
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical transfer in E. coli ribonucleotide reductase: a NH2Y731/R411A-α mutant unmasks a new conformation of the pathway residue 731†
Müge Kasanmascheff‡
 ab,
Wankyu Lee‡c,
Thomas U. Nicka,
JoAnne Stubbe*c and
Marina Bennati*ab
ab,
Wankyu Lee‡c,
Thomas U. Nicka,
JoAnne Stubbe*c and
Marina Bennati*ab
aMax Planck Institute for Biophysical Chemistry, 37077 Göttingen, Germany. E-mail: marina.bennati@mpibpc.mpg.de
bDepartment of Chemistry, University of Göttingen, 37077 Göttingen, Germany
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: stubbe@mit.edu
First published on 9th December 2015
Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of ribonucleotides to deoxyribonucleotides in all living organisms. The catalytic cycle of E. coli RNR involves a long-range proton-coupled electron transfer (PCET) from a tyrosyl radical (Y122˙) in subunit β2 to a cysteine (C439) in the active site of subunit α2, which subsequently initiates nucleotide reduction. This oxidation occurs over 35 Å and involves a specific pathway of redox active amino acids (Y122 ↔ [W48?] ↔ Y356 in β2 to Y731 ↔ Y730 ↔ C439 in α2). The mechanisms of the PCET steps at the interface of the α2β2 complex remain puzzling due to a lack of structural information for this region. Recently, DFT calculations on the 3-aminotyrosyl radical (NH2Y731˙)-α2 trapped by incubation of NH2Y731-α2/β2/CDP(substrate)/ATP(allosteric effector) suggested that R411-α2, a residue close to the α2β2 interface, interacts with NH2Y731˙ and accounts in part for its perturbed EPR parameters. To examine its role, we further modified NH2Y731-α2 with a R411A substitution. NH2Y731˙/R411A generated upon incubation of NH2Y731/R411A-α2/β2/CDP/ATP was investigated using multi-frequency (34, 94 and 263 GHz) EPR, 34 GHz pulsed electron–electron double resonance (PELDOR) and electron–nuclear double resonance (ENDOR) spectroscopies. The data indicate a large conformational change in NH2Y731˙/R411A relative to the NH2Y731˙ single mutant. Particularly, the inter-spin distance from NH2Y731˙/R411A in one αβ pair to Y122˙ in a second αβ pair decreases by 3 Å in the presence of the R411A mutation. This is the first experimental evidence for the flexibility of pathway residue Y731-α2 in an α2β2 complex and suggests a role for R411 in the stacked Y731/Y730 conformation involved in collinear PCET. Furthermore, NH2Y731˙/R411A serves as a probe of the PCET process across the subunit interface.
Introduction
Coupling of electron and proton transfers between donors and acceptors in proteins are ubiquitous in biology and can occur in a stepwise or concerted fashion. The concerted case avoids high energy intermediates and is designated as proton coupled electron transfer (PCET).1 The mechanisms of these couplings are fundamental to our understanding of photosynthesis, respiration, synthesis of DNA building blocks, and many other processes. Unresolved issues describing these mechanisms have been articulated in several recent comprehensive reviews, with different mechanisms dictated by transfer distances, protein environment and dynamics.2,3 When the proton and electron donor and acceptor are distinct, the mechanism involves orthogonal PCET; when the donor and acceptor are the same, it involves collinear PCET.4,5 A different mechanism in which a proton is transferred through water chains over long distances in concert with electron transfer (ET) has also been recently studied and discussed extensively in model systems.6,7 In all mechanistic cases, since the electrons and protons have very different masses, electrons tunnel over large distances (10–15 Å) while proton tunnelling is restricted to shorter distances, on the order of hydrogen bond lengths.1,8,9 This distance dependence complicates the issue of proton management. One important representative of the diversity of PCET mechanisms in proteins is found in the class I ribonucleotide reductases (RNRs). These enzymes catalyze the conversion of nucleotides to deoxynucleotides, the monomeric precursors required for DNA replication and repair in all eukaryotic and some prokaryotic organisms.10,11 In this paper, we use the Escherichia coli (E. coli) class Ia RNR as a model system to interrogate the PCET process across the interface of the two subunits of this enzyme, proposed to involve two redox active protein tyrosine residues, one on each subunit, and a water interface between the subunits.12The E. coli RNR consists of two homodimeric subunits, α2 and β2.13 The enzyme is active when a transient α2β2 complex is formed.14 α2 contains the active site for nucleotide reduction and two allosteric effector binding sites that regulate the specificity and the rate of reduction.15–19 β2 harbors the essential di-iron tyrosyl radical cofactor (FeIII2–Y122˙).20,21 During each turnover, Y122˙-β2 oxidizes C439-α2 to a thiyl radical, which subsequently initiates dNDP production.11 There are X-ray structures of the individual subunits, and a docking model of the α2β2 complex places Y122˙ at a distance of about 35 Å from C439.22,23 These initial studies led to the first formulation of radical transfer (RT) in RNR via a radical hopping mechanism involving a pathway of conserved amino acids (Y122 ↔ [W48?] ↔ Y356 in β2 to Y731 ↔ Y730 ↔ C439 in α2). Biochemical24 and biophysical (EPR,25,26 SAXS,27 and cryoEM28) studies confirmed that the docking model provides a reasonable representation of E. coli RNR in its transient, active form and led to a detailed mechanism of RT over such a long distance.4,14,24 Nevertheless, in wild-type (wt) E. coli RNR, the rate limiting step, conformational change(s) upon substrate and allosteric effector binding to α2, has prevented spectroscopic detection of any intermediates in this process.29 The recent development of methods to site-specifically incorporate tyrosine analogs with altered pKas and reduction potentials has permitted the detection of pathway radical intermediates30–32 and, combined with state-of-the-art EPR spectroscopy,12,26,33,34 has started to reveal the molecular basis of the long-range RT in RNR.14
