
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of the CuII-catalyzed benzylic oxygenation of (aryl)(heteroaryl)methanes with oxygen†
Hans
Sterckx
 a,
Johan
De Houwer
a,
Carl
Mensch
a,
Ignacio
Caretti
b,
Kourosch Abbaspour
Tehrani
a,
Wouter A.
Herrebout
a,
Sabine
Van Doorslaer
b and
Bert U. W.
Maes
*a
a,
Johan
De Houwer
a,
Carl
Mensch
a,
Ignacio
Caretti
b,
Kourosch Abbaspour
Tehrani
a,
Wouter A.
Herrebout
a,
Sabine
Van Doorslaer
b and
Bert U. W.
Maes
*a
aDepartment of Chemistry, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium. E-mail: bert.maes@uantwerpen.be
bDepartment of Physics, University of Antwerp, Universiteitsplein 1, B-2610 Antwerp, Belgium
First published on 29th September 2015
Abstract
A mechanistic study of the copper-catalyzed oxidation of the methylene group of aryl(di)azinylmethanes was performed. Initial reaction rates were measured making use of in situ IR reaction monitoring and a kinetic analysis of the reaction was executed. The reaction proved to be first order in oxygen concentration. For substrate and acid concentration, saturation kinetics due to O2 mass transfer limitation were observed. The occurrence of mass transfer limitation was further confirmed by examining the effect of the stirring rate on the initial reaction rate. Interestingly, the effect of the concentration of the catalyst on the rate shows that higher loadings result in a maximal initial rate, followed initially by a steady decrease and subsequently a rate plateau when the concentration is increased further. Mass transfer limitation and increased concentration of dinuclear catalytically active species rationalizes this hitherto unprecedented rate behavior. Continuous-wave and pulsed electron paramagnetic resonance methods were used to characterize the catalytic species present in the solution during the reaction and confirmed the presence of both mono- and dinuclear copper species. Analysis of a diverse substrate scope points towards imine–enamine tautomerization as a crucial process in the oxidation reaction. DFT calculations of these equilibrium constants (pKeq) provided us with a qualitative tool to predict whether or not a substrate is viable for oxidation under the reaction conditions developed.
Introduction
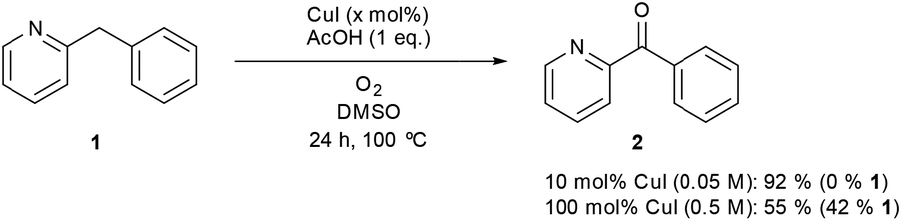
Two decades ago benzylic oxidation reactions made use of unsustainable oxidants such as potassium permanganate or chromium acid derivatives. This produced vast quantities of hazardous waste.1 While MnII and CrIII are present as trace elements in the human body as essential nutrients, CrVI is a known carcinogen and exposure to MnVII can lead to ‘manganism’, a central nervous system disorder.2 Nowadays these classical oxidants are mostly shunned and a major shift has taken place towards less toxic and sustainable oxidants like peroxides or molecular oxygen preferably in combination with a cheap transition-metal catalyst.3 Recently substantial progress has been made in the development of preparative base metal-catalyzed aerobic oxidation reactions of benzylic alcohols.4 The direct benzylic methylene oxygenation is however far less studied.5 Notable examples using peroxides as the terminal oxidant on active methylenes are reported by the groups of Bolm, Punniyamurthy, Chen and Sasson using respectively FeCl3, a CuII Salen-H4, a Mn(OAc)2·4H2O complex and CuCl2 as the catalyst.6,7 Methylene oxidations using copper and iron with molecular oxygen as the terminal oxidant are of special interest. After all, these metals are abundant, feature a low toxicity, and oxygen is the most sustainable oxidant on earth.8 There is a great number of important articles available on the structure and reactivity of various Cu- and, to a lesser degree, Fe-complexes with molecular oxygen.9 The stoichiometric study of these complexes provides a simplified mimic of the active site of several important metalloproteins (for instance: tyrosinase, dopamine β-monooxygenase, galactose oxidase or cytochrome P450) and are therefore easier to study. These complexes are usually very well defined and are formed by reaction of a reduced metal center (CuI and FeII) with O2 to form the reactive intermediate, which reacts directly with the substrate. Alternatively, they can also be formed by using metals in a higher oxidation state (CuII and FeIII) in combination with H2O2. A stabilizing nitrogen-containing ligand (bidentate or higher) is a prerequisite for these systems to be stable enough to be studied. Furthermore the ligand provides a mimic of the polypeptide chain around the metal center and can be used to tune the reactivity of the complex.In comparison to the reports describing well characterized model complexes in stoichiometric reactions, preparative protocols for oxygenation of active methylenes involving in situ formed copper and iron catalysts with oxygen as the stoichiometric oxidant are still limited.10,11 In 2012 our group published a communication describing a synthetic protocol for the copper- and iron-catalyzed oxidation of the methylene group of aryl(di)azinylmethanes with acetic acid as promoter (Scheme 1).11a
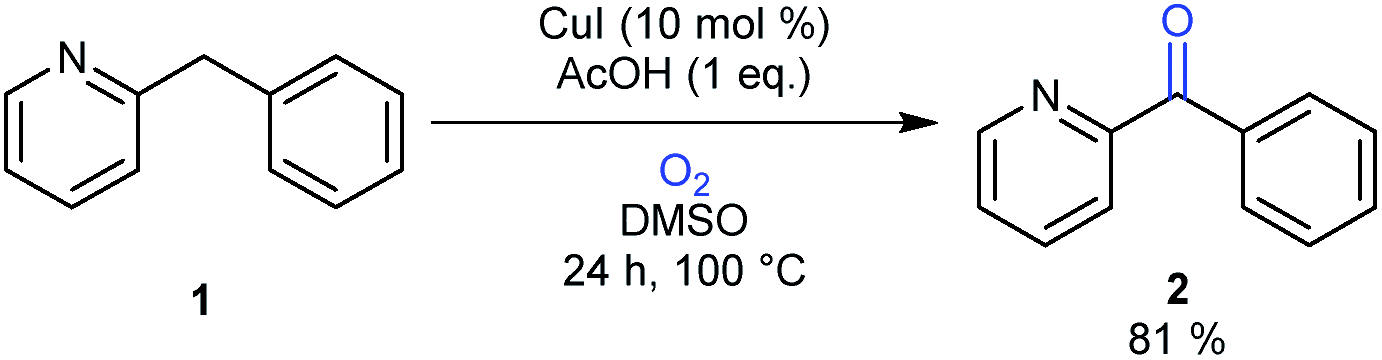 | ||
| Scheme 1 Oxidation of model substrate 2-benzylpyridine (1) to 2-benzoylpyridine (2) under the standard reaction conditions. | ||
Several groups have made creative use of these reaction conditions and some extended them to other substrate systems. Kappe successfully translated our protocol into an efficient flow process allowing scale up and continuous production.12 By slightly modifying our disclosed reaction conditions Touré achieved selective oxidation of C–H bonds in drug candidates and APIs delivering drug metabolites. The oxidation protocol could also be applied to oxygenate carbons alpha to a tertiary amine and therefore mimic or complement cytochrome P450.13 Very recently Zhuo and Lei disclosed a similar oxidation protocol in which ethyl chloroacetate was used as a promoter instead of acetic acid.14 A final interesting and notable example of an extension towards other functionalizations is the copper-catalyzed α-methylenation of benzylpyridines using DMA as a one-carbon source.15 When Na2S2O8 was used as an oxidant instead of O2, chemoselective methylation occurred over oxygenation of the methylene. Although there are several publications describing preparative methylene oxidation protocols based on copper or iron and O2, no detailed mechanistic studies (involving kinetics) have been performed yet to support the proposed catalytic cycles. As copper and iron salts easily form polynuclear species, the nature of the catalyst in these reactions remains also unknown. To allow further developments in this field, knowledge of the catalytically active species and the catalytic cycle is crucial. In this paper we disclose the results of mechanistic studies on the oxidation of 2-benzylpyridine under copper catalysis with oxygen as oxidant as model system (Scheme 1), leading to an insight into the actual reaction mechanism of this intriguing transformation. DMSO was chosen as the solvent for this mechanistic study to avoid solubility problems considering the large ranges of concentrations of the different reaction components that had to be used.
