
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A case study of proton shuttling in palladium catalysis†
Julien
Monot
ab,
Paul
Brunel
ab,
Christos E.
Kefalidis
c,
Noel Ángel
Espinosa-Jalapa
ab,
Laurent
Maron
*c,
Blanca
Martin-Vaca
*ab and
Didier
Bourissou
*ab
aUniversité de Toulouse, UPS, 118 route de Narbonne, F-31062 Toulouse, France
bCNRS, LHFA UMR5069, F-31062 Toulouse, France. E-mail: bmv@chimie.ups-tlse.fr; dbouriss@chimie.ups-tlse.fr
cUniversité de Toulouse, INSA, UPS, LCPNO, CNRS, UMR 5215 CNRS-UPS-INSA, 135 avenue de Rangueil, 31400 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 7th December 2015
Abstract
The mechanism of alkynoic acid cycloisomerization with SCS indenediide Pd pincer complexes has been investigated experimentally and computationally. These studies confirmed the cooperation between the Pd center and the backbone of the pincer ligand, and revealed the involvement of a second molecule of substrate. It acts as a proton shuttle in the activation of the acid, it directs the nucleophilic attack of the carboxylic acid on the π-coordinated alkyne and it relays the protonolysis of the resulting vinyl Pd species. A variety of H-bond donors have been evaluated as external additives, and polyols featuring proximal hydroxyl groups, in particular catechol derivatives, led to significant catalytic enhancement. The impact of 4-nitrocatechol and 1,2,3-benzenetriol is particularly striking on challenging substrates such as internal 4- and 5-alkynoic acids. Endo/exo selectivities up to 7.3/1 and 60-fold increase in reactivity were achieved.
Introduction
The interplay between experiment and theory is extremely powerful for the study of the mechanism of catalytic transformations, to understand the factors influencing their efficiency and selectivity, and finally to optimize their performance.1 From an experimental point of view, mechanistic proposals are typically drawn on the basis of kinetic studies,2 isotopic labeling experiments3 and characterizations of intermediates.4 On the other hand, the advent of DFT methods has revolutionized the ability of computational methods to describe chemical systems and nowadays virtually all molecular compounds, including transition metal complexes, can be investigated in extenso. Accordingly, accurate and valuable information can be gained on the entire reaction profile. Geometric and electronic structures of key intermediates and transition states can be analyzed, different mechanistic scenarios can be probed and compared, kinetic data and selectivities can be estimated.5 Combining and comparing the experimental and theoretical data is a major source of information and often helps to identify and understand the very mechanism of catalytic transformations.In light of our activities on pincer complexes, we recently reported SCS indenediide Pd complexes featuring ligand non-innocent character.6 These indenediide complexes proved very efficient in the catalytic cycloisomerization of alkynoic acids as well as N-tosylalkynylamides (Chart 1).7 They do not require the presence of an external base and represent a rare example of metal–ligand cooperative catalysis involving Pd.8 The catalytic system can be recycled without deactivation up to ten times and it is efficient for a large variety of substrates, giving access to 5-, 6- and even 7-membered alkylidene lactones and lactams with high selectivity (generally in favor of the exo product). The SCS indenediide Pd complexes have clearly enhanced the efficiency and extended the scope of such cycloisomerizations, but there is certainly room for further improvement. In particular, the cyclization of substrates featuring internal alkynes remains challenging. It requires much higher temperatures and longer reaction times, and the exo/endo selectivity is often modest.
 | ||
| Chart 1 Cycloisomerization of alkynoic acids and N-tosylalkynylamides promoted by palladium indenediide pincer complexes. | ||
Concerning the mechanism of cycloisomerization, first insights have been obtained experimentally.7 The active participation of the SCS pincer ligand to the activation of substrate has been substantiated (non-innocent character by protonation of electron-rich indenediide backbone). In addition, analysis of the stereochemical outcome of the reaction (with internal alkynes or via D-labeling of terminal alkynes) indicated the exclusive formation of Z products, in line with anti addition of the carboxylic acid/amide to the Pd-coordinated alkyne. But the exact ways the substrate is activated and the cyclization proceeds are still to be determined.
These two aspects (precise understanding of the mode of action of the Pd indenediide pincer complexes and further catalytic improvement) prompted us to perform detailed investigations combining theory and experiment. Here, we report DFT calculations and kinetic studies that shed light onto the mechanism of the cycloisomerization. The reaction is proposed to involve a second molecule of alkynoic acid acting as a proton shuttle, and this paved the way for catalytic enhancement. A broad variety of H-bond donor additives have been screened and catechol derivatives were found to improve very significantly both activity and selectivity. The combination of Pd-ligand cooperative catalysis and proton shuttling was successfully applied to the cycloisomerization of internal 4- and 5-alkynoic acids.
Results and discussion
DFT studies
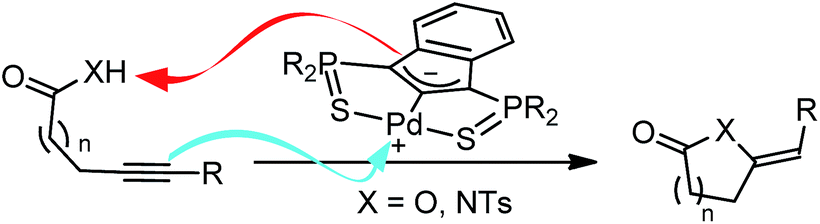
The mode of action of the Pd indenediide pincer complex in the cycloisomerization of alkynoic acids was first investigated computationally using DFT theory (B3PW91 functional).9 Calculations were performed on the real Pd system without simplification (phenyl groups were introduced at the P atoms, as in the first reported complexes7a). A mononuclear T-shape SCS complex with a vacant coordination site was considered as starting species since the nature of the Pd pre-catalyst (co-ligand at Pd and mono vs. polynuclear character) was found to only marginally influence catalytic performance.7The reaction profile for the cyclization of 4-pentynoic acid involving one molecule of substrate per Pd center was explored first (Fig. 1). The “active catalyst” Cat-1 is formed by side-on coordination of the alkyne to Pd. The carboxylic acid is readily activated (with a barrier of only 12.2 kcal mol−1) and the proton is transferred to the indene backbone. Cyclization then occurs by nucleophilic attack of one oxygen atom to the alkyne affording Int-1B (the corresponding transition state TS1B lies 24.2 kcal mol−1 above Cat-1). The catalytic cycle is closed by protonolysis of the vinyl-Pd species (back transfer of the proton from the indene backbone viaTS1C) and substrate/product exchange at Pd. This reaction profile follows the mechanistic scenario commonly proposed to account for the metal-catalyzed cycloisomerization of alkynoic acids,10 but two inconsistencies makes it very unlikely. First, the cyclization occurs by syn addition to the π-coordinated alkyne (O attacks cis to Pd) while anti addition is observed experimentally (as shown by D-labeling experiments).11 Second, the activation barrier for the proton back-transfer leading to Int-1D (54.4 kcal mol−1 from Int-1B) is prohibitively high.