These experiments have led to the current model illustrated in Fig. 1, which involves orthogonal PCET35 steps within subunit β2 and collinear PCET steps within the α2 subunit.12,33 However, the mechanism of the PCET process at the subunit interface between Y356 in β2 and Y731 in α2 remains elusive, as structural information on the C-terminal 35 amino acids of β2, including a putative proton acceptor E350 and Y356 (Fig. 1), is missing.22
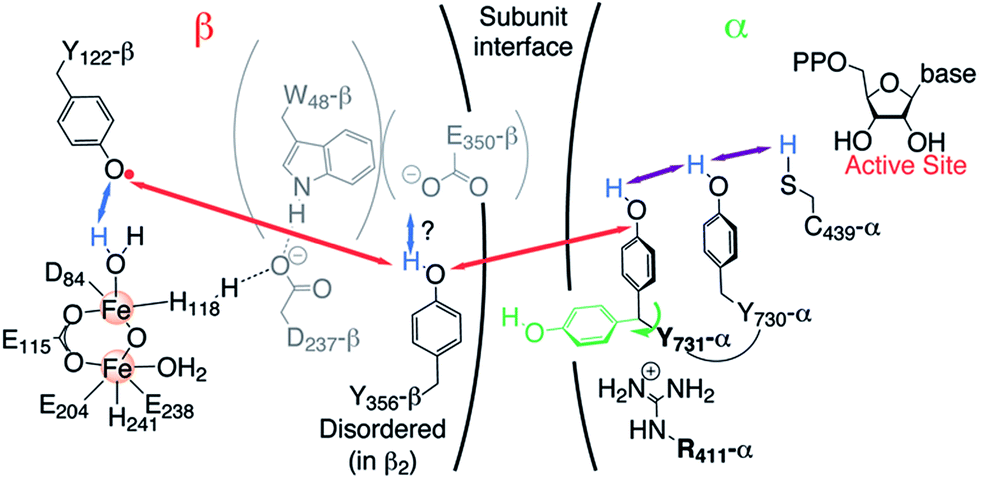 | ||
| Fig. 1 Current model of the long-range PCET in E. coli RNR. R411 is shown along with the redox active amino acid residues that are involved in this process (Y122 ↔ [W48?] ↔ Y356 in β2 ↔ Y731 ↔ Y730 ↔ C439 in α2). W48, D237 and E350 are shown in grey, because currently there is no evidence for their role in this process.14 The locations of Y356 and E350 are unknown, as they are within the flexible C-terminal tail (35 amino acids) of β2. Red, blue and purple arrows represent electron transfer, proton transfer and collinear PCET pathways, respectively. | ||
Our recent high-field (HF) EPR/ENDOR and DFT investigations using the 3-aminotyrosine mutants NH2Y730-α2 and NH2Y731-α2, which generate the corresponding NH2Y˙ upon incubation with β2, CDP (substrate) and ATP (allosteric effector), established that an unusual stacked conformation of residues 730 and 731, observed in some X-ray structures of α2 (ref. 23 and 36) (see ESI, Fig. S1†), occurs in the α2β2 complex.12,33 However, the X-ray structure of NH2Y730-α2 (PDB 2XO4) alone exhibited multiple conformations for Y731-α2, with one rotated away from NH2Y730-α2 toward the α2β2 subunit interface.30 This “flipped” conformation was accompanied by reorientations of R411 and N733 in α2. Further comparison of NH2Y730˙-α2, NH2Y731˙-α2 and NH2Y356˙-β2 by HF EPR indicated that the electrostatic environment of all three transient NH2Y˙s is strongly perturbed and that their hydrogen bond interactions are intrinsically different.12,33 Interestingly, one of our DFT models of the protein environment for NH2Y731˙-α2 required R411-α2 to explain the perturbed gx value observed and suggested that R411-α2 approaches to NH2Y731˙-α2 within 2.6 Å (Fig. S1†).12 Therefore, to examine the role of R411-α2 during the PCET process in E. coli RNR, we generated two mutants: R411A-α2 and the double mutant NH2Y731/R411A-α2. Here, we report the incubation of NH2Y731/R411A-α2 with β2/CDP and ATP, which generates the NH2Y731˙/R411A-α2β2 complex. Using advanced EPR methods, including 263 GHz pulse EPR and 34 GHz PELDOR/DEER (pulsed electron–electron double resonance) and ENDOR (electron–nuclear double resonance) spectroscopies, we have provided evidence for a new conformation of NH2Y731˙/R411 that is “flipped” towards the subunit interface in the α2β2 complex. This is the first time an alternative conformation of any pathway tyrosine (NH2Y731˙) has been observed and it provides a new probe of the PCET mechanism across the subunit interface, which remains unknown.
Experimental
Materials
4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes) was purchased from EMD Bioscience. Adenosine-5′-triphosphate (ATP), cytidine-5′-diphosphate (CDP), reduced β-nicotinamide adenine dinucleotide phosphate (NADPH), hydroxyurea (HU), kanamycin (Km), chloramphenicol (Cm), 2XYT media, M9 Minimal Salts, L-arabinose (ara), β-mercaptoethanol (β-ME), streptomycin sulfate and NH2Y were purchased from Sigma-Aldrich. Isopropyl-β-D-thiogalactopyranoside (IPTG) and 1,4-dithiothreitol (DTT) were purchased from Promega. Tris(2-carboxyethyl)phosphine (TCEP) hydrochloride was purchased from Thermo Scientific. Nucleotide primers were purchased from Invitrogen, and Pfu Ultra II polymerase was purchased from Stratagene.Site-directed mutagenesis to generate R411A-α2 and NH2Y731/R411A-α2
The Quikchange kit (Stratagene) was used to generate each mutant according to the manufacturer's protocol. The templates pET28a-nrdA and pET28a-nrdA Y731Z30 were amplified with primer 5′-G CAG GAA CGT GCG TCT ACC GGT![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ATC TAT ATT CAG AAC GTT GAC-3′ and its reverse complement and used to insert a GCG (Ala) at position 411. The sequences were confirmed by QuintaraBio Laboratory. All constructs contain an N-terminal (His)6-tag with a 10 amino acid linker.30
ATC TAT ATT CAG AAC GTT GAC-3′ and its reverse complement and used to insert a GCG (Ala) at position 411. The sequences were confirmed by QuintaraBio Laboratory. All constructs contain an N-terminal (His)6-tag with a 10 amino acid linker.30
Expression, purification and activity assays of R411A-α2 and NH2Y731/R411A-α2
(His)6-wt-α2 (2750 nmol min−1 mg−1) and wt-β2 (7000 nmol min−1 mg−1), and 1.2 Y˙/β2 were expressed and purified by standard protocols.30,37,38 All α2 mutants were pre-reduced with 30 mM DTT and 15 mM HU before use.29E. coli thioredoxin (TR, 40 U mg−1) and thioredoxin reductase (TRR, 1800 U mg−1) used in assays were isolated as previously described.39,40 (His)6-NH2Y731-α2 was purified as previously described.30 Expression and purification of R411A-α2 and NH2Y731/R411A-α2 followed previous protocols,30 except that the purification buffer (50 mM Tris, 5% glycerol, 1 mM PMSF, pH 7.6) for NH2Y731/R411A-α2 contained 1 mM TCEP. The yields of purified R411A-α2 and NH2Y731/R411A-α2 were 10–12 mg g−1 and 6–7 mg g−1 cell paste, respectively. The activity of R411A-α2 (0.2 μM) and NH2Y731/R411A-α2 (1 μM) was determined in the presence of 50-fold excess of wt-β2 with 3 mM ATP, 1 mM [3H]-CDP (4850 cpm nmol−1), 30 μM TR, 0.5 μM TRR, and 1 mM NADPH in assay buffer (50 mM HEPES, 1 mM EDTA, 15 mM MgSO4, pH 7.6). The amount of dCDP was determined by the method of Steeper and Steuart.41 For single turnover experiments, NH2Y731/R411A-α2 (5 μM) was incubated with wt-β2 (5 μM), 3 mM ATP, and 1 mM [3H]-CDP (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cpm nmol−1) in assay buffer. The dissociation constant (Kd) for R411A-α2 and wt-β2 was determined in H2O and D2O buffers by the competitive inhibition assay42 (SI-2, Fig. S2†).
000 cpm nmol−1) in assay buffer. The dissociation constant (Kd) for R411A-α2 and wt-β2 was determined in H2O and D2O buffers by the competitive inhibition assay42 (SI-2, Fig. S2†).
