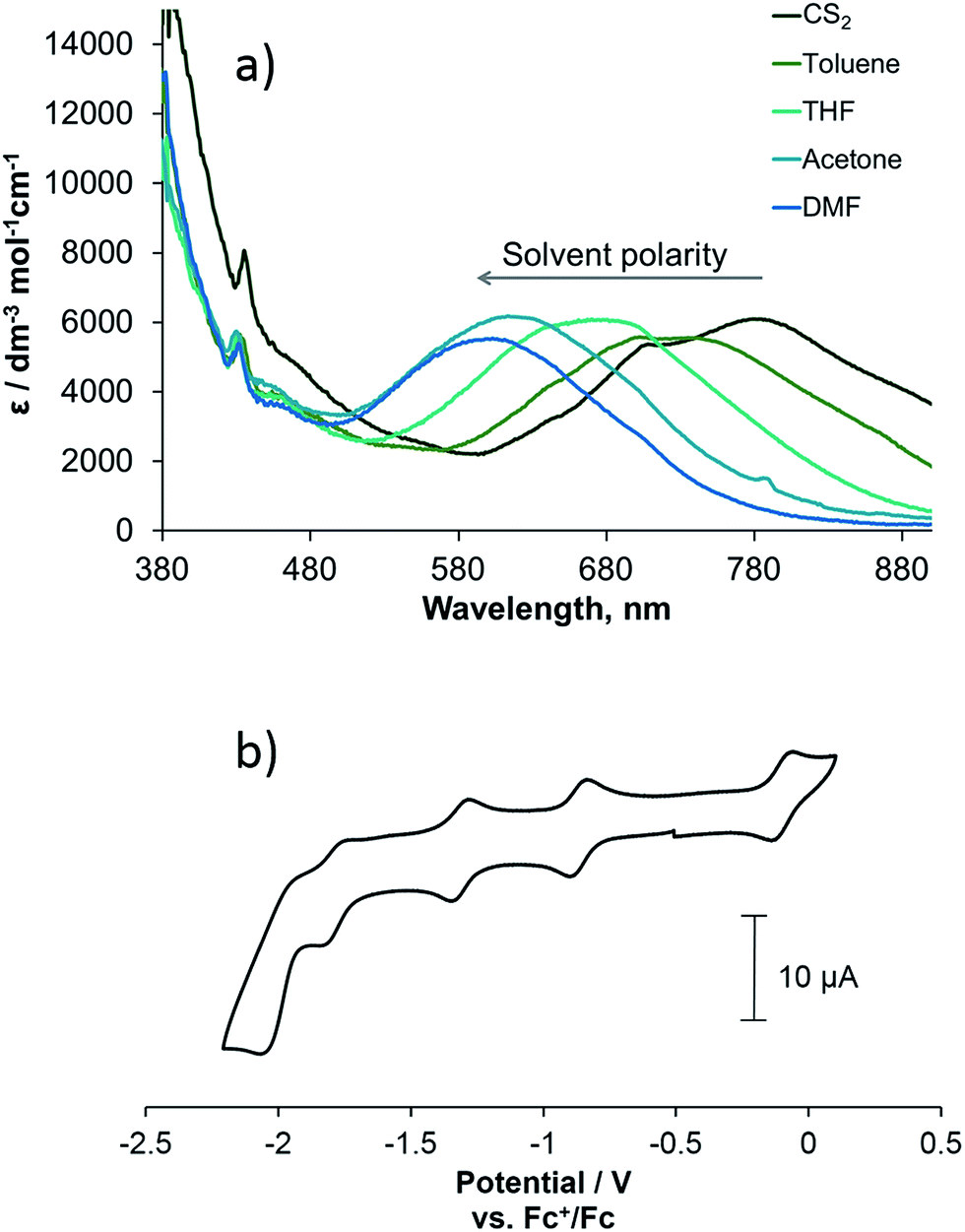 | ||
| Fig. 1 (a) UV/vis absorption spectra of complex 3 recorded in solvents of different polarity and (b) the cyclic voltammogram of 3 in DMF with [nBu4N][BF4] (0.2 M) as supporting electrolyte at a scan rate of 0.1 V s−1. | ||
The redox properties of compounds 1–3 were studied by cyclic voltammetry. The cyclic voltammogram of complex 3 shows three quasi-reversible and one irreversible reduction processes and one quasi-reversible oxidation process (Fig. 1b):
Comparison of the redox potentials of the fullerene-bipyridine ligand 1, (C60-bpy)PtCl2 complex 2 and (C60-bpy)Pt(cat) complex 3 (Table 1) suggests that the one-electron reduction processes occurring at potentials of −0.87/−0.85 V, −1.31 V and −1.98/−2.00 V in all complexes correspond to the sequential fullerene cage reductions C60/C601−, C601−/C602− and C602−/C603− respectively, as these values are virtually identical for all the complexes studied. The process at −1.67 V in 2 and −1.79 V in 3, which is not observed in uncoordinated 1, most likely corresponds to the one-electron reduction of the bpy-ligand coordinated to the Pt(II) centre, although a metal-based redox process cannot be discounted. This reduction potential is somewhat affected by the co-ligands in 2 and 3 and is shifted to a more negative value in 3 due to the electron donating nature of the catecholate moiety.40 The oxidation process for 3 at −0.10 V is not observed in 1 or 2, and most likely corresponds to formation of the semiquinone form of catecholate;49 this potential is very close to that of analogous Pt(diimine)(cat).38
| Compound | E 1/2ox | E 1/2red1 | E 1/2red2 | E 1/2red3 | E 1/2red4 |
|---|---|---|---|---|---|
| a Potentials (E1/2 = (Eap + Ecp)/2) in V quoted to the nearest 0.01 V. All potentials are reported against the Fc+/Fc couple for 0.5–1.0 mM solutions in DMF containing 0.2 M [nBu4N][BF4] as the supporting electrolyte. The anodic/cathodic peak separation (ΔE = Eap − Ecp) is given in brackets. | |||||
| 1 | — | −0.85 (0.07) | −1.31 (0.07) | — | −1.98 (0.07) |
| 2 | — | −0.87 (0.06) | −1.31 (0.06) | −1.67 (0.06) | −2.00 (0.06) |
| 3 | −0.10 (0.07) | −0.87 (0.06) | −1.31 (0.06) | −1.79 (0.07) | −2.00 (0.09) |
| (bpy)Pt(cat)44 | −0.14 | — | — | −1.74 | — |
To elucidate the localization of electron density in the one-electron reduced and oxidised species, and to determine the nature of the frontier orbitals in 3, UV/vis and EPR spectroelectrochemistry on 3 was performed. Electrolysis of a solution of complex 3 in DMF containing [nBu4N][BF4] (0.2 M) as supporting electrolyte was performed at 243 K by applying the potentials corresponding to the first, second and third reductions and the first oxidation of 3. UV/vis and EPR spectra of the corresponding reduced/oxidised states and intermediates were recorded, allowing the nature, reversibility and stability of each process/state to be assessed.
The spectra corresponding to the individual redox states of 3 are presented in Fig. 2, and the absorption data and simulated EPR parameters are summarised in Table 2. The first reduction and first oxidation processes were found to be chemically reversible under the conditions of the experiment. However, the second, third and fourth reduction processes are found to be unstable and chemically irreversible under these conditions. The UV/vis spectrum of 31−, generated by applying a potential of −1.09 V to the neutral sample (Fig. 3a), shows the disappearance of the absorption band at 436 nm, assigned to the functionalised fullerene, and an increase in absorbance in the 700–900 nm region which is characteristic of a fullerene C60˙− monoanion.51 Fullerene monoanions show absorption bands in the NIR region at approximately 1000–1100 nm,4,52,60 which was outside the spectral range accessible in our experiments. The solution X-band EPR spectrum of 31− at room temperature (Fig. 2a, inset) shows an isotropic signal with giso = 2.001 which is also characteristic for fulleropyrrolidine monoanions53,54 and confirms that the SOMO of the radical-anion 31−, and therefore the likely LUMO of 3, is localised on the fullerene cage. The two-electron reduced 32− species, generated by applying a potential of −1.57 V to the neutral sample, possesses a UV/vis spectrum similar to 31− with a further increase in the strength of the absorbance in the 700–900 nm region, consistent with the formation of a fullerene dianion (Fig. 2b).46 The absence of a strong signal in the EPR spectrum of 32−, with only a very weak residual signal corresponding to the presence of trace amounts of the 31−, is consistent with EPR data previously reported for fulleropyrrolidine dianions55 and confirms the formation of a species in which the C60 moiety is doubly reduced with the two electrons paired on the LUMO of the fullerene. The UV/vis spectrum of 33−, generated by applying a potential of −1.91 V to the neutral sample, is drastically different from the spectra of 32− (Fig. 2c) and shows new absorption bands at 523 nm, 487 nm, 453 nm and 368 nm with high extinction coefficients. Such bands have been observed previously for one-electron reduced bipyridyl complexes55 implying that the third reduction of 3 occurs on the bipyridine unit. Additionally, the frozen solution X-band EPR spectrum of the 33− recorded at 77 K (Fig. 2c, inset) shows a rhombic signal with g-values and hyperfine coupling which are very similar to the EPR spectra reported previously for related bipyridyl-platinum systems.55 The intense central feature is assigned to an organic radical formed as a result of decomposition of the sample and has been observed previously in bulk electrolysis experiments of solutions containing fullerene.56 We were not able to determine the nature of the 4th reduction process due to the low stability of the multi-electron reduced species.