 | ||
| Fig. 1 Reaction profile (kcal mol−1) computed for the cyclization of 4-pentynoic acid involving one molecule of substrate per Pd center. The phenyl substituents on the P atoms are omitted for clarity. | ||
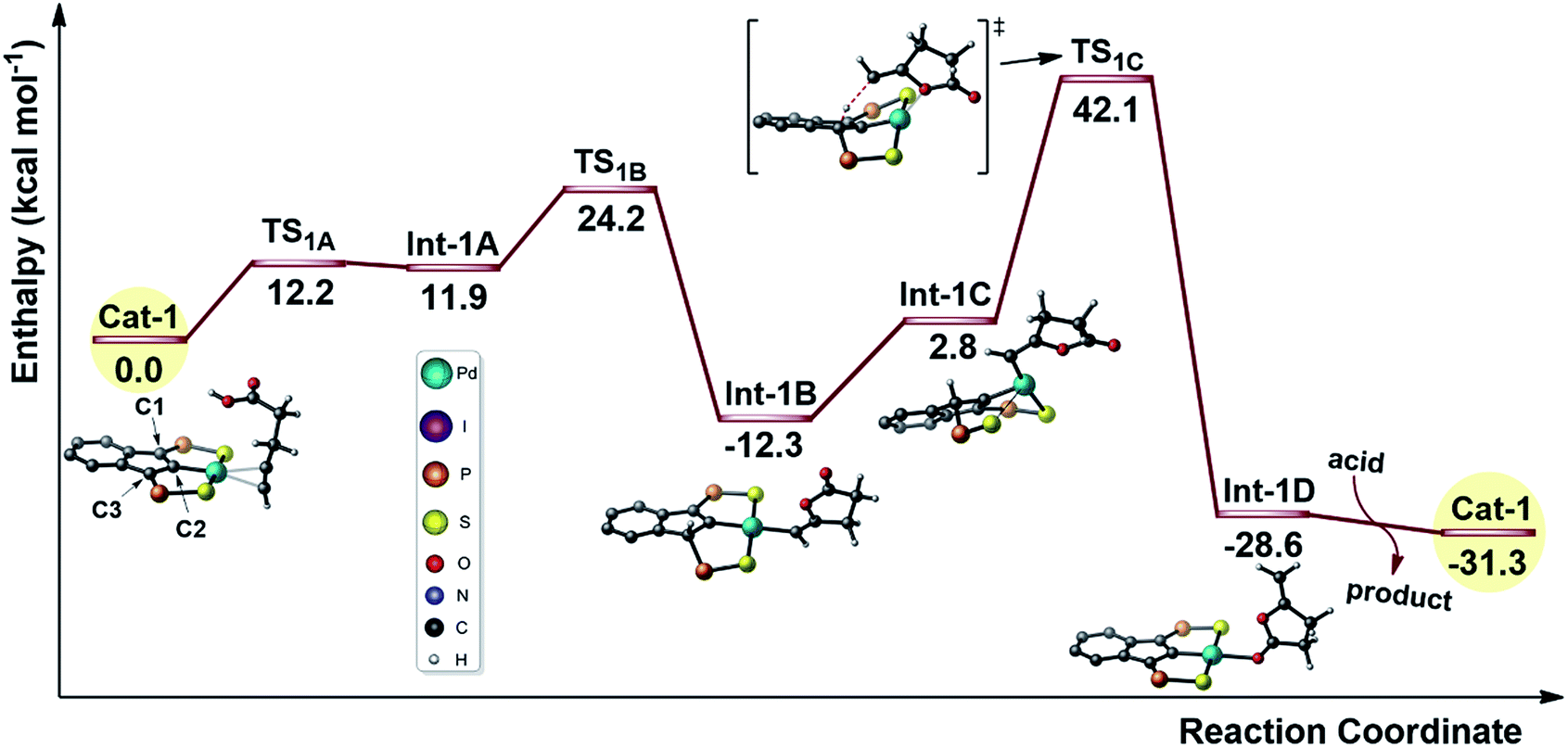
Given the results of the kinetic studies (vide infra), we envisioned to refine our model by involving a second molecule of alkynoic acid in the cycloisomerization process. Interaction of Cat-1 with 4-pentynoic acid is downhill in energy and leads to Cat-2 (Fig. 2). The alkyne bonded to Pd is oriented perpendicular to the coordination plane and its carboxylic acid moiety is engaged in H-bonding. The second molecule of substrate approaches perpendicularly and its acidic proton lies in close proximity to the indene backbone. Accordingly, activation of the acid moiety is very easy (activation barrier of only 5.1 kcal mol−1) and involves the “external” substrate as a proton shuttle between the π-bonded alkynoic acid and the electron-rich carbon atom C3 (transition state TS2A). The formation of Int-2A substantiates the non-innocent character of the indenediide backbone which is temporarily protonated. Cyclization then occurs by nucleophilic attack of one oxygen atom to the internal carbon atom of the alkyne (Ci). The reaction is directed by an “external” substrate molecule which remains H-bonded during the whole process. It involves the rotation of the alkyne and its slippage (in TS2B, it is essentially σ-bonded via the terminal carbon atom Ct). The cyclization requires important geometric changes, but it now proceeds via anti addition (in the resulting vinyl Pd complex Int-2B, the O atom is trans to Pd) and the corresponding activation barrier is accessible (27.8 kcal mol−1 from Cat-2). The external substrate molecule weakly interacts with the proton at C3 in Int-2B, and subsequently relays the protonolysis of the vinyl Pd species. The formation of the alkylidene lactone is strongly exothermic (Int-2C lies 27.5 kcal mol−1 below Cat-2), and thanks to this proton shuttling, the associated activation barrier is readily accessible (only 13.5 kcal mol−1). Product to substrate exchange at Pd closes the cycle and regenerates Cat-2.
 | ||
| Fig. 2 Reaction profile (kcal mol−1) computed for the cyclization of 4-pentynoic acid involving two molecules of substrate per Pd center. The phenyl substituents on the P atoms are omitted for clarity, the key carbon atoms of the indene backbone are labelled C1/C2/C3 and the carbons atoms of the alkynyl substrate are labelled Ct/Ci. | ||
Thus, involving two molecules of 4-pentynoic acid, it is possible to propose a catalytic cycle which is consistent with the experimental observations. The second molecule of substrate acts as a proton shuttle in the acid activation and in the protonolysis of the key vinyl Pd intermediate. It also directs the cyclization to anti addition.