Results and discussion
Substrate scope: structural requirements
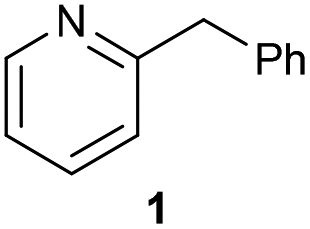
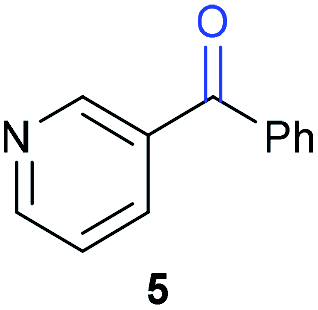
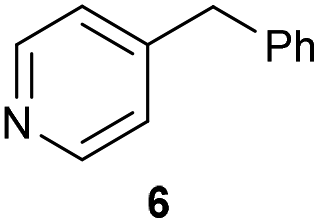
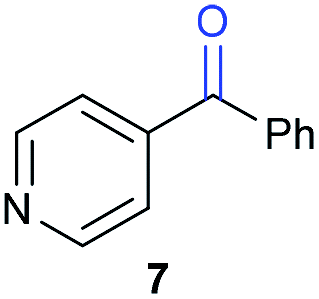
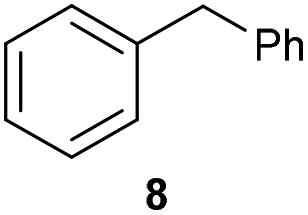
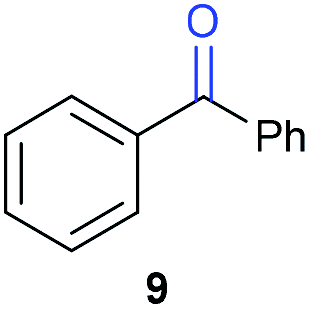
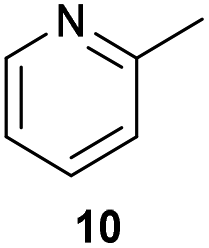
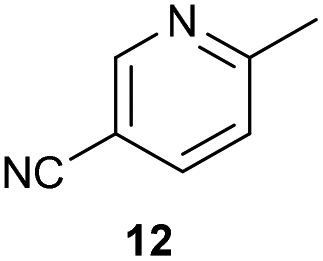
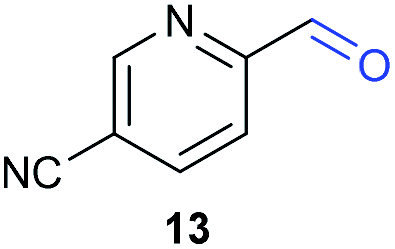
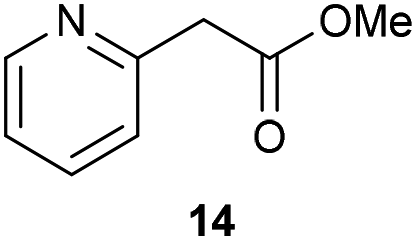
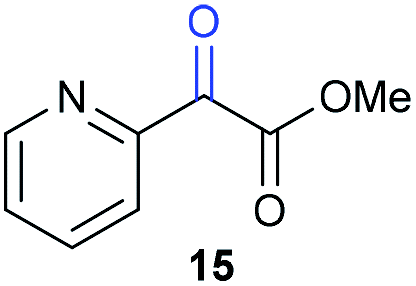
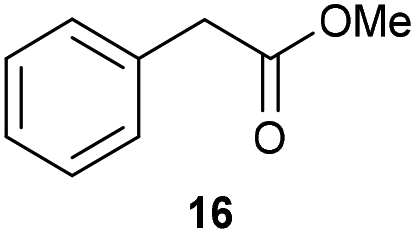
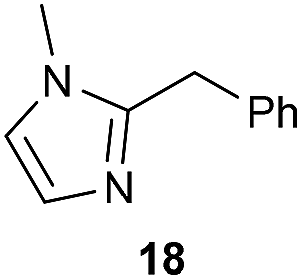
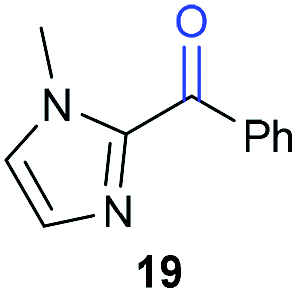
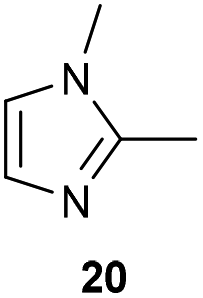
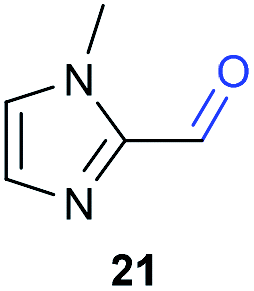
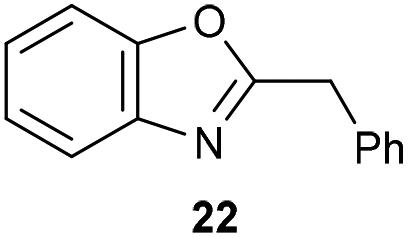
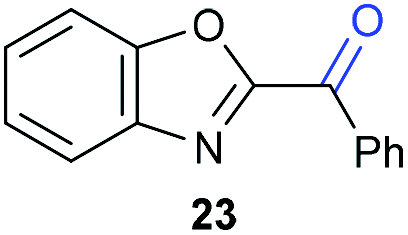
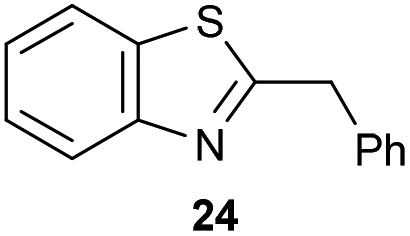
The previously developed reaction conditions (Scheme 1) allowed to oxidize 2- (1) and 4-benzylpyridine (6), while no oxidation of 3-benzylpyridine (4) and diphenylmethane (8) occurred.11a From this was concluded that the oxidation is not a directed reaction since coordination of the metal catalyst to the nitrogen in 4-benzylpyridine would bring the metal too far away from the benzylic position to promote an intramolecular reaction. Instead, these observations point to an imine–enamine tautomerization as a prerequisite for the reaction to proceed. Besides, the acidity of the methylene protons is also important since 2-methylpyridine (10) could not be oxidized. Further support that a combination of these two factors is necessary was gathered by performing experiments on a selected set of substrates (Table 1). When the acidity of the methylene in 10 is increased by introducing an electron-withdrawing group, either on the pyridine ring or on the methylene group, the substrate can be oxidized again as exemplified by methyl 2-(pyridin-2-yl)acetate (14) and 6-methylnicotinonitrile (12). A control experiment with methyl 2-phenylacetate (16) proves that the imine moiety is still necessary and that activation by the methyl ester alone is not sufficient for this reaction to proceed. To test whether this oxidation protocol can also be used beyond azine type systems it was applied to azoles; 2-benzyl-1-methyl-1H-imidazole (18) and 1,2-dimethylimidazole (20). The phenyl-activating group appears to still be a necessary structural element since we were only able to oxygenate the former substrate, similarly to what has been observed for 10. Other 2-benzylazoles can also be used, as shown by the oxidation of 2-benzylbenzoxazole (22) and 2-benzylbenzothiazole (24), further underlining the general applicability of the oxidation method.| Entry | Substrate | Product | pKeq | Yielda (%) |
|---|---|---|---|---|
| a Substrate (0.5 mmol), CuI (10 mol%) AcOH (1 eq.), DMSO (1 mL), O2 (balloon), 100 °C, 24 h. b At 120 °C. c In n-BuOAc (1 mL) with addition of molecular sieves. d Compound reported in our communication, see ref. 11a. | ||||
| 1 |
|
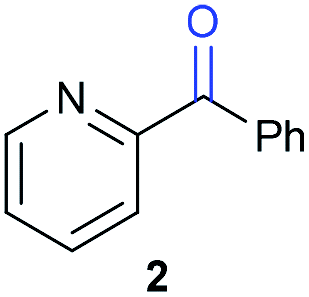
|
11.5 | 92d |
| 2 |
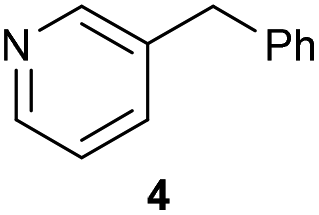
|
|
20.4 | 0d |
| 3 |
|
|
11.0 | 89d |
| 4 |
|
|
21.7 | 0d |
| 5 |
|
|
15.9 | 0d |
| 6 |
|
|
13.0 | 27 (60)b |
| 7 |
|
|
6.2 | 23 (83)c |
| 8 |
|
|
18.8 | 0 |
| 9 |
|
|
13.9 | 34 (85)b |
| 10 |
|
|
19.0 | 0 |
| 11 |
|
|
9.1 | 98 |
| 12 |
|
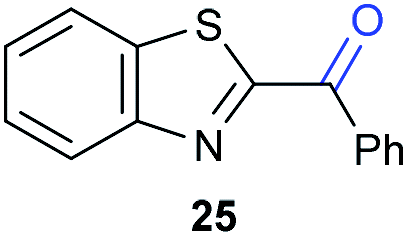
|
8.1 | 99 |
Imine–enamine tautomerization: computational study
The structural features of substrates suitable for oxidation under our protocol (vide supra) indicated that the mechanism of the oxidation seems to revolve around an (initial) imine–enamine tautomerization. The imine–enamine tautomerization can be thermodynamically quantified by an equilibrium constant Keq (Fig. 1). To gain more insight in these tautomerizations, the pKeq values for the different experimentally tested substrates were calculated using density functional theory (DFT, Table 1).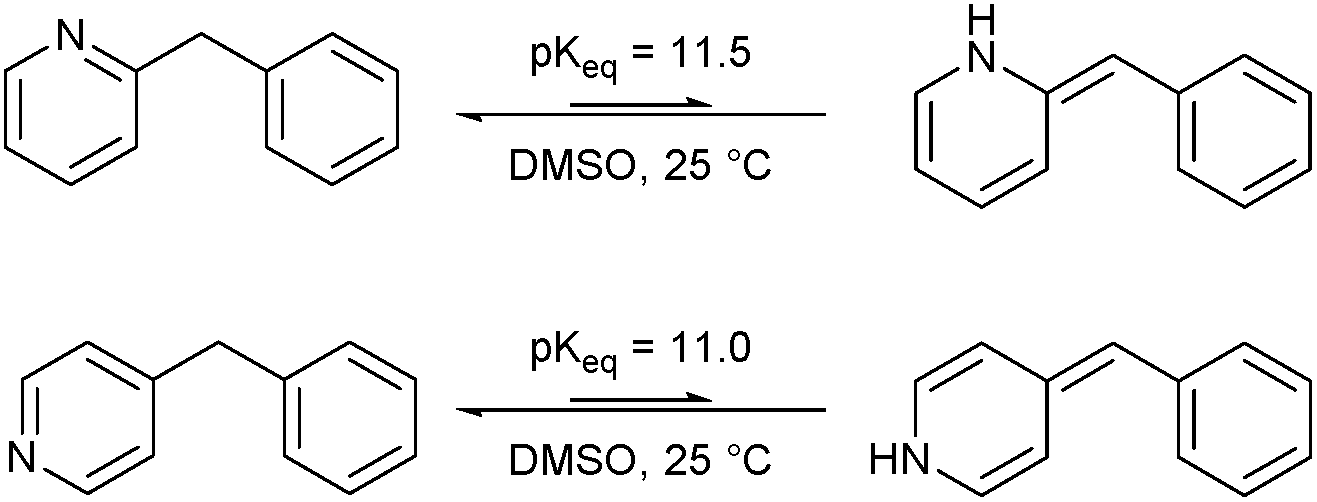 | ||
| Fig. 1 Imine–enamine equilibrium of 2- and 4-benzylpyridine. | ||
First, the geometries of the different conformations of both the imine- and the enamine-form were optimized at the B3PW91/aug-cc-pVDZ level of theory. Due to the rather large dipole moment of the molecules and the difference in dipole moment of the imine and the respective enamine tautomer (Table S5†), the self-consistent reaction field (scrf) model was used to account for solvent–solute interactions with DMSO. In a second phase, the most stable imine and respective enamine conformer were used to calculate the Gibbs free energy (ΔG°) and the desired equilibrium constant Keq (see ESI† for more details). All calculations were performed using the Gaussian09 program. In Table 1 the results of these calculations for the different experimentally tested benzylic systems are summarized. These calculated values do not have a direct absolute meaning but can be compared relatively between different substrates.
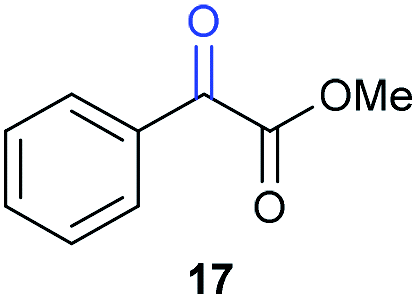
Benzylpyridines (1 and 6) have a pKeq value of 11.5 and 11.0, respectively, which is up to 9 orders of magnitude lower than 3-benzylpyridine (4), with a pKeq value of 20.4. This is because in 3-benzylpyridine stabilization by an enamine tautomeric form is not possible, as is also the case for diphenylmethane (8). Furthermore, without the second aromatic ring attached to the methylene center the pKeq rises with about 4 orders of magnitude, as exemplified by 2-methylpyridine (10). Introducing an electron withdrawing group either on the pyridine or on the methylene moiety results in a higher equilibrium constant (12 and 14). Although a keto–enol tautomerization can be drawn for the methylester functionality of methyl 2-phenylacetate (16) it still provides a relatively high pKeq value (18.8) (see Table S4† for more details on the tautomerization equilibria).
Interestingly, from Table 1, a clear correlation exists between the calculated pKeq values and whether oxidation occurs or not for each substrate. Only the substrates with a sufficiently low pKeq or high Keq value are reactive enough to be oxidized. Empirically, based on Table 1 and the pKeq values calculated for substrates previously published, one can deduce the maximum pKeq value for the reaction to proceed under our standard reaction conditions to be around 15.
Homolytic benzylic C–H bond dissociation energies (BDE) of the molecules in Table 1 were also calculated (see Table S6†). If a correlation exists this would be an indication for a mechanism in which the active catalyst abstracts a hydrogen atom from the substrate before quenching with O2. However, no correlation could be found since the bond dissociation energies of diphenylmethane (8), 2-, 3-, and 4-benzylpyridine (1, 4, and 6) are similar while there is a major difference in their reactivity in our oxidation protocol. DFT calculations thus provide an interesting qualitative tool to predict whether or not a substrate will be viable for oxygenation under our reaction conditions and strongly suggest that the oxidation reaction proceeds via the enamine tautomer as the reactive species.