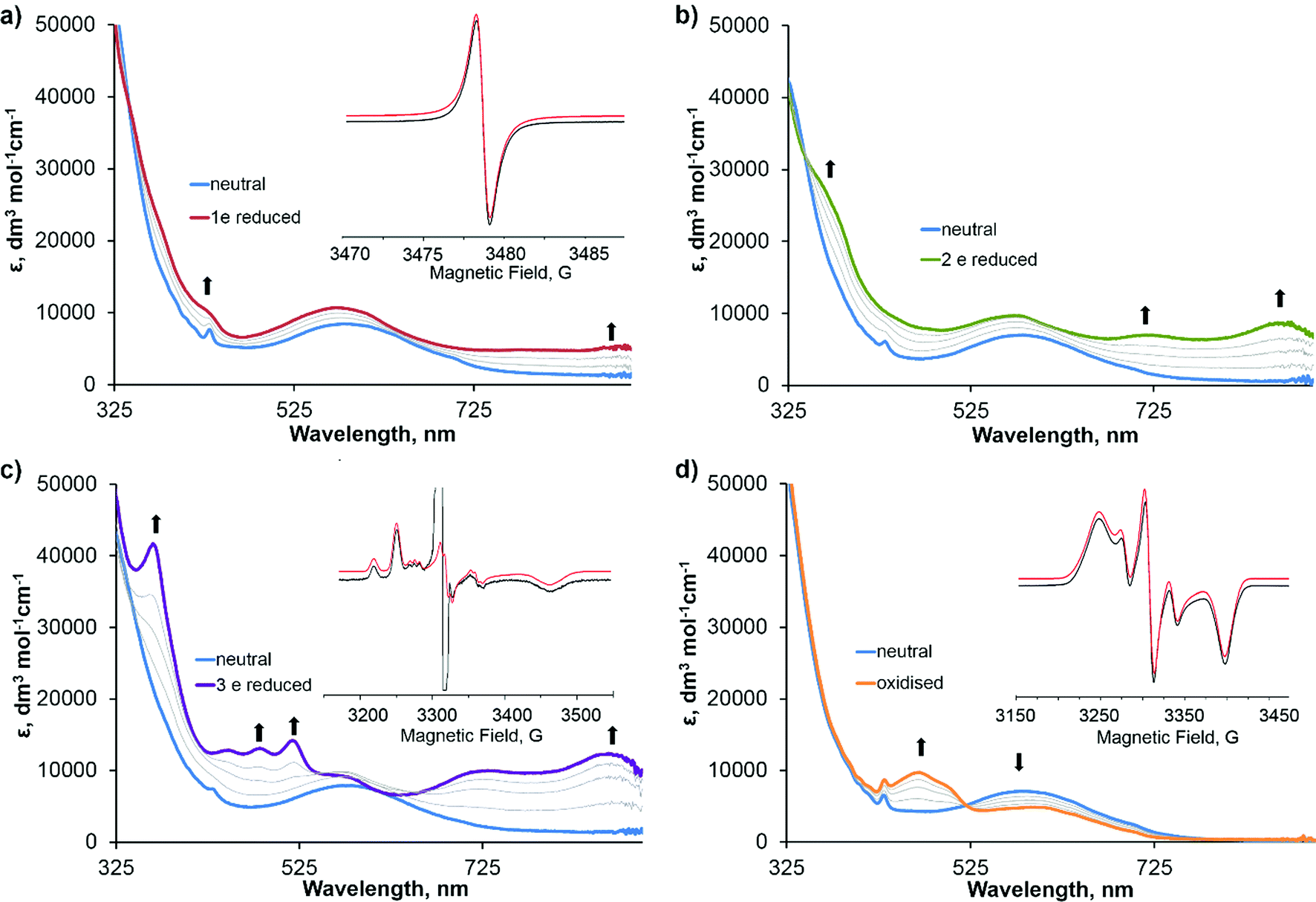 | ||
| Fig. 2 UV/vis spectra of 3 (blue line in all cases) and the different electrochemically generated radical ion species for the formation of (a) 31− (red line), (b) 32− (green line), (c) 33− (purple line) and (d) 31+ (orange line) with transitional spectra included for all processes (grey lines). All spectra were recorded in DMF solution containing 0.2 M [nBu4N][BF4] as supporting electrolyte. The X-band EPR spectra (black line, inset) and corresponding simulated EPR spectra (red line, inset) of each electrochemically generated species were recorded as (a) a fluid solution spectra for 31− at 298 K, (c) a frozen solution spectra of 33− at 77 K, and (d) a frozen solution spectra for 31+ at 77 K. | ||
| Compound | UV/vis: λ, nm (ε × 10−3, dm3 mol−1 cm−1) | EPRa | ||
|---|---|---|---|---|
| g values | A × 10−4 cm−1 (a × 10−4 cm−1) | Linewidth, G | ||
| a g-Values, A constants and linewidths are obtained from the simulation of the experimental spectra using WINEPR SimFonia, Shareware version 1.25, Bruker Analytische Messtechnik GmbH; Lorentzian lineshape for simulation of the fluid solution EPR spectra and Gaussian lineshape for simulation of the frozen solution EPR spectra. | ||||
| 3 | 587 (6.88), 436 (6.64) | — | — | — |
| 31− | 893 (5.49), 785 (4.82), 580 (10.62) | g iso 2.001 | — | 0.85 |
| 32− | 871 (8.48), 725 (6.96), 577 (9.44), 363 (26.32) | — | — | — |
| 33− | 871 (12.16), 725 (10.00), 577 (9.68), 523 (13.60), 487 (12.88), 453 (12.72), 368 (41.20) | g xx 1.998 | A xx −78.3 (a2H 5.6) | 4.5 |
| g yy 2.040 | A yy −60.0 | 9 | ||
| g zz 1.915 | A zz −28.6 (a2H 12.0) | 15 | ||
| 31+ | 605 (4.82), 502 (7.54), 473 (9.59), 434 (8.51) | g xx 2.042 | A xx 33.4 | 20 |
| g yy 2.004 | A yy 52.4 | 9 | ||
| g zz 1.951 | A zz 18.0 | 13 | ||
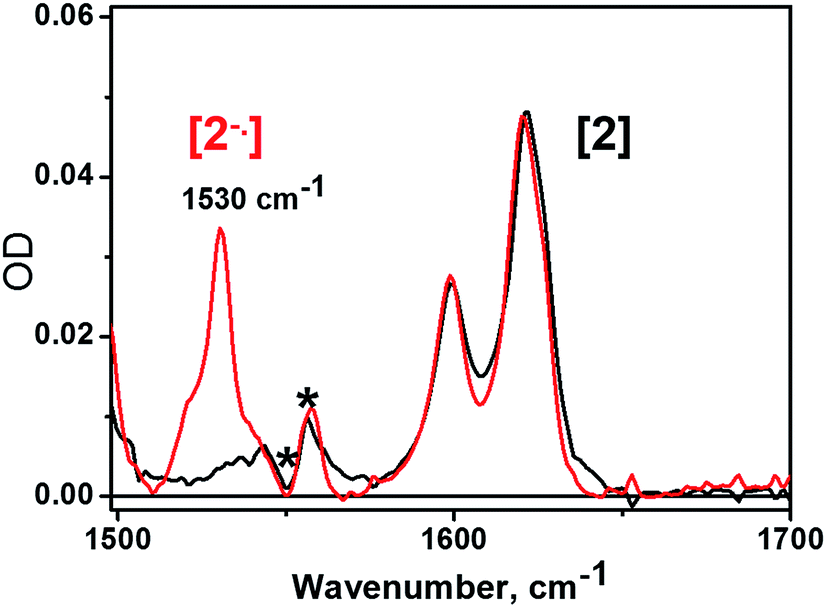 | ||
| Fig. 3 IR spectra of 2 (black line) and electrochemically generated 21− (red line) recorded in CH2Cl2 solutions containing 0.2 M [nBu4N][BF4] electrolyte. The asterisks refer to incomplete solvent subtraction. | ||
The UV/vis spectrum of the one electron oxidised 31+, generated by applying a potential of 0.09 V to the solution of 3, shows the depletion of the LLCT band at 587 nm and formation of several new bands at 430–510 nm and in the 580–620 nm region. These bands can be assigned to absorption due to semiquinone radical cation bound to PtII.55 The frozen solution EPR spectrum of 31+ recorded at 77 K (Fig. 2d, inset) can be assigned to a radical cation in which the unpaired electron is localised predominantly on the catecholate ligand with a small contribution from Pt, suggesting that the HOMO of 3 is localised mainly on the catecholate moiety. The hyperfine splitting constants for 31+ are very similar to those observed in fullerene-free analogous compounds, indicating a similar noteworthy contribution of Pt orbitals in the SOMO.40,55
Therefore, based on UV/vis and EPR spectroelectrochemical data, complex 3 is a donor–acceptor–stronger acceptor (D–A1–A2) assembly with significant charge storing capacity via formation of multiple redox states (Scheme 3). The HOMO is located on the catecholate moiety with a small contribution from Pt, and the low-lying LUMO is located on the fullerene cage. No ground-state electronic intramolecular interactions have been observed in complex 3. However the HOMO–LUMO gap of approx. 0.77 eV as estimated from electrochemical data is relatively small, and therefore intramolecular charge-transfer processes can be expected in the excited state upon absorption of visible light.