Kinetic studies
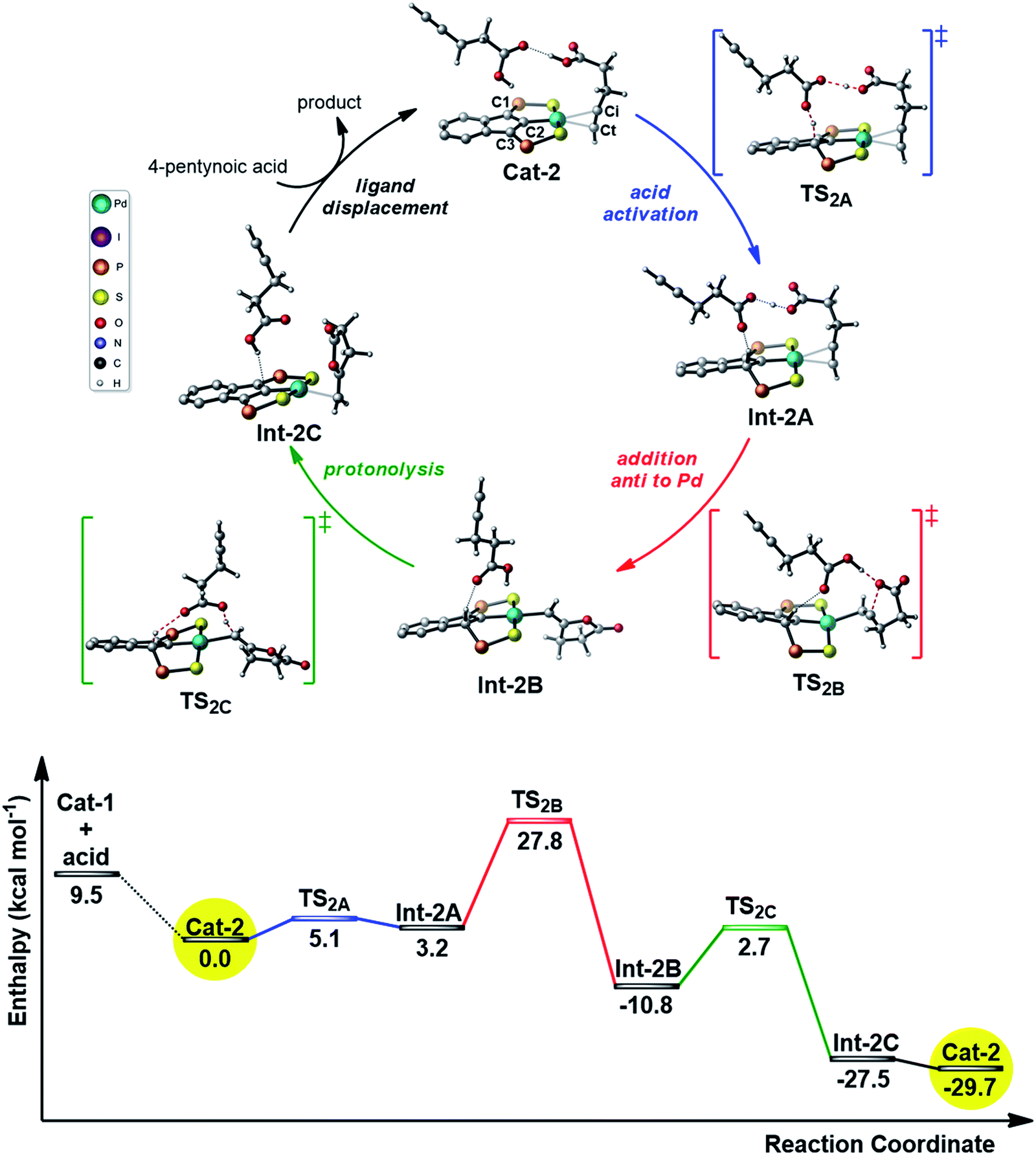
In parallel with the DFT calculations, kinetic studies were performed. The partial orders in palladium and substrate were determined using the initial rate method. The iPr-substituted palladium dimer I (Fig. 3) was chosen as pre-catalyst (rather than the Ph-substituted iodo/chloropalladate complexes or the corresponding neutral trimer) because of practical reasons (better solubility and stability, absence of ammonium counter-cations).7b The cyclization of 5-hexynoic acid 1a was selected as model reaction and it proceeded within hours at 90 °C in deuterated chloroform and could thus be conveniently monitored by NMR using a pressure tube. Under these conditions, the Pd precatalyst and substrate are fully soluble and no induction period is observed. 1H NMR monitoring shows the concomitant consumption of 1a and formation of methylene-valerolactone 2a, and no intermediate or side-product is detected. The initial rates were determined at different Pd loadings (3, 5, 7.5, 10 mol% in [Pd], Fig. S1†) and at different substrate concentrations (0.14–0.75 M, Fig. S4†). The initial rates of the cycloisomerization reaction were determined by plotting the evolution of the methylene-valerolactone 2a concentration versus reaction time at the various concentrations of catalyst or substrate (Fig. S2 and S5†). Each experiment was run in triplicate for a better accuracy and the averaged values were linearly fitted to equations ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) v0 = aln[Pd] + b (Fig. 3a) and lnv0 = cln[1a] + d (Fig. 3b) for palladium and 5-hexynoic acid variations, respectively. Accordingly, the rate law can be expressed as follows: v = k[Pd]1/2[Sub]3/2.
v0 = aln[Pd] + b (Fig. 3a) and lnv0 = cln[1a] + d (Fig. 3b) for palladium and 5-hexynoic acid variations, respectively. Accordingly, the rate law can be expressed as follows: v = k[Pd]1/2[Sub]3/2.
 | ||
| Fig. 3 Dependence of the initial rate of 5-hexynoic acid cyclization on the concentration of palladium (a) and substrate (b). The rates were averaged over three independent measurements. The error bars represent the standard deviation of the results from the three independent measurements. | ||
The partial order of 0.5 in [Pd] clearly indicates that the dinuclear complex I is not the active species. It is in fact the resting state of the precatalyst, which dissociates and coordinates the substrate to give the active species. The dinuclear form is favored at room temperature (the SCS pincer ligand is dissymmetric and two distinct signals are observed by 31P NMR), but tends to dissociate upon heating (the 31P NMR signals progressively broaden and coalesce at ca. 90 °C, Fig. S24†). Similar situations (square root order in [Pd] and off-cycle equilibrium of the active mononuclear species with an inactive dimeric form) have been encountered in Pd-catalyzed Heck couplings and C–H acetoxylation of benzene.12
The partial order of 1.5 in [1a] also deserves comment. It is unexpected and reveals that the reaction is not a simple unimolecular process, as may be anticipated for a cycloisomerization. The way the substrate reacts is apparently more complicated and in line with the reaction profile computed by DFT, and it is proposed that a second molecule of alkynoic acid is involved in the catalytic cycle. The partial order in [1a] determined experimentally at 90 °C is not 2 but 1.5, probably due to the tendency of carboxylic acids to form dimers in solution.13 This self-association phenomenon tends to reduce the partial order in substrate. It has been tracked by IR and a dimer to monomer ratio of 7.5 has been determined for 1a at 25 °C and 0.14 mol L−1.14
According to calculations, the second molecule of substrate participates via H-bonding and acts as a proton shuttle. We were thus intrigued about the possibility to use H-bond donors as additives in these cycloisomerization processes. These additive could substitute the “external” molecule of substrate, and optimizing the structure of the additive may afford the opportunity to enhance the catalytic performance of the Pd complex in terms of both activity and selectivity.