Samples for HF EPR and PELDOR spectroscopy
NH2Y731/R411A-α2 and wt-β2 were mixed 1:1 to a final concentration of 160–180 μM in D2O assay buffer as previously described.32,34 These protein concentrations resulted in >95% binding between subunits. The reaction was initiated at room temperature by adding CDP and ATP to final concentrations of 1 and 3 mM, respectively. The reactions were manually freeze-quenched in liquid N2 within 10–23 s. The PELDOR sample was prepared by adding glycerol-(OD)3 to a final concentration of 10% (v/v) 16 s after the initiation of the reaction. This reaction was manually freeze-quenched after 56 s as just described. The NH2Y731˙ accounted for 30–33% of the total spin for all the samples used in this work, which was similar to the yields reported previously.30,32
HF pulsed EPR spectroscopy
Echo-detected (ESE: π/2 – τ – π – echo) EPR spectra at 263 GHz were recorded on a Bruker Elexsys E780 quasi optical spectrometer using a single mode (TE011) cylindrical resonator (E9501610 – Bruker BioSpin) with a typical quality factor of 500–1000. The maximum microwave power coupled to the resonator was about 15 mW. Samples for 263 GHz EPR were inserted in capillaries (0.33 mm OD, Vitrocom CV2033S) with typical volumes of ca. 50 nL. 94 GHz ESE spectra were recorded on a Bruker E680 spectrometer with a 400 mW W-band power setup (Bruker power upgrade – 2). Samples for 94 GHz ESE contained typical volumes of 2 μL in 0.84 mm OD capillaries (Wilmad S6X84). All manually freeze-quenched samples were immersed in liquid N2 and loaded into pre-cooled EPR cryostats.34 GHz PELDOR spectroscopy
34 GHz ESE and PELDOR spectra were recorded on a Bruker E580 X/Q-band spectrometer equipped with a Bruker EN 5107D2 pulse EPR/ENDOR resonator. The spectrometer was power-upgraded with a Q-band TWT amplifier, providing about 170 W output power at 34.1 GHz. PELDOR experiments were recorded with an overcoupled resonator. The center of the mode was chosen for the pump frequency for measurements at 20 K. However, for measurements at 50 K the detection frequency was set in the center of the cavity mode to enhance detection sensitivity. Q-band samples contained typical volumes of 10 μL in 1.6 mm OD capillaries (Wilmad 222T-RB).Processing and simulation of EPR spectra
Spectra were processed by phasing and baseline correction. Derivatives of the absorption spectra were obtained by fitting every four points with a second order polynomial and differentiating the function in MATLAB_R2014b.43 EPR spectra were simulated using the EasySpin-4.5.5 “pepper”-routine which was run in MATLAB.44DFT calculations
DFT calculations were performed with the ORCA 3.0.0 program package.45 The geometry optimization of the neutral NH2Y˙ was performed using the unrestricted B3LYP46–48 hybrid density functional in combination with the def2-TZVPP basis set and def2-TZVPP/JK auxiliary basis set.49,50 To take into account the electrostatic environment of the radical intermediate at the protein interface, a solvation model (COSMO51) with the polarity of ethanol (ε = 24) was used. Otherwise, Grimme's dispersion correction52,53 and RIJCOSX54 approximations were employed. The energy converged to 10−9 Eh. The hyperfine couplings and g values were calculated using NH2Y˙-C4 as the gauge origin.55,56 The def2-TZVPP basis set was consistent with the geometry optimization step.50 The C2–C1–Cβ–Cα dihedral angle of the NH2Y˙ was changed stepwise with a geometry optimization for each step. The xyz coordinates for one of the optimized models are given in the ESI.†PyMOL models
The docking model refers to the α2β2 complex structure generated from the individual wt-α2 and wt-β2 X-ray structures.22,23 In order to predict distances, the mutant E. coli RNR structure (PDB 2XO4)30 was overlaid with the wt-α2 structure in the docking model23 using PyMOL, which first performs a sequence alignment and then aligns the structures to minimize the root mean square deviation between the structures.Results and discussion
Preparation and characterization of R411A-α2, NH2Y731/R411A-α2 and ND2Y731˙/R411A-α
Our recent studies on NH2Y731-α2 (ref. 12) suggested that R411 might interact with NH2Y731˙, partially accounting for the measured EPR and ENDOR parameters. To investigate this proposal, R411A-α2 was generated and characterized. Because the mutation is proposed to be at the interface of α2 and β2, the dissociation constant (Kd) for subunit interactions was also examined and was determined to be 0.94 ± 0.33 μM (Fig. S2A†), ∼5 fold higher than that for wt-α2 (0.18 μM).42 Under these conditions, this mutant was shown to have a specific activity of 467 ± 22 nmol min−1 mg−1, 17% of that of the wt enzyme (2750 nmol min−1 mg−1). The reduced activity and weaker subunit binding suggest that R411 plays a functional role.Furthermore, we characterized the role of R411 in the oxidation of Y731-α2 by generating the double mutant NH2Y731/R411A-α2. The Kd for subunit interactions between NH2Y731/R411A-α2 and wt-β2 was determined to be 8 ± 1 nM (Fig. S2C†), which is consistent with the formation of a tight complex when a NH2Y˙ is generated.28 Its specific activity was 13 ± 3 nmol min−1 mg−1, 0.4% of the specific activity of wt-RNR and in the range of contaminating wt-α2 activity.32 A more sensitive, one turnover assay was then employed to determine if this double mutant could generate any dCDP. When pre-reduced NH2Y731/R411A-α2 was mixed with wt-β2, CDP, and ATP for 5 min, only 0.036 ± 0.018 dCDP/α2 was observed, consistent with contaminating wt-α2. Thus, the double mutant is unable to make detectable dCDP, which is not unexpected, given the specific activities of the R411A and the NH2Y731-α2 mutants (see also SI-3 and Fig. S3†).
We next investigated whether NH2Y731˙ could be generated by NH2Y731/R411-α2, despite its inability to make dCDPs. NH2Y731/R411A-α2, wt-β2, CDP and ATP were studied by stopped-flow (SF) spectroscopy and the reaction was monitored at 320 nm, the absorption feature associated with the NH2Y˙ (Fig. S4,† red). The data were split into two time domains: 5 ms to 6 s and 25 s to 100 s. In the first time domain, NH2Y731˙ formation was fit to a double exponential with kfast of 3.6 ± 0.5 s−1 (amplitude 8%) and kslow of 0.47 ± 0.03 s−1 (amplitude 21%) (Table S1†). The rate constants for NH2Y731˙ in the single mutant control were similar: kfast of 9.6 ± 0.6 s−1 and kslow of 0.8 ± 0.1 s−1. However, in this case, the fast phase accounted for 27% and the slow phase accounted for 13% of the NH2Y731˙. The biphasic kinetics of NH2Y731˙ formation in both cases is attributed to multiple conformations that give rise to NH2Y731˙.32 From 25 s to 100 s, NH2Y731˙ in the double mutant reaction disappeared with a kobs of 0.02 ± 0.003 s−1, while with the single mutant, disappearance occurred with a kobs of 0.005 ± 0.002 s−1. Analysis of the Y122˙-β disappearance kinetics was unsuccessful at early time points due to the detection limits, as described in SI-4.
Given the distinct kinetics of our double mutant relative to the NH2Y731-α2, the 9 GHz EPR spectrum of the sample generated from the reaction of NH2Y731/R411A-α2 with wt-β2, ATP, and CDP quenched after 25 s was recorded and is shown in Fig. S5A and C.† Subsequent to subtraction of Y122˙, 32% of the total spin is associated with NH2Y731˙/R411A-α2 with no spin loss. This result is similar to that of the single mutant, NH2Y731˙.30,32 A comparison of their spectra, as shown in Fig. S5B,† revealed substantial differences in their hyperfine interactions, suggesting that further characterization of this radical might provide insight into the function of R411. Therefore, the role of R411 in the RT pathway was further studied with advanced EPR spectroscopy.