Kinetic experiments
In the oxidation reaction of 1, small amounts of phenyl(pyridin-2-yl)methanol (3) were observed besides the desired 2-benzoylpyridine (2).11a This raised the question whether formation of the 2-benzoylpyridine (2) proceeds via phenyl(pyridin-2-yl)methanol (3) as an intermediate (Fig. 2, pathway 1a/1b) or via a pathway directly from 2-benzylpyridine (1) (Fig. 2, pathway 2), in which case the alcohol is merely a formed side product.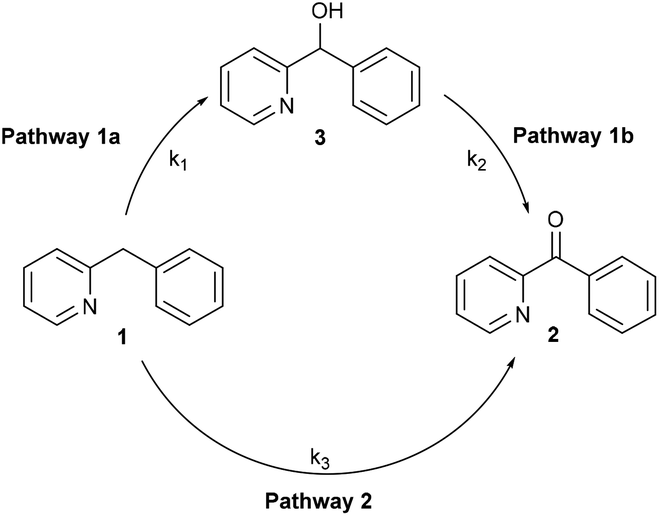 | ||
| Fig. 2 Two possible reaction pathways in the oxidation of 1. | ||
As β-hydride elimination of copper alkoxide complexes is known but rare, we first tested whether alcohol 3 could be oxidized under our reaction conditions.16 When the optimal reaction conditions were applied (Scheme 2), ketone 2 was isolated in 77% yield with 12% recovery of 3.
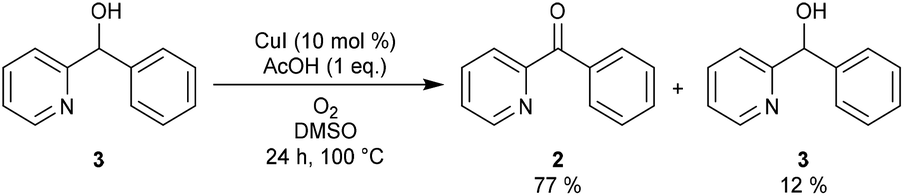 | ||
| Scheme 2 Copper-catalyzed oxidation reaction of 3 under standard conditions. | ||
To gain more insight we performed a kinetic study using in situ IR monitoring. The concentration of ketone 2 was monitored over time (CO stretch frequency) and the initial reaction rate was determined. Both the oxidation of 1 and 3 (Fig. 3) were followed in function of time. For each of these transformations the results were compared to a similar reaction where 1 equivalent of the radical scavenger 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) was added to the reaction mixture. Fig. 3 (left) shows that the presence of TEMPO has almost no influence on the formation of ketone 2, starting from 1. However, the oxidation of alcohol 3 is significantly accelerated in the presence of the radical scavenger (Fig. 3, right). Stahl already reported that smooth oxidation of alcohols can be achieved by the use of a radical catalyst, such as TEMPO, in combination with a copper catalyst and oxygen as the stoichiometric oxidant, which accounts for these observations.17 Assuming that the reaction proceeds exclusively via pathway 1a/1b the first step (1a) should be rate determining to account for a benzylic oxidation in 1 to ketone 2 which is not influenced by a radical scavenger, while the oxidation of alcohol 3 (pathway 1b) to the same product is significantly accelerated by its addition. Contradictorily, a comparison between the initial rate of oxidation of 1 and 3, shows that the oxidation of the latter is in fact slower than the oxidation of the former. On the other hand, if the second step (pathway 1b) would be rate determining, one expects a build-up of alcohol in the reaction mixture. This is not the case since only small amounts are present during the reaction course (vide supra). These contradictions imply that an indirect pathway 1a/1b can be excluded and our reaction proceeds via a direct pathway 2 with the formation of 3 as side compound. In addition to the kinetic experiments using TEMPO we also tried to isolate TEMPO-adducts to find out whether stable radical species were formed during the reaction.18 No such adducts could be observed via LC-MS analysis. When using 1,1-diphenylethylene as an alternative radical scavenger the oxidation reaction was also not inhibited and 1 was fully converted into ketone 2 and 1,1-diphenylethylene was quantitatively recovered.19a Next to these soluble radical scavengers activated carbon was used as a solid scavenger. Also in this case no inhibition could be detected (Fig. S27†).19b These results suggest that no stable organic radical species are formed in solution during the course of this reaction. This is also supported by EPR analysis (vide infra).
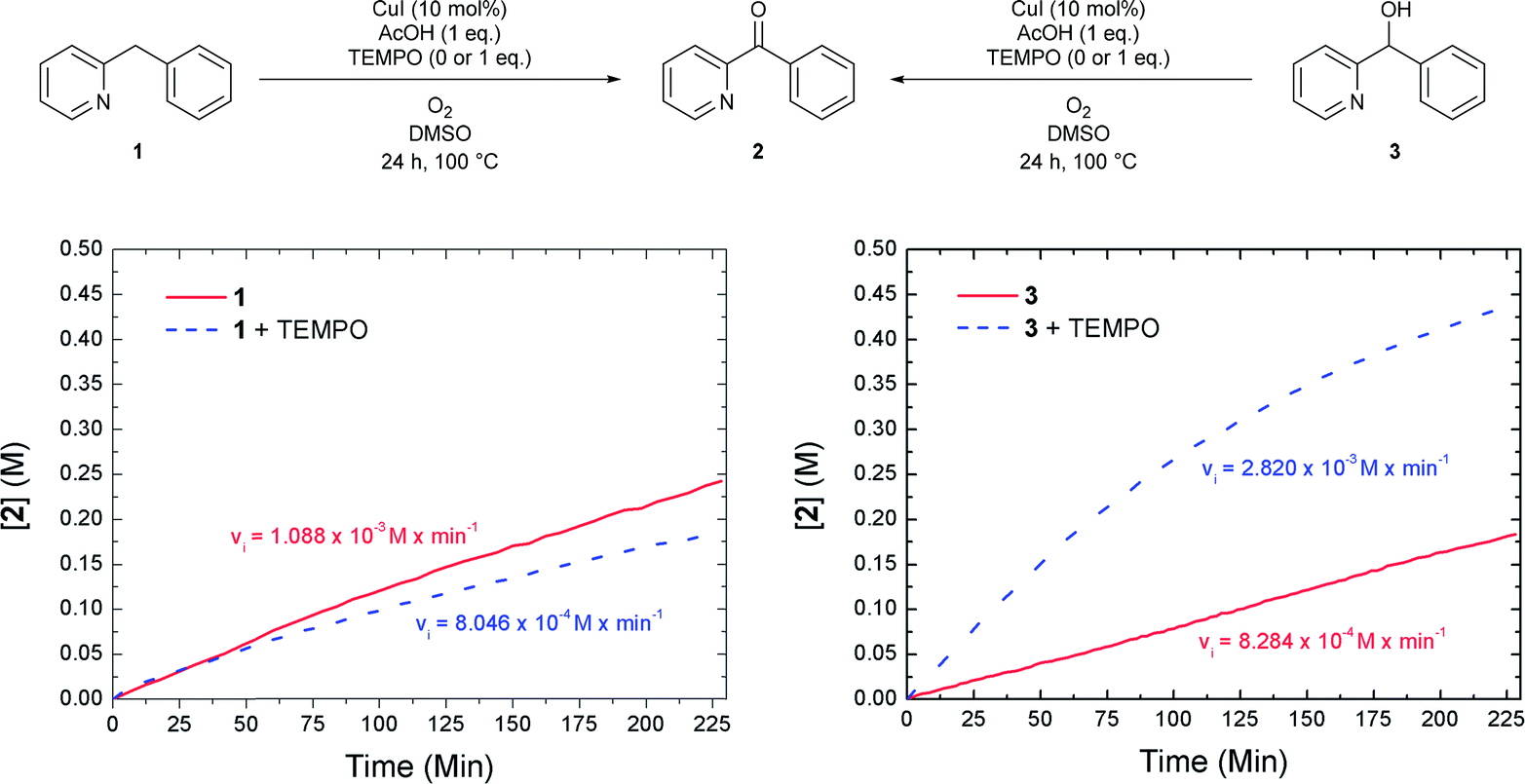 | ||
| Fig. 3 Effect of TEMPO on the rate of the oxidation reaction of 1 and 3. | ||
Considering that TEMPO, 1,1-diphenylethylene and activated carbon are bimolecular probes and oxygen is also a radical trap, we synthesized a substrate which can act as an intramolecular probe for a benzylic radical. 2-(2-Allylbenzyl)pyridine (26) was selected for this purpose. While benzylic radical clocks are known to be relatively slow due to the inherent stability of the benzylic radical, the reported unimolecular rate constant (k = 3 × 102 s−1) is still a factor of 107 faster than our overall maximum reaction rate (2 × 10−5 M s−1) (vide infra).19c (2-Allylphenyl)(pyridin-2-yl)methanone (27) was formed and no ring closed products due to radical cyclization could be detected in the reaction mixture (Scheme 3). Formation of 27 further supports that a free benzylic radical is not involved in our reaction protocol.
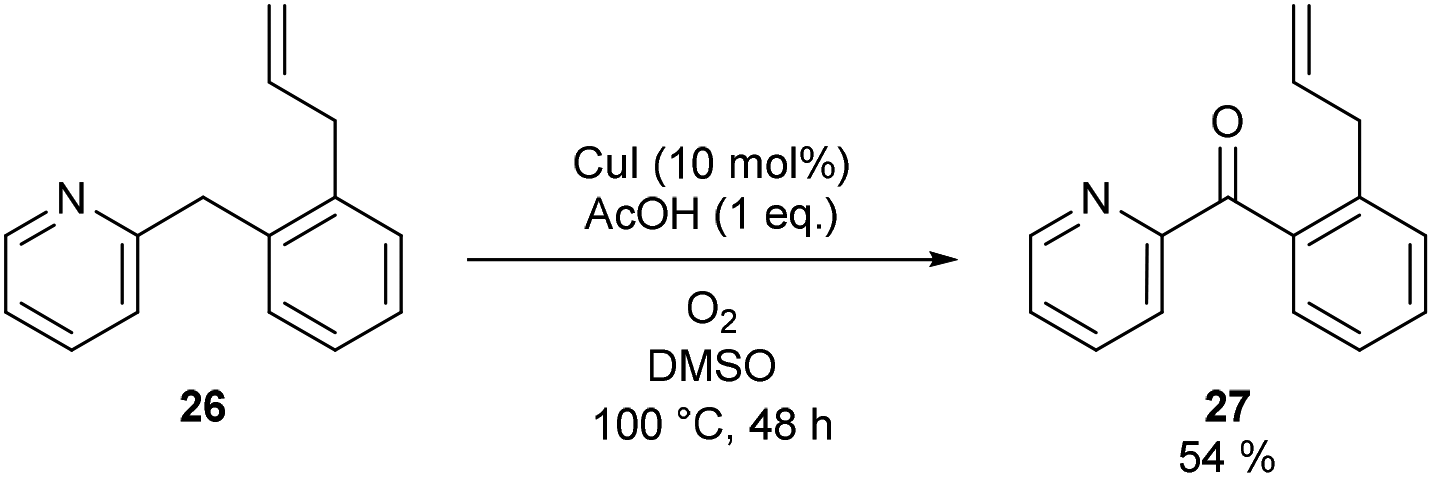 | ||
| Scheme 3 Benzylic radical clock reaction. | ||
It is important to determine whether mass-transfer effects are in play since O2 is a gaseous reagent.20 The effect of the stirring on the initial reaction rate was examined at 0.2 M of catalyst (Fig. 4). It was found that up until 550 rpm there is a first order relation of the stirring rate on the initial rate of the reaction. At higher stirring rates the vi becomes independent. From this we can conclude that at a certain reaction rate (vmax) the uptake of O2 becomes rate limiting. From the stirring rate experiment we see that the value of vmax is approximately 1.2 mM min−1. Under our standard reaction conditions we work at a stirring rate of 700 rpm and thus no mass-transfer effects are in play.