Screening of H-bond donor additives
The influence of additives was first investigated on 4-pentynoic acid 1b, since its cyclization is easier to perform (it proceeds within hours at rt with complex I). We started the screening with aliphatic and aromatic carboxylic acids 3a–d (30 mol%) (Scheme 1), but in all cases, the conversion of 1b was essentially unaffected (∼75% after 1 h, ∼86% after 2 h, Table S9†).15 Stronger acids such as methane sulfonic acid 3e and diphenylphosphoric acid 3f completely inhibited the reaction, probably due to the protonation of the indenediide backbone, giving inactive indenyl species. | ||
| Scheme 1 Additives having no impact (3a–d) or inhibiting (3e,f) the cyclization of 4-pentynoic acid 1b. | ||
Then, we turned our attention to alcohols, starting with catechol (4a) which has already been used as additive in organo-catalyzed aldol reactions.16 The addition of 30 mol% of 4a significantly speeds up the cycloisomerization of 1b (complete conversion required only 30 min instead of 5 h without additive). To confirm the impact of 4a, it was then tested on the cyclization of 5-hexynoic acid 1a (which is much less reactive than 1b), and again the reaction time was significantly reduced (from 10 h to 30 min to achieve complete conversion at 90 °C). These encouraging results prompted us to screen a broad variety of alcohols, diols and triols with aliphatic and aromatic skeletons. Overall, 24 compounds (4a–x) were tested on the cyclization of 5-hexynoic acid 1a. Selected results are displayed in Fig. 4 (see Fig. S26 and S27† for more details).
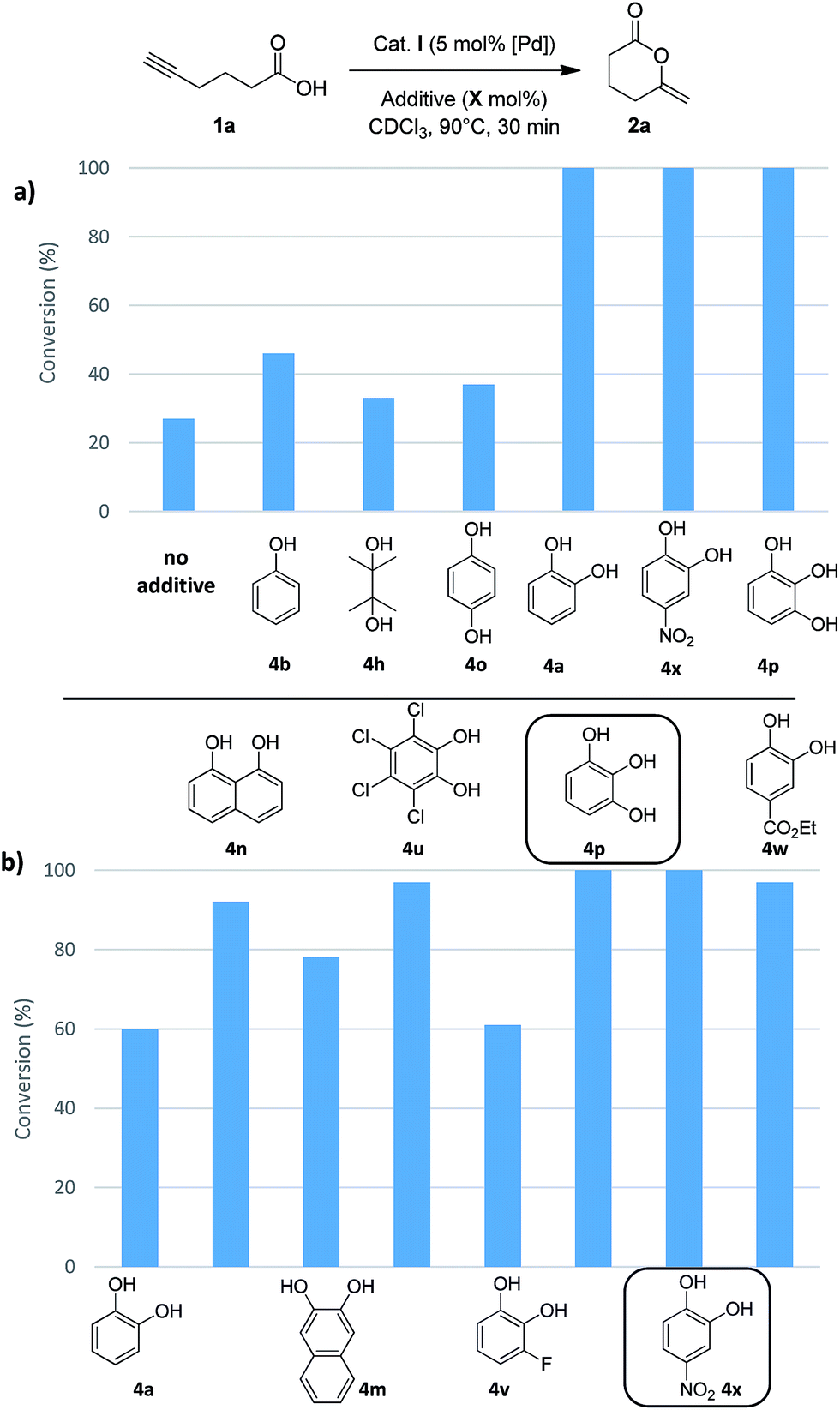 | ||
| Fig. 4 Impact of H-donor additives on the cyclization of 5-hexynoic acid 1a. The reaction is performed with 30 or 10 mol% of additives (a and b, respectively) and the conversion of 1a after 30 min is reported. | ||
Simple alcohols have little or no impact, but diols and triols were found to significantly improve the conversion, in particular the aromatic ones.17 After 30 min, the conversion of 1a is only of 27% when the Pd complex I is used alone, but it is complete when 8 additives are adjoined: catechol 4a, its derivatives substituted with electron-withdrawing groups (4u, 4v, 4w and 4x), 1,2,3-benzenetriol 4p, 1,8-dihydroxynaphtalene 4m and 2,3-dihydroxynaphthalene 4n. The most efficient additives are those featuring proximal hydroxyl groups, to ensure optimal proton transfer. This is particularly apparent when comparing hydroquinone 4o (which has only little effect) and catechol.18 The proton transfer may involve the two hydroxyl groups (proton shuttling via H-bonding, Scheme 2a) or only one (Scheme 2b, the other hydroxyl group can participate in adjacent H-bonding).