HF EPR of ND2Y731˙/R411A-α2
To examine the generated ND2Y731˙/R411A-α2, we took advantage of the proximity of Y122˙ to the di-iron cluster and its altered relaxation properties. Pulsed EPR spectra of ND2Y731˙/R411A-α2 at 34, 94 and 263 GHz were recorded in D2O buffer at 70 K and are shown in Fig. 2A. The use of D2O considerably simplifies the EPR spectra due to the absence of 1H hyperfine (hf) splittings arising from the amino protons. The ND2Y731˙/R411A-α2 EPR spectrum at 34 GHz is mainly dominated by the large hf couplings with the deuterons of the amino group and the two Cβ-methylene protons.34 On the other hand, the 94 and 263 GHz EPR spectra are dominated by g-anisotropy, and the relative contributions of g- and hf-anisotropy are strongly dependent on the operating magnetic field. The g values of ND2Y731˙/R411A-α2 are best resolved at 263 GHz and are consistent with the values from our previous ND2Y˙ studies.12,33 The 94 GHz spectra reveal differences in the hf splitting of the Cβ-methylene protons (Fig. 2A, marked with an arrow): the large hf splitting of the Cβ-methylene proton visible in the central line of ND2Y731˙-α2 (red) is missing in ND2Y731˙/R411A-α2 (black). This splitting is also absent in the 263 GHz spectrum. The EPR spectra were simulated iteratively to find a global solution for the contributing hf couplings. All of the EPR data and simulations, in which the previously reported34 hf coupling for 14N is used, are consistent with the NH2Y731˙ generated in the NH2Y731˙/R411A-α2/β2 complex being a single, well-oriented radical species with one set of magnetic parameters, which are listed in Table 1 (see also Fig. S7†). This finding is not self-evident, as our previous experiments with other double mutants, NH2Y731˙/Y730F-α2 and NH2Y730˙/C439A-α2, showed distributions in g values indicative of multiple radical environments and/or molecular orientations.12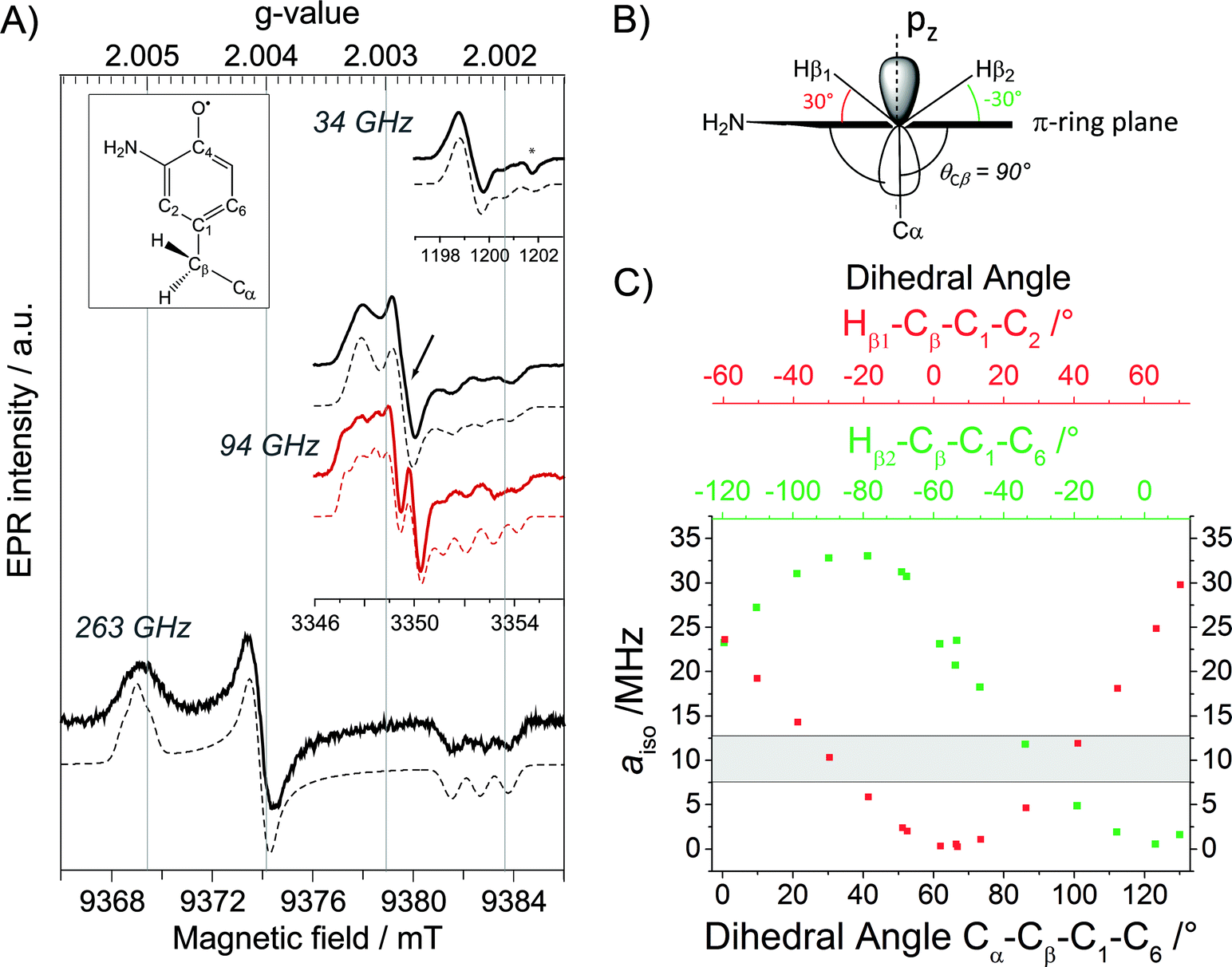 | ||
| Fig. 2 (A) Derivative EPR spectra (solid black lines) at 34 GHz (top), 94 GHz (middle) and 263 GHz (bottom) of ND2Y731˙/R411A-α2 with the corresponding simulations (dashed black lines). The 94 GHz EPR spectrum of ND2Y731˙-α2 in the single mutant (solid red line) with the corresponding simulation (dashed red line) is shown for comparison. The difference between the two spectra around gy is marked with an arrow. The glass signal is marked with an asterisk. Exp. conditions (34 GHz): π/2 = 6 ns, τ = 280 ns, shot repetition time = 6 ms, shots/point = 80, number of scans = 10; (94 GHz): π/2 = 30 ns, τ = 280 ns, shot repetition time = 5 ms, shots/point = 100, number of scans = 50–100; (263 GHz): π/2 = 40 ns, τ = 270 ns, shot repetition time = 6 ms, shots/point = 250, number of scans = 36. Structure of NH2Y˙ is shown in the inset. (B) Orientation of the Cβ-methylene protons with respect to the phenol ring, as extracted from the observed hyperfine couplings. (C) aiso as a function of the dihedral angle for each Cβ-methylene proton, calculated from a DFT model for NH2Y˙ (Fig. S6†). | ||
| Sample | g values | a iso (MHz) | ||||
|---|---|---|---|---|---|---|
| g x | g y | g z | Hβ1 | Hβ2 | 14N | |
| a Uncertainties in the g values and hf couplings are about 0.00005 and up to 10%, respectively, as obtained from the spectral simulations. b g values and hf couplings were reported in ref. 12 and 34, respectively. | ||||||
| ND2Y731˙/R411A-α | 2.0051 | 2.0041 | 2.0022 | 10 | 10 | 12 |
| ND2Y731˙-αb | 2.0051 | 2.0041 | 2.0022 | 22 | 9 | 12 |
Interestingly, we do not observe changes in the g values between ND2Y731˙/R411A-α and ND2Y731˙-α2. This is unexpected because the gx value is affected by the electrostatic environment of a radical,57 and the R411A mutation has changed the local environment of ND2Y731˙, as demonstrated by the substantial changes in the Cβ-methylene 1H couplings (Table 1). These couplings are related to the dihedral angle θCβ between the Cβ–H bond and the pz orbital axis of C1 (Fig. 2B), and therefore provide information on the molecular orientation of the tyrosyl and 3-aminotyrosyl radicals.34 The dihedral angle can be extracted from the McConnell equation (aiso(Cβ–H) = B1 × ρC1 × cos2θCβ),58 which provides a semi-empirical relationship for the observed isotropic constant aiso. The C2–C1–Cβ–Cα angle of ND2Y731˙/R411A-α2 is estimated to be ≈90° by using B1 of 162 MHz (ref. 59) for tyrosyl radicals, an electron spin density ρC1 of 0.214,12 and an isotropic Cβ-methylene proton hf coupling aiso = 10 ± 1 MHz (Table 1). This dihedral angle is indeed consistent with the hf couplings of the two Cβ-methylene 1H resonances being indistinguishable, as reported in Table 1 and seen in Fig. 2B and C. This result was confirmed by DFT calculations on the observed hf couplings of NH2Y˙, in which the ring orientation was modeled with respect to the backbone and showed a symmetric orientation relative to the pz orbital axis of C1 (Fig. 2B). In this calculation, a θCβ angle of 90° corresponds to aiso = 9 ± 3 MHz (grey area in Fig. 2C) for both Cβ-methylene protons, Hβ2/1.