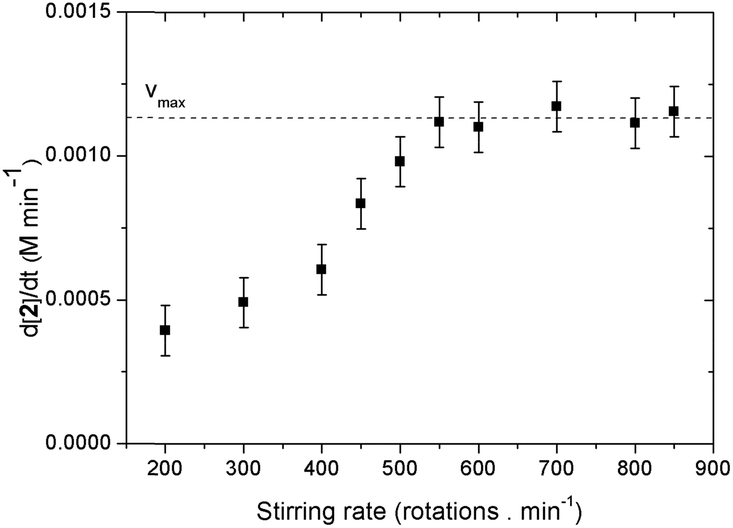 | ||
| Fig. 4 Effect of the stirring rate on the initial reaction rate. | ||
A first order dependence was found for the O2 partial pressure (Fig. 5, upper right, solid line). Since this first order relation might also derive from the observed mass transfer limitation effects we also examined the influence of the O2 partial pressure at a very low catalyst concentration (6.25 mM versus 50 mM) to make sure that no mass transfer limitation is in play. In this instance again a first order relation was found (dashed line). From this we can conclude that the observed first order relation derives directly from the involvement of O2 in or before the rate determining step.
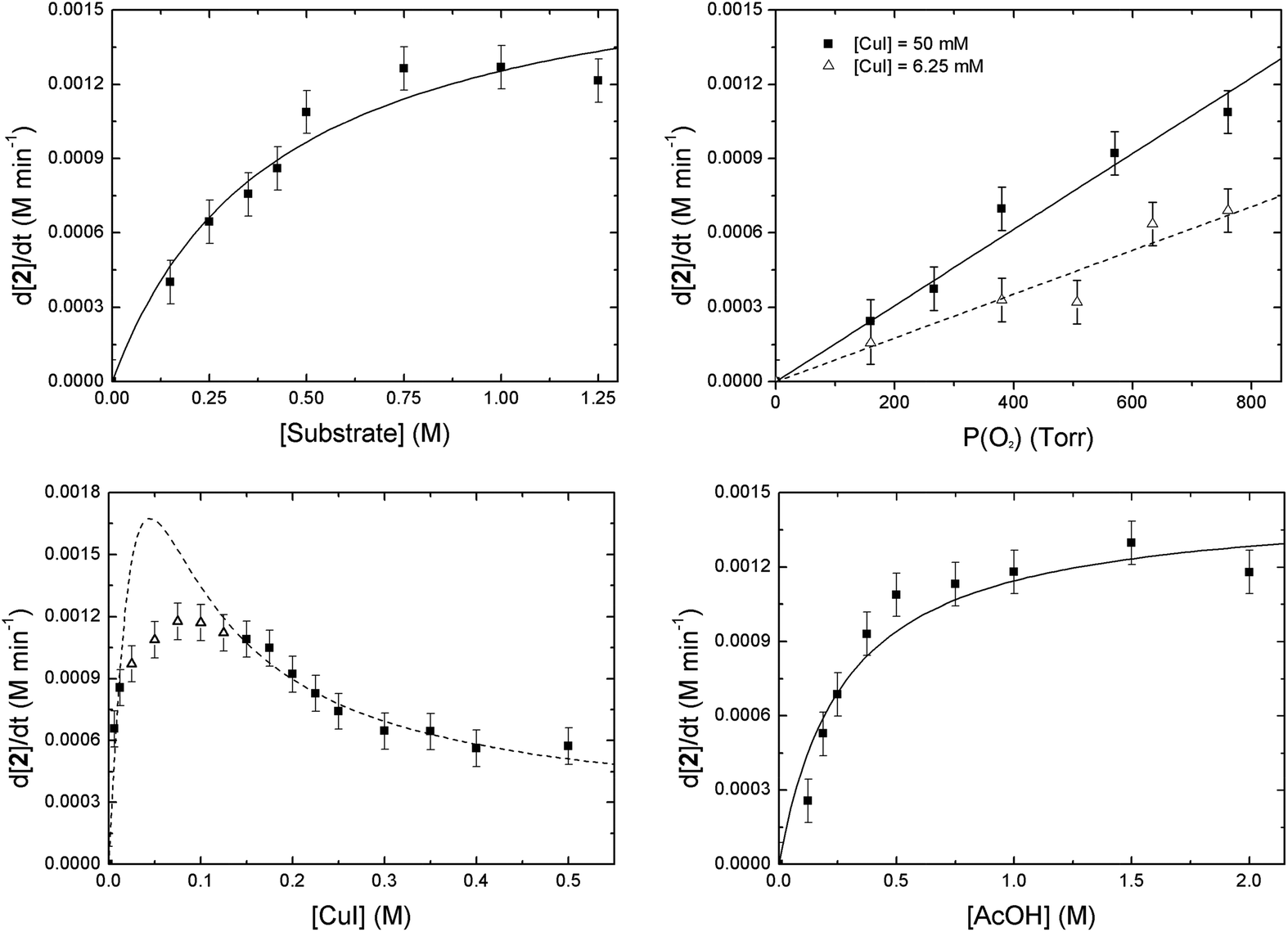 | ||
| Fig. 5 Dependence of the copper-catalyzed oxidation of 2-benzylpyridine (1) on [1] (upper left), O2 partial pressure (upper right), [Cu] (lower left) and [AcOH] (lower right) on the initial reaction rate. The dashed curve for the [CuI] (lower left) is a fit of the following equation: d[2]/dt = (c1[Cu]2 + c2[Cu])/(c3[Cu]2 + c4[Cu] + c5) to the datapoints (■). The data points for the [AcOH] and [1] respectively fit to the following equations: d[2]/dt = c1[AcOH]/(c2[AcOH] + c3) and d[2]/dt = c1[1]/(c2[1] + c3). Reaction conditions: 1 (5 mmol), CuI (10 mol%), AcOH (1 eq.), O2 (balloon), DMSO (10 mL), 100 °C. | ||
For the substrate (1) as well as for the acetic acid concentration saturation kinetics are seen. A quasi linear (first order) dependence is observed when less than 1 equivalent (0.5 M) of acetic acid or 1 is used (Fig. 5, upper left and lower right). When higher concentrations are used the reaction approaches zeroth order for the acid and 1 concentrations. This saturation behavior with vmax at approximately 1.2 mM min−1 is due to the occurrence of O2 mass transfer limitation conditions as was the case for the stirring experiments (vide supra).20 The measured data points are in accordance with hyperbolic functions (see ESI† for derivations). An alternative explanation for the saturation behavior might be the formation of a pre-equilibrium between substrate, AcOH and catalyst before rate determining oxygenation.17b This scenario, however, can be excluded. After all, when the concentration of these three components was raised simultaneously, thereby keeping their respective ratios constant, the initial rate again showed the same vmax value (Fig. S16†).
The dependence of the reaction rate on the catalyst concentration follows a more complex pattern. At very low catalyst concentrations the rate increases very fast in a hyperbolic fashion. At a rate of 1.2 mM min−1 it also reaches a maximum and after that the reaction rate becomes invariant of the catalyst concentration. The value of this maximum is determined by the O2 mass transfer limitation and corresponds to the vmax that was observed for [AcOH] and [1]. However, when the catalyst loading is further increased, the reaction rate diminishes significantly (Fig. 5, lower left) to finally settle at a constant rate independent of the catalyst concentration. As the occurrence of mass transfer limitation would result in a hyperbolic curve approaching 1.2 mM min−1 as vmax, the significant drop in initial rate cannot be explained by the occurrence of mass transfer limitation and another factor has to be taken into account. Other examples of non-linear dependence on [catalyst] for aerobic oxidations have previously been reported by the groups of Stahl and Sheldon.17b,21 We attribute the continuous drop in rate to the existence of dinuclear Cu-species. Trapping the active catalyst in a dinuclear species can inhibit the reaction since adding more catalyst will only result in the formation of more dimer via a second order process in [catalyst]. The non-zero reaction rate at very high catalyst loadings can only be explained by considering that these dinuclear species are also catalytically active (rate constant kd), however substantially less reactive than the mononuclear species (rate constant km) (km > kd). This is interesting as usually non-reactive dinuclear complexes are considered. A rate law (eqn (1)) was derived (see ESI†) which takes into account reactive di-nuclear complexes but no mass transfer limitation.22 A very good fit could be obtained for the data points before and after mass transfer limitation (dashed line). Between a copper concentration from 25 mM to 125 mM (triangles) where O2 diffusion is at play the simulated rate is much higher than the measured data points as expected.
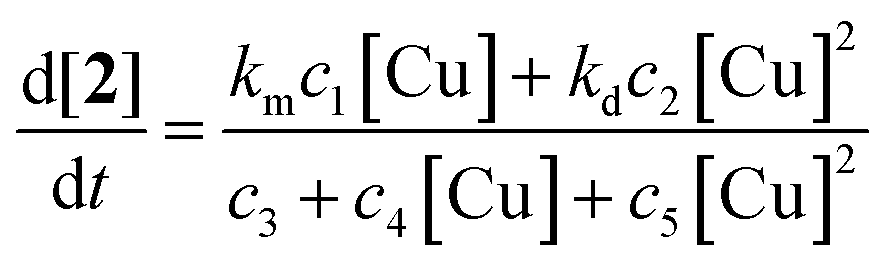 | (1) |
To support the catalytic activity at very high catalyst loading an experiment was performed with a stoichiometric catalyst loading. Significant conversion and a moderate yield was seen after 24 hours (Scheme 4) which is in accordance with the kinetic measurements. No significant side reactions take place at high catalyst loading as the reaction gave a good mass balance (>90%).
 | ||
| Scheme 4 Control experiments with (super) stoichiometric amounts of catalyst. | ||
To exclude the possibility of any catalyst degradation the reaction was monitored with in situ IR and, after the reaction was complete, another batch of substrate (1) was added. The second reaction proceeded with no significant loss in reaction rate (see Fig. S21†). Work-up of this reaction mixture provided ketone 2 in 92% yield with no starting material remaining. From this we conclude that no significant degradation of the catalyst to inactive (dimeric) species is occurring.
Structure of the catalytic species: an EPR-study
In order to identify the copper species present during the reaction, continuous-wave (CW) and pulsed EPR experiments were performed on the oxidation reaction of the model substrate 1 shown in Scheme 1.23 In the initial screening of catalysts it was shown that the reaction worked well for most copper sources, irrespective of the oxidation state (Cu0, CuI, CuII).11a As we are working under O2 atmosphere in the presence of acetic acid, the active catalytic species is likely to contain a CuII center and the reaction therefore involves initial oxidation when salts at lower oxidation state are used as precatalyst (eqn (2)).24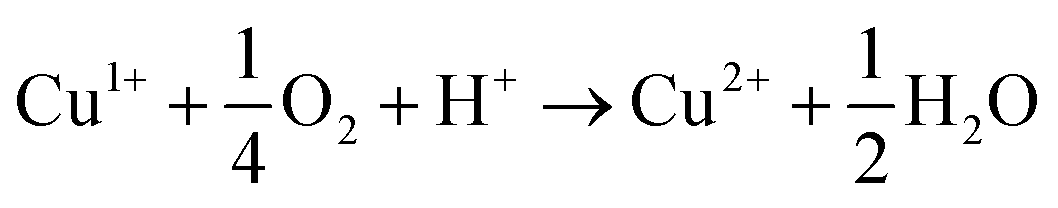 | (2) |
The oxidation of CuI in the presence of pyridine to hydroxide-bridged polynuclear complexes has been well-documented.25 The situation in our case is different: since we are able to start from a CuII source. This would require the use of H2O2 in stead of O2 to end up with the same reactive species. Possible anions for the CuII species are iodide, acetate and hydroxide. The latter is a result from the oxidation of CuI while the former two are deliberately added to the reaction. Since no ligand is added it is likely that DMSO will ligate (S- or O-ligation) to the copper center to stabilize it in solution.26 Alternatively the substrate (1) or reaction product (2) can also act as a ligand. The intermediate reacting with O2 likely contains a CuII–C(sp3) bond with the methylene carbon (Fig. 10, B and B′). As stated above this bond can be formed through initial tautomerization of substrate followed by reaction with copper species. However, since the equilibrium of this tautomerization is highly shifted towards the imine tautomer we expect the concentration of this intermediate to be very low during the course of the reaction.