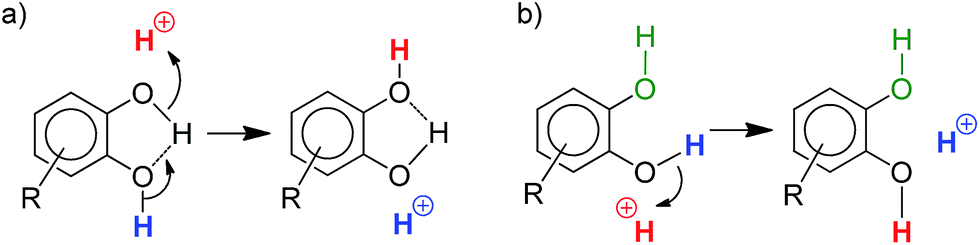 | ||
| Scheme 2 Schematic representation of two different modes of proton transfer with catechol derivatives. | ||
In order to discriminate the additives enabling complete conversion of 1a after 30 min, their loading was reduced, first to 10 mol% (Fig. 4b) and then to 5 mol% (Table S10†). Under these conditions, conversions of 90% or more were also achieved. The best results were obtained with 4-nitrocatechol 4x which gave full conversion of 1a within 30 min, even at 5 mol%. With this leading additive, we sought to optimize the reaction conditions (Table 1). Using 5 mol% of Pd and 5 mol% of 4x, the reaction temperature could be significantly decreased (entries 2 and 3): at 60 °C, 5-hexynoic acid 1a was fully consumed in 30 min, and even at 40 °C, full conversion was reached in only 1 h, while 10 h of reaction at 90 °C are needed without additive. The catalyst loading could also be significantly reduced. Using 5 mol% of 4-nitrocatechol 4x and only 1 mol% of Pd, 1a was completely cycloisomerized within 30 min at 90 °C (entry 4). Full conversion could also be achieved with only 0.2 mol% of Pd and 1 mol% of 4-nitrocatechol 4x after 36 h at 90 °C (entry 5).
| Entrya | Mol% Pd | Mol% additive | T (°C) | Time | Conversionb |
|---|---|---|---|---|---|
| a Catalytic reactions performed under argon using 0.1 mmol of 1a (0.14 M in CDCl3) and dimer I (5 mol% [Pd]). b Conversion were determined by 1H NMR analysis. | |||||
| 1 | 5 | 5 | 90 | 30 min | >99% |
| 2 | 5 | 5 | 60 | 30 min | >99% |
| 3 | 5 | 5 | 40 | 1 h | >99% |
| 4 | 1 | 5 | 90 | 40 min | >99% |
| 5 | 0.2 | 1 | 90 | 36 h | >99% |
The impact of the additive on the kinetics of the reaction, and in particular on the partial order in substrate, was then explored. Tetrachlorocatechol 4u was chosen as the additive for solubility issues, and its loading was varied (catechol to Pd ratio of 0.2, 1, 2 and 4). The reactions were carried out at 25 °C to ensure accurate NMR monitoring.9 A strong impact of the catechol was observed, with a significant decrease of the partial order in substrate (from 1.33 without additive, to 0.62 for a catechol to Pd ratio of 0.2, and even to 0.26 for a catechol to Pd ratio of 4). Self-association of the carboxylic acid as well as crossed association with the catechol probably comes into play here and it is not surprising that fractional orders below 1 are obtained.
In order to gain insight into the effect of the additive, the reaction profile for the cyclization of 4-pentynoic acid in the presence of catechol (chosen for symmetry reasons) was computed at the DFT level (Fig. 5).
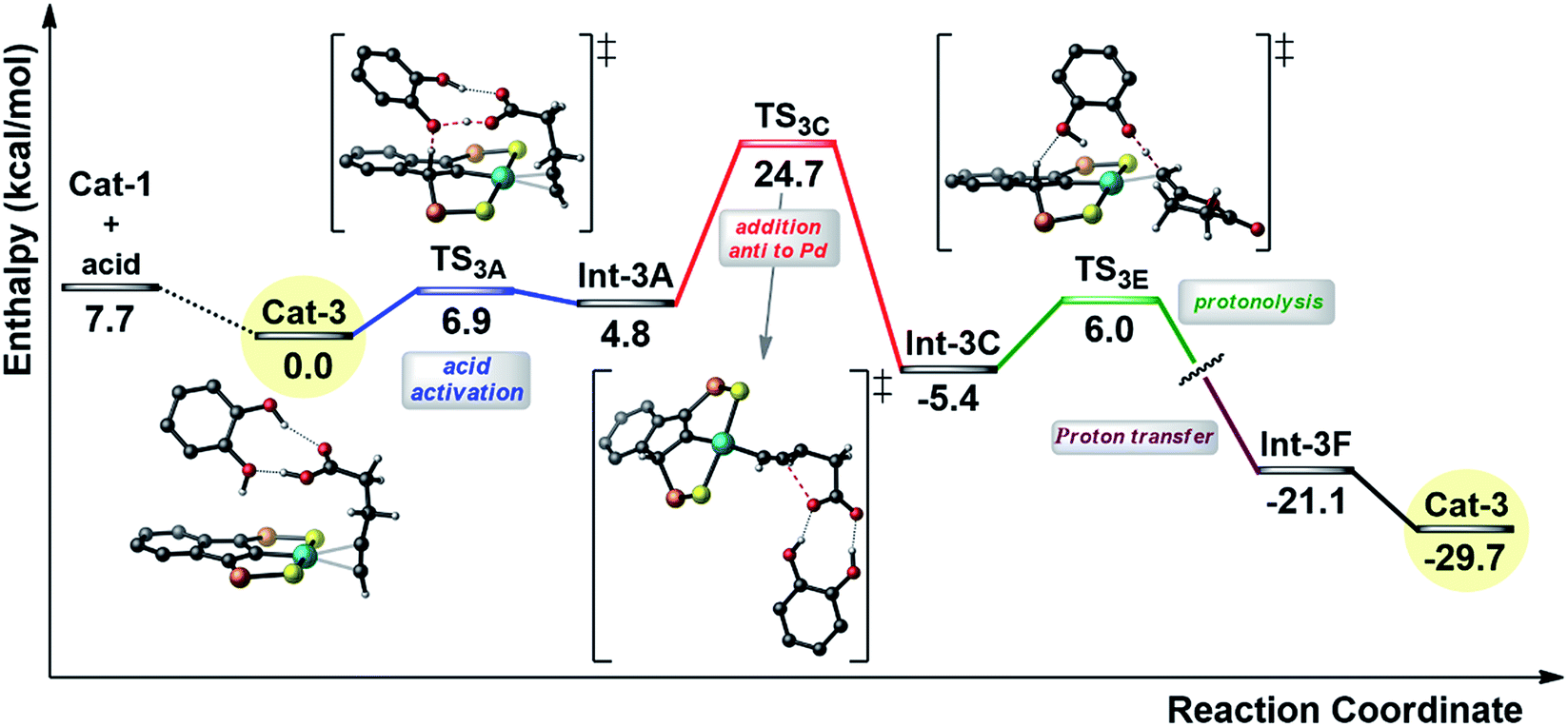 | ||
| Fig. 5 Simplified reaction profile (kcal mol−1) computed for the cyclization of 4-pentynoic acid in the presence of catechol. The phenyl substituents on the P atoms are omitted for clarity. | ||
As for the mechanism involving two carboxylic acid molecules, the first step corresponds to the activation of the carboxylic acid with protonation of the indenyl backbone (transition state TS3A). In this case, it is assisted by the catechol molecule. One hydroxyl group acts as a relay for the proton transfer (from the substrate to the indenyl) while the second hydroxyl is involved in hydrogen bonding with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the carboxylic acid (cf.Scheme 2b). Then, the cyclization occurs via anti-attack of the carboxylate, which is engaged in double hydrogen bonding with the catechol (TS3C). Finally, the proton borrowed by the indenyl backbone is transferred back and the alkylidene lactone is released. In this case, this is a two-step process, with protonation of the vinyl Pd species by catechol and then proton transfer from the indenyl to the catechol (see ESI†). The catechol acts here a proton shuttle (see Scheme 2a), in a similar way to the “external” molecule of carboxylic acid. Note that the rate-determining step is again the cyclization and that the corresponding activation barrier is about 3 kcal mol−1 lower in energy than that computed for the mechanism involving two carboxylic acid molecules, in line with the rate improvement observed experimentally.