ENDOR for detection of hydrogen bonds to ND2Y731˙/R411A-α2
Given that the R411A mutation had little effect on gx, 2H ENDOR spectroscopy was used to further examine a possible correlation of the observed gx value (gx = 2.0051) with the hydrogen bonding environment. Fig. 3 illustrates the 2H Mims ENDOR spectra of ND2Y731˙-α2 and ND2Y731˙/R411A-α2. Both spectra contain a broad signal that extends over ±2 MHz, arising from the strongly coupled amino deuterons, which is a common feature of ND2Y˙ Mims ENDOR spectra.12,33 However, we observe that the 2H hf tensor previously assigned to the moderately strong hydrogen bond between Y730 and Y731 in ND2Y731˙-α2, which is almost perpendicular to the tyrosine ring plane,12 is absent in the ND2Y731˙/R411A-α2 spectrum. Therefore, the hydrogen bonding environment of NH2Y731˙/R411A-α2 is distinct from that of the single mutant, consistent with the different side chain conformations observed by HF EPR spectroscopy. Note that almost the complete EPR line of ND2Y731˙/R411A-α2 can be excited at 34 GHz by using very short microwave pulses, and thus hf couplings cannot be missed due to orientation selective effects.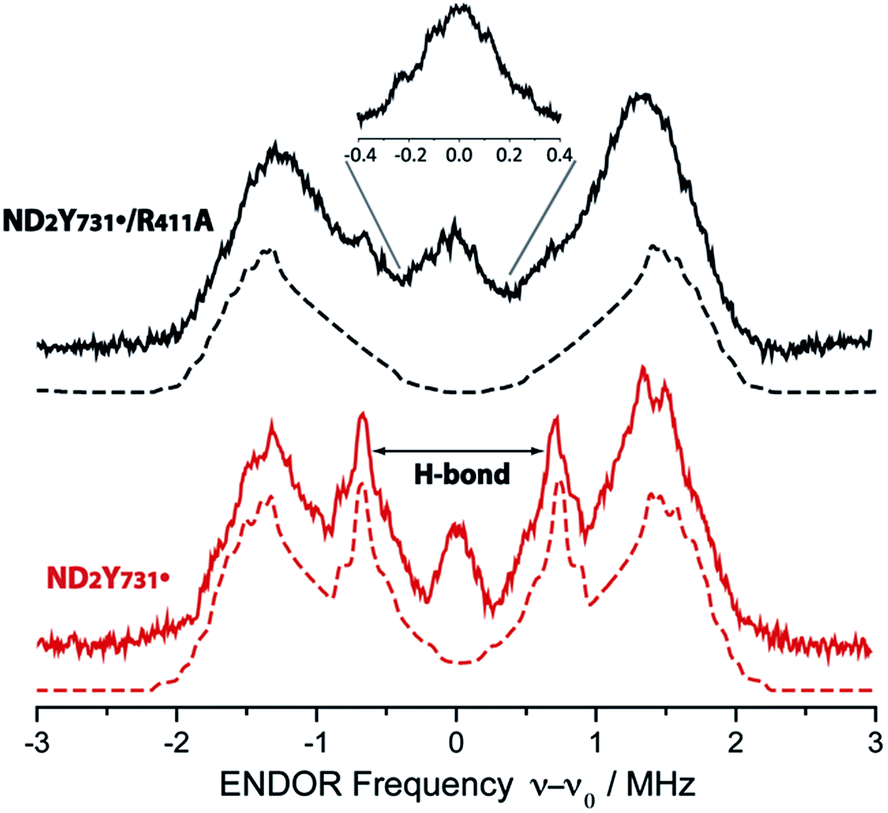 | ||
| Fig. 3 34 GHz 2H ENDOR spectra of ND2Y731˙/R411A-α2 (solid black line) and ND2Y731˙-α2 (solid red line). Simulations are shown with dashed lines. The hydrogen bond peaks, “H-bond”, assigned to a deuteron located between Y730-α2 and ND2Y731˙-α2 (ref. 12), are not observed for ND2Y731˙/R411A-α2. The inset shows the structured and broad matrix line of ND2Y731˙/R411A-α2. Exp. conditions: Mims ENDOR with π/2 = 6 ns, τ = 200 ns and 320 ns for the inset, shot repetition time = 15 ms, random RF acquisition60 with 1 shot/point, acquisition time = 15–20 h, T = 70 K. Excitation in the EPR line is set to B0||gy. ENDOR spectra are centered at the Larmor frequency of 2H, ν0 = 7.9 MHz at 1.2 T. | ||
Although no exchangeable moderately strong hydrogen bonds (rO–H ∼ 1.7–2 Å) to ND2Y731˙/R411A-α2 are observed, the ENDOR spectrum of ND2Y731˙/R411A-α2 exhibits a broad and structured matrix line, which is associated with weak hf interactions of the radical with distant nuclei61 (see Fig. 3, inset). The structure in this matrix line suggests the presence of weakly coupled deuterons that cannot be resolved from the matrix ones (matrix line). We note that the ENDOR spectrum of ND2Y731˙/R411A-α2 is reminiscent of the one previously observed for ND2Y356˙-β2, also located at the subunit interface and likely surrounded by a defined hydrogen bonded network of water molecules.12 The similarity between the ENDOR spectra of ND2Y356˙-β2 and ND2Y731˙/R411A-α2 suggests a similar origin for the gx values in these two mutants, which is distinct from that in ND2Y731˙-α2. As noted above, in the case of ND2Y356˙-β2 the gx value was also strongly shifted (NH2Y356˙: gx = 2.0049 vs. free NH2Y˙: gx = 2.0061 (ref. 33)). Therefore, we propose that the gx-shift in NH2Y731˙/R411A-α2, as well as in ND2Y356˙-β2, arises from weakly coupled hydrogen bonds observed in the 0.3 MHz region of the ENDOR spectrum. The complexity of the g tensor interpretation was underlined by our recent DFT calculations, in which three distinct models for NH2Y731˙-α2 resulted in similar g-shifts.12 Overall, these data clearly indicate that the molecular orientation of ND2Y731˙/R411A-α2 is different to that of ND2Y731˙-α2 and is affected by R411A-α2 substitution.
