
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure elucidation of a complex CO2-based organic framework material by NMR crystallography†
Julien
Leclaire
*ab,
Guillaume
Poisson
ab,
Fabio
Ziarelli
c,
Gerard
Pepe
d,
Frédéric
Fotiadu
b,
Federico M.
Paruzzo
f,
Aaron J.
Rossini
ef,
Jean-Nicolas
Dumez‡
e,
Bénédicte
Elena-Herrmann
*e and
Lyndon
Emsley
f
aUniv Lyon, Université Claude Bernard, CNRS, INSA, CPE, ICBMS UMR 5246, 69622 Villeurbanne, France. E-mail: julien.leclaire@univ-lyon1.fr
bAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397 Marseille, France
cAix-Marseille Université, Fédération des Sciences Chimiques, Spectropôle, 13397 Marseille, France
dAix-Marseille Université, CNRS, UMR 7325 CINaM, 13288 Marseille, France
eUniversité de Lyon, Institut des Sciences Analytiques, Centre de RMN à très hauts champs, CNRS/ENS Lyon/UCBL, 69100 Villeurbanne, France. E-mail: benedicte.elena@ens-lyon.fr
fInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
First published on 22nd March 2016
Abstract
A three-dimensional structural model of a complex CO2-based organic framework made from high molecular weight, self-assembled, flexible and multi-functional oligomeric constituents has been determined de novo by solid-state NMR including DNP-enhanced experiments. The complete assignment of the 15N, 13C and 1H resonances was obtained from a series of two-dimensional through space and through bond correlation experiments. MM-QM calculations were used to generate different model structures for the material which were then evaluated by comparing multiple experimental and calculated NMR parameters. Both NMR and powder X-ray diffraction were evaluated as tools to determine the packing by crystal modelling, and at the level of structural modelling used here PXRD was found not to be a useful complement. The structure determined reveals a highly optimised H-bonding network that explains the unusual selectivity of the self-assembly process which generates the material. The NMR crystallography approach used here should be applicable for the structure determination of other complex solid materials.
Introduction
The last decades have seen an explosive growth in the production of rationally designed supramolecular materials resulting from directed self-assembly of molecular building units. Among these self-assembled molecular networks displaying promising properties, one can cite metal organic frameworks (MOFs),1 obtained by connecting metal containing secondary units with organic linkers, and organic supramolecular solids2 resulting from the connection of carbon-based building blocks. Their synthesis is based on attractive one-pot self-assembly processes which illustrate the concept of molecular tectonics.3–5 Self-organization results from the structure of and the interactions between the rigid building blocks.6 Short-range, strong anisotropic interactions drive self-organization across length scales from the molecular and nano-scale level7 up to the micro- and milli-scale levels.8–11 The well-defined building units, coupled with the thermodynamic control of the self-assembly process, often lead to long range structural order i.e. to highly crystalline solids.12 X-ray diffraction can frequently be used to characterize the structural features of these self-assembled networks if they are sufficiently regular.Such supramolecular materials can have tunable physical and chemical properties, with cavities whose size and shape are easily manipulated.13 However, the complete structural characterization of increasingly complex systems, that is essential to determine structure–activity relations and to then rationally improve properties, remains a considerable challenge. This notably often prevents exploration of molecular assemblies based on more flexible objects involving multiple functionalities.
In this context we have recently synthesized and described a self-assembled supramolecular solid which results from the one-pot thermodynamically controlled simultaneous reaction of two organic building blocks and CO2 (Scheme 1).14 Notably, this reversible CO2-based organic framework is produced by aggregation of flexible self-complementary oligomeric objects. As illustrated in Scheme 1 these oligomeric building blocks are themselves assembled in situ from the basic components, including CO2. The covalent assembly process of the oligomers consists in the simultaneous and reversible formation of 20 carbon–nitrogen bonds of three different chemical types (imine, aminal and carbamate). Since the final material results from the packing of oligomers that were themselves spontaneously assembled from elementary units through the reversible formation of covalent bonds, this supramolecular solid is in the class of what have been dubbed “dynamic materials” by Lehn.15–17 This particular dynamic material possesses original properties: both the material and its constitutive oligomers dissociate upon CO2 departure in a cooperative manner, and the assembly process is found to be extremely selective in terms of building block incorporation.18,19 Both features (reversibility and selectivity) are desirable in the context of CO2 capture and use, as well as for selective extraction of valuable molecules from waste.20 However, the material precipitates from solution as an insoluble nano-crystalline powder, which is not amenable to standard molecular level characterization. To address this problem we previously used solid-state Nuclear Magnetic Resonance (NMR) to propose basic structural features for this supramolecular system.14
 | ||
| Scheme 1 Double self-assembly process leading to the discovery of a 3-component dynamic material based on CO2. (a) Elementary building blocks such as polyamines n (brown connector) and polyaldehydes X (orange hexagon) self-assemble in methanol at RT under air to yield 2-component oligomers (b), mainly X2n3 capsules. In the case X = A and n = 2 in the same conditions, exposure to CO2 (blue arch) as a third building block spontaneously yields the 3-component soluble “oligodynablock” A429(CO2)9. This self-assembled species then itself spontaneously further self-assembles into the corresponding insoluble dynamic material DMA2(CO2) as a nanocrystalline powder (electron micrograph, white bar: 100 nm) (c). Heating to around 50 °C induces CO2 release and disassembly of the dynamic material which converts back into A223. | ||
Here we further explore the three-dimensional molecular structure of the nano-crystalline dynamic material using an NMR crystallography approach,21,22 and we evaluate the pertinence of powder X-ray diffraction (PXRD). Solid-state NMR spectroscopy complements diffraction methods and has recently developed into a versatile tool to determine the structures of both inorganic and molecular solids.23–34 In combination with crystal structure prediction methods and DFT chemical shift calculations, NMR chemical shifts have recently been used to determined de novo crystal structures.23 In particular in the context of supramolecular solids, solid-state NMR and DFT calculations have been used to explore hydrogen bonding and molecular structure in organic supramolecular systems35,36 and to characterise structural features in MOFs, including determination of conformation changes upon adsorption or to observe the dynamic events that govern host–guest interactions.37–42 Most recently, the superstructure of a substituted zeolitic imidazolate framework (ZIF) type MOF was elucidated by combining solid-state NMR, PXRD, and DFT calculations of NMR parameters from potential model structures.43
Notably very rapid progress has recently been made by combining sophisticated spectral assignment strategies with state-of-the-art Density Functional Theory (DFT) chemical shift calculation methods. Today, this provides a platform for analysis of both 13C and 15N spectra,44–46 as well as of 1H shifts.22–24,47–51
Following this general approach, here MM-QM (molecular mechanics-quantum mechanics) calculations were used to generate several different model structures of the dynamic material. The model structures were then evaluated by comparing multiple experimental and calculated NMR and PXRD parameters. A detailed structural model is proposed for the individual oligomeric constituents of the material including their exact sequence, conformation, and potential packing.
Results and discussion
Synthesis of the dynamic material
Simple molecular building blocks such as polyamines and polyaldehydes are well-known to spontaneously interconnect with each other in mild conditions (room temperature, no catalyst, open air, undistilled solvents) through the formation of reversible C–N bonds (single: aminal; double: imine) to generate dynamic combinatorial libraries.The objects assembled from these two classes of components using reversible or dynamic covalent bonds52 are referred to as “oligo-dynablocks”53–55 as they can themselves be building blocks (or dynamic tectons) to generate supramolecular structures. Here, this second level of assembly can be triggered by exposure to CO2 which can post-functionalize the two-component oligomers. CO2 reversibly reacts with primary or secondary amines yielding ammonium carbamate ion pairs. CO2 exposure hence converts neutral objects into zwitterionic species. The CO2-postfunctionalization can perturb the pre-equilibrated 2-component system and induce a redistribution of the populations toward one or several stable 3-component objects. Formation of a solid network is then a potential driving force that can displace the equilibria and select a self-complementary post-functionalized species.56
Twenty-eight libraries corresponding to the various combinations of polyamines n(2–8) and polyaldehydes X(A–D) were screened for the formation of a dynamic material induced by reversible CO2-postfunctionalization (Scheme 1). In the absence of CO2, the 2-component systems were dominated by low-molecular weight entropically favoured soluble oligomers (objects involving two aromatic units at most), with the capsule made from two aldehydes and three polyamine units noted X2n3 being the major adduct as indicated by ESI-MS analysis (Scheme 1a and b).57 For all combinations except one, this composition remained unchanged when CO2 was introduced. In contrast, polyamine n = 2 and polyaldehyde X = A lead to the quantitative formation of a single 3-component dynamic material which precipitated upon CO2 exposure as a nano-crystalline solid and re-dissolved upon gentle heating (Scheme 1b and c). As reversible linkages were used to assemble the building blocks, their average stoichiometry within the material was precisely determined by titration (1H solution NMR for A and 2; volumetric analysis for CO2) after acid hydrolysis of the dried powder. While 1D solid-state NMR confirmed that trialdehyde A and CO2 were both covalently connected to the diethylenetriamine building block 2, the titration revealed that the oligo-dynablocks constituting the materials were mostly linear oligomers made of four aromatic units A condensed with nine diethylenetriamine chains 2 each bearing one molecule of CO2 (noted A429(CO2)9, Scheme 1b). Thermogravimetric analysis coupled to mass spectrometry measurements additionally revealed the presence of substantial amounts of water (15 percent in mass). The water is presumably adsorbed in layers rather than integrated into the framework of the zwitterionic material as it could be entirely expulsed by heating at 70 °C under atmospheric pressure for several minutes without modification of the solid-state 13C CP-MAS solid-state NMR spectrum. No high molecular weight 3-component objects were detected in the mother liquor, nor was the A429 precursor present in the parent 2-component library before CO2 exposure. The oligo-dynablocks A429(CO2)9 are therefore clearly stabilized by their intermolecular packing within the solid network formed.
When the stoichiometry between the building blocks A and 2 was changed, their titration after cleavage of the resulting material revealed that the average measured length of the oligomers constituting the material changed correspondingly, in agreement with equilibrium displacement rules. As a consequence, we conclude that the chain length in the objects that make up the dynamic material may display some degree of dispersity around the major chain length of 4. The analysis of the structure of these objects that are intrinsically unstable in solution (noted A429(CO2)9) and of their packing mode into the assembled dynamic material (noted DMA2(CO2)) can therefore only be performed in the solid state.58 However, the precipitation of DMA2(CO2) as a nano-crystalline solid with some disorder in chain length and water content makes it challenging to both perform basic molecular level characterization or to determine a crystal structure. It also hampers the understanding of the precise intra- and intermolecular interactions that drive the assembly of DMA2(CO2). We have therefore used solid-state NMR spectroscopy, which is not necessarily hampered by this type of disorder, in combination with quantum chemical calculations to propose a three-dimensional structural model of the dynamic material DMA2(CO2).
Determining constitutional isomers
We have previously identified14 some of the functional groups borne by the oligo-dynablocks A429(CO2)9 from basic one-dimensional magic angle spinning 15N and 13C solid-state NMR spectroscopy (Fig. 1a). It is worth noting at this stage that the 13C CP-MAS NMR spectrum of DMA2(CO2) remained unchanged when the stoichiometry of the molecular constituents was varied by tuning the initial ratio between building blocks A and 2, strongly suggesting that variations in the chain length can be accommodated by the crystal structure of the material. In order to explore the connectivity between individual heavy atoms (carbon and nitrogen), a partially 13C and fully 15N enriched dynamic material (referred to as DMA2(CO2)*) was produced by using fully 13C-labeled CO2 (to label carbamates) and 15N-labeled diethylenetriamine for the assembly process.59 A 2D double cross-polarization (DCP) 15N–13C HETCOR60 spectrum of DMA2(CO2)* allowed us to assign most of the carbon nuclei directly bonded to nitrogen centers (Fig. 1c, black/nitrogen to red/carbon labels) by analogy with known shifts (N1′ and N2′ are characterized by broad peaks of low intensity and failed to provide detectable correlations with neighboring carbons except for N1′–C14). This experiment confirmed the presence of unusual chemical functionalities stabilized by the solid environment, such as the aminal carbamate moiety (carbon atom 11 and nitrogen 3′, Fig. 1b). However, the similarity of the 13C chemical shifts of several carbon atoms in the ethyl bridges prevented complete 13C assignment (Fig. 1c, purple labels). A DNP enhanced 13C–13C refocused INADEQUATE spectrum61 (Fig. 1d and e) was then recorded on the natural abundance DMA2(CO2) to complete the carbon-13 assignment and to elucidate the substitution pattern of the three carbamate-diethylenetriamine side arms borne by the phenolic core of the monomeric unit. | ||
| Fig. 1 (a) Building block content and molecular patterns previously identified.14 (b) Structure of the oligo-dynablock proposed here based upon solid-state NMR analysis. (c) 2D DCP 15N–13C HETCOR spectrum for partially 13C and fully 15N enriched DMA2(CO2)*. Correlations between nitrogen atoms (black labels) and carbon atoms (red labels) are indicated by dashed lines. (d) 100 K DNP-enhanced 2D refocused INADEQUATE 13C–13C correlation spectrum (aromatic region only) acquired from natural isotopic abundance DMA2(CO2) impregnated with a 16 mM solution of TEKPol in 1,1,2,2-tetrachloroethane. Correlations/connectivity are indicated by solid orange lines and provide iterative assignment of green labels. (e) Aliphatic region of the 2D refocused INADEQUATE 13C–13C correlation spectrum (provides unambiguous assignments of purple labels). By combining the information in the 2D DCP 15N–13C HETCOR spectrum and the 2D refocused INADEQUATE 13C–13C correlation spectrum it is possible to completely assign the spectra and propose the model chemical structure for the oligo-dynablock shown in (b). Carbon resonances are numbered on the spectra and assigned to the proposed structure of DMA2(CO2). | ||
The 13C–13C refocused INADEQUATE spectrum (Fig. 1d and e) indicates the direct connections between the neighboring sp2 carbon atoms (C1–C10 region) of the polyaldehyde building block, including the carbonyl groups engaged in three chemically distinct types of linkages (imine, enamine and aminal). Note that we used high field dynamic nuclear polarization (DNP)62,63 to enhance the sensitivity of the solid-state NMR experiments to enable acquisition of a refocused INADEQUATE 13C–13C correlation spectrum in a reasonable experimental time.64,65 In order to perform DNP experiments the powdered DMA2(CO2) was impregnated with a solution of the nitroxide biradical polarizing agent TEKPol66 in 1,1,2,2-tetrachloroethane (TCE).67 TCE was chosen since it is a non-solvent for DMA2(CO2). In these experiments the protons within the DMA2(CO2) crystallites are remotely polarized by proton spin diffusion.64,68,69 DNP provides a spectacular increase in the signal-to-noise ratio as compared with analogous room temperature experiments (ESI, Fig. S6†), revealing well resolved individual scalar coupling correlations between all the carbons in the 13C–13C refocused INADEQUATE spectrum. This leads to the full assignment of the carbon nuclei (C13–C19) in the aliphatic region (Fig. 1e). In conclusion, this analysis allows us to propose the chemical structure for DMA2(CO2) shown in Fig. 1b.
Determining tautomers
2D 1H–13C dipolar HETCOR (Fig. 2a) and 1H–15N solid-state refocused INEPT70 (Fig. 2b) experiments were performed in order to identify hydrogen atoms bound to carbon and nitrogen atoms on the monomeric unit. These experiments also allowed us to explore intermolecular connections between oligo-dynablocks. | ||
| Fig. 2 Heteronuclear correlation (HETCOR) spectra for assignment of 1H chemical shifts and determination of the protonation state and intermolecular binding modes of the oligodynablocks. (a) The carbonyl region of the 1H–13C dipolar HETCOR spectrum of DMA2(CO2). (b) Full 1H–15N solid-state refocused INEPT spectrum of DMA2(CO2)*; eDUMBO-1221H homonuclear decoupling was applied during t1 evolution to obtain high 1H resolution.71 (c) Corresponding identified intermolecular binding modes. | ||
By determining the protonation state of heteroatoms it was possible to identify the tautomeric forms of the various possible carbamic acid-amine and/or ammonium carbamate and imine-phenol or enamine-quinone combinations. The 1H–15N solid-state refocused INEPT spectrum (Fig. 2b) clearly shows that of all the nitrogen atoms, only N1′ and N6′ are protonated. Comparison of the 1H–13C and 1H–15N 2D correlation spectra indicates that both N6′ (likely an ammonium nitrogen) and C11 (a carbamate carbon) show correlations to the same 1H nuclei with a shift of 9 ppm typical of ammonium protons. This suggests that the aminoethylaminal N6′–C11 side chain exists as an ammonium – aminal carbamate ion pair (Fig. 2c).
In contrast, the diethylenetriamine central spacer is involved on its extremities in enamine and imine linkages with the aromatic units. N5′ can only be post-functionalized into a neutral carbamic acid. The 1H–13C dipolar HETCOR experiment also provided some information about the intermolecular bonding between oligo-dynablocks. In this spectrum, both C10 and C11 showed correlations to 1H nuclei with a chemical shift of ca. 13.7 ppm. This suggests that the anionic carbamate (C11) site engages in hydrogen bonds with the neutral carbamate sites (C10). Considering that C11 also engages in a hydrogen bond to the ammonium group, we can conclude that it is a pivotal anchoring group, acting as a hydrogen bond acceptor both with the ammonium center and with the carbamic acid groups of two different neighboring oligo-dynablock within the solid network (Fig. 2c).
Determining conformers
To explore the conformation of the flexible zones (namely the dihedral angles C12–C13, C14–C15, C16–C17 on the main chain and the C4–C9 dihedral angle on the bicyclic aminal-phenol system), a DNP-enhanced Lee-Goldberg (LG) CP HETCOR experiment72 was conducted to probe short range contacts between 1H and 13C nuclei (Fig. 3a). This experiment confirmed the regioisomeric assignment and the intermolecular pairing mode through the observation of correlations between C4–![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C5; C6–C5; C6–C3; C2–C3; C2–C7; C1–N1; C11–C19; C11–C5 and C10–C19 (Fig. 3b). Besides a C4–C9 contact, two correlations (C1–C9) and (C11–C5) between the aminal carbamate side chain and the central aromatic core could be observed (Fig. 3a and b). A statistical analysis of the Cambridge structural database indicates that for benzylic aminal structures the planes of the six and five membered rings are systematically almost perpendicular in such molecular patterns.14 While this orientation fulfills the requirements for the inter-molecular ionic pairing between oligo-dynablocks, the aforementioned contacts between nuclei from these adjacent rings additionally tend to indicate that C9–H and C1–O bonds are almost coplanar.
C5; C6–C5; C6–C3; C2–C3; C2–C7; C1–N1; C11–C19; C11–C5 and C10–C19 (Fig. 3b). Besides a C4–C9 contact, two correlations (C1–C9) and (C11–C5) between the aminal carbamate side chain and the central aromatic core could be observed (Fig. 3a and b). A statistical analysis of the Cambridge structural database indicates that for benzylic aminal structures the planes of the six and five membered rings are systematically almost perpendicular in such molecular patterns.14 While this orientation fulfills the requirements for the inter-molecular ionic pairing between oligo-dynablocks, the aforementioned contacts between nuclei from these adjacent rings additionally tend to indicate that C9–H and C1–O bonds are almost coplanar.
 | ||
| Fig. 3 Coupled configurational/conformational exploration of the junction between main and side chain around the connective center C9 through (a) a short range (τCP = 500 μs) DNP-enhanced 1H–13C LG-CP HETCOR spectrum revealing spatial contacts. (b) Structural model with black arrows indicating correlations observed within the rigid backbone, and red arrows indicating observed correlations between flexible parts. This analysis leads to two possible enantiomeric monomeric units with opposite absolute coupled configuration/conformation on the C9 center (c). | ||
The perpendicular orientation of the primary and side chains, which are both non-symmetrical, results in axial chirality around the C4–C9 linkage. As C9 is itself a stereogenic center, both axial and central chirality are inter-related. In the present case, only (S,P) or (R,M) combinations of configuration and conformation are in agreement with the LG-CP HETCOR experiment (Fig. 3c).
Elucidation of tacticity and crystal structure
From the above, for a given oligomeric primary chain, two opposite absolute configurations corresponding to reversed orientations of the N3′-carbamated diethylenetriamine can be envisaged, leading to structures of varying tacticity.To proceed further to an atomic-level model of the structure, we modelled six different trial structures of the oligo-dynablocks (shown schematically in Scheme 2 (1–6)), which differed in terms of stereochemistry. For each type of structure trial crystal structures were generated using the direct-space genetic algorithm (GA) technique incorporated in the program Genmol.73–75 Among these six trial architectures, objects 4, 5 and 6 were true potential candidates whose sequence and structure were in full agreement with the data from the previous solid-state NMR experiments. Objects 1, 2 and 3 were reference compounds with erroneous configurations and/or conformations on part or all of the monomeric units which were deliberately included (all disagree with the analysis of the LG-CP HETCOR spectrum, in Fig. 3a). They were included to validate the ability and sensitivity of the chemical shifts to discriminate diastereoisomers on high molecular weight multi-component architectures and simultaneously elucidate their packing.
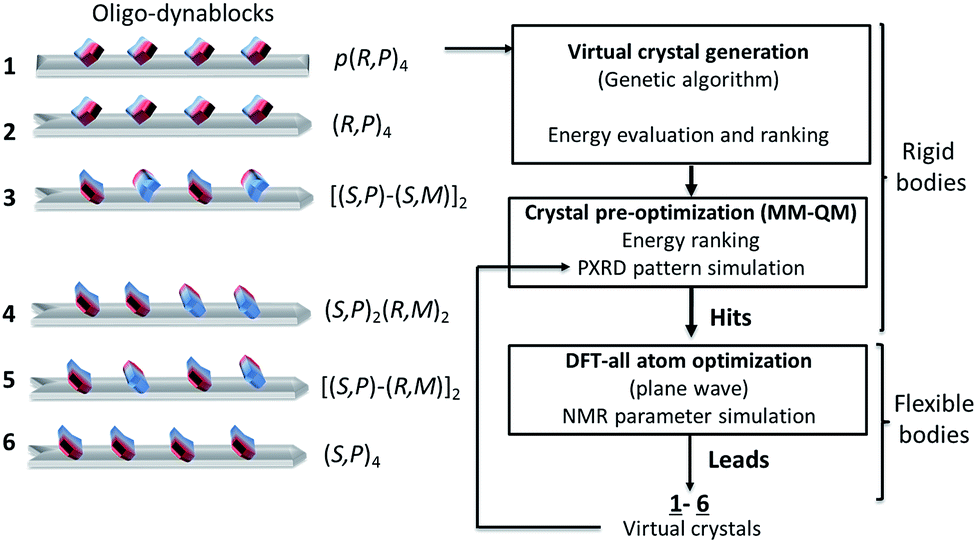 | ||
| Scheme 2 Crystal structure prediction process on oligo-dynablocks 1–6 with their Cahn–Ingold–Prelog configurations. | ||
In more detail, 6 consists of isotactic (S,P)4 oligo-dynablocks, while 5 and 4 are their syndiotactic [(S,P)-(R,M)]2 and block (made of meso dyads) (S,P)2-(R,M)2 diastereoisomer respectively. 3 is also syndiotactic [(S,P)-(S,M)]2 but displays erroneous configurations (with respect to 5) or conformations (with respect to 6) on non-adjacent monomeric units. 2 is isotactic (R,P)4 with erroneous configurations/conformations on every unit. Finally, 1 is the regioisomer of 2 where all phenol groups are localized in para position with respect to the aminal ring (configuration referred to as: [p-(R,P)]4) and corresponds to the structure previously suggested for DMA2(CO2).14
DMA2(CO2) is well beyond the scope of comprehensive crystal structure prediction today, as it has 130 heavy atoms and each of the four monomeric units of the oligo-dynablocks has at least 13 degrees of conformational freedom. In addition the models we use here neglect the presence of water in the structure, and do not consider disorder in the chain lengths. We are therefore looking for structural models that will certainly not be exact, but which will reproduce the qualitative features of the conformation and intermolecular packing in the crystal structure. The models used to generate the trial crystal structures were thus built from tetramers whose configuration and conformation agreed with most or all NMR experiments (anti conformation was chosen for the main chain and the bicyclic dihedral angle set to 90°, in agreement with NMR data). These structures were then treated as rigid bodies during the process of crystal generation and preliminary minimization. Candidate crystal structures for each of the six oligodynablock structures 1–6 were generated in the P1 space group. For each candidate the three hundred most stable lattices were selected from a randomly generated pool and then pre-optimized by energy minimization with MM + QM calculations (see Section S1.5.1. in ESI for details†).
In the case of 2 and 6, respectively, two and three hits (labelled a–c) having a high predicted stability (lattice energy lower than −180 kcal mol−1) were obtained and conserved. 1, 3, and 5 all yielded a most stable predicted structure with lattice energies lower than −105 kcal mol−1. Block oligomers 4 (made of meso dyads) failed to yield stable crystal packing (lattice energies were all higher than −100 kcal mol−1) and were therefore excluded.
All-atom DFT geometry optimization was performed on the eight candidates obtained (1, 2a,b, 3, 5, 6a–c) and the chemical shift tensors of the resulting leads were calculated using the GIPAW (Gauge Including Projector Augmented Wave)44,76,77 method in CASTEP.78 To assess the quality of the proposed structures 1–3 and 5–6, they were ranked based on the combination of predicted lattice energy and four different NMR chemical shift parameters (Fig. 4; ESI, Table S18†). For the chemical shifts, root-mean-squared deviations (rmsd) between experimental and calculated values of isotropic chemical shifts (1H, 15N and 13C), as well as 13C chemical shift anisotropy (Fig. 4; ESI, Table S18†) were computed.24,27 Experimental 13C chemical shift tensor parameters were determined from a DNP-enhanced 13C magic angle turning (MAT) spectrum of DMA2(CO2) (Fig. S9†).
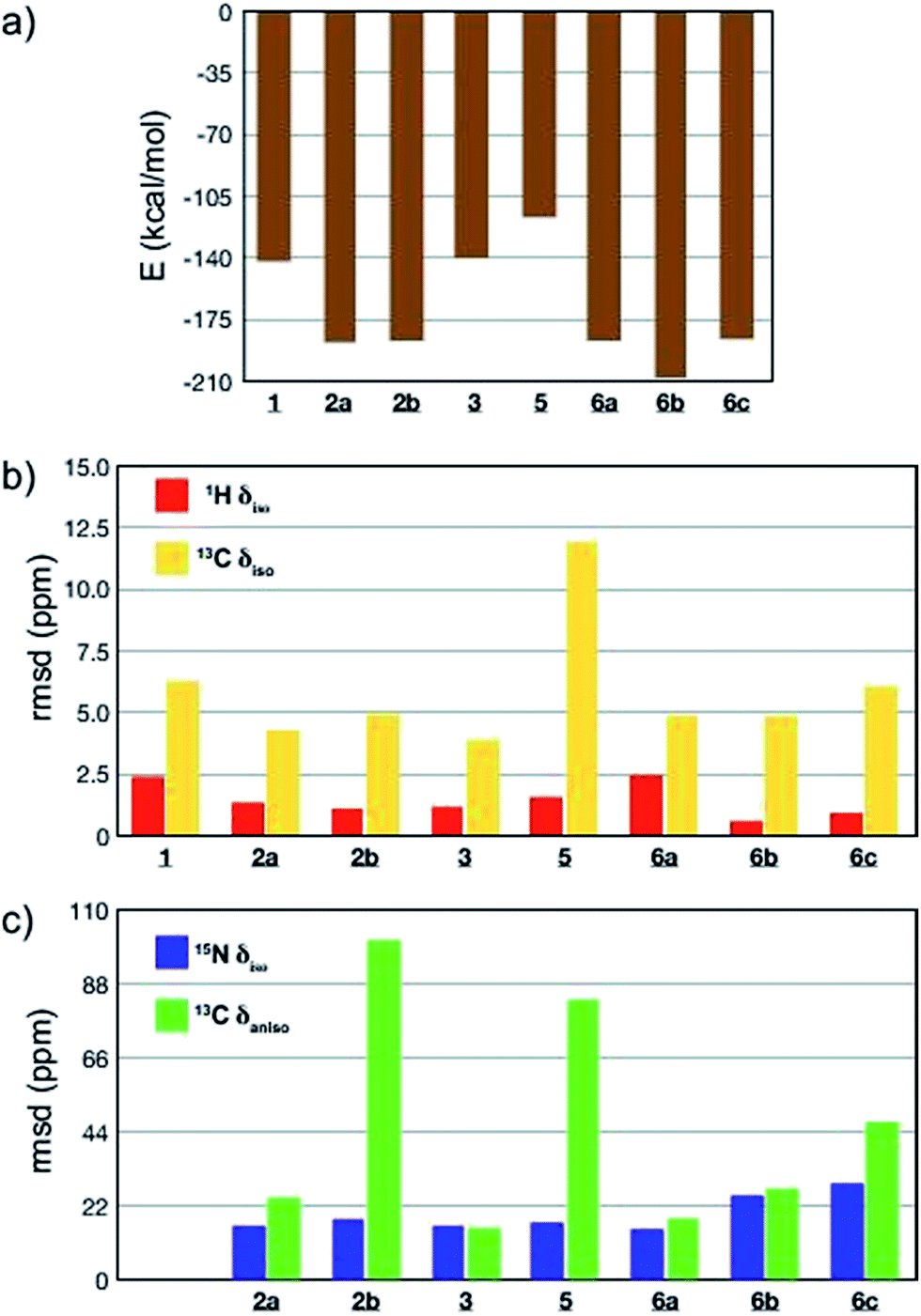 | ||
| Fig. 4 Comparison of agreement between calculated and experimental data for the predicted crystal structures 1–6. (a) Predicted crystal lattice energies; (b) root-mean-square deviation (rmsd) between experimental and calculated 1H (red) and 13C (yellow) isotropic chemical shifts; (c) rmsd between experimental and calculated 15N isotropic chemical shifts (blue) and 13C chemical shift anisotropy (green). | ||
Computed crystal energy, 1H and 13C isotropic chemical shift are today reasonably well understood, and were hence chosen as quantitative indicators (Fig. 4a–c). In the particular case of 1H chemical shifts the “cut-off value” of rmsd under which a virtual crystal is considered to be in good agreement with the experiment is usually around 0.6 ppm.23,49 Since they have so far been less frequently used, 15N isotropic chemical shift and 13C chemical shift anisotropy were considered in Fig. 4c as qualitative indicators of potential discrepancy when abnormally high values of rmsd were obtained.
The results of the comparisons are summarized in Fig. 4a–c, and ESI Table S18.† Structures 1, 3 and 5 are predicted to be significantly less stable than the others, with 1 and 5 also showing significant deviations in the chemical shift parameters, and can be discounted. The case of oligo-dynablock 1 highlights the difference between the molecular and the supramolecular point of view: the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O positioning on C3 obviously induces a decrease in steric hindrance locally with the five-membered aminal ring but globally yields a packing of poor stability. Similarly, syndiotactic oligo-dynablocks 3 and 5 with alternate conformations and configurations on adjacent monomeric units, respectively yield structures with higher predicted energy compared to isotactic oligo-dynablocks 6. Structures 2a and 2b, which differ in the conformation of the flexible side chains of their constituents and their relative position in the structures, display contrasted scores for qualitative NMR indicators. Lattices, 2 and 6 which are formed from isotactic oligo-dynablocks are indeed predicted to be significantly more stable. Structure 2b can be discounted since it has an abnormal disagreement in the 13C CSA parameters. When comparing structures 2a, 6a, 6b and 6c, we see that 6c has overall only qualitative agreement. 2a, 6a and 6b all have better agreement, but we see that among them only 6b is in good agreement with the 1H chemical shifts, with a 1H rmsd value below 0.6 ppm. At this point we recall that structure 2 does not agree with the NMR correlation data. We therefore tentatively conclude that structure 6b, which is predicted to be the most stable, is the structure that is in best overall agreement with the ensemble of experimental data from NMR, including chemical shifts and correlation spectra. While we are certain that this is not an exact representation of the lattice (due to the disorder in chain length and solvation discussed above), we hypothesise that it is likely to contain the essential structural features of the solid.
O positioning on C3 obviously induces a decrease in steric hindrance locally with the five-membered aminal ring but globally yields a packing of poor stability. Similarly, syndiotactic oligo-dynablocks 3 and 5 with alternate conformations and configurations on adjacent monomeric units, respectively yield structures with higher predicted energy compared to isotactic oligo-dynablocks 6. Structures 2a and 2b, which differ in the conformation of the flexible side chains of their constituents and their relative position in the structures, display contrasted scores for qualitative NMR indicators. Lattices, 2 and 6 which are formed from isotactic oligo-dynablocks are indeed predicted to be significantly more stable. Structure 2b can be discounted since it has an abnormal disagreement in the 13C CSA parameters. When comparing structures 2a, 6a, 6b and 6c, we see that 6c has overall only qualitative agreement. 2a, 6a and 6b all have better agreement, but we see that among them only 6b is in good agreement with the 1H chemical shifts, with a 1H rmsd value below 0.6 ppm. At this point we recall that structure 2 does not agree with the NMR correlation data. We therefore tentatively conclude that structure 6b, which is predicted to be the most stable, is the structure that is in best overall agreement with the ensemble of experimental data from NMR, including chemical shifts and correlation spectra. While we are certain that this is not an exact representation of the lattice (due to the disorder in chain length and solvation discussed above), we hypothesise that it is likely to contain the essential structural features of the solid.
Note that candidates 6b and 6c were the only model structures in the series whose constituents have a zwitterionic protonation state (ammonium carbamate) on the C11–N6′ chain, in agreement with experimental observations. Although the other model crystal structures were systematically generated from zwitterionic oligo-dynablocks, proton exchange during all atom DFT optimization resulted in neutral structures. The correct protonation state on the hetero-nuclei partially accounts for the lower rmsd of the 1H chemical shift values in the structures 6b and 6c. As mentioned above, when 6b and 6c are compared on the basis of the other major structural parameters, 6b appears to be the best candidate. Note that, 6b is not in the best agreement among the set in terms of calculated and experimental isotropic 15N chemical shifts. This is particularly true for the sp2 nitrogen centers N1′ and N2′ (with rmsd of 47 and 32 ppm respectively). However, these nuclei systematically yielded poor correlation for all candidate structures (the average difference along the series 5 and 6 being 35 and 33 ppm respectively). While the chemical shift of sp3 nitrogens seem to display little sensitivity to their protonation state (a 10 ppm shift on average was observed when comparing candidates 1, 2a, 2b, 5 and 6a with 6b and 6c), sp2 nitrogens in salicylic systems are reported to be extremely sensitive to the proton transfer from the phenol group (from −50 to −220 ppm) and to the exact position of the proton between the oxygen and the nitrogen N1′ centers,79 and other work has shown strong sensitivity of nitrogen shifts to the migration of protons across hydrogen bonds.80 It is therefore expected that a very slight discrepancy in the calculated position of the N1′–⋯O proton could results in large shifts in the resonance of the N1′ and to a lesser extent on N2′ which is directly conjugated to N1′ through the aromatic ring. As a result we choose the disregard these shifts in the evaluation. We note that recent reports of calculated 15N isotropic chemical shifts in the literature include both cases of excellent agreement81,82 or systematic overestimation with respect to experimental data.83
Intermolecular H-bonding drives selectivity
Structure 6b, which appears as the most credible model from among the idealised structures proposed here, is characterized by a significantly lower calculated energy than the other potential candidates 5, 6a and 6c. Examination of the inter-molecular binding modes in the lattice of 6b (Fig. 5b and c) reveals an optimized network of hydrogen bonds. 6b is the only structure whose ammonium and carbamate/carbamic acid moieties are engaged in three and two hydrogen bonds (Fig. 5d), respectively (vs. one and two at most in all other structures). This optimization of the hydrogen bond network not only provides subsequent stabilization to the molecular network but also presumably accounts for the significantly improved agreement between calculated and experimental data for structure 6b with respect to the remaining candidates. Importantly, this hydrogen bond network would appear to strongly depend on the nature of the constituent building blocks. In fact, polyamines of increased length or steric hindrance would compromise this optimal pairing. Therefore, the choice of the building block incorporated into the DMA2(CO2) controls the packing efficiency, which in turn drives the self-assembly process. The new picture that emerges here from a simplified model appears to explain the singular selectivity previously observed for the formation of DMA2(CO2). | ||
| Fig. 5 Crystal structure 6b which shows the best overall agreement with the experimental data. (a) Comparison of the structure of oligomer 6b in the crystal before (blue) and after all-atom geometry optimization with CASTEP (red). Packing between nearest neighbours along (b) z axis and (c) x axis. (d) Partial view of the intermolecular main and side chain binding. | ||
Powder X-ray diffraction
Powder X-ray diffraction and solid-state NMR are both powerful techniques which have been used independently or in tandem for crystal structure solution.21,24,29,31,35,84–86 We therefore did assess the possible contribution of PXRD to the structural modelling developed here in parallel to the NMR measurements. Fig. 6 shows the PXRD pattern obtained from the nanocrystalline material, and which is found to have quite high resolution with reflections beyond 30° indicative of a structure that has long range periodicity and well defined regular structural features. At first sight, it may thus be possible to index this pattern and determine the structure. Indexing of the experimental PXRD spectrum was first attempted using the McMaille program. Despite a thorough screening of the possible Bravais lattices and cell parameters, the procedure failed, leading to several possible solutions, even when combining two polymorphic structures. The main reason for failure appeared to be the lack of well-resolved peaks at large angles.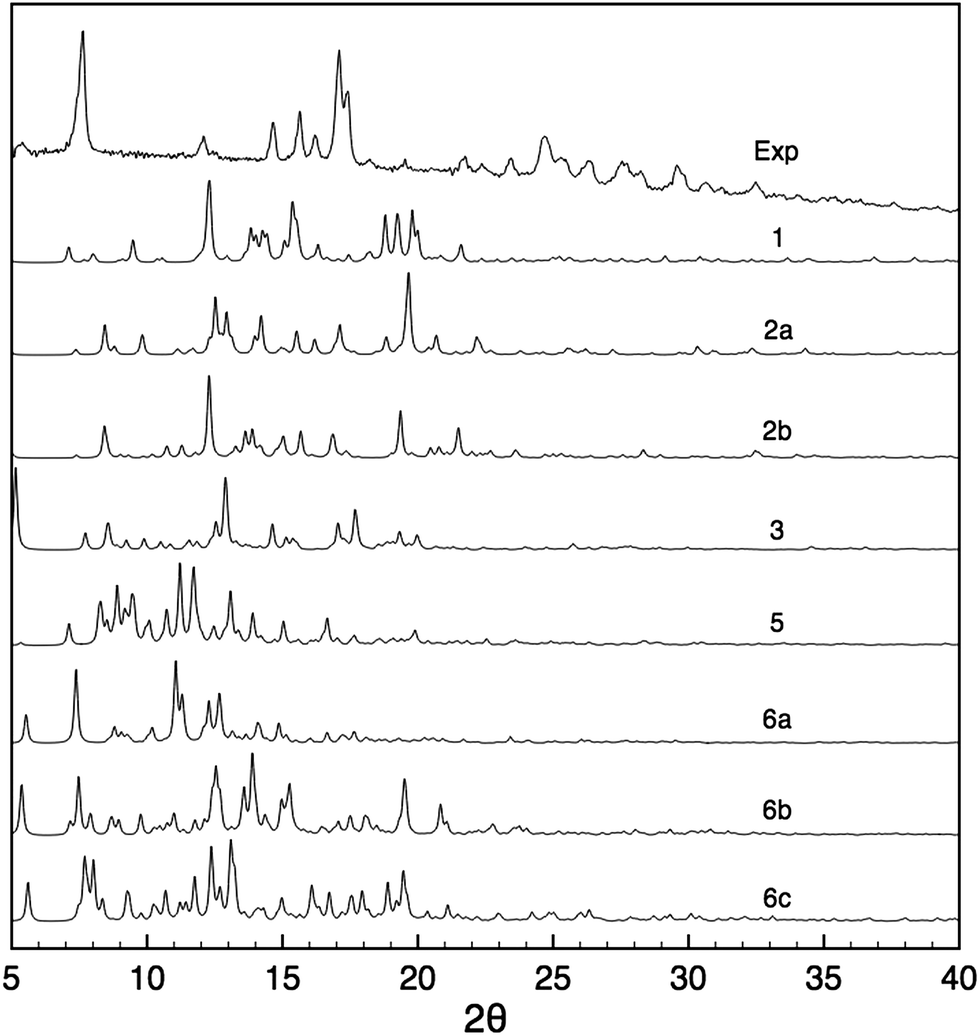 | ||
| Fig. 6 Comparison between the experimental PXRD pattern and the simulated spectra for structures 1–6 after all-atom DFT optimization. | ||
PXRD spectra were then simulated for all the candidate 1–6. It can be seen from Fig. 6 that the resulting patterns displayed very little similarity with the experimental spectrum, whether the oligomeric constituents were true candidates or not. Furthermore, the changes observed in the PXRD patterns predicted for structures before and after DFT geometry optimisation (Fig. S2†) are of the same order of magnitude as the difference between the candidate structures even though the structural changes induced by optimisation are relatively minor.
This highlights the fact that PXRD patterns are sensitive to very small structural changes, such that if the candidate structure is not within a few fractions of an angstrom in rmsd from the correct structure then poor agreement will always be obtained.85 Notably, changes in the unit cell parameters induce large changes in the PXRD line positions and intensities, and since indexing failed here it is unlikely that the candidate structures have exactly correct cell parameters. This is especially true if we recall that that the materials here may have some dispersion in the chain lengths and the hydration states that would almost certainly induce changes with respect to the model cells predicted here. We have seen that removing some empty space (which may contain water) from the structures by changing the unit cell dimensions while keeping the atomic coordinates fixed produces large changes in the predicted PXRD patterns (Fig. S24†), while including water molecules in the same empty space in the candidate structures also changes the predicted PXRD intensities.
For well ordered crystalline materials perfect agreement with PXRD can often be obtained and would be the gold standard in validating a final structure. However, in this case of a well ordered but slightly disperse material, and where indexing fails, we see that PXRD is not a useful indicator, whereas high resolution NMR data can still be obtained and directly related to the average properties of the structures. This is an excellent example of the complimentary nature of the two methods.
Conclusions
Structural elucidation of organized nano-crystalline powdered solids made of complex molecular ingredients represents an unresolved challenge to date. Here we have applied state of the art NMR crystallography methods to obtain a proposal for the structure of a complex three-component dynamic oligomeric material by using a series of solid-state MAS NMR experiments, including DNP enhanced spectroscopy. It has allowed to obtain the complete assignment of 1H, 13C and 15N resonances, and to determine a number of spatial proximities between atoms thereby providing strong constraints on possible structures. We used comparisons between experimental and calculated NMR chemical shifts to select the best model structures. This is one of the most complex systems for which solid-state NMR has been able to provide a de novo structure, starting only with the molecular formula, to date.Since the material contains disorder both in chain length and solvation the structure proposed here is certainly not exact (for example in terms of the predicted unit cell parameters), but we suggest it does capture the essential structural features. Dispersity in terms of oligomer length, and the integration of hydration, is well beyond the current scope of the prediction tools used here. However, this did not hamper the application of NMR and the extraction of quantitative constraints on the structure, whereas they did render comparison with PXRD of no use.
These results highlight the increasing capabilities of the combination of solid-state NMR, crystal structure prediction, periodic DFT plane wave calculations (and powder X-ray diffraction) for the determination of crystal structures when single crystal X-ray diffraction is not possible. The structure determined from NMR notably suggests the presence of an optimised intermolecular hydrogen bonding network that explains why the self-assembly of the material is so selective in terms of building blocks. Small changes in the building blocks would prevent formation of the H-bond network.
Acknowledgements
We are grateful to Prof. P. Tordo, Dr O. Ouari and Dr G. Casano (Aix-Marseille Université, France) for providing the biradicals used in the DNP NMR experiments. Financial support from the Agence Nationale de la Recherche (ANR-12-JS07-0011-01), the TGIR-RMN-THC FR3050 CNRS, and Swiss National Science Foundation grant no. 160112 for conducting the research is gratefully acknowledged. This work was supported by the LABEX iMUST (ANR-10-LABX-0064) of Université de Lyon, within the program “Investissements d'Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR).Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. O. Lee, D. M. Shacklady, M. J. Horner, S. Ferlay, M. W. Hosseini and M. D. Ward, Cryst. Growth Des., 2005, 5, 995–1003 CAS.
- M. W. Hosseini, in Applications of supramolecular chemistry, ed. H.-J. Schneider, CRC Press, 2012 Search PubMed.
- S. Ferlay and M. W. Hosseini, in Functional supramolecular architectures for organic electronics and nanotechnology, ed. P. Samori and F. Cacialli, Wiley-VCH, 2010 Search PubMed.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- E. F. Bres, S. Ferlay, P. Dechambenoit, H. Leroux, M. W. Hosseini and S. Reyntjens, J. Mater. Chem., 2007, 17, 1559–1562 RSC.
- P. Dechambenoit, S. Ferlay and M. W. Hosseini, Cryst. Growth Des., 2005, 5, 2310–2312 CAS.
- P. Dechambenoit, S. Ferlay, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2009, 1559–1561 RSC.
- G. Marinescu, S. Ferlay, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2013, 49, 11209–11211 RSC.
- M. W. Hosseini, Chem. Commun., 2005, 5825–5829 RSC.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- J. Leclaire, G. Husson, N. Devaux, V. Delorme, L. Charles, F. Ziarelli, P. Desbois, A. Chaumonnot, M. Jacquin, F. Fotiadu and G. Buono, J. Am. Chem. Soc., 2010, 132, 3582–3593 CrossRef CAS PubMed.
- The term was first coined by J-M. Lehn refering to a polymeric material spontaneously obtained from monomers assembled with reversible covalent and non covalent bonds.
- E. Kolomiets and J. M. Lehn, Chem. Commun., 2005, 1519–1521 RSC.
- E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC.
- F. Fotiadu, M. Jacquin, J. Leclaire, A. Methivier, P. A. Bouillon and P. Desbois, FR Pat., 2 969 504, 2012.
- F. Fotiadu, M. Jacquin, J. Leclaire, A. Methivier, P. Desbois and P. A. Bouillon, FR Pat., 2 969 503, 2012.
- J. Leclaire, G. Canard, F. Fotiadu and G. Poisson, World Pat., 2014188115 A1, 2014.
- R. K. Harris, Solid State Sci., 2004, 6, 1025–1037 CrossRef CAS.
- C. J. Pickard, E. Salager, G. Pintacuda, B. Elena and L. Emsley, J. Am. Chem. Soc., 2007, 129, 8932–8933 CrossRef CAS PubMed.
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed.
- M. Baias, C. M. Widdifield, J.-N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein, A. Lesage, G. M. Day and L. Emsley, Phys. Chem. Chem. Phys., 2013, 15, 8069–8080 RSC.
- D. H. Brouwer, R. J. Darton, R. E. Morris and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 10365–10370 CrossRef CAS PubMed.
- B. Elena, G. Pintacuda, N. Mifsud and L. Emsley, J. Am. Chem. Soc., 2006, 128, 9555–9560 CrossRef CAS PubMed.
- K. Kalakewich, R. Iuliucci and J. K. Harper, Cryst. Growth Des., 2013, 13, 5391–5396 CAS.
- T. Loiseau, L. Lecroq, C. Volkringer, J. Marrot, G. Ferey, M. Haouas, F. Taulelle, S. Bourrelly, P. L. Llewellyn and M. Latroche, J. Am. Chem. Soc., 2006, 128, 10223–10230 CrossRef CAS PubMed.
- L. Mafra, S. M. Santos, R. Siegel, I. Alves, F. A. Almeida Paz, D. Dudenko and H. W. Spiess, J. Am. Chem. Soc., 2012, 134, 71–74 CrossRef CAS PubMed.
- E. Salager, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, Phys. Chem. Chem. Phys., 2009, 11, 2610–2621 RSC.
- D. V. Dudenko, P. A. Williams, C. E. Hughes, O. N. Antzutkin, S. P. Velaga, S. P. Brown and K. D. M. Harris, J. Phys. Chem. C, 2013, 117, 12258–12265 CAS.
- X. Filip and C. Filip, Solid State Nucl. Magn. Reson., 2015, 65, 21–28 CrossRef CAS PubMed.
- D. Lüdeker and G. Brunklaus, Solid State Nucl. Magn. Reson., 2015, 65, 29–40 CrossRef PubMed.
- M. Sardo, S. M. Santos, A. A. Babaryk, C. López, I. Alkorta, J. Elguero, R. M. Claramunt and L. Mafra, Solid State Nucl. Magn. Reson., 2015, 65, 49–63 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- Z. Zujovic, A. L. Webber, J. Travas-Sejdic and S. P. Brown, Macromolecules, 2015, 48, 8838–8843 CrossRef CAS.
- Y. Jiang, J. Huang, B. Kasumaj, G. Jeschke, M. Hunger, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 2058–2059 CrossRef CAS PubMed.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- S. Devautour-Vinot, G. Maurin, C. Serre, P. Horcajada, D. P. da Cunha, V. Guillerm, E. d. S. Costa, F. Taulelle and C. Martineau, Chem. Mater., 2012, 24, 2168–2177 CrossRef CAS.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed.
- C. R. Murdock, N. W. McNutt, D. J. Keffer and D. M. Jenkins, J. Am. Chem. Soc., 2014, 136, 671–678 CrossRef CAS PubMed.
- J. Xu, V. V. Terskikh, Y. Chu, A. Zheng and Y. Huang, Chem. Mater., 2015, 27, 3306–3316 CrossRef CAS.
- M. Baias, A. Lesage, S. Aguado, J. Canivet, V. Moizan-Basle, N. Audebrand, D. Farrusseng and L. Emsley, Angew. Chem., Int. Ed., 2015, 54, 5971–5976 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- A. C. Dedios, J. G. Pearson and E. Oldfield, Science, 1993, 260, 1491–1496 CAS.
- J. C. Facelli and D. M. Grant, Nature, 1993, 365, 325–327 CrossRef CAS PubMed.
- N. Mifsud, B. Elena, C. J. Pickard, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2006, 8, 3418–3422 RSC.
- A. Rapp, I. Schnell, D. Sebastiani, S. P. Brown, V. Percec and H. W. Spiess, J. Am. Chem. Soc., 2003, 125, 13284–13297 CrossRef CAS PubMed.
- E. Salager, G. M. Day, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, J. Am. Chem. Soc., 2010, 132, 2564–2566 CrossRef CAS PubMed.
- J. R. Yates, S. E. Dobbins, C. J. Pickard, F. Mauri, P. Y. Ghi and R. K. Harris, Phys. Chem. Chem. Phys., 2005, 7, 1402–1407 RSC.
- J. R. Yates, T. N. Pham, C. J. Pickard, F. Mauri, A. M. Amado, A. M. Gil and S. P. Brown, J. Am. Chem. Soc., 2005, 127, 10216–10220 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- N. Jouault, R. Nguyen, M. Rawiso, N. Giuseppone and E. Buhler, Soft Matter, 2011, 7, 4787–4800 RSC.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS PubMed.
- R. Nguyen, E. Buhler and N. Giuseppone, Macromolecules, 2009, 42, 5913–5915 CrossRef CAS.
- D. M. Rudkevich and H. Xu, Chem. Commun., 2005, 2651–2659 RSC.
- P. T. Corbett, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 9390–9392 CrossRef CAS PubMed.
- P. Jackson, K. J. Fisher and M. I. Attalla, J. Am. Soc. Mass Spectrom., 2011, 22, 1420–1431 CrossRef CAS PubMed.
- Z. J. Guo, P. J. Sadler and E. Zang, Chem. Commun., 1997, 27–28 RSC.
- J. Schaefer, E. O. Stejskal, J. R. Garbow and R. A. McKay, J. Magn. Reson., 1984, 59, 150–156 CAS.
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K.-N. Hu, C.-G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A. van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211, DOI:10.1063/1.2833582.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwalder, D. Gajan, C. Coperet, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- H. Takahashi, B. Viverge, D. Lee, P. Rannou and G. De Paëpe, Angew. Chem., Int. Ed., 2013, 52, 6979–6982 CrossRef CAS PubMed.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwaerzwalder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Coperet, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- A. Zagdoun, A. J. Rossini, D. Gajan, A. Bourdolle, O. Ouari, M. Rosay, W. E. Maas, P. Tordo, M. Lelli, L. Emsley, A. Lesage and C. Coperet, Chem. Commun., 2012, 48, 654–656 RSC.
- P. C. A. van der Wel, K.-N. Hu, J. Lewandowski and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 10840–10846 CrossRef CAS PubMed.
- A. C. Pinon, A. J. Rossini, C. M. Widdifield, D. Gajan and L. Emsley, Mol. Pharm., 2015, 12, 4146–4153 CrossRef CAS PubMed.
- B. Elena, A. Lesage, S. Steuernagel, A. Bockmann and L. Emsley, J. Am. Chem. Soc., 2005, 127, 17296–17302 CrossRef CAS PubMed.
- B. Elena, G. de Paepe and L. Emsley, Chem. Phys. Lett., 2004, 398, 532–538 CrossRef CAS.
- B.-J. van Rossum, C. P. de Groot, V. Ladizhansky, S. Vega and H. J. M. de Groot, J. Am. Chem. Soc., 2000, 122, 3465–3472 CrossRef CAS.
- A statistical analysis has shown that there is a strong correlation between the geometrical feature of the molecules and the space group of their crystalline phases. For more details, see ref. 74 and 75.
- G. Pepe, S. Fery-Forgues and P. Jouanna, J. Cryst. Growth, 2011, 333, 25–35 CrossRef CAS.
- G. Pepe, R. Perbost, J. Courcambeck and P. Jouanna, J. Cryst. Growth, 2009, 311, 3498–3510 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101 CrossRef.
- T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1–20 CrossRef CAS PubMed.
- http://www.castep.org .
- W. Schilf, B. Kamieński, B. Kołodziej and E. Grech, J. Mol. Struct., 2004, 708, 33–38 CrossRef CAS.
- H. E. Kerr, L. K. Softley, K. Suresh, A. Nangia, P. Hodgkinson and I. R. Evans, CrystEngComm, 2015, 17, 6707–6715 RSC.
- S. A. Joyce, J. R. Yates, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 12663–12670 CrossRef CAS PubMed.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, CrystEngComm, 2012, 14, 2654–2659 RSC.
- N. Mercadal, S. P. Day, A. Jarmyn, M. B. Pitak, S. J. Coles, C. Wilson, G. J. Rees, J. V. Hanna and J. D. Wallis, CrystEngComm, 2014, 16, 8363–8374 RSC.
- K. D. M. Harris, M. Tremayne and B. M. Kariuki, Angew. Chem., Int. Ed., 2001, 40, 1626–1651 CrossRef CAS.
- A. Meden and I. Radosavljevic Evans, Cryst. Res. Technol., 2015, 50, 747–758 CrossRef CAS.
- R. K. Harris, Analyst, 2006, 131, 351–373 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra, modelled data and structures. See DOI: 10.1039/c5sc03810c |
| ‡ Present address: Institut de Chimie des Substances Naturelles (CNRS UPR 2301), 91190 Gif-sur-Yvette, France. |
| This journal is © The Royal Society of Chemistry 2016 |