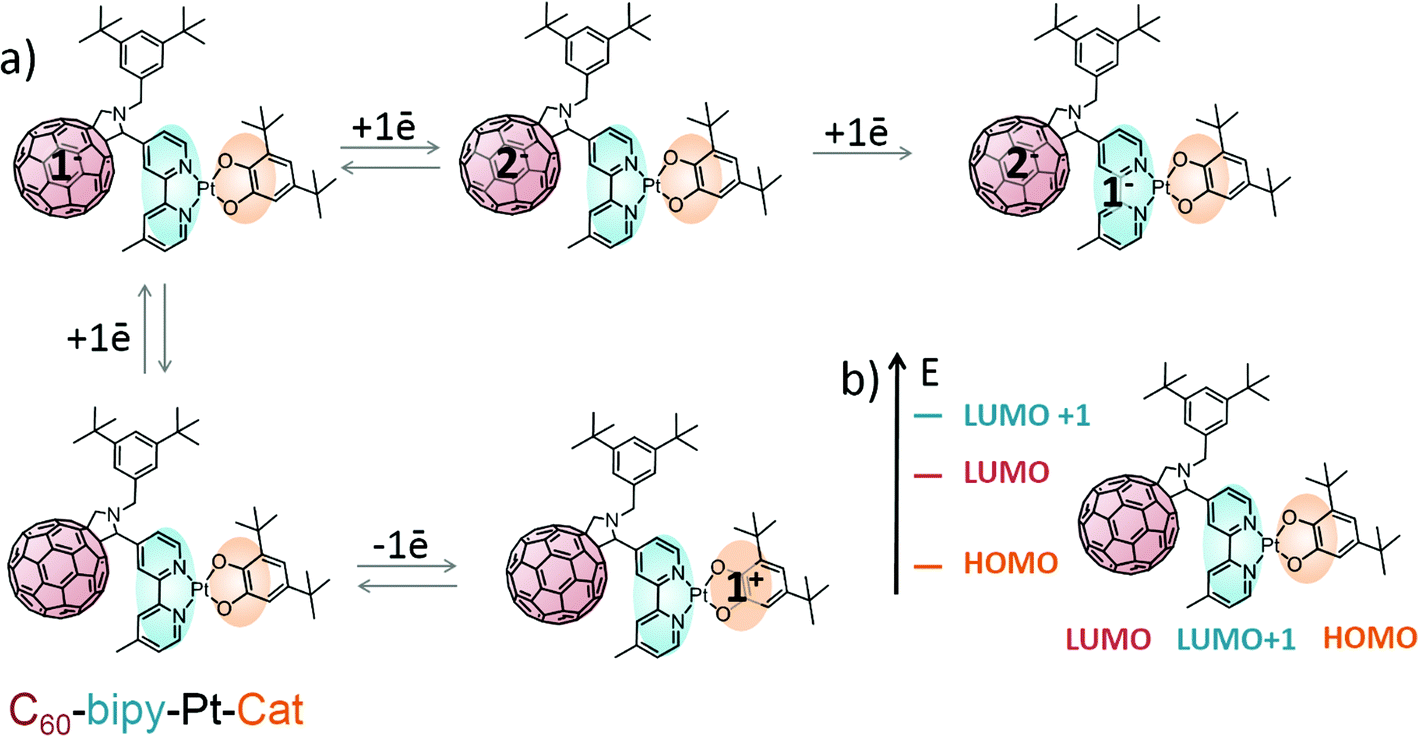 | ||
| Scheme 3 (a) Schematic representation of the sequential electrochemical processes occurring in complex 3 upon reduction/oxidation and (b) corresponding qualitative energy level diagram for the frontier molecular orbitals deduced from cyclic voltammetry, spectroelectrochemical and EPR measurements (the HOMO, LUMO and LUMO+1 are located predominantly on the catecholate, fullerene and bipyridine moieties of the complex 3 respectively). | ||
Photoinduced charge separation
The nature and dynamics of the excited states derived from 3 have been resolved by femtosecond time-resolved IR studies, aided by transient absorption spectroscopy. The IR spectra of C60 and its mono- and di-anions in both the gas phase and solid state display several major features including vibrational bands at 1530 and 1408 cm−1 for the C60˙− monoanion.57 However, to the best of our knowledge no experimental or theoretical reports of the IR spectra of fulleropyrrolidine anions in solution have been described. We therefore performed IR spectroelectrochemical studies on a (C60-bpy)PtCl2, 2, in CH2Cl2 solution in the presence of [nBu4N][BF4] as supporting electrolyte, and recorded the IR spectra of the electrochemically generated one- and two-electron reduced species, 21− and 22− respectively. Due to strong IR absorbancies of the electrolyte solution below 1500 cm−1, IR spectra were investigated over the range from 1500–1650 cm−1. A strong band at 1530 cm−1 developed upon electrochemical generation of 21−; this is not present for 2 and disappears upon further reduction to 22− (Fig. 3). This band is not present in the spectra of radical-anions of fullerene-free 10.39This result is consistent with the observed spectra of C60˙− monoanion in a Ne matrix,58 and to the best of our knowledge is the first reported example of the characteristic IR signature of a C60-fulleropyrrolidine monoanion in solution. The 1530 cm−1 band can serve a spectroscopic marker for C60˙− in solution.
TRIR spectroscopy of 3 was performed in CD2Cl2 solution under 400 nm, ∼50 fs excitation (Fig. 4, 5). Upon excitation, bleaching of the ground state IR bands is observed, along with the formation of multiple transient bands which can be assigned by comparison with model dyads (Fig. 5). The appearance of a transient band at 1530 cm−1 in the TRIR spectrum of 3 which does not occur for the control fullerene-free dyad39 (red spectrum, Fig. 5b) is of particular importance: as discussed above, this band is assigned to the skeletal vibration of the fulleropyrrolidine monoanion, directly confirming formation of C60˙− upon photoexcitation in 3.
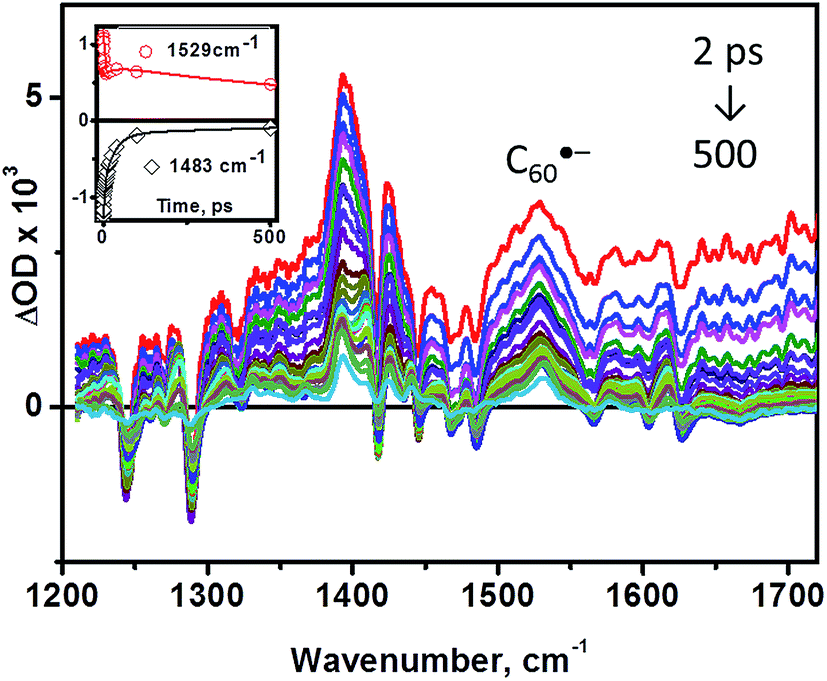 | ||
| Fig. 4 TRIR spectra of 3 in CD2Cl2 solution at 298 K upon laser excitation at 400 nm, ∼50 fs pulse, at different time delays. Inset: kinetics at selected frequencies. Symbols are experimental points, and the lines are multiexponential fits to the data using the parameters obtained by a global fit, which yielded lifetimes values of 2.5 (±0.3) ps, 31 (±0.4) ps, and 890 (±17) ps. | ||
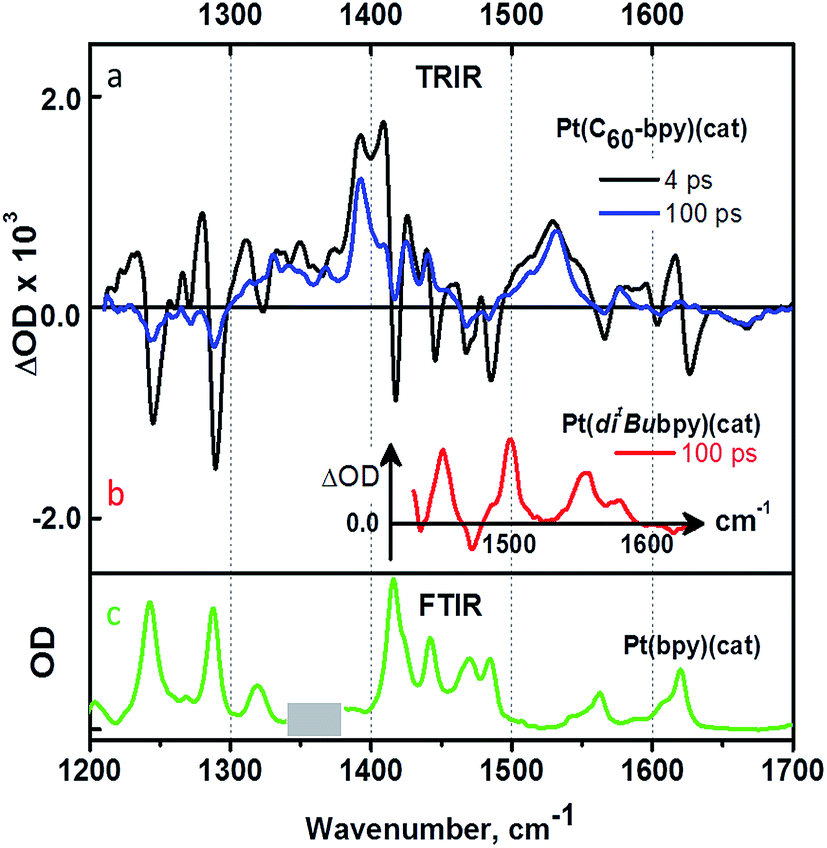 | ||
| Fig. 5 (a). TRIR spectra of 3 in CD2Cl2 at two selected time delays; (b) TRIR spectrum of a model compound 10 (no band at 1530 cm−1 observed) obtained under identical conditions to (a); (c) FTIR spectrum of compound 10 in CH2Cl2. | ||
The bleaching of the IR bands corresponding to the cathecholate moiety at 1243, 1288, 1470 and 1484 cm−1 is also observed which confirms participation of the catecholate in the excited state.38 The catecholate-based bleaches persist at later times, and decay concomitantly with the 1530 cm−1 feature assigned to C60˙−. Thus, we conclude that the final excited state in 3 is a C60˙−-bpy-Pt-Cat˙+ radical-ion pair with an energy of 0.77 eV as determined from the difference in the first oxidation and reduction potentials of 3 by cyclic voltammetry (see above). This state is significantly lower in energy than 3C60* (1.51 eV) or 3MLCT/LLCT* states (estimated to be in the range of 1.98–2.55 eV as reported for structurally similar systems28,35), and returns to the ground state through charge-recombination with a lifetime of 890 ps (see ESI†).