PELDOR gives evidence for a conformational change in ND2Y731˙/R411A-α2
Our previous PELDOR studies26 have demonstrated that half sites reactivity of E. coli RNR allows for the detection of the diagonal inter-spin distance between Y122˙ in one αβ pair and any radical trapped in the second αβ pair (Fig. 4A).25,62 To gain insight into the location of NH2Y731˙/R411A-α2, three sets of PELDOR experiments were recorded using broadband excitation with a high-power Q-band set up at different excitation positions in the EPR line63–66 (see Fig. 4B and S8†). The recorded time traces are displayed in Fig. 4C and show substantial differences in modulation depth (10 to 50%), which is typical for orientation selection effects. Trace D1 also shows a higher frequency component that arises from the parallel component of a dipolar Pake pattern (Fig. S8†). For this reason, the background corrected PELDOR time traces from the three sets of experiments were summed and the resulting trace was analyzed as shown in Fig. 4C and D. Additional comparison of the Fourier-transformed traces (Fig. S8†) shows that the sum trace leads to an almost complete Pake pattern. Distance distribution analysis revealed a clear dominant peak at 35 Å with a distance distribution of Δr = ±2.7 Å. We note that the error in the peak distance is much less than the distribution and is estimated to be ≤ ±0.5 Å. The width of the distance distribution is slightly larger than in previous measurements within the E. coli RNR α2β2 complex,25,26,62 suggesting more conformational heterogeneity for ND2Y731˙/R411A-α2, consistent with the observed flexibility of this residue. Nevertheless, the results clearly indicate that the R411 mutation induces a conformational change of ND2Y731˙ into a new well-defined conformation.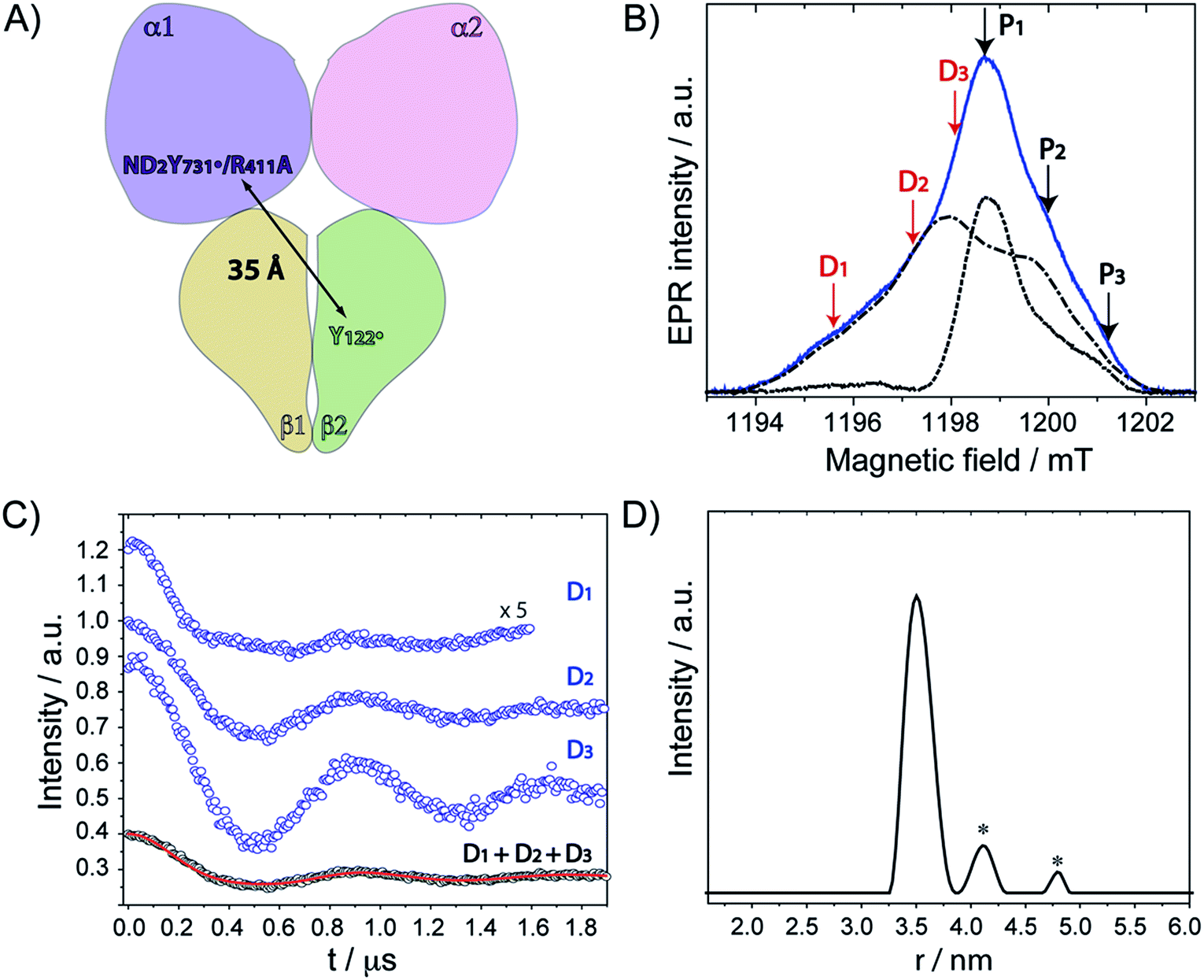 | ||
| Fig. 4 (A) Diagonal distance between Y122˙ in β2 and ND2Y731˙/R411A-α2. (B) The ESE spectrum of ND2Y731˙/R411A-α2 is composed of the Y122˙-β2 spectrum (dashed and dotted black line) and the ND2Y731˙-α2 spectrum (dotted black line). Detect (D) and pump (P) frequency positions for each PELDOR measurement are displayed by red and black arrows, respectively. Exp. conditions (EPR): π/2 = 6 ns, τ = 280 ns, shot repetition time = 120 ms, shots/point = 10, 4 scans, T = 20 K. (C) Background- and phase-corrected, normalized 34 GHz PELDOR time traces of three experimental setups (D1, D2, D3). The sum of the three traces (D1 + D2 + D3) was analyzed by DeerAnalysis 2015 (ref. 67) and is shown in black with the fitting overlaid in a solid red line. Detect/pump π pulse lengths for D1, D2 and D3 were 30 ns/12 ns, 30 ns/12 ns and 20 ns/14 ns, respectively. The frequency separation between detect and pump pulses was 80 MHz for all data sets. (D) Distance distribution obtained from the analysis in (C). Asterisks indicate artifacts attributed to the analysis procedure. | ||
The peak distance of 35.0 Å has never been observed between any radicals formed in this pathway before, and it is 3 Å shorter than that previously measured for ND2Y731˙-α2.26 This distance might appear to be rather close to the initial distance (prior to turnover) between the two stable Y122˙s, that is 33.1 ± 0.2 Å.62 To confirm our assignment, we recorded PELDOR experiments at higher temperature (50 K), in which the Y122˙-β2 contribution to the re-focused echo is filtered and ND2Y731˙-α2 is the only radical species detected (Fig. S9†). However, Y122˙-β2 can still be excited by the pump pulse and contributes to the PELDOR signal. Under these conditions, any distance observed in the PELDOR experiments at 50 K is related to Y122˙–ND2Y731˙ and cannot be associated with the Y122˙–Y122˙ distance, as the latter radical is not detected. The distance distribution analysis of the 50 K measurements yielded a peak distance of 35.3 Å with a distribution of Δr = ± 2.0 Å, and thus validated our assignment (see Fig. S9†).