Firstly, control CW-EPR measurements at 100 K were performed on a solution of 0.05 M CuI in DMSO heated to 100 °C during 10 minutes under O2 atmosphere (Fig. S1(a)†). The CuII complex observed (species I) (Table 2, entry 1) has g and copper hyperfine values that are similar to those reported earlier for CuBr2·2H2O in DMSO and is in accordance with the complexation of CuII with DMSO ligands.27 The same experiment was performed, but now in the presence of 0.5 M AcOH (Fig. S1(b)†); copper(II) acetate is known to exist mainly in a dinuclear form Cu2(OAc)4 in acetic acid and other solvents, with only a small amount of mononuclear Cu(OAc)2 complex present.28 Both types of CuII species (monomeric and multimeric) can be distinguished by EPR. Indeed, two CuII mononuclear complexes (species I′ and II) are detected (Table 2, entry 2, Fig. S1(b)†), together with high-field signals from CuII dimers (Fig. S2†). Species I′ has parameters similar to those of the complex observed without acetic acid and can be assigned to the same complex. The small shifts in the parameters are induced by the change in the dielectric constant of the mixture. Species II features EPR parameters similar to those observed earlier for Cu(OAc)2·2H2O in different solvents.29 With regard to the dinuclear species, the strong antiferromagnetic coupling within the pair of copper ions in Cu2(OAc)4 generally results in a triplet state that lies approximately 250–300 cm−1 (=|J|) above the singlet state, with typical zero-field splitting (D) for the triplet of the order of 0.34 cm−1 and characteristic EPR spectra.30 Fig. S2† shows the temperature dependence of one of the high-field signals of this contribution, from which the exchange coupling J can be determined (J = −274 cm−1), in agreement with what was earlier reported for Cu2(OAc)4(H2O)2, namely J = −269 cm−1 in frozen acid solution.31 In the EPR spectra of the mixture without addition of acetic acid (Table 2, entry 1), no such dinuclear copper complex could be found, in accordance with the absence of bridging ligands.
| Entry | [CuI] (M) | Heating time | g x ± 0.002 | g y ± 0.002 | g z ± 0.001 | |Ax| (MHz) ± 15 | |Ay| (MHz) ± 15 | |Az| (MHz) ± 5 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a The relative contributions of the different species to the experimental spectra are given in the ESI. b For the simulation of the contribution of species III in reaction mixture with starting concentration [CuI] = 0.05 M, the interaction with two 14N nuclei with a isotropic hyperfine coupling of 40 ± 5 MHz had to be taken into account. c The 14N hyperfine interaction was not resolved due to the increased linewidth, but was considered in the simulation. | ||||||||||
| 1 | CuI/DMSO | 0.05 | 10′ | I | 2.082 | 2.087 | 2.405 | 45 | 55 | 385 |
| 2 | CuI/DMSO/AcOH | 0.05 | 10′ | I′ | 2.081 | 2.083 | 2.403 | 26 | 25 | 375 |
| II | 2.080 | 2.080 | 2.374 | 30 | 26 | 430 | ||||
| 3 | CuI/DMSO/AcOH/(1) | 0.05 | 5′ | IIIb | 2.070 | 2.070 | 2.333 | 25 | 25 | 440 |
| IV | 2.062 | 2.063 | 2.286 | 35 | 35 | 510 | ||||
| 4 | CuI/DMSO/AcOH/(1) | 0.05 | 10′ | IIIb | 2.070 | 2.070 | 2.333 | 25 | 25 | 440 |
| IV | 2.062 | 2.063 | 2.286 | 35 | 35 | 510 | ||||
| 5 | CuI/DMSO/AcOH/(1) | 0.05 | 1 h | IV | 2.063 | 2.063 | 2.285 | 30 | 30 | 510 |
| 6 | CuI/DMSO/AcOH/(1) | 0.05 | 4 h | IV′ | 2.063 | 2.063 | 2.290 | 30 | 30 | 505 |
| V | 2.067 | 2.067 | 2.262 | 35 | 35 | 560 | ||||
| 7 | CuI/DMSO/AcOH/(1) | 0.25 | 10′ | IV | 2.066 | 2.066 | 2.286 | 35 | 35 | 510 |
| IIIc | 2.072 | 2.072 | 2.330 | 25 | 25 | 450 | ||||
| 8 | CuI/DMSO/AcOH/(1) | 0.25 | 4 h | IV | 2.062 | 2.063 | 2.285 | 35 | 35 | 510 |
| IIIc | 2.075 | 2.072 | 2.334 | 25 | 25 | 455 | ||||
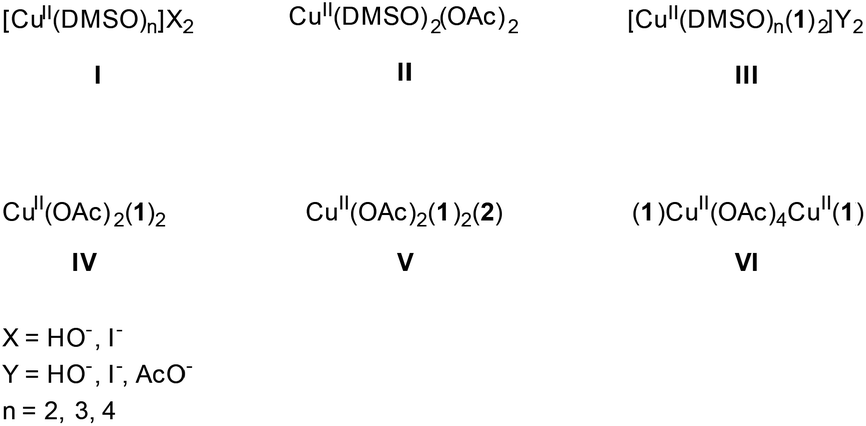 | ||
| Chart 1 Mono- and dinuclear copper species present in the reaction. | ||
Next, EPR measurements were done on reaction mixtures. Fig. 6A shows the EPR spectra observed after heating a reaction mixture for 5 min, 10 min, 1 h and 4 h. The field area specific for mononuclear CuII complexes is shown for the spectra recorded at 100 K. None of the EPR spectra showed signals typical of organic radicals. Instead, a clear evolution of mononuclear CuII complexes is observed in function of the reaction time, with different species contributing to the spectra, as is best visible in the low-field range (Fig. 6B). At short reaction times (5 and 10 min), the EPR spectra revealed two species, labeled III and IV, with the contribution of III decreasing over time. After 1 h, only component IV is left and as the reaction progresses, a contribution of a new species V starts to emerge. The EPR parameters of species IV are slightly changing with time (species IV′), which may be related to changes in the direct neighborhood of the complex. The EPR spectrum of species III can be obtained by a weighted subtraction of the spectra recorded after 5 and 10 minutes (bottom spectrum in Fig. 6A). In the high-field region, clearly resolved hyperfine couplings are visible that can be satisfactorily simulated considering the interaction of the unpaired electron with the copper nucleus and with two 14N surrounding nuclei. The hyperfine coupling of the latter interaction (40 MHz) is similar to that obtained for the hyperfine value of the pyridine nitrogen nuclei in Cu(py)4 complexes.32 Given that species III is observed at an early stage of the reaction it can be ascribed to an initial complexation of the copper ion with two 2-benzylpyridine (1) molecules, [Cu(DMSO)n(1)2]Y2 (Fig. 6), where n = 2, 3 or 4 and Y = −OH, −I or −OAc. Moreover, when the same reaction is carried out in toluene instead of DMSO, the CW-EPR signal after 5 minutes reaction (Fig. S12†) shows only species with g and A parameters similar to species IV (see Table S3†), without contribution from DMSO-related species III. The EPR parameters of species IV are very close to those reported for a powder of Cu(O2CCPh3)2·2py and for a frozen solution of CaCu(OAc)4·6H2O in pyridine.33,34 Addition of pyridine to dinuclear copper carboxylates has been shown to yield dinuclear complexes in equilibrium with mononuclear species of the form Cu(O2CR)2py2.31 The structure of IV can therefore be assigned to Cu(OAc)2(1)2 (Fig. 6). This is further supported by the decrease of gz and increase of |Az| compared to complex I and II, which is expected if more N-bases are ligated to the CuII than in the former case. The pulsed EPR analysis also confirmed the involvement of 2-benzylpyridine (1) ligation in the mononuclear species IV (see ESI†). The influence of acetic acid on the N-coordination of 1 is negligible (Fig. S13 and S14†). Furthermore, it has been reported that substitution of one N by one C in copper(II) porphyrins significantly lowers the gz value and increases |Ax,y| to values significantly different from those observed for species III and IV.35 Hence, ligand to catalyst coordination through a Cu–C bond can be discarded in species III and IV.
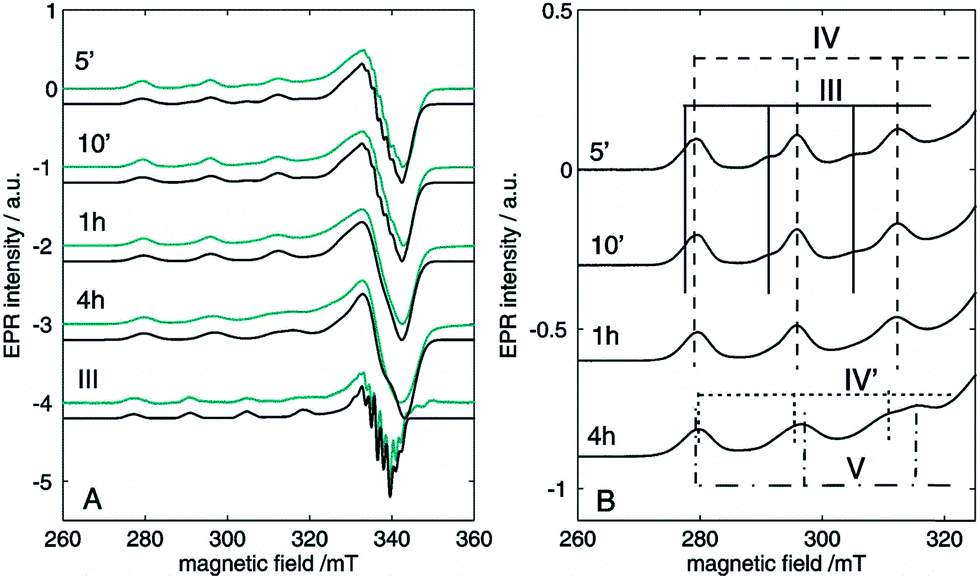 | ||
| Fig. 6 Experimental (cyan) and simulated (black) CW-EPR spectra of the reaction mixture with 0.05 M CuI and 0.5 M 2-benzylpyridine (1), 0.5 M acetic acid and DMSO heated to 100 °C under O2 for 5 min, 10 min, 1 h and 4 h. The spectra were recorded at 100 K with a microwave power of 0.47 mW and a microwave frequency of 9.73 GHz. (A) The spectrum of species III, obtained by subtraction of the spectra obtained after 5 and 10 minutes is shown at the bottom with its corresponding simulated spectrum. (B) Zoom on the low-field components revealing the time-dependent evolution of the different species. The experimental spectra are normalized for comparison. | ||
Additionally, EPR signals in agreement with dinuclear copper species could be found in the reaction mixture at all times during the reaction. Fig. 7 shows a temperature analysis of one of the high-field EPR signals from copper dimers displayed in the inset. The EPR intensity variation can be fit using the Bleaney–Bower equation (eqn (3)), yielding a value for the exchange coupling J of −306 cm−1.36
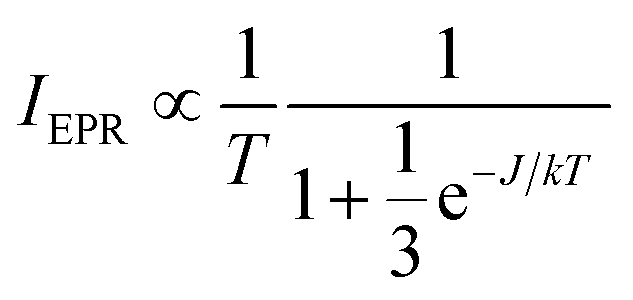 | (3) |
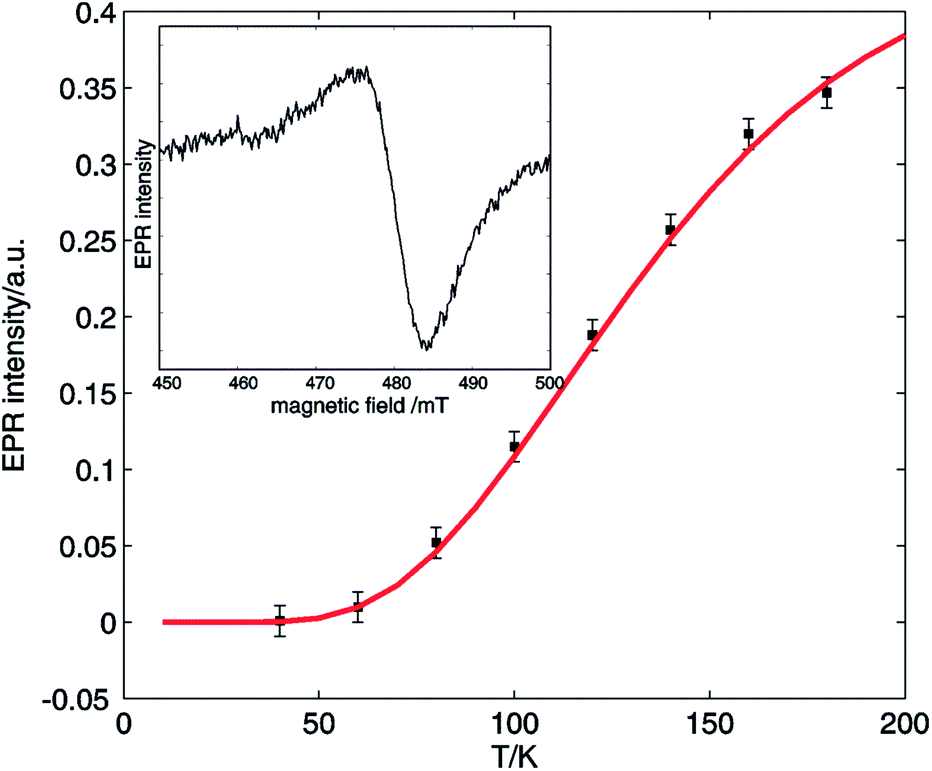 | ||
| Fig. 7 Temperature evolution of the EPR intensity of one of the high-field EPR signals typical for copper dimers (■) formed in the reaction mixture: 2-benzylpyridine (1) 0.5 M, 0.05 M CuI, 0.5 M acetic acid and DMSO heated to 100 °C under O2 for 1 h. The red curve shows the fit using eqn (3). The inset shows the high-field EPR signal used for the evaluation. The spectra were recorded using a microwave frequency of 9.73 GHz and a microwave power of 0.047 mW. | ||
This is larger in absolute value than the one found for the copper acetate dimer, agreeing with the earlier observation that addition of pyridine to copper carboxylates yields dinuclear Cu2(O2CR)4py2 complex with increased |J| value.31 Furthermore, slight changes in the position of EPR peaks are observed, indicative of a change in the D values of the triplet state. The changes in J and D prove the presence of dinuclear copper species with ligation of 2-benzylpyridine (1), in accordance with Cu2(OAc)4(1)2 (species VI).
Interestingly, mononuclear complexes III and IV are also found in the EPR spectra of the reaction mixture using a higher copper concentration of 0.25 M (Fig. S5†). However, here the contribution of species III dominates the EPR spectrum even after 4 h of reaction (Table 2, entries 7 and 8, spectrum not shown), while it disappears within the first hour in the reaction mixture with a 0.05 M copper concentration (Fig. 6B, Table 2, entries 3–6). This can be rationalized when considering that the Cu(OAc)2(1)2 complex (IV) is readily formed for high [1]/[CuII] and [AcOH]/[CuII] ratios, as is the case for our standard reaction conditions. However, when the copper concentration is increased, the relative amount of acetate per copper is lower and mononuclear copper complexes involving 1 and/or DMSO with I− or HO− as anions, instead of 1 and acetate may be formed at higher relative quantities.
Complex V is only resolved in the EPR spectra of the reaction mixture with low copper content after a longer (4 h) reaction time (Table 2, entry 6, Fig. 6B). Since the [2]/[CuII] ratio can already be significant at this stage of the reaction, the ligation of 2 to the copper complex becomes competitive. In the reaction with high copper content, the [2] may be too small for the complex to be formed. Unfortunately, the copper amount was too high at this stage of the reaction for both mixtures to allow for pulsed EPR analyses to verify this possibility (too fast electronic relaxations). However, CW-EPR spectra recorded of a reference mixture of CuI, AcOH and ketone 2 revealed the presence of a type-V species (see ESI, Fig. S6–S7†) supporting our hypothesis. The trend in the EPR parameters suggests a higher number of nitrogen ligands in the case of species V than for species III and IV, suggesting an extra ligation of 2 to a copper ion that is already ligated to two other nitrogen ligands 1.37 The assignment of species I–VI is summarized in Chart 1
In order to check the influence of the measurement temperature on the occurrence of the different copper species, CW-EPR experiments were performed at different temperatures. Fig. 8 and 9 show the room-temperature CW-EPR spectra of the copper-catalyzed oxidation of 2-benzylpyridine (1) with starting concentrations of 0.05 M and 0.25 M CuI, respectively, recorded on aliquots of the reaction mixture taken at different times in the reaction. Again, none of the spectra show contributions of organic radicals, confirming the low-temperature EPR results and the kinetic experiments using radical scavengers. In line with the low-temperature EPR (Fig. 6 and S5†), a significant variation of the EPR spectrum can be seen during the course of the reaction involving a lower copper loading due to contributions of species III and IV (Fig. 8), whereas a negligible variation is seen when a higher amount of copper is used (Fig. 9) (see also detailed comparison in ESI, Fig. S8 and S9†). Comparison of the latter spectra with the EPR spectrum of the solution without substrate 1 (bottom Fig. 9) suggests a strong contribution of CuII complexes that are related to species I and II. This is surprising, since no contributions of these species were observed in the spectra at 100 K (Fig. S5†). This indicates that the equilibrium of reaction (4) shifts to the right as temperature decreases.
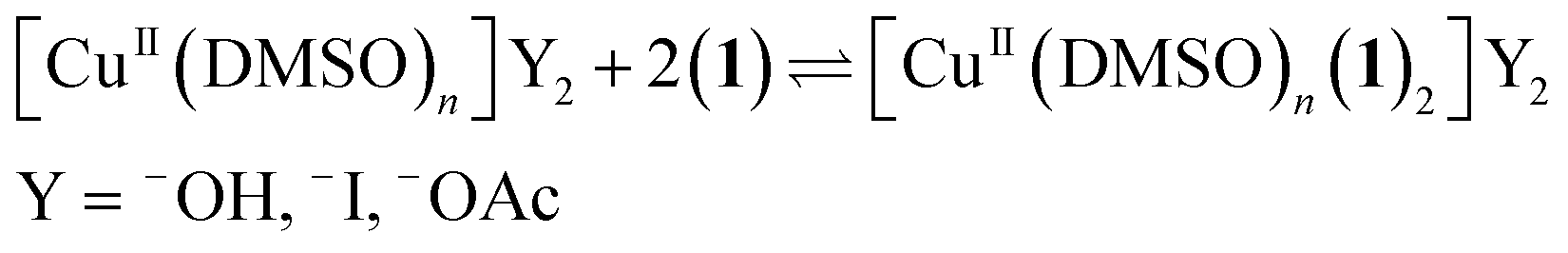 | (4) |
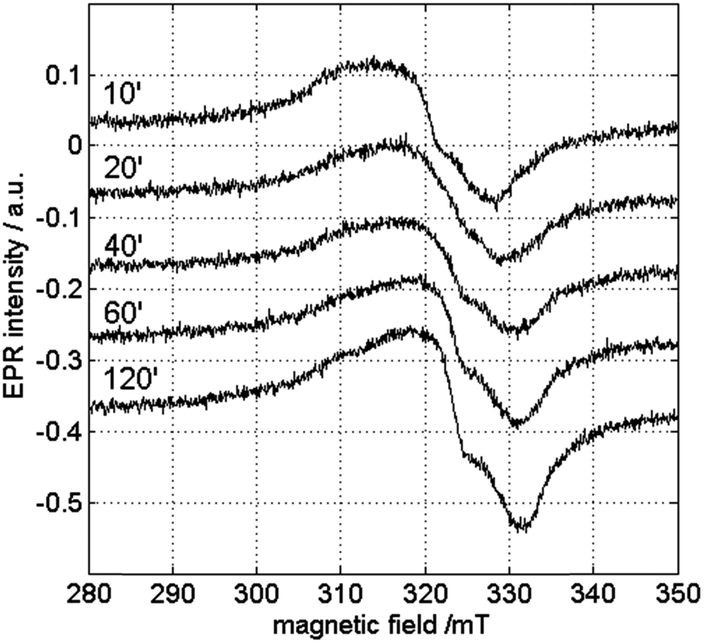 | ||
| Fig. 8 Room-temperature X-band CW-EPR spectra of the copper-catalyzed oxidation of 1 with 0.05 M CuI recorded in function of time. The spectra are recorded with a microwave power of 0.047 mW and a microwave frequency of 9.73 GHz. | ||
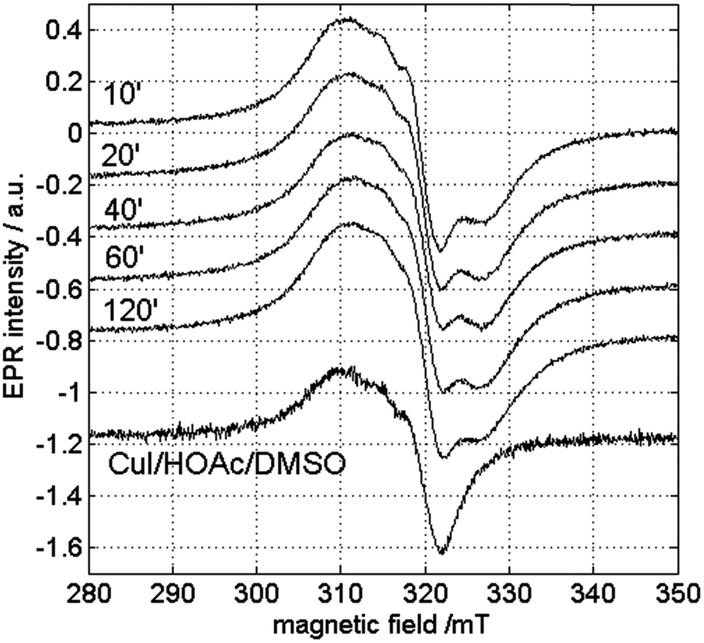 | ||
| Fig. 9 Room-temperature X-band CW-EPR spectra of the copper-catalyzed oxidation of 1 with 0.25 M CuI in function of time. At the bottom, the corresponding spectrum of the reaction mixture without the substrate 1 is shown. The spectra are recorded with a microwave power of 0.047 mW and a microwave frequency of 9.73 GHz. | ||
A high [1]/[CuII] ratio evidently also pushes the equilibrium to the right, explaining why the contribution of species III and IV can still be seen and I and II are less dominant in the room-temperature spectra of the reaction mixture at lower CuI concentration (Fig. 8).
Although clear evidence of the presence of dinuclear species in the reaction mixture with 0.05 M CuI (Table 2, entries 3–6) and the CuI/DMSO/AcOH system (Table 2, entry 2) is found at low temperatures (Fig. S2† and 7), no signals due to dimers are observed in the EPR spectra at room temperature (Fig. S10†). This is not due to the absence of the dimers, but is simply due to spectral broadening effects as reported earlier by others for dinuclear copper acetate species.38 Large CuII concentrations are required to be able to observe the typical EPR features stemming from dinuclear copper species at room temperature (Fig. S11,† signals indicated with arrows). The amount of dinuclear species is clearly increasing during the reaction time. Furthermore, the signal change at a later stage of the experiment (see for instance the appearance of a peak around 480 mT) is indicative of variation in the types of dinuclear complexes that are formed under these circumstances. This points to a variation in the axial ligands L of Cu2(OAc)4L2; 1, 2 or solvent molecules. Further proof for these acetate bridged dinuclear CuII species was obtained using infrared spectroscopy. When working at increasing concentrations of added CuI, new peaks started to arise in the infrared spectrum of the reaction mixture. By deconvolution of the overall IR spectrum a new IR spectrum could be obtained. The resulting spectrum matches with that of Cu(OAc)2·2H2O in DMSO which is known to exist as a dinuclear species and a reported dinuclear acetate bridged CuII complex (see Fig. S23 and S24†).39 Furthermore, increasing amounts of CuI cause a decrease in the concentration of free AcOH in the reaction mixture. This can be monitored by following the decrease in area of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibration of AcOH (around 1718 cm−1). At the same time an increase in intensity is observed for the vibration appearing at 1623 cm−1 (see Fig. S25 and S26†). The lowering of the wavenumber of AcOH can be rationalized by its complexation, bridging two copper centers, hereby weakening it's CO bond.
O bond vibration of AcOH (around 1718 cm−1). At the same time an increase in intensity is observed for the vibration appearing at 1623 cm−1 (see Fig. S25 and S26†). The lowering of the wavenumber of AcOH can be rationalized by its complexation, bridging two copper centers, hereby weakening it's CO bond.
Catalytic cycle
Based on all the insight gathered a catalytic cycle is proposed in Fig. 10. Aerobic oxidation of the added CuI brings the catalyst to the active +II oxidation state (complexes I–IV). An initial acid-catalyzed imine–enamine tautomerization, characterized by the equilibrium constant Keq transforms the substrate 1 into the enamine tautomer A. Nucleophilic attack of A on an electrophilic CuII center (monomer IV or dimer VI) produces a C–CuII bond (B or B′). Acetic acid acts as a catalyst for the tautomerization. Although adding an acid does not change the equilibrium between 1 and A, it will lower the activation energy, thereby increasing the rate by which enamine A is replenished. All these steps are reversible and in rapid equilibrium characterized by their respective equilibrium constants. Their relative concentrations depend on the initial reaction conditions (the ratios between all the reaction components: substrate, catalyst, acid and solvent). In the next step the CuII center that is bound to the substrate molecule will be oxidized by O2 to a highly reactive CuIII-superoxide species (C or C′).40,41 The oxidation of B/B′ is thought to be the rate determining step and it is herein that lies the difference in reaction rate for mononuclear (km) versus dinuclear (kd) catalyst species (km > kd). The superoxide species will quickly rearrange via a radical pathway with an electron transfer to the catalyst producing the CuII organoperoxide D or D′. Heterolytic dissociation of these organoperoxides by deprotonation of the remaining benzylic proton produces the reaction product 2 and recovers the catalyst with a hydroxide rather than an acetate bound (E or E′).42E and E′ can be converted back to IV and VI by anion exchange with acetate, hereby producing one equivalent of water.43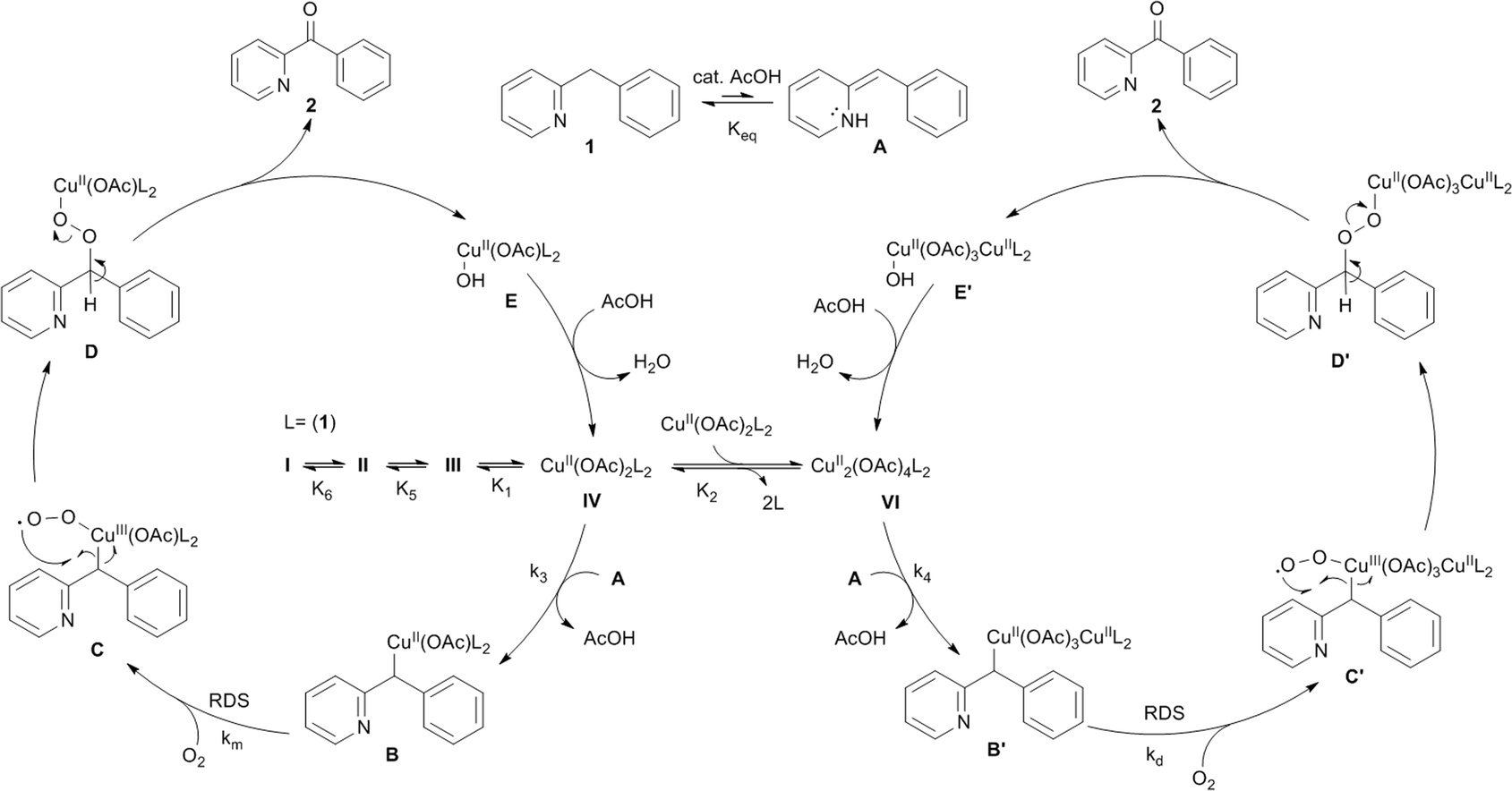 | ||
| Fig. 10 Mechanistic proposal for the aerobic Cu-catalyzed oxidation of 1 into 2. | ||
The amount of O2 required to make one molecule of ketone was determined by performing gas uptake experiments. It was found that when using CuI (10 mol%) as the catalyst the O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate stoichiometry is ∼1 (see ESI†). These results confirm that in accordance with our mechanistic proposal for each molecule of product one molecule of water is formed and that no extra oxygen is required to oxidize the catalyst during the catalytic cycle.
:substrate stoichiometry is ∼1 (see ESI†). These results confirm that in accordance with our mechanistic proposal for each molecule of product one molecule of water is formed and that no extra oxygen is required to oxidize the catalyst during the catalytic cycle.
For our proposed catalytic cycle a kinetic isotope effect is expected even though C–H/D bond cleavage is not included in the rate-determining step. As the C–H bond cleavage (via imine–enamine tautomerization) is reversible and its k−1 is much faster than the k2 of the RDS (oxygenation), the observed KIE is the equilibrium isotope effect.44 When comparing the initial rates of the reaction on 1 using AcOH with that of 1-d2 using AcOD-d4 a KIE value of 4.28 was found (Fig. S19†). As an additional experiment we tested if deuterium could be build-in via tautomerization under our reaction conditions. When mixing 2-benzylpyridine with d4-AcOD in deuterated DMSO in the absence of catalyst and O2 the singlet of the benzylic protons started to disappear in 1H-NMR and a broad triplet resulting from 1-d1 started to appear further supporting the imine–enamine tautomerization (see ESI Fig. S20†). In contrast, diphenylmethane (8) could not be deuterated in the same manner as expected.
The EPR experiments (Table 2) indicate that the catalyst resting state is species IV with even at low [catalyst] a significant contribution of dinuclear species VI. When the [catalyst] is increased the equilibrium is significantly shifted towards the dinuclear species VI. Because the equilibrium between the imine (1) and enamine (A) tautomers is highly shifted towards the imine (1) the concentration of species B and B′ is expected to be very low which explains why these species were never directly observed by EPR measurements. The reason why the dinuclear species oxidize slower than the mononuclear is unclear. The simplest explanation is the increased steric bulk of the catalyst (4 bridging acetates). However it is also possible that the second CuII in C′ reacts with the formed CuIII-superoxide species producing a less reactive dioxygen bridged species.
Formation of side product 3 can occur via protonation of intermediate D or D′ producing a hydroperoxide that can undergo a Fenton like CuI-catalyzed homolytical cleavage of the O–O bond yielding alcohol 3 and a hydroxyl radical. This radical is known to be very reactive and thus very short-lived.45 Alcohol 3 is formed in such small quantities so that an in-depth study of the mechanism of its formation was not possible. Alcohol 3 can be transformed into 2 by copper-catalyzed β-hydride elimination (Scheme 2).16
Conclusions
This research has provided deeper insight into the mechanism of CuII-catalyzed aerobic oxygenations. Although the mechanism of Cu-catalyzed aerobic alcohol oxidation has been thoroughly studied, kinetic studies on the corresponding methylene oxidation are unprecedented. Only reactions on stoichiometric Cu-complexes with oxygen have been reported. In this study we have shown that the substrates are activated via an initial acid catalyzed imine–enamine tautomerization. DFT calculations can be used as useful method to predict the equilibrium constant (Keq) and can act as a tool to qualitatively predict whether or not a substrate can be oxidized. Alcohol 3 was excluded as an intermediate in this reaction and is a side product presumably generated by the homolytical breaking of the O–O bond in the hydroperoxide formed from complex D/D′via protonation. Kinetic studies revealed O2 mass transfer limitation effects limiting the turnover of the reaction at high concentrations of substrate and acetic acid. A complex kinetic behavior was observed when increasing the catalyst loading. At intermediate loadings mass transfer is again occurring resulting in a maximum rate. The continued decrease of the reaction rate at higher concentrations of catalyst however can't be explained by the occurrence of mass transfer alone. When using transition metal catalysis in combination with (acetate) bridging ligands dinuclear species need to be considered. Although these are usually considered to be related to catalyst deactivation this study shows this is not necessarily the case as they explain the further reduction in rate observed at a higher concentration than at which the mass transfer determined vmax occurs. The formation of a dinuclear catalytically active species which is less reactive than the mononuclear catalyst explains the non-zero reaction rate at increased concentration of added catalyst. Such behavior is to the best of our knowledge unprecedented in literature. In addition to these kinetic experiments we performed an in-depth EPR-study to characterize the catalytic species present in the reaction. These measurements confirmed the presence of both different mono- and dinuclear CuII-species. The results presented in this paper can directly be used in the mechanistic study of other active methylene oxygenations where the methylene moiety is activated by a tautomerization step.Acknowledgements
This work was supported by the Research Foundation Flanders (FWO-Flanders), the Agency for Innovation by Science and Technology (IWT-Flanders), the University of Antwerp (BOF, IOF) and the Hercules Foundation. The authors thank Philippe Franck, Heidi Seykens and Norbert Hancke for technical assistance.Notes and references
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC; (b) A. Wells, M. Teria and T. Eve, Biochem. Soc. Trans., 2006, 34, 304–308 CrossRef CAS.
- D. Avila, R. Puntel and M. Aschner, in Interrelations Between Essential Metal Ions and Human Diseases, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer, Netherlands, 2013, vol. 13, pp. 199–227 Search PubMed.
- (a) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (b) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC; (c) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- (a) M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612–2616 CrossRef CAS; (b) I. Karthikeyan, S. K. Alamsetti and G. Sekar, Organometallics, 2014, 33, 1665–1671 CrossRef CAS; (c) B. Join, K. Möller, C. Ziebart, K. Schröder, D. Gördes, K. Thurow, A. Spannenberg, K. Junge and M. Beller, Adv. Synth. Catal., 2011, 353, 3023–3030 CrossRef CAS PubMed.
- (a) Y.-F. Wang, F.-L. Zhang and S. Chiba, Synthesis, 2012, 44, 1526–1534 CrossRef CAS; (b) B. Orlińska, Tetrahedron Lett., 2010, 51, 4100–4102 CrossRef PubMed; (c) N. Komiya, T. Naota, Y. Oda and S. I. Murahashi, J. Mol. Catal. A: Chem., 1997, 117, 21–37 CrossRef CAS; (d) B. Gutmann, P. Elsner, D. Roberge and C. O. Kappe, ACS Catal., 2013, 3, 2669–2676 CrossRef CAS.
- (a) M. Nakanishi and C. Bolm, Adv. Synth. Catal., 2007, 349, 861–864 CrossRef CAS PubMed; (b) S. Velusamy and T. Punniyamurthy, Tetrahedron Lett., 2003, 44, 8955–8957 CrossRef CAS PubMed; (c) J. F. Pan and K. M. Chen, J. Mol. Catal. A: Chem., 2001, 176, 19–22 CrossRef CAS.
- (a) The group of Sasson reported the oxidation of alcohols and benzylic methylenes using CuCl2 in combination with t-BuOOH. They also provide a detailed mechanistic study of this reaction where they propose a dimeric (Cl−) bridged Cu-species to be the active catalyst while another dimeric species is proposed to cause deactivation: G. Rothenberg, Y. Yatziv and Y. Sasson, Tetrahedron, 1998, 54, 593–598 CrossRef CAS; (b) For a study involving allylic oxidations see: G. Rothenberg, L. Feldberg, H. Wiener and Y. Sasson, J. Chem. Soc., Perkin Trans. 2, 1998, 2429–2434 RSC.
- A. J. Hunt, T. J. Farmer and J. H. Clark, in Element Recovery and Sustainability, The Royal Society of Chemistry, 2013 Search PubMed.
- For some recent reviews and notable examples see: (a) P. Haack and C. Limberg, Angew. Chem., Int. Ed., 2014, 53, 4282–4293 CrossRef CAS PubMed; (b) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed; (c) I. V. Korendovych, S. V. Kryatov and E. V. Rybak-Akimova, Acc. Chem. Res., 2007, 40, 510–521 CrossRef CAS PubMed; (d) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed; (e) H. R. Lucas, L. Li, A. A. N. Sarjeant, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 3230–3245 CrossRef CAS PubMed; (f) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS PubMed; (g) L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- (a) F. M. Moghaddam, Z. Mirjafary, H. Saeidian and M. J. Javan, Synlett, 2008, 892–896 CrossRef CAS; (b) C. Zhang, Z. Xu, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11088–11092 CrossRef CAS PubMed; (c) A. Ilangovan and G. Satish, Org. Lett., 2013, 15, 5726–5729 CrossRef CAS PubMed; (d) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., 2013, 125, 11513–11517 CrossRef PubMed.
- (a) J. De Houwer, K. Abbaspour Tehrani and B. U. W. Maes, Angew. Chem., Int. Ed., 2012, 51, 2745–2748 CrossRef CAS PubMed; (b) J. De Houwer and B. U. W. Maes, Synthesis, 2014, 46, 2533–2550 CrossRef CAS.
- B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320–324 RSC.
- J. Genovino, S. Lütz, D. Sames and B. B. Touré, J. Am. Chem. Soc., 2013, 135, 12346–12352 CrossRef CAS PubMed.
- J. Liu, X. Zhang, H. Yi, C. Liu, R. Liu, H. Zhang, K. Zhuo and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1261–1265 CrossRef CAS PubMed.
- M. Itoh, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2050–2053 CrossRef CAS PubMed.
- G. Franc and A. Jutand, Dalton Trans., 2010, 39, 7873–7875 RSC.
- (a) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed; (b) J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- D. Liu, Y. Li, X. Qi, C. Liu, Y. Lan and A. Lei, Org. Lett., 2015, 17, 998–1001 CrossRef CAS PubMed.
- Next to a small amount of benzophenone which was probably formed by hydroperoxides present during the reaction in combination with our transition metal catalyst (Cu and Fe). See: (a) M. M. Hossain and S.-G. Shyu, Tetrahedron, 2014, 70, 251–255 CrossRef CAS PubMed; Activated carbon can be used as a solid radical scavenger to extract a fraction of radicals in solution, see: (b) E. V. Ramos-Fernandez, N. J. Geels, N. R. Shiju and G. Rothenberg, Green Chem., 2014, 16, 3358–3363 RSC; (c) M. Newcomb, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, 2012 Search PubMed.
- B. A. Steinhoff and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 4348–4355 CrossRef CAS PubMed.
- (a) B. A. Steinhoff, S. R. Fix and S. S. Stahl, J. Am. Chem. Soc., 2002, 124, 766–767 CrossRef CAS PubMed; (b) B. A. Steinhoff, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 11268–11278 CrossRef CAS PubMed; (c) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Adv. Synth. Catal., 2002, 344, 355–369 CrossRef CAS.
- The inhibition of a reaction by formation of dinuclear species has some resemblance to substrate-inhibition in enzyme kinetics: (a) H. Lineweaver and D. Burk, J. Am. Chem. Soc., 1934, 56, 658–666 CrossRef CAS; (b) M. C. Reed, A. Lieb and H. F. Nijhout, BioEssays, 2010, 32, 422–429 CrossRef CAS PubMed.
- S. van Doorslaer and D. M. Murphy, Top. Curr. Chem., 2012, 321, 1–40 CrossRef CAS.
- Y. Deng, G. Zhang, X. Qi, C. Liu, J. T. Miller, A. J. Kropf, E. E. Bunel, Y. Lan and A. Lei, Chem. Commun., 2015, 51, 318–321 RSC.
- (a) P. Capdevielle and M. Maumy, Tetrahedron Lett., 1990, 31, 3891–3892 CrossRef CAS; (b) P. Capdevielle, A. Lavigne, D. Sparfel, J. Baranne-Lafont, K. C. Nguyen and M. Maumy, Tetrahedron Lett., 1990, 31, 3305–3308 CrossRef CAS; (c) P. Capdevielle, A. Lavigne and M. Maumy, Synthesis, 1989, 453–454 CrossRef CAS.
- T. Diao, P. White, I. Guzei and S. S. Stahl, Inorg. Chem., 2012, 51, 11898–11909 CrossRef CAS PubMed.
- E. J. Ukpong, N. W. Akpanudo and J. Prasad, Afr. J. Pure Appl. Chem., 2010, 4, 38–43 CAS.
- (a) J. K. Kochi and R. V. Subramanian, Inorg. Chem., 1965, 4, 1527–1533 CrossRef CAS; (b) H. Grasdalen, J. Chem. Soc., Faraday Trans., 1973, 69, 462–470 RSC; (c) P. Sharrock and M. Melník, Can. J. Chem., 1985, 63, 52–56 CrossRef CAS.
- (a) H. Grasdalen, J. Magn. Reson., 1973, 9, 166 CAS; (b) P. Sharrock and M. Melník, Can. J. Chem., 1985, 63, 52–56 CrossRef CAS.
- (a) J. R. Wasson, C.-I. Shyr and C. Trapp, Inorg. Chem., 1968, 7, 469–473 CrossRef CAS; (b) P. Sharrock and M. Melník, Can. J. Chem., 1985, 63, 52–56 CrossRef CAS.
- W. J. Newton, C. Oldham and B. J. Tabner, J. Chem. Soc., Dalton Trans., 1980, 1379–1382 RSC.
- A. Pöppl, M. Hartmann, W. Böhlmann and R. Böttcher, J. Phys. Chem. A, 1998, 102, 3599–3606 CrossRef.
- O. W. Steward, M. Kato, S.-C. Chang, M. Sax, C.-H. Chang, C. F. Jury, Y. Muto, T. Tokii, T. Taura, J. F. Pletcher and C. S. Yoo, Bull. Chem. Soc. Jpn., 1991, 64, 3046–3058 CrossRef CAS.
- C. Ramachary and V. G. Krishnan, J. Mol. Struct., 1980, 64, 263–267 CrossRef CAS.
- C. Calle, A. Schweiger and G. Mitrikas, Inorg. Chem., 2007, 46, 1847–1855 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CrossRef CAS.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 163, 691–708 CrossRef.
- P. Sharrock and M. Melník, Can. J. Chem., 1985, 63, 52–56 CrossRef CAS.
- B. K. Koo, Bull. Korean Chem. Soc., 2001, 22, 113–116 CAS.
- For examples of CuIII in catalysis see: (a) A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301–2318 RSC; (b) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068–12073 CrossRef CAS PubMed.
- For related mechanistic proposals see: (a) H. Wang, Y. Wang, D. Liang, L. Liu, J. Zhang and Q. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5678–5681 CrossRef CAS PubMed; (b) L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2011, 13, 1622–1625 CrossRef CAS PubMed; (c) C. Menendez, S. Gau, S. Ladeira, C. Lherbet and M. Baltas, Eur. J. Org. Chem., 2012, 409–416 CrossRef CAS PubMed.
- (a) N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880–881 CrossRef CAS; (b) S. J. Blanksby, G. B. Ellison, V. M. Bierbaum and S. Kato, J. Am. Chem. Soc., 2002, 124, 3196–3197 CrossRef CAS PubMed.
- M. Devereux, D. O'shea, A. Kellett, M. McCann, M. Walsh, D. Egan, C. Deegan, K. Kędziora, G. Rosair and H. Müller-Bunz, J. Inorg. Biochem., 2007, 101, 881–892 CrossRef CAS PubMed.
- This case is mentioned in a review dealing with kinetic isotope effects, see: E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- J. Edwards and R. Curci, Catalytic Oxidations with Hydrogen Peroxide as Oxidant, 1992, vol. 9, pp. 97–151 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03530a |
| This journal is © The Royal Society of Chemistry 2016 |