O group of the carboxylic acid (cf.Scheme 2b). Then, the cyclization occurs via anti-attack of the carboxylate, which is engaged in double hydrogen bonding with the catechol (TS3C). Finally, the proton borrowed by the indenyl backbone is transferred back and the alkylidene lactone is released. In this case, this is a two-step process, with protonation of the vinyl Pd species by catechol and then proton transfer from the indenyl to the catechol (see ESI†). The catechol acts here a proton shuttle (see Scheme 2a), in a similar way to the “external” molecule of carboxylic acid. Note that the rate-determining step is again the cyclization and that the corresponding activation barrier is about 3 kcal mol−1 lower in energy than that computed for the mechanism involving two carboxylic acid molecules, in line with the rate improvement observed experimentally.
Cycloisomerization of internal alkynoic acids in the presence of catechol additives
The cyclization of alkynoic acids bearing internal alkynes is notoriously challenging. These substrates are much less reactive than terminal alkynoic acids, and exo/endo selectivity issues are frequently met.11,19,20 We were thus very eager to assess the influence of catechol additives in such difficult cycloisomerizations. Catalytic runs were performed at 90 °C with 5 mol% of Pd (dimer I) and 30 mol% of additive (either 4-nitrocatechol 4x or 1,2,3-benzenetriol 4p) on four representative substrates (Table 2).| Entrya | Alkynoic acid | Lactone | Additive | Time | Convb (%) | Exo/endo |
|---|---|---|---|---|---|---|
| a Catalytic reactions performed at 90 °C under argon atmosphere using 0.1 mmol of the corresponding alkynoic acid 1a–h (0.1 M in CDCl3) and dimer I (5 mol% [Pd]). b Conversions were determined by 1H NMR analysis. | ||||||
| 1 |
|
|
— | 1.5 h | 99 | 1/1.2 |
| 4x (30%) | 10 min | 99 | 1/5.3 | |||
| 4p (30%) | 10 min | 99 | 1/7.3 | |||
| 2 |

|
|
— | 28 h | 98 | 1.5/1 |
| 4x (30%) | 30 min | 99 | 1/1.5 | |||
| 4p (30%) | 30 min | 99 | 1/2.3 | |||
| 3 |
|
|
— | 5 h | 99 | 1/0 |
| 4x (5%) | 30 min | 99 | 1/0 | |||
| 4 |
|
|
— | 23 h | n.r. | 1/0 |
| 4x (30%) | 60 h | 99 | 1/0 | |||
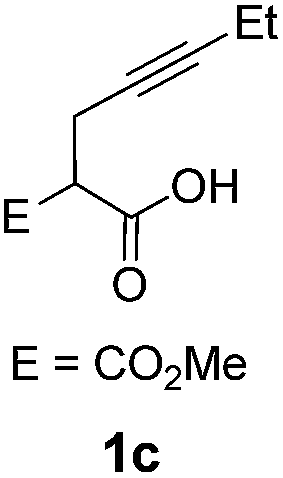
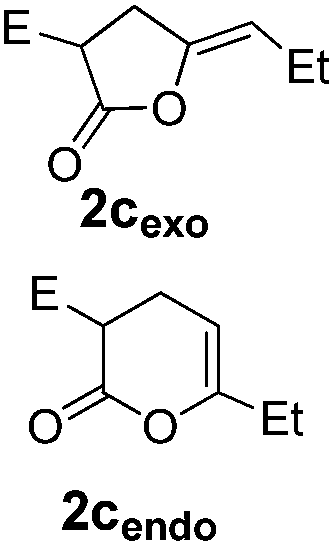
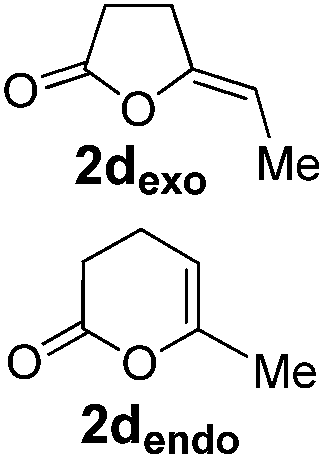
Pd complexes have rarely been used to catalyze the cycloisomerization of internal 4-alkynoic acids.19a,b Compounds 1c and 1d were tested and in both cases, the additives spectacularly shortened the reaction time (entries 1 and 2). The cyclization of 1c could be completed within 10 min (instead of 1.5 h) and that of 1d required only 30 min (instead of 28 h), representing speeds up by approximately 10 to 60 times. The additives also noticeably influenced the regioselectivity of the cyclization and favored the formation of 6-membered lactones (6-endo vs. 5-exo cyclization). Starting from 1c, a 1/1.2 mixture of 2cexo/2cendo products was obtained with Pd dimer I alone, while in the presence of the additives, 2cendo largely prevailed (the selectivity reached 1/7.3 with 4p). In the case of 4-hexynoic acid 1d, the catechol additives even switched the selectivity and the 6-endo cyclized product 2dendo could be obtained with a selectivity of up to 1/2.3 in the case of 1,2,3-benzenetriol 4p.
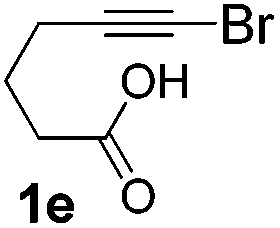
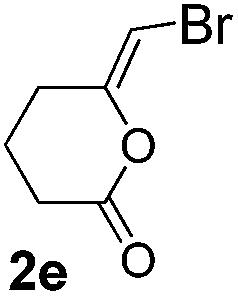
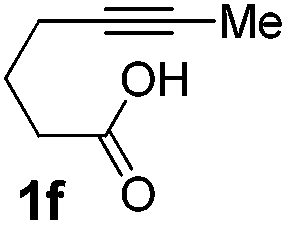
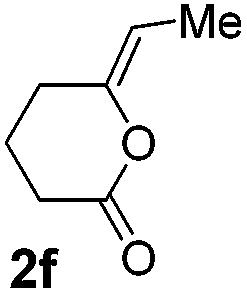
We then investigated the cycloisomerization of internal 5-alkynoic acids which has been scarcely achieved, using gold complexes.20 The Pd dimer I was previously shown to efficiently cycloisomerize 6-bromo-5-hexynoic acid 1e, a relatively activated substrate, and the corresponding 6-membered alkylidene-lactone 2e could be quantitatively obtained within 5 h (entry 3). In the presence of 4-nitrocatechol 4x, the reaction time was again drastically decreased. Full conversion was achieved in only 20 min using 5 mol% of the additive. Note that 2e is obtained exclusively in its Z form as the result of anti addition of the carboxylate to the Pd-coordinated alkyne. The catechol additive has an even more striking influence on the cyclization of 5-heptynoic acid 1f (entry 4). No reaction occurred with Pd dimer I alone, even after prolonged heating, but in the presence of 4-nitrocatechol (30 mol%), complete conversion was achieved within 60 h. Under these conditions, simple hydration of the alkyne tends to compete with the cyclosiomerization,9 but the targeted 6-membered alkylidene-lactone 2f was obtained in good yield (70%), again as a unique isomer (Z).
Conclusions
The precise mode of action of SCS indenediide Pd complexes in the cycloisomerization of alkynoic acids has been determined thanks to DFT and kinetic studies. Cooperation between the Pd center and the backbone of the pincer ligand has been confirmed computationally. In addition, the mechanistic study revealed the involvement of a second molecule of alkynoic acid all along the catalytic cycle: it acts as a proton shuttle in the activation of the acid and in the protonolysis of the intermediate vinyl Pd species, it also directs the nucleophilic attack of the carboxylic acid on the π-coordinated alkyne (anti addition). The possibility of replacing the second substrate molecule by an additive was investigated. A variety of H-bond donors were screened and polyols featuring proximal hydroxyl groups led to significant catalytic enhancement. Catechol derivatives substituted by electron-withdrawing groups proved highly beneficial on both activity and selectivity. The impact of 4-nitrocatechol 4x and 1,2,3-benzenetriol 4p on the cyclization of internal alkynoic acids is spectacular (endo/exo selectivities up to 7.3/1 and 60-fold increase in reactivity were achieved).This work also illustrates the interest to perform mechanistic studies on catalytic transformations. It enables us to understand the key factors influencing their efficiency and/or selectivity, and paves the way for further improvement. In this study, the combination of metal–ligand cooperation and proton shuttling by a second molecule of substrate has been pointed out. The ability of protic substrates, solvents or water molecules to act as proton shuttles in transition-metal catalyzed transformations has been occasionally proposed and supported by DFT calculations.21 We have shown here that this role can be transferred to an H-bond donor additive, and that optimizing the structure of this additive can significantly enhance the catalytic activity and selectivity. Proton shuttling is relatively well-established in organic synthesis and polymerization catalysis.22 We believe it is also highly relevant in transition-metal catalysis23 and can be applied to a variety of transformations.
Acknowledgements
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, and the Agence Nationale de la Recherche (ANR CE6-CYCLOOP) is acknowledged. The authors are grateful to CalMip (CNRS, Toulouse, France) for calculation facilities. L. M. thanks the Institut Universitaire de France.Notes and references
- (a) D. Lupp, N. J. Christensen and P. Fristrup, Dalton Trans., 2014, 43, 11093 RSC; (b) A. J. Nett, W. Zhao, P. M. Zimmerman and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 7636 CrossRef CAS PubMed.
- For selected references, see: (a) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef PubMed; (b) S. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 1617 CrossRef CAS PubMed; (c) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef PubMed; (d) P. M. Pérez Garcia, P. Ren, R. Scopelliti and X. Hu, ACS Catal., 2015, 5, 1164 CrossRef.
- G. C. Lloyd-Jones and M. P. Muñoz, J. Labelled Compd. Radiopharm., 2007, 50, 1072 CrossRef.
- For selected references, see : (a) O. R. Luca, J. D. Blakemore, S. J. Konezny, J. M. Praetorius, T. J. Schmeier, G. B. Hunsinger, V. S. Batista, G. W. Brudvig, N. Hazari and R. H. Crabtree, Inorg. Chem., 2012, 51, 8704 CrossRef PubMed; (b) T. Stahl, P. Hrobarik, C. D. Konigs, Y. Ohki, K. Tatsumi, S. Kemper, M. Kaupp, H. F. T. Klare and M. Oestreich, Chem. Sci., 2015, 6, 4324 RSC.
- For selected references, see: (a) A. C. Tsipis, Coord. Chem. Rev., 2014, 272, 1 CrossRef CAS; (b) H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895 CrossRef CAS; (c) G. J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y. D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed; (d) G. Jindal, H. K. Kisan and R. B. Sunoj, ACS Catal., 2014, 5, 480 CrossRef.
- (a) P. Oulié, N. Nebra, N. Saffon, L. Maron, B. Martin-Vaca and D. Bourissou, J. Am. Chem. Soc., 2009, 131, 3493 CrossRef PubMed; (b) N. Nebra, J. Lisena, N. Saffon, L. Maron, B. Martin-Vaca and D. Bourissou, Dalton Trans., 2011, 40, 8912 RSC; (c) P. Oulié, N. Nebra, S. Ladeira, B. Martin-Vaca and D. Bourissou, Organometallics, 2011, 30, 6416 CrossRef; (d) N. Nebra, N. Saffon, L. Maron, B. Martin−Vaca and D. Bourissou, Inorg. Chem., 2011, 50, 6378 CrossRef CAS; (e) N. Nebra, S. Ladeira, L. Maron, B. Martin-Vaca and D. Bourissou, Chem.–Eur. J., 2012, 18, 8474 CrossRef CAS PubMed; (f) J. Lisena, J. Monot, S. Mallet-Ladeira, B. Martin-Vaca and D. Bourissou, Organometallics, 2013, 32, 4301 CrossRef CAS.
- (a) N. Nebra, J. Monot, R. Shaw, B. Martin-Vaca and D. Bourissou, ACS Catal., 2013, 3, 2930 CrossRef CAS; (b) N. A. Espinosa-Jalapa, D. Ke, N. Nebra, L. Le Goanvic, S. Mallet-Ladeira, J. Monot, B. Martin-Vaca and D. Bourissou, ACS Catal., 2014, 4, 3605 CrossRef CAS.
- A. Scharf, I. Goldberg and A. Vigalok, J. Am. Chem. Soc., 2013, 135, 967 CrossRef CAS PubMed.
- See ESI† for details.
- (a) D. M. T. Chan, T. B. Marder, D. Milstein and N. J. Taylor, J. Am. Chem. Soc., 1987, 109, 6385 CrossRef CAS; (b) T. B. Marder, D. M. T. Chan, W. C. Fultz, J. C. Calabrese and D. Milstein, Chem. Commun., 1987, 1885 RSC.
- Cycloisomerization with syn-addition has been occasionally observed, see: S. Elgafi, D. Leslie, A. Barbara and A. Messerle, J. Organomet. Chem., 2000, 607, 97 CrossRef CAS.
- (a) G. P. F. v. Strijdonck, M. D. K. Boele, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 1999, 1999, 1073 CrossRef; (b) T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848 CrossRef CAS PubMed; (c) A. Sud, R. M. Deshpande and R. V. Chaudhari, J. Mol. Catal. A: Chem., 2007, 270, 144 CrossRef CAS; (d) A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef CAS PubMed.
- For a representative example see: F. Ragaini, M. Gasperini, S. Cenini, L. Arnera, A. Caselli, P. Macchi and N. Casati, Chem.–Eur. J., 2009, 15, 8064 CrossRef CAS PubMed.
- For sake of consistency, the partial order in 1a was also measured at 25 °C. It is even lower than at 90 °C (1.33 vs. 1.46), in line with an increase of the dimer to monomer ratio for entropic reasons.
- The progress of the reaction was followed by 1H NMR spectroscopy by integrating the resonance signal of the vinylic proton of the lactone 2b (δ = 4.30 ppm) and the acetylenic proton of the starting material (δ = 1.99 ppm).
- For catechol as additive, see: (a) C. Ji, Y. Peng, C. Huang, N. Wang, Z. Luo and Y. Jiang, J. Mol. Catal. A: Chem., 2006, 246, 136 CrossRef CAS; (b) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed; (c) J. Luo, R. Tan, Y. Kong, C. Li and D. Yin, Chin. J. Catal., 2012, 33, 1133 CrossRef CAS. For catechol derivatives as additives, see: (d) T. J. Peelen, Y. Chi and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 11598 CrossRef CAS PubMed; (e) M. J. Campbell and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 10370 CrossRef CAS PubMed; (f) C. M. Filloux, S. P. Lathrop and T. Rovis, Proc. Natl. Acad. Sci. Unit. States Am., 2010, 107, 20666 CrossRef CAS PubMed; (g) X. Chen, X. Fang and Y. R. Chi, Chem. Sci., 2013, 4, 2613 RSC.
- Control experiments with catechol 4a and 4-nitrocatechol 4x showed that no reaction occurs in the absence of palladium complex I.
- Similarly, 1,2,3,-benzenetriol 4p gave better results than 1,2,4- and 1,3,5-benzenetriols 4q and 4r.
- For Pd-catalyzed cycloisomerization of internal 4-alkynoic acids, see: (a) J. Garcia-Alvarez, J. Diez and C. Vidal, Green Chem., 2012, 14, 3190 RSC; (b) V. Sridharan, L. Fan, S. Takizawa, T. Suzuki and H. Sasai, Org. Biomol. Chem., 2013, 11, 5936 RSC; For cycloisomerization of internal 4-alkynoic acids catalyzed by other transition metals, see: (c) P. Pale and J. Chuche, Tetrahedron Lett., 1987, 28, 6447 CrossRef CAS; (d) T. Wakabayashi, Y. Ishii, K. Ishikawa and M. Hidai, Angew. Chem., Int. Ed. Engl., 1996, 35, 2123 CrossRef CAS; (e) I. Takei, Y. Wakebe, K. Suzuki, Y. Enta, T. Suzuki, Y. Mizobe and M. Hidai, Organometallics, 2003, 22, 4639 CrossRef CAS; (f) E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J. P. Genêt and V. Michelet, J. Am. Chem. Soc., 2006, 128, 3112 CrossRef CAS PubMed; (g) J. Aleman, V. del Solar and C. Navarro-Ranninger, Chem. Commun., 2010, 46, 454 RSC; (h) E. Tomas-Mendivil, P. Y. Toullec, J. Diez, S. Conejero, V. Michelet and V. Cadierno, Org. Lett., 2012, 14, 2520 CrossRef CAS PubMed; (i) E. Tomas-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet and V. Cadierno, ACS Catal., 2013, 3, 3086 CrossRef CAS; (j) L. C. Lee and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 5579 CrossRef CAS PubMed; (k) M. J. Rodriguez-Alvarez, C. Vidal, J. Diez and J. Garcia-Alvarez, Chem. Commun., 2014, 50, 12927 RSC; (l) X. Z. Shu, S. C. Nguyen, Y. He, F. Oba, Q. Zhang, C. Canlas, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7083 CrossRef CAS PubMed.
- For cycloisomerization of internal 5-alkynoic acids, see: (a) H. Harkat, J. M. Weibel and P. Pale, Tetrahedron Lett., 2006, 47, 6273 CrossRef CAS; (b) H. Harkat, A. Y. Dembelé, J. M. Weibel, A. Blanc and P. Pale, Tetrahedron, 2009, 65, 1871 CrossRef CAS.
- (a) F. Q. Shi, X. Li, Y. Xia, L. Zhang and Z. X. Yu, J. Am. Chem. Soc., 2007, 129, 15503 CrossRef CAS PubMed; (b) L. Bellarosa, J. Díez, J. Gimeno, A. Lledos, F. J. Suárez, G. Ujaque and C. Vicent, Chem.–Eur. J., 2012, 18, 7749 CrossRef CAS PubMed; (c) U. Gellrich, J. R. Khusnutdinova, G. M. Leitus and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4851 CrossRef CAS PubMed; (d) R. Yuan and Z. Lin, ACS Catal., 2015, 5, 2866 CrossRef CAS; (e) S. Qu, Y. Dang, C. Song, M. Wen, K. W. Huang and Z. X. Wang, J. Am. Chem. Soc., 2014, 136, 4974 CrossRef CAS PubMed.
- For an example in organic catalysis, see: (a) M. Dryzhakov, M. Hellal, E. Wolf, F. C. Falk and J. Moran, J. Am. Chem. Soc., 2015, 137, 9555 CrossRef CAS PubMed; For examples in polymerization, see: (b) M. Terada, Chem. Commun., 2008, 4097 RSC; (c) N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581 CrossRef CAS PubMed; (d) D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249 RSC.
- (a) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120 CrossRef CAS PubMed; (b) W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668 CAS; (c) B. R. Goldsmith, T. Hwang, S. Seritan, B. Peters and S. L. Scott, J. Am. Chem. Soc., 2015, 137, 9604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data; computational studies. See DOI: 10.1039/c5sc04232a |
| This journal is © The Royal Society of Chemistry 2016 |