To gain more insight into the conformation of NH2Y731˙/R411A-α2 and the role of R411, we examined the available X-ray structures of E. coli α2s in the R411 region. In the structure of E. coli NH2Y730-α2 (2XO4),30 Y731 is flipped away from NH2Y730, as shown in Fig. 5. This altered conformation is compared with a second α in the unit cell, in which the Y731 is not flipped. To match the 35 Å distance observed by PELDOR spectroscopy, the aromatic ring of NH2Y731 must rotate away from Y730 toward the β2 subunit, as observed for Y731 in the E. coli Y730NH2Y-α2 structure (Fig. 5).
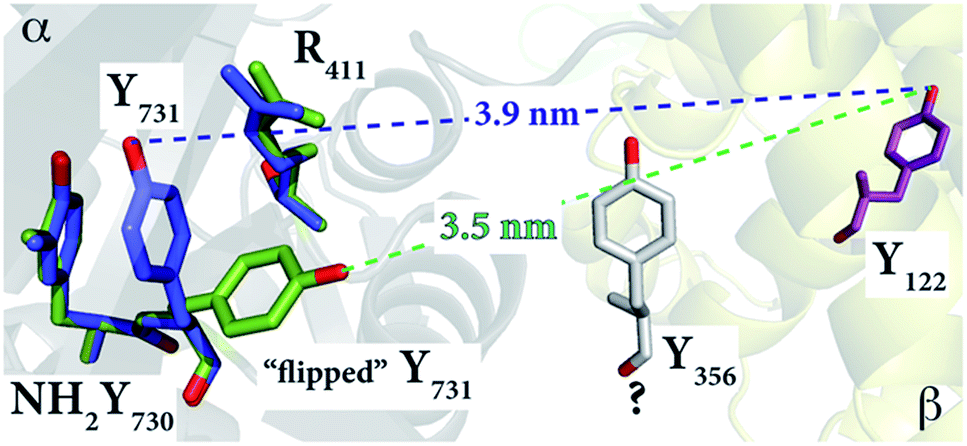 | ||
| Fig. 5 The E. coli Y730NH2Y-α2 structure (2XO4)30 in green shows the reoriented Y731 overlaid with the stacked Y731 in a different monomer (blue) of the unit cell. The diagonal distances between the “flipped” and non-flipped Y731 and Y122 are 3.5 nm and 3.9 nm, respectively. These distances, which are between two phenolic oxygen atoms of the tyrosine residues, are based on the alignment with the E. coli α2β2 docking model. Residue Y356 is shown in grey with a “?” because its position is unknown. | ||
This reorientation is also supported by the ENDOR data, which indicate that the stacked conformation between NH2Y731˙ and Y730 with a shared, perpendicular hydrogen bond is absent in NH2Y731˙/R411A-α2, and that the radical is instead surrounded by weakly coupled hydrogen bonds, likely water molecules at the α2β2 subunit interface. The exposure of NH2Y731˙/R411A-α2 to the interface and the buffer in this new conformation might be the origin of the instability of the radical as compared to the single mutant (Table S1†).
We have also examined another possible conformation, in which the amino group of NH2Y731-α2 moves to occupy the vacancy created by the mutation of arginine to alanine. This conformation is displayed in Fig. S10.† However, in this case the expected distance between the oxygen atoms of NH2Y731 and Y122 exceeds the observed distance by ≥2 Å. We note that the “flipped” conformation has not been observed in the single mutant NH2Y731-α2 or in the double mutant NH2Y731/Y730F-α2, in which Y731 lacks its hydrogen bonding partner,12 suggesting the importance of R411 in stabilizing the stacked conformation. This change between a flipped and non-flipped conformation of the interface Y might play an active role in the PCET process between Y731 and Y356 in wt RNR, the mechanism of which is still not understood. With the wt enzyme, this conformational change is kinetically masked by physical gating, which rate-limits RNR, and is too fast to be detected based on the recently measured rate constants for electron transfer (ET) (104 to 105 s−1) at the interface by photo-RNRs that unmask this gating.68,69 Thus, the R411A mutation might have fortuitously allowed detection of this movement at the subunit interface.
While the lack of structural information at the subunit interface poses a challenge for a mechanistic understanding of interfacial PCET, the detection of the NH2Y731˙/R411 provides us with a spectroscopic probe of this interface. Mutagenesis and site-specific isotopic labeling of interface residues could provide us with additional insight into how this step is controlled. Finally, the mechanism of PCET across the subunit interface observed with the E. coli RNR is likely to be conserved in all class I RNRs based on their subunit structures and the conserved weak subunit associations dictated by the C-terminal tail of β2.70,71 The pathway for oxidation is conserved between RNR classes Ia, Ib and Ic, as is the regulation of the pathway by NDP/effector binding.72 Thus, while the “details” of the radical transfer mechanism might be different in the individual class I RNRs, general principles will likely emerge from the studies on E. coli RNR, given all of the evolutionarily conserved features.
Conclusions
This study has revealed that the E. coli RNR double mutant NH2Y731/R411A-α2 unmasks a new conformation of pathway residue 731 in the α2β2 complex. This is the first experimental evidence for the flexibility of this pathway or any pathway residue in the active enzyme. The results have provided insight into the mechanisms of PCET within α2, as well as through the α2β2 interface. First, R411 appears to play a role in the stabilization of the stacked conformation of Y731 and Y730, and thus in the facilitation of collinear PCET within the α2 subunit. Second, the new conformation is consistent with Y731 pointing toward the subunit interface, in the direction of the adjacent pathway residue Y356, located in the flexible C-terminal tail of subunit β2. The flexibility of these two contiguous pathway residues, which have been suggested to communicate during PCET,69 might be the key to driving the RT chemistry at the subunit interface through water clusters.6,7 This opens up a new hypothesis for the PCET mechanism between residues Y731–α2 and Y356–β2, which could involve a gated conformational change in Y731–α2 in wt RNR on a fast time scale, not observable without the R411A mutation. While this hypothesis remains to be proven, the present results will serve as a basis to design new experiments aimed at detecting a possible combined role of Y731–α2 and Y356–β2 in PCET through the subunit surface.Acknowledgements
We acknowledge Igor Tkach for the help with technical aspects of the HF EPR spectrometers. MK thanks Karin Halbmair for the assistance with PELDOR measurements. We gratefully acknowledge financial support for this work from Deutsche Forschungsgemeinschaft DFG-IRTG 1422 (GRK 1422 to MK and MB) and DFG-SPP 1601, the Max Planck Society and NIH (GM29595 to JS).References
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- D. N. Beratan, C. Liu, A. Migliore, N. F. Polizzi, S. S. Skourtis, P. Zhang and Y. Zhang, Acc. Chem. Res., 2015, 48, 474–481 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. Y. Reece, J. M. Hodgkiss, J. Stubbe and D. G. Nocera, Philos. Trans. R. Soc., B, 2006, 361, 1351–1364 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, C. Louault, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2011, 133, 6668–6674 CrossRef CAS PubMed.
- J. M. Savéant, Annu. Rev. Anal. Chem., 2014, 7, 537–560 CrossRef PubMed.
- R. I. Cukier and D. G. Nocera, Annu. Rev. Phys. Chem., 1998, 49, 337–369 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS PubMed.
- A. Jordan and P. Reichard, Annu. Rev. Biochem., 1998, 67, 71–98 CrossRef CAS PubMed.
- J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed.