Analysis of the early stages of the time dynamics of the TRIR spectra reveals considerable spectral shape evolution, which is complete by ca. 100 ps. Multiple bleaches and corresponding transient bands which are present only at early times (Fig. 5a) largely correspond to bpy-localised vibrational modes (for example, the bleach at ∼1628 cm−1), clearly confirming participation of the bipyridyl moiety in the initially populated excited state. The fast decay components resolved by global fit analysis, 2.5 and 31 ps, can be assigned to vibrational cooling of the initially formed LLCT state [C60(bpy˙−)Pt(cat˙+)], concomitant with the electron transfer from bpy-anion to the fullerene, forming [C60˙−(bpy)Pt(cat˙+)]. The presence of the 1530 cm−1 band attributed to C60˙− at the earliest detection times, ∼400 fs, indicates that another, ultrafast, channel of photoreduction of fullerene independent of electron-transfer from the bpy-anion also exists in 3. This parallel channel may originate from the population of 1C60* concurrent with that of Pt-based excited states upon 400 nm excitation; 1C60* then engages in oxidative quenching of the [(bpy)Pt(cat)] (ΔG = −0.93 eV), which clearly outcompetes intersystem crossing to 3C60*.‡
The extremely broad feature appearing as a background offset across the entire IR detection range, but being more pronounced towards higher energies, has dynamics nearly identical to that of the 1530 cm−1 transient band, and can be attributed to the residual electronic absorbance of an unpaired electron delocalised over the C60-cage in the charge-separated state, of which the major absorbance occurs in the NIR region.4,59,60
The results of the photoinduced dynamics of 3 deduced from TRIR data are further corroborated by picosecond transient absorption spectroscopy (Fig. 6, THF, 355 nm, ∼27 ps excitation pulse). The spectra reconstructed using global fit in a sequential model demonstrate the presence of two excited states with lifetimes of ca. 15 ps and 890 ps. Bleaching of the cat-to-bpy LLCT band at 675 nm is observed, together with the growth of a new broad transient absorption band at 400–500 nm, as well as absorption developing towards the NIR region of the spectrum. Importantly, the absorption of the fullerene triplet state58 expected at ca. 700 nm was not observed.
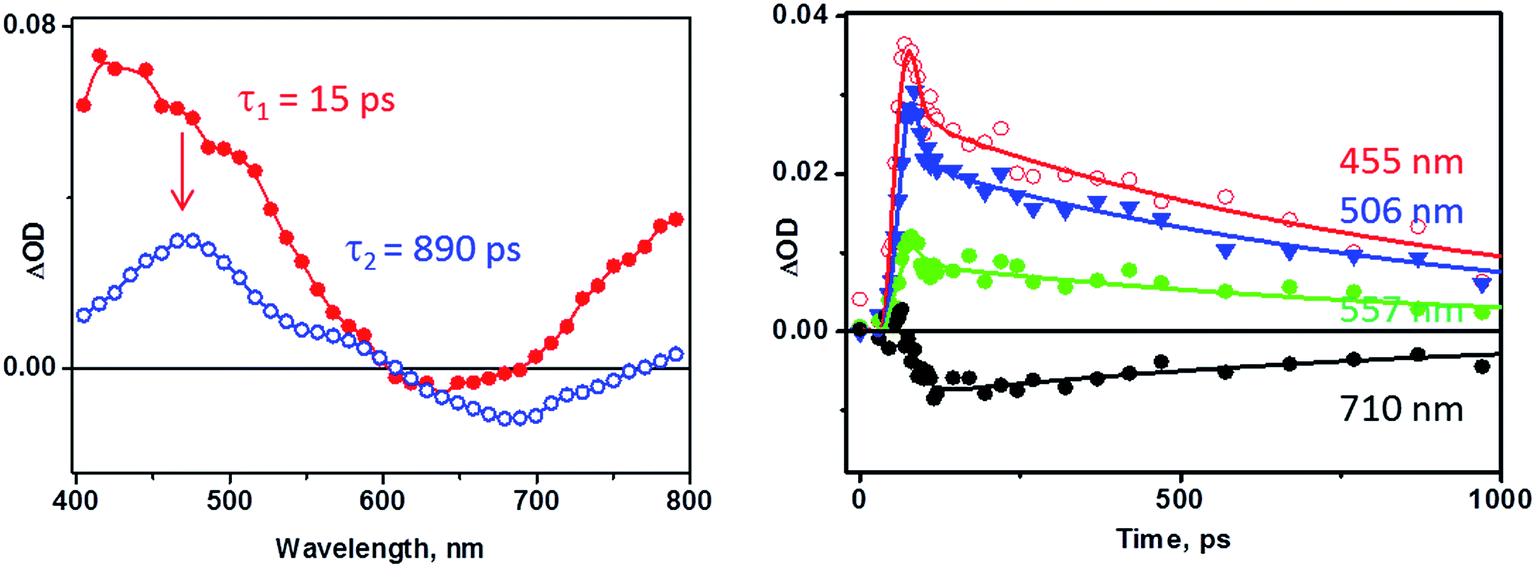 | ||
| Fig. 6 Transient absorption spectra of 3 in THF solution observed upon laser excitation (355 nm) at room temperature. Left: the decay associated spectra obtained by global fit to the data using a cascade model A → B → C, (C = ground state); τ1 15 (±6) ps, τ2 890 (±80) ps (see text for detail). Right: kinetic traces at selected wavelength (symbols) with the solid lines representing the two-exponential fit to the data using the parameters above. | ||
The decay of the transient features and the recovery of the bleach occur synchronously on the sub-nanosecond timescale, and are complete by 3800 ps. The transient band in the 400–500 nm region corresponds to the absorption of the coordinated catechol radical-cation (ca. 470 nm, Fig. 2d), as is anticipated when a charge-transfer excited state is formed. The evolution of both the position of the bleach and the shape of the transient spectra at early times indicate complex dynamics. Such dynamics may involve several convoluted processes including formation of the singlet excited state of the fullerene,4 as well as initial formation of the bipyridine radical-anion expected in the LLCT state (implied by the bleaching of the LLCT absorption band), which then engages in an electron-transfer process to the fullerene acceptor. The characteristic transient spectra of the LLCT states observed for model compounds 10 and 11 are shown in Fig. 7a and b. Comparison between the transient spectra of 10, 11 and 3 (Fig. 7c) reveals that characteristic bipyridyl radical-anion absorption signatures (Fig. 2c, 7a–c) are barely distinguishable in the spectrum of 3 at comparatively early times (Fig. 6, deconvolved from the ca. 27 ps instrument response) and are absent from the late-time spectra (blue spectrum, Fig. 6 and 7c). This observation is consistent both with the presence of two independent channels of fullerene-anion population, and with the ∼30 ps timescale of electron transfer from the bipyridyl radical anion formed in the LLCT state to the fullerene, as estimated from the TRIR data. Comparison of the extinction coefficients of 1 and 3 at 400 nm suggests an approximately 1:1 population of either channels under 400 nm excitation; however, quantitative information on the relative contribution of each pathway cannot be extracted reliably due to time-resolution limitations. The fulleropyrrolidine radical-anion absorbs over the whole visible range with relatively low extinction coefficients (Fig. 2a).
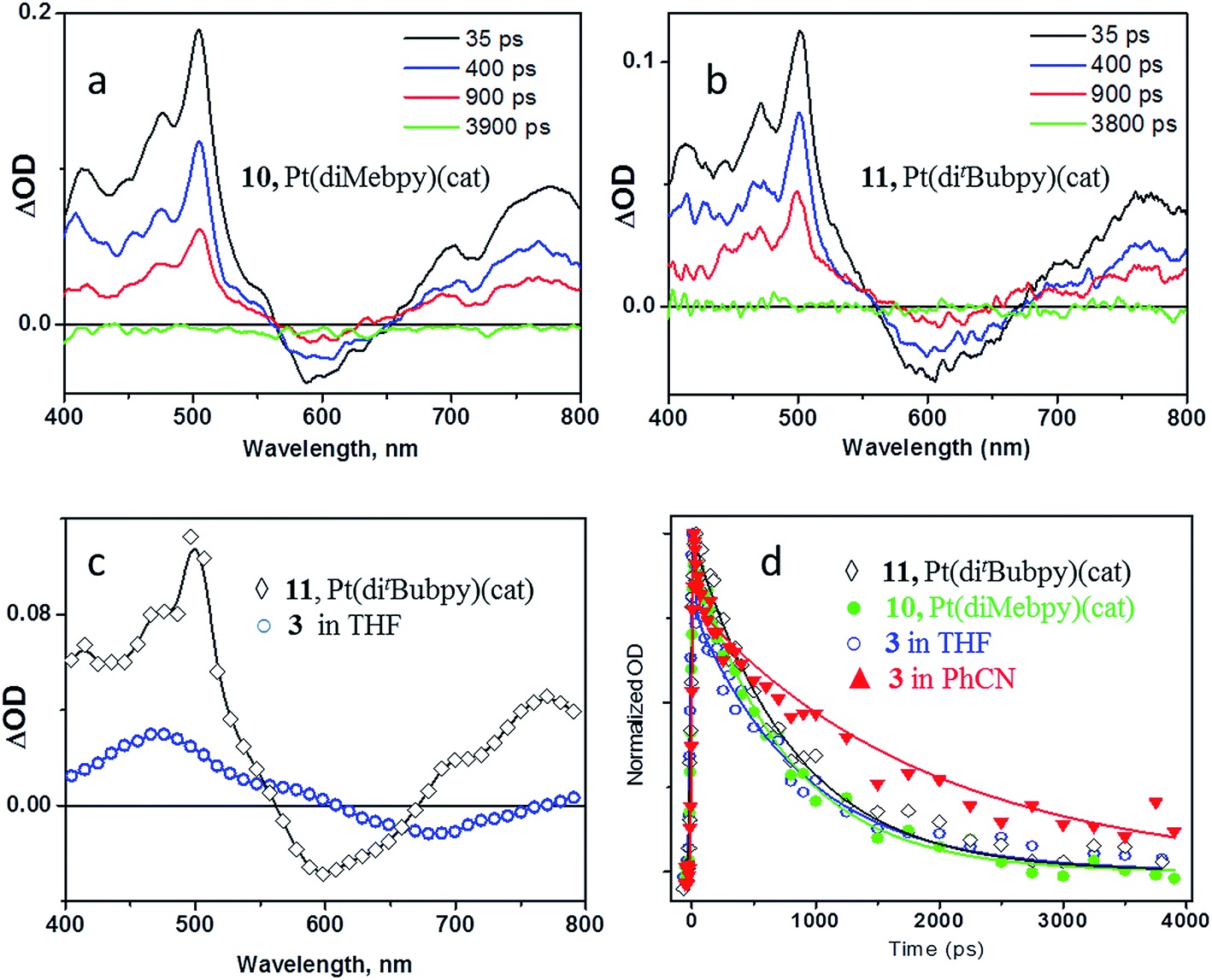 | ||
| Fig. 7 Comparison of the transient absorption spectra obtained for model dyads (a) 10, (b) 11 and (c) for the lowest excited state of 3. (d) Kinetic traces (symbols) and multi-exponential fits to the data with the parameters specified in the text (solid lines) for 3 and model compounds. | ||
The lifetimes of different processes determined by transient absorption and TRIR methods are mutually consistent. The 2.5 and 31 ps components obtained in the TRIR experiments (∼50 fs excitation pulse) were not resolved in the transient absorption experiments due to the longer pulse length (ca. 27 ps instrument response), and appear as one component with a 15 ps lifetime. The 890 ± 80 ps lifetime of the lowest excited state determined by global fit analysis of the transient absorption data is a good match with the value obtained from the TRIR data. Performing the experiment in a more polar solvent, PhCN, yielded similar results on the timescale >100 ps, but with considerable elongation of the lifetime of the final, charge-separated, excited state to ca. 1.9 ns (see Fig. 7d).
The presence of C60-anion in the excited state of 3, detected by TRIR, should also be evident in the NIR region of the spectrum. The absorption maximum attributed to C60-anion has been reported in the region from 1005–1110 nm,4,59 with the exact position being strongly dependent on the solvent, and on the chemical functionalisation of the fullerene cage.60 Femtosecond NIR transient absorption experiments were performed on 3 in DCM, under 400 nm, ∼50 fs excitation, using an adaptation of the setup used for the TRIR studies. The spectra were recorded in several sets of ∼40 nm windows between 1020 and 1200 nm, which permitted analysis of excited state dynamics associated with this spectral region. The NIR range of the spectrum (800–1200 nm) is congested: the bipyridyl radical anion formed in the LLCT excited state, (bpy˙−)-Pt-(cat˙+) has intense absorption in this region, which would decay as fullerene-anion is being populated, adding complexity to the dynamics. Global fit to the data, consistent between four different experiments at different laser powers and concentrations of 3, revealed a three-component dynamics, with the rate constants similar to those derived from TRIR experiments. The longest decay component has a lifetime of ca. 1 ns, which matches the decay of the catecholate-cation based absorptions observed in the visible region of the spectrum (∼470 nm, Fig. 2d and 6). Thus the NIR data are consistent with the proposed formation of the charge-separated excited state, [(C60˙−-bpy)-Pt-(cat˙+)] as the lowest excited state of 3.
Comparison of the photophysical properties of 3 with the control fullerene-free bipyridine-Pt-catechol dyads (Mebpy)Pt(cat) 10, and (tBubpy)Pt(cat) 11, enables the effect of introducing a fullerene cage to the complex to be quantified. Transient absorption experiments reveal the formation of a (bpy˙−)Pt(cat˙+) radical-ion pair in both 10 and 11, which have energies of approx. 1.64 eV.55 Both dyads show rapid formation of a vibrationally hot charge-separated state upon photoexcitation, which, following vibrational cooling with a ca. 5 ps lifetime, decays to the ground state with the lifetime of 750 ps for 10, and 800 ps for 11 (Fig. 7d). It is worth noting the sub-picosecond ISC has been reported for many small transition metal complexes,61 and the lowest detected excited state in Pt(II) diimine catecholates has been assigned as a 3MLCT/LLCT in the previous works.38,62,63 It has also been proposed that S1 → T1 conversion in (bpy)Pt(cat) may be comparatively slow with regard to the IC deactivation of S1 to the ground state if C2v symmetry is maintained.64 Since the early time spectra (∼0.5 ps), electronic and vibrational, of the model (diimine)Pt(cat) compounds and the asymmetric 3 are very similar, and no stimulated emission was detected in the transient absorption spectra, we propose that the first detected excited state as 3CT, (C60-bpy˙−)-Pt-(cat˙+) in its nature, and that electron transfer from bpy-anion to C60-moiety occurs in the triplet manifold.
Although the fullerene cage is an excellent electron acceptor and the energy of the LUMO in 3 is significantly lower than in the control fullerene-free dyad, the lifetime of the charge-separated state in 3 is only slightly different to that of the control dyads, which implies that the rate of the charge recombination is similar in all compounds. A possible explanation comes from comparison of the geometries of 3 and the control dyads. In the fullerene-free dyads the centres of the donor and acceptor moieties are separated by approximately 4.5 Å in the solid state (Fig. 8c and d).38 In the fullerene-containing complex 3 the donor catechol moiety and the fullerene cage are separated by a significantly larger number of bonds. However, molecular mechanics (MM) geometry optimisation (please see details of MM in the ESI file†) of the shape of the molecule reveals the presence of close intramolecular contacts between the bipyridyl group and the C60 fullerene cage resulting in so-called ‘cup-and-ball’ type interactions typical for metalloporphyrin-fullerene complexes (Fig. 8a).65 The centres of the two pyridine rings in the bipyridine group are located at a distance of 3.3 and 3.7 Å from the corresponding double bonds of the fullerene cage which is characteristic of π–π interactions (Fig. 8b).66 These intramolecular interactions stabilise the conformation of 3 in which the Pt-catecholate moiety is in relatively close proximity to the fullerene cage resulting in the shortest through-space distance between the centre of the Pt-catecholate ring and the fullerene cage being approx. 5.7 Å (Fig. 8a). This means that the distance between the donor and acceptor moieties in 3 and control fullerene-free dyads are similar with a difference of only approx. 1 Å. This results in similar charge separated state lifetimes for the fullerene-containing and fullerene-free compounds, as in all cases tunnelling is anticipated as the mechanism of charge recombination.
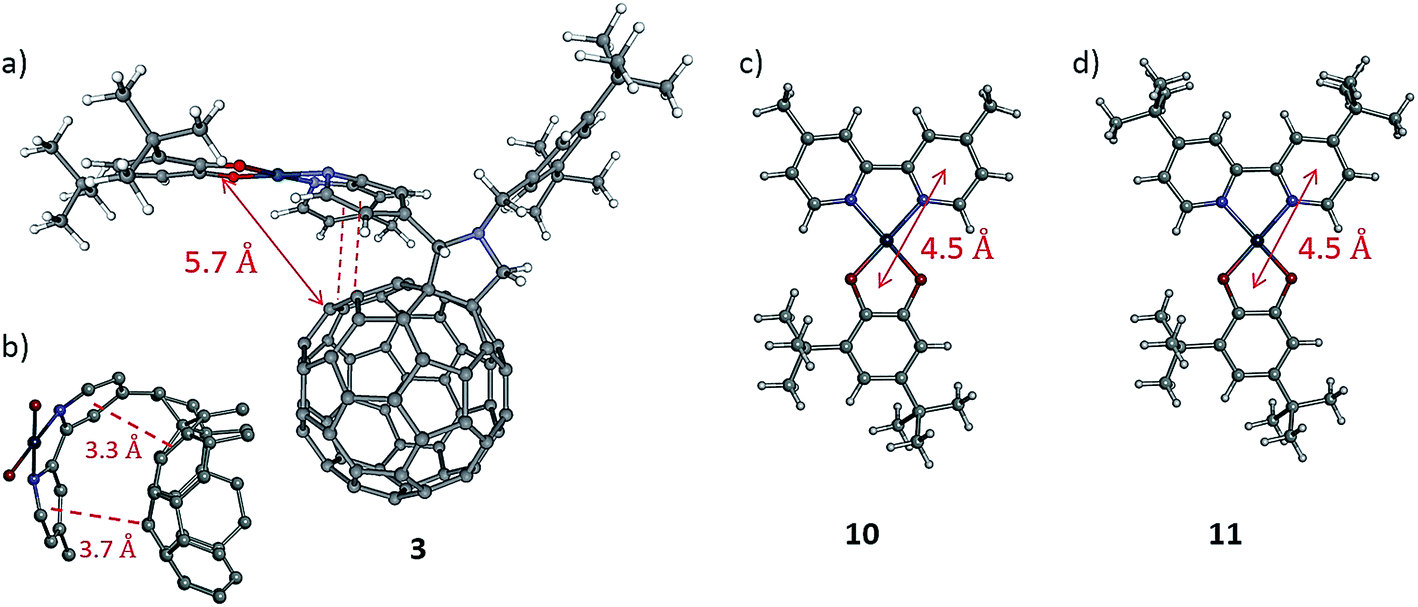 | ||
| Fig. 8 (a) Molecular mechanics optimised geometry of 3 indicating a distance of 5.7 Å between the centre of the donor moiety and the nearest C-atom of the fullerene cage, and (b) view of a fragment of 3 highlighting the complementary cup–ball interactions between the bipyridyl group and the fullerene cage resulting in close contacts with distances of 3.3 and 3.7 Å, respectively. Geometries of (c) the (Mebpy)Pt(cat) dyad 10, and (d) the (tBubpy)Pt(cat) dyad 11 for which a distance of 4.5 Å between the centres of the donor and acceptor moieties have also been calculated. | ||
The overall scheme of excited state processes in the (C60-bpy)Pt(cat) triad is summarised in Fig. 9. Two parallel pathways of forming the desired charge separated excited state are outlined. We note that the population of the 3C60* is not observed.
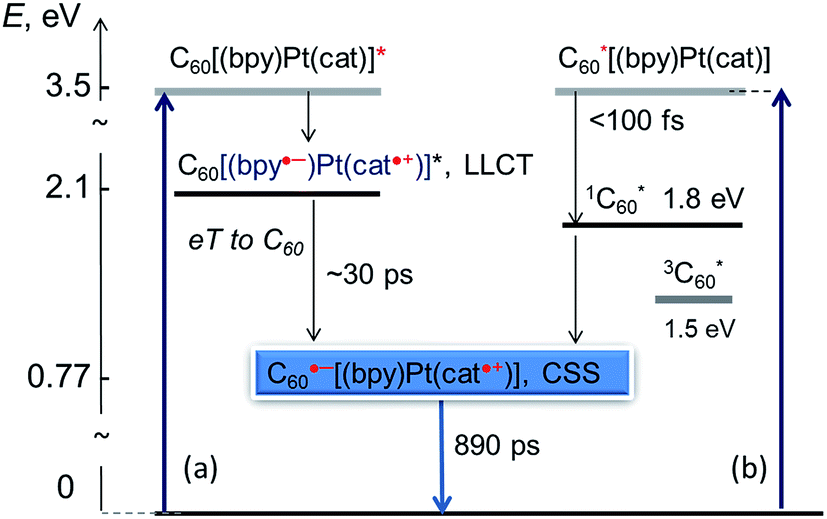 | ||
| Fig. 9 Schematic representation of light-induced processes in the fullerene-bipy-Pt(II)-cat donor–acceptor assembly. The energies of the excited states are estimated from electrochemical data. | ||
The lifetime of 890 ps in THF (NB a lifetime of 1.9 ns in PhCN) achieved for 3 in this study is to the best of our knowledge one of the longest observed for a C60-donor assembly where the donor is in close proximity of the fullerene cage (see Table S1† and ref. 67), and the first observation of the lowest energy CSS in a transition metal complex-fullerene assembly.
Conclusions
This work demonstrates the design of a fullerene donor–acceptor complex assembly in which photoreduction of the fullerene has been achieved through photosensitisation with a transition metal chromophore, and in which the charge-separated state has been stabilised. In this donor–acceptor assembly, C60 fullerene has been successfully incorporated into the (diimine)Pt(cat) moiety which acts both as a chromophore, and as an excited state electron donor. A new synthetic route has been developed which allows the donor catechol moiety to be incorporated into the Pt(II) centre first, followed by coordination to the aldehyde functionalised bipyridine moiety and subsequent Prato cycloaddition reaction with C60. This methodology avoids the problems associated with unwanted nucleophilic attack on the fullerene cage.The resulting fullerene-containing donor–acceptor assembly combines several essential characteristics for efficient light harvesting and energy storage: (i) panchromatic absorption in the UV/vis region due to a combination of the electronic transitions of the fullerene cage and a strongly solvatochromic catechol-to-diimine charge-transfer transition; (ii) several redox states readily accessible within the visible light energy gap, leading to the capacity of storing multiple redox equivalents; (iii) an incorporation of C60 as a primary electron acceptor with the LUMO located on the fullerene cage with the HOMO located on the catecholate/Pt unit. The small, 0.8 eV HOMO–LUMO energy gap ensures that the energy of the charge-separated state is lower than that of the 3C60* (ca. 1.51 eV), thus overcoming the typical problem of this latter state acting as an energy sink in fullerene-containing assemblies.
The interrogation of light-induced properties of compound 3 by transient absorption and TRIR spectroscopy establishes the formation of the C60˙−-bpy-Pt-cat˙+ radical-ion pair as the lowest excited state. TRIR spectroscopy revealed formation of a 1530 cm−1 band in the lowest excited state, which was assigned to the formation of a fulleropyrrolidine monoanion by complementary IR (spectro)electrochemistry of the monoreduced species. This is the first report of the IR signature of fulleropyrrolidine monoanion in solution, and the first application of time-resolved infrared spectroscopy for the study of fullerene-based donor–acceptor assemblies. The charge-separated state in 3 is comparatively long-lived with a lifetime of 890 ps in THF, which is significantly longer than the lifetimes exhibited by the majority of the isostructural D–A dyads in which the donor moiety is located in close proximity to the fullerene cage.
A wide range of other donor groups can be introduced to the Pt–diimine moiety (or to other transition metal–diimine units) and attached to the fullerene cage using the methodology reported herein; where for example the final CSS can still lie below 3C60, but possesses higher energy than the system reported here, thereby potentially increasing the excited state lifetime in accordance with the energy gap law. We, therefore, regard this as a general route to the preparation of this type of multicomponent, redox active, donor–acceptor systems. The methodologies reported herein can be translated readily to other fullerene-containing assemblies, potentially leading to new structural motifs based on the hybrid systems which combine the advantages of the carbon-based nanosized fullerene cage, a transition metal chromophore, and the lowest energy charge-separated excited state, for photocatalysis, molecular electronics and photovoltaics. We also demonstrate that the light-induced properties of such fullerene-containing systems can be investigated by TRIR spectroscopy, monitoring the unique IR absorption features of the fullerene monoanion, which can be readily transferred to a variety of fullerene-containing systems, including donor–acceptor assemblies, fullerene-containing films and fullerene/polymer blends for solar energy conversion.
Experimental
Experimental section detailing the compounds synthesis and characterisation, TA and TRIR measurements, electrochemistry, and spectroelectrochemistry experiments is available in the ESI file.†Acknowledgements
This work was supported by the University of Nottingham, Engineering and Physical Sciences Research Council (EPSRC) (M.A.L., T.W.C. and A.N.K.) and European Research Council (ERC) (M.A.L., T.W.C. and A.N.K.), an ERC Starting Grant, EPSRC Career Acceleration Fellowship, and New Directions for EPSRC Research Leaders Award (E.B.), and EPSRC and ERC Advanced Grant (M.S.). We thank EPSRC and STFC for program access to beam time, and the University of Sheffield and its strategic equipment fund (M.D., P.A.S., I.V.S. and J. A. W.).References
- (a) J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560–15564 CrossRef CAS PubMed; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed; (b) T. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS; (c) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- (a) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed; (b) V. Bandi, F. P. D'Souza, H. B. Gobeze and F. D'Souza, Chem. Commun., 2016, 52, 579–581 RSC.
- (a) G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS; (b) N. Armaroli, Photochem. Photobiol. Sci., 2003, 2, 73–87 RSC; (c) M. Rudolf, S. V. Kirner and D. M. Guldi, Chem. Soc. Rev., 2016, 45, 612–630 RSC; (d) H. Imahori, Org. Biomol. Chem., 2004, 2, 1425–1433 RSC; (e) T. Konishi, A. Ikeda and S. Shinkai, Tetrahedron, 2005, 61, 4881–4899 CrossRef CAS.
- (a) P. A. Troshin, R. Koeppe, A. S. Peregudov, S. M. Peregudova, M. Egginger, R. N. Lyubovskaya and N. S. Sariciftci, Chem. Mater., 2007, 19, 5363–5372 CrossRef CAS; (b) D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolski and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC; (c) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- (a) J. N. Clifford, G. Accors, F. Cardinali, J.-F. Nierengarten and N. Armaroli, C. R. Chim., 2006, 9, 1005–1013 CrossRef CAS; (b) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913–4928 RSC; (c) R. Chitta and F. D'Souza, J. Mater. Chem., 2008, 18, 1440–1471 RSC.
- (a) C. Villegas, J. L. Delgado, P.-A. Bouit, B. Grimm, W. Seitz, N. Martín and D. M. Guldi, Chem. Sci., 2011, 2, 1677–1681 RSC; (b) G. de Miguel, M. Wielopolski, D. I. Schuster, M. A. Fazio, O. P. Lee, C. K. Haley, A. L. Ortiz, L. Echegoyen, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 13036–13054 CrossRef CAS PubMed; (c) N. V. Tkachenko, H. Lemmetyinen, J. Sonoda, K. Ohkubo, T. Sato, H. Imahori and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 8834–8844 CrossRef CAS; (d) A. Kahnt, J. Kärnbratt, L. J. Esdaile, M. Hutin, K. Sawada, H. L. Anderson and B. Albinsson, J. Am. Chem. Soc., 2011, 133, 9863–9871 CrossRef CAS PubMed.
- (a) G. Bottari, A. Kahnt, D. M. Guldi and T. Torres, ECS J. Solid State Sci. Technol., 2013, 2, M3145–M3150 CrossRef CAS; (b) J.-J. Cid, A. Kahnt, P. Vázquez, D. M. Guldi and T. Torres, J. Inorg. Biochem., 2012, 108, 216–224 CrossRef CAS PubMed; (c) M. Ince, A. Hausmann, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, Chem. Commun., 2012, 48, 4058–4060 RSC.
- (a) M. Maggini, D. M. Guldi, S. Mondini, G. Scorrano, F. Paolucci, P. Ceroni and S. Roffia, Chem.–Eur. J., 1998, 4, 1992–2000 CrossRef CAS; (b) S. Karlsson, J. Modin, H.-C. Becker, L. Hammarström and H. Grennberg, Inorg. Chem., 2008, 47, 7286–7294 CrossRef CAS PubMed.
- V. Bandi, M. E. El-Khouly, K. Ohkubo, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem.–Eur. J, 2013, 19, 7221–7230 CrossRef CAS PubMed.
- (a) N. Martín, L. Sánchez and M. A. Herranz, J. Phys. Chem. A, 2000, 104, 4648–4657 CrossRef; (b) N. Martín, L. Sánchez, M. Á. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef PubMed; (c) M. C. Díaz, M. A. Herranz, B. M. Illescas and N. Martín, J. Org. Chem., 2003, 68, 7711–7721 CrossRef PubMed.
- (a) D. M. Guldi, M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1997, 119, 974–980 CrossRef CAS; (b) D. González-Rodríguez, E. Carbonell, G. de Miguel Rojas, C. A. Castellanos, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2010, 132, 16488–16500 CrossRef PubMed.
- D. I. Schuster, K. Li, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973–15982 CrossRef CAS PubMed.
- M. Wielopolski, M. G. de Rojas, C. van der Pol, L. Brinkhaus, G. Katsukis, M. R. Bryce, T. Clark and D. M. Guldi, ACS Nano, 2010, 4, 6449–6462 CrossRef CAS PubMed.
- Y. Matsuo, M. Maruyama, S. S. Gayathri, T. Uchida, D. M. Guldi, H. Kishida, A. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 12643–12649 CrossRef CAS PubMed.
- M. Ince, M. V. Martínez-Díaz, J. Barberá and T. Torres, J. Mater. Chem., 2011, 21, 1531–1536 RSC.
- A. Mateo-Alonso, C. Sooambar and M. Prato, C. R. Chim., 2006, 9, 944–951 CrossRef CAS.
- D. I. Schuster, K. Li and D. M. Guldi, C. R. Chim., 2006, 9, 892–908 CrossRef CAS.
- R. R. Hung and J. J. Grabowski, J. Phys. Chem., 1991, 95, 6073–6075 CrossRef CAS.
- (a) Z. Zhou, G. H. Sarova, S. Zhang, Z. Ou, F. T. Tat, K. M. Kadish, L. Echegoyen, D. M. Guldi, D. I. Schuster and S. R. Wilson, Chem.–Eur. J., 2006, 12, 4241–4248 CrossRef CAS PubMed; (b) D. M. Guldi, M. Maggini, E. Menna, G. Scorrano, P. Ceroni, M. Marcaccio, F. Paolucci and S. Roffia, Chem.–Eur. J., 2001, 7, 1597–1605 CrossRef CAS PubMed; (c) F. Chaignon, J. Torroba, E. Blart, M. Borgström, L. Hammarström and F. Odobel, New J. Chem., 2005, 29, 1272–1284 RSC.
- (a) J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed; (b) S. V. Kirner, D. Arteaga, C. Henkel, J. T. Margraf, N. Alegret, K. Ohkubo, B. Insuasty, A. Ortiz, N. Martín, L. Echegoyen, S. Fukuzumi, T. Clark and D. M. Guldi, Chem. Sci., 2015, 6, 5994–6007 RSC.
- S. V. Kirner, C. Henkel, D. M. Guldi, J. D. Megiatto Jr and D. I. Schuster, Chem. Sci., 2015, 6, 7293–7304 RSC.
- J. B. Kelber, N. A. Panjwani, D. Wu, R. Gómez-Bombarelli, B. W. Lovett, J. J. L. Morton and H. L. Andreson, Chem. Sci., 2015, 6, 6468–6481 RSC.
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 256, 2530–2561 CrossRef CAS.
- M. Chergui, Dalton Trans., 2012, 41, 13022–13029 RSC.
- E. A. Glik, S. Kinayyigit, K. L. Ronayne, M. Towrie, I. V. Sazanovich, J. A. Weinstein and F. N. Castellano, Inorg. Chem., 2008, 47, 6974–6983 CrossRef CAS PubMed.
- I. V. Sazanovich, M. A. H. Alamiry, J. Best, R. D. Bennett, O. V. Bouganov, E. S. Davies, V. P. Grivin, A. J. H. M. Meijer, V. F. Plyusnin, K. L. Ronayne, A. H. Shelton, S. A. Tikhomirov, M. Towrie and J. A. Weinstein, Inorg. Chem., 2008, 47, 10432–10445 CrossRef CAS PubMed.
- K. Base, M. T. Tierney, A. Fort, J. Muller and M. W. Grinstaff, Inorg. Chem., 1999, 38, 287–289 CrossRef CAS.
- P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J.-M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15388–15389 CrossRef CAS PubMed.
- E. A. M. Geary, L. J. Yellowlees, L. A. Jack, I. D. H. Oswald, S. Parsons, N. Hirata, J. R. Durrant and N. Robertson, Inorg. Chem., 2005, 44, 242–250 CrossRef CAS PubMed.
- Y. Zhang, K. D. Ley and K. S. Schanze, Inorg. Chem., 1996, 35, 7102–7110 CrossRef CAS PubMed.
- L. Chassot, A. von Zelewsky, D. Sandrini, M. Maestri and V. Balzani, J. Am. Chem. Soc., 1986, 108, 6084–6085 CrossRef CAS PubMed.
- C. C. Chang, B. Pfennig and A. B. Bocarsly, Coord. Chem. Rev., 2000, 208, 33–45 CrossRef CAS.
- S.-H. Lee, C. T.-L. Chan, K. M.-C. Wong, W. H. Lam, W.-M. Kwok and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 10041–10052 CrossRef CAS PubMed.
- S.-H. Lee, C. T.-L. Chan, K. M.-C. Wong, W. H. Lam, W.-M. Kwok and V. W.-W. Yam, Dalton Trans., 2014, 43, 17624–17634 RSC.
- J. A. Weinstein, D. C. Grills, M. Towrie, P. Matousek, A. W. Parker and M. W. George, Chem. Commun., 2002, 382–383 RSC.
- J. Best, I. V. Sazanovich, H. Adams, R. D. Bennett, E. S. Davies, A. J. H. M. Meijer, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, M. D. Ward and J. A. Weinstein, Inorg. Chem., 2010, 49, 10041–10056 CrossRef CAS PubMed.
- (a) R. D. Pensack, K. M. Banyas, L. W. Barbour, M. Hegadorn and J. B. Asbury, Phys. Chem. Chem. Phys., 2009, 11, 2575–2591 RSC; (b) R. D. Pensack, K. M. Banyas and J. B. , Asbury, J. Phys. Chem. C, 2010, 114, 5344–5350 CrossRef CAS.
- J. Weinstein, M. Tierney, E. S. Davies, K. Base, A. Robeiro and M. Grinstaff, Inorg. Chem., 2006, 45, 4544–4555 CrossRef CAS PubMed.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- G. Strouse, J. Schoonover, R. Duesing, S. Boyde, W. Jones and T. Meyer, Inorg. Chem., 1995, 34, 473–487 CrossRef CAS.
- B. Farrington, M. Jevrich, G. Rance, A. Ardavan, A. N. Khlobystov, A. G. Briggs and K. Porfyrakis, Angew. Chem., Int. Ed., 2012, 51, 3587–3590 CrossRef CAS PubMed.
- D. Mancel, M. Jevrich, E. S. Davies, M. Schröder and A. N. Khlobystov, Dalton Trans., 2013, 42, 5056–5067 RSC.
- J. Price, A. Williamson, R. Schramm and B. Wayland, Inorg. Chem., 1972, 11, 1280–1284 CrossRef CAS.
- T. W. Chamberlain, E. S. Davies, A. N. Khlobystov and N. R. Champness, Chem.–Eur. J., 2011, 17, 3759–3767 CrossRef CAS PubMed.
- M. A. Lebedeva, T. W. Chamberlain, E. S. Davies, D. Mancel, B. E. Thomas, M. Suyetin, E. Bichoutskaia, M. Schröder and A. N. Khlobystov, Chem.–Eur. J., 2013, 19, 11999–12008 CrossRef CAS PubMed.
- A. Hirsh, T. Grösser, A. Siebe and A. Soi, Chem. Ber., 1993, 126, 1061–1067 CrossRef.
- P. Ghosh, A. Begum, D. Herebian, E. Bothe, K. Hildebrand, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 2003, 42, 563–567 CrossRef CAS PubMed.
- S. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS.
- (a) L. Kavan and L. Dunsch, ChemPhysChem, 2007, 8, 974–998 CrossRef CAS PubMed; (b) T. Gareis, O. Kötche and J. Daub, Eur. J. Org. Chem., 1998, 1549–1557 CrossRef CAS.
- D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem., 1995, 99, 9380–9385 CrossRef CAS.
- M. Brustolon, A. Zoleo, G. Agostini and M. Maggini, J. Phys. Chem. A, 1998, 102, 6331–6339 CrossRef CAS.
- I. Nuretdinov, V. Yanilkin, V. Morozov, V. Gubskaya, V. Zverev, N. Nastapova and G. Fazleeva, Russ. Chem. Bull., 2002, 51, 263–268 CrossRef CAS.
- N. Shavaleev, E. S. Davies, H. Adams, J. Best and J. Weinstein, Inorg. Chem., 2008, 47, 1532–1547 CrossRef CAS PubMed.
- M. A. Lebedeva, T. W. Chamberlain, E. S. Davies, B. E. Thomas, M. Schröder and A. N. Khlobystov, Beilstein J. Org. Chem., 2014, 10, 332–343 CrossRef PubMed.
- B. Kern, D. Strelnikov, P. Weis, A. Böttcher and M. M. Kappes, J. Phys. Chem. A, 2013, 117, 8251–8255 CrossRef CAS PubMed.
- N. Armaroli and G. Accorsi, Light-induced processes in Fullerene multicomponent systems, in Fullerenes: Principles and Applications, ed. F. Langa and J.-F. Nierengarten, RSC Publishing, 2007, ch. 4 Search PubMed.
- (a) M. Quintiliani, A. Kahnt, T. Wölfle, W. Hieringer, P. Vázquez, A. Görling, D. M. Guldi and T. Torres, Chem.–Eur. J., 2008, 14, 3765–3775 CrossRef CAS PubMed; (b) Z. Gasyna, L. Andrews and P. N. Schatz, J. Phys. Chem., 1992, 96, 1525–1527 CrossRef CAS; (c) V. Brezová, A. Staško and P. Rapta, J. Phys. Chem., 1995, 99, 16234–16241 CrossRef.
- (a) H. Imahori, H. Yamada, D. M. Guldi, Y. Endo, A. Shimomura, S. Kundu, K. Yamada, T. Okada, Y. Sakata and S. Fukuzumi, Angew. Chem., Int. Ed., 2002, 41, 2344–2347 CrossRef CAS; (b) F. DʼSouza, R. Chitta, K. Ohkubo, M. Tasior, N. K. Subbaiya, M. E. Zandler, M. K. Rogacki, D. T. Gryko and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 14263–14272 CrossRef PubMed; (c) D. M. Guldi, R. E. Huie, P. Neta, H. Hungerbühler and K.-D. Asmus, Chem. Phys. Lett., 1994, 223, 511–516 CrossRef CAS; (d) D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem., 1995, 99, 9380–9385 CrossRef CAS; (e) A. Staško, V. Brezová, P. Rapta, K.-D. Asmus and D. M. Guldi, Chem. Phys. Lett., 1996, 262, 233–240 CrossRef; (f) D. M. Guldi, Res. Chem. Intermed., 1997, 23, 653–673 CrossRef CAS; (g) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS PubMed; (h) D. M. Guldi, H. Hungerbühler and K.-D. Asmus, J. Phys. Chem. A, 1997, 101, 1783–1786 CrossRef CAS; (i) D. M. Guldi, J. Phys. Chem. A, 1997, 101, 3895–3900 CrossRef CAS; (j) K. G. Thomas, V. Biju, M. George, D. M. Guldi and P. V. Kamat, J. Phys. Chem. A, 1998, 102, 5341–5348 CrossRef CAS; (k) A. Polese, S. Mondini, A. Bianco, C. Toniolo, G. Scorrano, D. M. Guldi and M. Maggini, J. Am. Chem. Soc., 1999, 121, 3446–3452 CrossRef CAS; (l) K. G. Thomas, V. Biju, D. M. Guldi, P. V. Kamat and M. George, J. Phys. Chem. B, 1999, 103, 8864–8869 CrossRef CAS; (m) D. M. Guldi, S. González, N. Martín, A. Antón, J. Garín and J. Orduna, J. Org. Chem., 2000, 65, 1978–1983 CrossRef CAS PubMed; (n) C. Luo, D. M. Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535–6551 CrossRef CAS; (o) M. A. Herranz, B. Illescas, N. Martín, C. Luo and D. M. Guldi, J. Org. Chem., 2000, 65, 5728–5738 CrossRef CAS PubMed; (p) A. Gégout, J.-F. Nierengarten, B. Delavaux-Nicot, C. Duhayon, A. Saquet, A. Listorti, A. Belbakra, C. Chiorboli and N. Armaroli, Chem.–Eur. J., 2009, 15, 8825–8833 CrossRef PubMed; (q) V. Bandi, H. B. Gobeze and F. D'Souza, Chem. Eur.–J., 2015, 21, 11483–11494 CrossRef CAS PubMed.
- (a) Z. Abedin-Siddique, T. Ohno and K. Nozaki, Inorg. Chem., 2004, 43, 663–673 CrossRef CAS PubMed; (b) G. Ramakrishna, T. Goodson III, J. E. Rogers-Haley, T. M. Cooper, D. G. McLean and A. Urbas, J. Phys. Chem. C, 2009, 113, 1060–1066 CrossRef CAS; (c) K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Chem. Sci., 2013, 4, 2630–2644 RSC; (d) G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. Ma, X. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819–4830 RSC; (e) P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed.
- P. A. Scattergood, P. Jesus, H. Adams, M. Delor, I. V. Sazanovich, H. D. Burrows, C. Serpa and J. A. Weinstein, Dalton Trans., 2015, 44, 11705–11716 RSC.
- N. Deibel, D. Schweinfurth, S. Hohloch, M. Delor, I. V. Sazanovich, M. Towrie, J. A. Weinstein and B. Sarkar, Inorg. Chem., 2014, 53, 1021–1031 CrossRef CAS PubMed.
- J. Yang, D. K. Kersi, L. J. Giles, B. W. Stein, C. Feng, C. R. Tichnell, D. A. Shultz and M. L. Kirk, Inorg. Chem., 2014, 53, 4791–4793 CrossRef CAS PubMed.
- L. Moreira, J. Calbo, B. M. Illescas, J. Aragó, I. Nierengarten, B. Delavaux-Nicot, E. Ortí, N. Martín and J.-F. Nierengarten, Angew. Chem., Int. Ed., 2015, 54, 1255–1260 CrossRef CAS PubMed.
- P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS PubMed.
- M. A. Lebedeva, T. W. Chamberlain and A. N. Khlobystov, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: A supporting information file is available containing all experimental details, 1H and 13C NMR spectra, UV/vis spectra, cyclic voltammetry and transient absorption data, as well as additional literature data for different fullerene containing D–A dyads. See DOI: 10.1039/c5sc04271b |
| ‡ The initial excited state(s) detected in the TRIR experiments will be vibrationally hot due to sub-100 fs loss of excess energy from the 400 nm, 3.1 eV, pump pulse. |
| This journal is © The Royal Society of Chemistry 2016 |