- T. U. Nick, W. Lee, S. Koßmann, F. Neese, J. Stubbe and M. Bennati, J. Am. Chem. Soc., 2015, 137, 289–298 CrossRef CAS PubMed.
- L. Thelander, J. Biol. Chem., 1973, 248, 4591–4601 CAS.
- E. C. Minnihan, D. G. Nocera and J. Stubbe, Acc. Chem. Res., 2013, 46, 2524–2535 CrossRef CAS PubMed.
- N. C. Brown and P. Reichard, J. Mol. Biol., 1969, 46, 25–38 CrossRef CAS PubMed.
- N. C. Brown and P. Reichard, J. Mol. Biol., 1969, 46, 39–55 CrossRef CAS PubMed.
- A. Hofer, M. Crona, D. T. Logan and B. M. Sjöberg, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 50–63 CrossRef CAS PubMed.
- J. A. Stubbe, J. Biol. Chem., 1990, 265, 5329–5332 CAS.
- M. Eriksson, U. Uhlin, S. Ramaswamy, M. Ekberg, K. Regnström, B. M. Sjöberg and H. Eklund, Structure, 1997, 5, 1077–1092 CrossRef CAS PubMed.
- A. Ehrenberg and P. Reichard, J. Biol. Chem., 1972, 247, 3485–3488 CAS.
- B. M. Sjoberg, P. Reichard, A. Graslund and A. Ehrenberg, J. Biol. Chem., 1978, 253, 6863–6865 CAS.
- P. Nordlund, B. M. Sjöberg and H. Eklund, Nature, 1990, 345, 593–598 CrossRef CAS PubMed.
- U. Uhlin and H. Eklund, Nature, 1994, 370, 533–539 CrossRef CAS PubMed.
- J. A. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Chem. Rev., 2003, 103, 2167–2201 CrossRef CAS PubMed.
- M. Bennati, J. H. Robblee, V. Mugnaini, J. Stubbe, J. H. Freed and P. Borbat, J. Am. Chem. Soc., 2005, 127, 15014–15015 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, C. T. Y. Chan, V. Mugnaini, J. Stubbe and M. Bennati, J. Am. Chem. Soc., 2007, 129, 15748–15749 CrossRef CAS PubMed.
- N. Ando, E. J. Brignole, C. M. Zimanyi, M. A. Funk, K. Yokoyama, F. J. Asturias, J. Stubbe and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 21046–21051 CrossRef CAS PubMed.
- E. C. Minnihan, N. Ando, E. J. Brignole, L. Olshansky, J. Chittuluru, F. J. Asturias, C. L. Drennan, D. G. Nocera and J. Stubbe, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3835–3840 CrossRef CAS PubMed.
- J. Ge, G. Yu, M. A. Ator and J. Stubbe, Biochemistry, 2003, 42, 10071–10083 CrossRef CAS PubMed.
- E. C. Minnihan, M. R. Seyedsayamdost, U. Uhlin and J. Stubbe, J. Am. Chem. Soc., 2011, 133, 9430–9440 CrossRef CAS PubMed.
- K. R. Ravichandran, E. C. Minnihan, Y. Wei, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2015, 137, 14387–14395 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, J. Xie, C. T. Y. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060–15071 CrossRef CAS PubMed.
- T. Argirević, C. Riplinger, J. Stubbe, F. Neese and M. Bennati, J. Am. Chem. Soc., 2012, 134, 17661–17670 CrossRef PubMed.
- M. R. Seyedsayamdost, T. Argirević, E. C. Minnihan, J. Stubbe and M. Bennati, J. Am. Chem. Soc., 2009, 131, 15729–15738 CrossRef CAS PubMed.
- B. Wörsdörfer, D. A. Conner, K. Yokoyama, J. Livada, M. Seyedsayamdost, W. Jiang, A. Silakov, J. Stubbe, J. M. Bollinger and C. Krebs, J. Am. Chem. Soc., 2013, 135, 8585–8593 CrossRef PubMed.
- K. Yokoyama, U. Uhlin and J. Stubbe, J. Am. Chem. Soc., 2010, 132, 8385–8397 CrossRef CAS PubMed.
- S. P. Salowe, M. A. Ator and J. Stubbe, Biochemistry, 1987, 26, 3408–3416 CrossRef CAS PubMed.
- S. P. Salowe and J. Stubbe, J. Bacteriol., 1986, 165, 363–366 CAS.
- P. T. Chivers, K. E. Prehoda, B. F. Volkman, B. M. Kim, J. L. Markley and R. T. Raines, Biochemistry, 1997, 36, 14985–14991 CrossRef CAS PubMed.
- M. Russel and P. Model, J. Bacteriol., 1985, 163, 238–242 CAS.
- J. R. Steeper and C. D. Steuart, Anal. Biochem., 1970, 34, 123–130 CrossRef CAS PubMed.
- S. Climent, B. M. Sjöberg and C. Y. Huang, Biochemistry, 1991, 30, 5164–5171 CrossRef PubMed.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- S. Kossmann, B. Kirchner and F. Neese, Mol. Phys., 2007, 105, 2049–2071 CrossRef CAS.
- F. Neese, in Multifrequency Electron Paramagnetic Resonance, ed. S. K. Misra, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, ch. 6, pp. 295–326 Search PubMed.
- S. Un, M. Atta, M. Fontecave and A. W. Rutherford, J. Am. Chem. Soc., 1995, 117, 10713–10719 CrossRef CAS.
- H. M. McConnell, J. Chem. Phys., 1956, 24, 764–766 CrossRef CAS.
- R. W. Fessenden and R. H. Schuler, J. Chem. Phys., 1963, 39, 2147–2195 CrossRef CAS.
- B. Epel, D. Arieli, D. Baute and D. Goldfarb, J. Magn. Reson., 2003, 164, 78–83 CrossRef CAS PubMed.
- A. V. Astashkin and A. Kawamori, J. Magn. Reson., 1998, 135, 406–417 CrossRef CAS PubMed.
- M. Bennati, A. Weber, J. Antonic, D. L. Perlstein, J. Robblee and J. Stubbe, J. Am. Chem. Soc., 2003, 125, 14988–14989 CrossRef CAS PubMed.
- V. P. Denysenkov, T. F. Prisner, J. Stubbe and M. Bennati, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13386–13390 CrossRef CAS PubMed.
- A. D. Milov, A. G. Maryasov and Y. D. Tsvetkov, Appl. Magn. Reson., 1998, 15, 107–143 CrossRef CAS.
- Y. Polyhach, A. Godt, C. Bauer and G. Jeschke, J. Magn. Reson., 2007, 185, 118–129 CrossRef CAS PubMed.
- G. Sicoli, T. Argirević, J. Stubbe, I. Tkach and M. Bennati, Appl. Magn. Reson., 2010, 37, 539–548 CrossRef.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- L. Olshansky, A. A. Pizano, Y. Wei, J. Stubbe and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 16210–16216 CrossRef CAS PubMed.
- D. Y. Song, A. A. Pizano, P. G. Holder, J. Stubbe and D. G. Nocera, Chem. Sci., 2015, 6, 4519–4524 RSC.
- M. Kolberg, K. R. Strand, P. Graff and K. K. Andersson, Biochim. Biophys. Acta, 2004, 1699, 1–34 CrossRef CAS.
- Y. Zhang, X. An, J. Stubbe and M. Huang, J. Biol. Chem., 2013, 288, 13951–13959 CrossRef CAS PubMed.
- P. Nordlund and P. Reichard, Annu. Rev. Biochem., 2006, 75, 681–706 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03460d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |